Back to Journals » Clinical Interventions in Aging » Volume 9
Type 2 diabetes as a risk factor for cognitive impairment: current insights
Authors Umegaki H ![]()
Received 17 March 2014
Accepted for publication 23 April 2014
Published 28 June 2014 Volume 2014:9 Pages 1011—1019
DOI https://doi.org/10.2147/CIA.S48926
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Hiroyuki Umegaki
Department of Community Healthcare and Geriatrics, Nagoya University Graduate School
of Medicine, Nagoya, Japan
Abstract: Type 2 diabetes mellitus (T2DM) is a risk factor for cognitive dysfunction and dementia in the elderly. T2DM has been thought to be associated with vascular diseases, eventually leading to vascular dementia, but recent studies have established that T2DM is also associated with Alzheimer’s disease (AD). With the increase in the number of elderly individuals with T2DM, the number of diabetic patients with cognitive dysfunction has been increasing. T2DM may accelerate AD-associated pathologies through insulin resistance. Vascular pathologies may also be associated with cognitive dysfunction and dementia in T2DM subjects. Several other mechanisms also seem to be involved in T2DM-related cognitive dysfunction. More investigations to clarify the association of T2DM with cognitive impairment are warranted. These investigations may help to increase our understanding of AD and open a new door to the development of therapeutics. Recent pharmaceutical advancement in T2DM treatment has resulted in the availability of a wide range of antidiabetics. Some evidence has suggested that antidiabetic therapies help to prevent cognitive dysfunction. At present, however, the optimal level of blood glucose control and the best combination of medications to achieve it in terms of cognitive preservation have not been established. More investigation is warranted. Cognitive dysfunction is an emerging new complication of T2DM that requires further study.
Keywords: insulin resistance, dementia, blood glucose, amyloid ß, tau, small vascular disease
Introduction
With the progressive aging of the population in developed countries, an increasing number of patients suffer from dementia. Dementia is a disease condition that precludes the activities of daily life and self-care behaviors, and it constitutes a great burden on the patients themselves, their careers, and society. The fundamental therapeutics of dementia have not yet been determined, although some therapies for treating symptoms are available. To establish disease-modifying therapies for dementia, a wide range of investigations to elucidate the mechanism of the development and progression of cognitive dysfunction in the elderly is needed.
Diabetes mellitus is a heterogeneous disorder that is manifested in the form of hyperglycemia and glucose intolerance due to relative insulin deficiency, impaired effectiveness of insulin action, or both. About 347 million people have diabetes worldwide, and the number is increasing.1 There are two types of diabetes mellitus, type 1 diabetes (T1DM) and type 2 diabetes (T2DM). These two types are differentiated on the basis of etiology and clinical presentation. T1DM is caused by the destruction of the insulin-producing cells of the pancreas, typically due to an autoimmune reaction. In T1DM, chronic hyperglycemia is caused by insulin deficiency. On the other hand, T2DM is characterized by insulin resistance (IR) and relative insulin deficiency. IR is defined as an inadequate response by insulin target tissues, such as skeletal muscle, liver tissue, and adipose tissue to the physiological effects of circulating insulin, and is often accompanied by raised insulin levels. T2DM is often, but not always, associated with obesity, which itself can cause IR and lead to elevated blood glucose levels.
The prevalence of T2DM has been rising in many regions of the world.1 Many studies have established that T2DM is a risk factor for cognitive dysfunction and dementia in the elderly. With the increase in the number of elderly individuals with T2DM, the number of diabetic patients with cognitive dysfunction has been increasing. Recent remarkable advances in pharmacological therapy have made a variety of interventions available. Patients with T2DM have benefitted from advancements in preventing and treating the classic microvascular and macrovascular complications.2 Cognitive dysfunction, however, has not been targeted by the current management strategies of T2DM. Cognitive impairment and dementia in patients with T2DM creates a large burden for patients, their families, and society. In the management of T2DM, self-care behavior is very important, but this behavior is impaired by cognitive dysfunction. The future development and implementation of diabetic treatments, especially for the elderly, should take brain protection into consideration.
T2DM and cognitive dysfunction or dementia
It has been established that T2DM is associated with cognitive dysfunction. While a wide range of cognitive domains was reportedly impaired in older patients with T2DM, one of the most frequently reported cognitive functional deficiencies in T2DM is impaired psychomotor speed.3
The incidences of both T2DM and dementia increase in later life, which increases the prevalence of the comorbidity of these pathologies. Moreover, recent studies have indicated that older people with T2DM have a higher risk of cognitive dysfunction or dementia.4 Ample evidence has indicated that T2DM is related not only to vascular dementia (VD) but also to the clinical diagnosis of Alzheimer’s disease (AD)-type dementia.5
One of the earliest findings from a large epidemiological study showing that T2DM patients had an increased risk for developing dementia came from the Rotterdam Study.6 Another study demonstrated a 65% increase of the risk for developing AD in T2DM subjects.7 A cohort of Japanese-Americans in Hawaii8 showed a 1.8-fold higher risk for developing AD and a 2.3-fold higher risk for vascular VD, and the Hisayama study reported similar results.9 The reported risks for total dementia, AD, or VD in T2DM patients are similar (Table 1). A recent comprehensive meta-analysis of population-based longitudinal studies showed that the pooled relative risk of AD in subjects with T2DM (a total of 506 subjects) was 1.46 (1.20–1.77) compared with the subjects without T2DM (36,191 subjects).10 For VD, the relative risk was 2.5 (2.1–3.0), based on ten studies including 3,519 subjects with T2DM and 23,026 subjects without.10
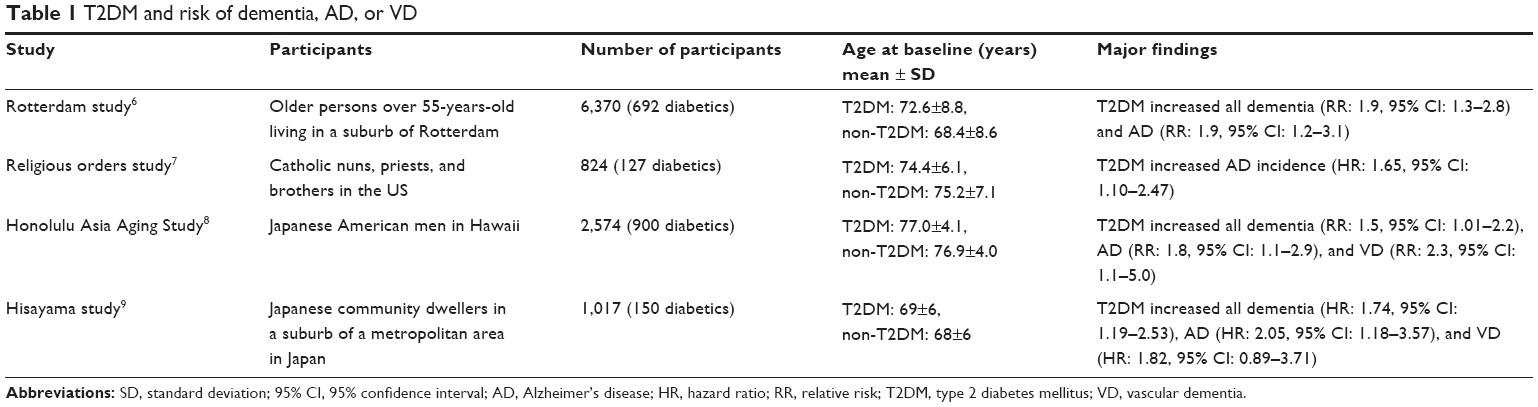 | Table 1 T2DM and risk of dementia, AD, or VD |
Clinical diagnosis of AD
Recently, with the advancements of biomarkers, newer clinical criteria for AD can include the positive features of AD, including abnormality in cerebrospinal fluid (CSF) markers and positive amyloid imaging by positron emission tomography (PET).11 However, the diagnosis of AD has depended on older clinical criteria, mostly those from the Diagnostic and Statistical Manual of Mental Disorders12 or the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) Alzheimer’s Criteria,13 in published research studies. It should be noted that the definitive diagnosis of AD is only made pathologically basically postmortem according to these criteria. The clinical diagnosis of AD in most of the previously published research studies was performed at least partly through the exclusion of diseases other than AD. The coexistence of a limited number of other pathologies including ischemic lesions does not prevent the diagnosis of AD. Indeed, more than 60% of autopsy-proven cases of AD had cerebrovascular lesions.14 Moreover, neuronal dysfunction and death not related to the amyloid-β peptide (Aβ) or to hyperphosphorylation of the microtubule-associated protein tau but rather to other causes may coexist in clinically diagnosed AD. Although the results of clinical and epidemiological studies largely agree with the increase in AD by comorbidity of T2DM, this does not necessarily correspond to an accelerated AD pathology in T2DM subjects. Many dementia subjects, especially older subjects, have multiple pathologies.15
Underlying mechanism of cognitive dysfunction in T2DM
The precise mechanisms underlying T2DM-related cognitive dysfunction or the development of dementia, especially AD-type dementia, remain to be elucidated; however, several hypothetical mechanisms have been proposed (Figure 1).
 | Figure 1 Mechanism of T2DM-associated cognitive dysfunction. |
Neurogenesis
Neurogenesis in the hippocampus plays a role in learning and memory, and age-associated decline in neurogenesis has been reported.16 Basic and animal experiments have indicated that a hyperglycemic environment induces the proliferation of adult neural progenitors, but is detrimental to their survival. The impaired neurogenesis in T2DM subjects may underlie an associated cognitive impairment and brain atrophy.17,18
Blood–brain barrier
The blood–brain barrier (BBB) consists of tight junctions between endothelial cells and astrocytic projections, which regulate paracellular and transcellular flow into the central nervous system (CNS), respectively.
Previous observations in brain tissue biopsied from AD subjects have indicated BBB breakdown in several respects.19,20 These include thinning of the endothelium, loss of mitochondria, and thickening of the basement membranes, the latter of which increase the accumulation of focal Aβ peptides. A break in the BBB also leads to potentially toxic substances and metabolites gaining access to the brain.21,22
Diabetes is associated with changes in both the barrier and transport functions of the cerebral microvessels.23 Dysfunction in the BBB may be associated with cognitive impairment and/or the incidence of dementia.
Hyperglycemia
Chronically higher blood glucose levels exert a negative influence on cognition and cause structural changes in the hippocampus.24 A recent longitudinal study over 6 years also reported higher glucose levels and an increased risk of the incidence of dementia in both diabetes and non-diabetes subjects.25
High glucose concentration, a major pathological characteristic of diabetes, may have toxic effects on neurons in the brain through several mechanisms. Osmotic insults and oxidative stress may be involved in the mechanism, and the maintenance of chronic high glucose leads to the enhanced formation of advanced glycation end products (AGEs),26 which have potentially toxic effects on neurons. Reportedly, AD patients with T2DM have increased levels of AGEs and microglial activation in the CNS compared to AD patients without T2DM. AGEs couple with free radicals and create oxidative damage, which in turn leads to neuronal injury.27 In addition to their direct toxicity, AGEs also activate microglia in the CNS. A wealth of evidence has demonstrated that microglia, the resident innate immune cells in the brain, can become deleterious and damage neurons.28 This process is implicated as an underlying mechanism in diverse neurodegenerative diseases, including AD. While microglial function is beneficial and mandatory for normal CNS functioning, unregulated overactivation of microglia causes damage to neurons. In diabetes, oxidative stress also increases because of reduced antioxidant capacity.29 Oxidative stress has been suggested to lead to neuronal injury through mitochondrial dysfunction.30
Inflammatory mechanism
Inflammation plays a role in the pathogenesis of IR and T2DM.31 It has also been suggested that inflammation is associated with the pathogenesis of AD. Chronic low-grade inflammation may be a contributor to the disease process of AD.32 Proinflammatory cytokines such as tumor necrosis factor alpha are known to be involved in the pathogenesis of both T2DM and AD.33 The activation of glia by inflammatory cytokines damages the neurons. Therefore, inflammation may be a link between T2DM and dementia, especially AD.
IR
Insulin is a hormone that regulates blood glucose levels. It is primarily secreted by the beta cells of the pancreas and is normally released into the systemic circulation through the portal vein in response to a rise in blood glucose. It is catabolized by insulin-degrading enzyme (IDE) in the liver, kidneys, and muscles.34,35 Insulin working within the brain is presumably of pancreatic origin, and it has passed the BBB through a saturable transporter mechanism from the systemic circulation, although there is debate about the amount of insulin that is produced de novo within the CNS.36 Insulin has multiple important functions in the brain including the control of food intake (via insulin receptors located in the olfactory bulb and thalamus) and effects on cognitive functions, including memory.37,38
Blood glucose abnormalities and IR may be associated with acetylcholine (ACh) synthesis. ACh transferase, which is an enzyme responsible for ACh synthesis, is expressed in insulin-receptor-positive cortical neurons, and insulin regulates ACh transferase expression. Because ACh is a critical neurotransmitter in cognitive function, it may be relevant to neurocognitive disorders in diabetics.39
The critical pathological mechanism in AD is an accumulation of Aβ. Overproduction of Aβ may be one of the mechanisms by which this accumulation occurs; however, the impairment of clearance of Aβ may also play a role. The desensitization of insulin receptors, ie, IR, reduces the synthesis of several proteins, including IDE. IDE degrades Aβ as well as insulin, and reduced amounts of IDE may result in greater amyloid deposition. Basic research has indicated that insulin controls the expression of IDE in the brain, and IR in the brain may downregulate IDE expression.40,41
Another critical pathology in AD is the hyperphosphorylation of tau. Insulin seems to be an important determinant of tau protein phosphorylation. Impaired insulin signaling can result in the inhibition of PI3K/Akt and the activation of glycogen synthase kinase-3β. The activation of glycogen synthase kinase-3β leads to the enhanced phosphorylation of tau protein and the formation of neurofibrillary tangles.42,43
A recent study by Talbot et al demonstrated that IR in neurons exists in AD.44 The ex vivo stimulation of insulin receptors in the brains of AD patients nicely showed that insulin signaling is greatly reduced where the phosphorylation of insulin receptor substrate 1 occurs at several serine residues, which has been known to be a feature of IR. Moreover, the levels of insulin receptor substrate 1 phosphorylation in hippocampal neurons were found to be negatively correlated with episodic and working memory. Another study investigated the critical role of the soluble Aβ oligomer in producing brain IR in AD, and successfully demonstrated the pharmacological manipulation of this pathway by a glucagon-like peptide 1 (GLP-1) agonist.45 Intranasal insulin reportedly improved memory and attention in humans.46 These achievements may hold promise for brain IR as a future therapeutic target in AD.
Vascular dysfunction
Excretion of even a very low amount of albumin in urine (microalbuminuria) is a good marker of cardiovascular events.47 Microalbuminuria, a marker of vascular dysfunction, predicted accelerated cognitive decline in T2DM subjects.48–51 These findings suggest that a deficit of vascular endothelial cells can lead to impairment of the functional coordination of the vascular supply in a timely response to the demand created by nervous activity. Neural activity requires a strong increase of cerebral blood flow and an acute increase in neuronal glucose. Hemodynamic neurovascular coupling coordinates these links (neurovascular units). Dysfunction of cerebral autoregulation with increasing age along with structural and functional alterations in cerebral blood vessels due to diabetes mellitus impairs the functioning of neurovascular units. These phenomena may induce functional deficits in neurons and increase neuronal degeneration and the susceptibility to hypoxia and ischemia.52 Impaired neurovascular units would also induce BBB leakage.52 Recently, the hypothesis that vascular dysfunction may impair the drainage pathways of Aβ from the brain parenchyma and thus increase the deposition of Aβ has been drawing interest.53 Vascular dysfunction could be associated with the progression of amyloid pathology.
Neurodegeneration, ischemic pathology, or functional alteration?
van Harten et al in their systematic review, reported consistent associations of T2DM with atrophy across many brain regions,54 and more recent reports have made similar findings.55–61 It was reported by den Heijer et al in their review that subjects with T2DM had more hippocampal and amygdalar atrophy on magnetic resonance imaging (MRI) compared to subjects without T2DM, and they speculated that this could suggest that T2DM directly influences the development of AD-related neuropathology.62 More recently, Willette et al reported that IR in an asymptomatic, late middle-aged cohort was associated with progressive atrophy in regions affected by early AD,63 and similar atrophy was found in older T2DM patients by Moran et al.64 A PET study demonstrated that subjects with prediabetes and early T2DM have Alzheimer-like reductions in regional cerebral glucose metabolism.65 These clinical studies may suggest that T2DM induces the AD-related pathology, and the acceleration of AD pathologies leads to neurodegeneration and brain atrophy.
In the Rotterdam study, a non-diabetic population cohort study, it was reported that IR assessed by homeostasis model assessment ratio was associated with the onset of AD within 3 years.66 A Japanese epidemiological study, the Hisayama study, reported that IR was associated with AD pathology.67 The results of pathological studies using autopsy samples, however, have not been consistent in terms of the association of T2DM with AD pathology. One study demonstrated that diabetics show significantly less AD-associated neuropathology,68 while another failed to show any relationship between diabetes and AD-associated neuropathology.69 The Baltimore Longitudinal Study of Aging (BLSA) recently reported that the association between AD-related pathology and IR could not be confirmed.70 Body mass index, serum insulin level, and IR level were very different between the Hisayama study and the BLSA study, which makes it difficult to interpret and compare the results of these two studies. A recently published study making use of amyloid PET imaging did not find differences in amyloid accumulation between AD subjects with or without T2DM.71
If T2DM has an impact on cognition only through accelerating AD pathology, the extent of the pathology should be similar when cases of cognitive impairment with similar levels of severity are compared cross-sectionally, either pathologically or using amyloid imaging. Thus, the rate of progression would also be considered when investigating the role of T2DM in increasing the incidence of AD by using amyloid PET or CSF biomarkers.
Another issue to be addressed in future studies is the Apo E genotype. Many studies reported that the Apo E4 genotype, which is a major genetic risk factor for AD,72 enhanced the association of AD with T2DM.73,74 Thus, considering the impact of the Apo E genotype is warranted in future pathological studies.
T2DM is an established risk factor for microvascular and macrovascular complications throughout the body, including brain stroke and small vessel disease.75 Therefore, vascular damage is likely to be one of the main reasons for the cognitive impairment in T2DM subjects, including VD subjects. Recent advances in MRI technology, specifically diffusion tensor imaging, have allowed subtle changes in white matter to be revealed. Some studies have reported T2DM to be associated with microstructural abnormalities in the white matter in various pathways in the brain independent of small vessel diseases, and these abnormalities are also associated with cognitive impairment.76,77 The combination of vascular lesions and AD pathology is very common in T2DM subjects, and cognitive dysfunction due to vascular pathology may lower the threshold of clinical diagnosis of dementia in early AD subjects.
A neurodegenerative mechanism other than AD pathology may contribute to the cognitive dysfunction in T2DM. Oxidative stress or neuroinflammation may be associated with neuronal death or synaptic dysfunction apart from Aβ or tau pathology.
The extent of the contribution of both neurodegeneration including AD pathology and vascular changes to T2DM-associated cognitive impairment should be further explored. Moreover, functional alteration of neurovascular units and synapses that are not necessarily associated with neuronal death may also contribute. In this case, imaging studies or pathological examination by autopsy might not find the differences between T2DM and non-T2DM subjects.
The effects of blood glucose control
Several studies have suggested that higher blood glucose levels as indicated by higher glycohemoglobin (HbA1c) levels were associated with worse cognitive performance.78–81
A large randomized controlled trial, the Action to Control Cardiovascular Risk in Diabetes-Memory in Diabetes (ACCORD-MIND) trial, aimed to compare the effects of intensive versus standard glycemic control on cognitive function and brain volume. Significant differences in total brain volumes favored the intensive treatment, although the cognitive outcomes were not different.82 Another randomized controlled trial demonstrated that the intervention was related to slower global cognitive decline in the intervention group. Improvements in HbA1c, but not in other clinical indices including blood pressure and lipid levels, mediated the effect of the intervention on cognitive decline.83 These results suggest that blood glucose control is important for the preservation of cognitive function in elderly diabetic patients. A recent prospective study by van den Berg et al however, reported that HbA1c levels at baseline had no effect in five cognitive domains.84,85 Another study found an inverse relationship between HbA1c and cognitive performance.86 Large prospective studies are warranted to establish the optimal blood glucose level for cognitive preservation. Moreover, with the advancement of pharmaceutical therapeutics, an increasing number of antidiabetics have become available. Each medicine has its own advantages in terms of pharmacological characteristics that may be associated with brain protection. The best combination of medicines for treating older T2DM patients should be explored. Another recent report suggested that a history of severe hypoglycemic episodes was associated with a greater risk of dementia.87 The merits of pharmacological blood glucose lowering should be balanced with the risk of hypoglycemia, especially in a frail elderly population.
Future directions
The elucidation of the mechanism of T2DM-associated cognitive dysfunction and dementia may lead to the development of therapeutics for this specific condition as well as for dementia, including AD, in general. Several basic studies have suggested that IR accelerates the AD-related pathology, and clinical evidence partly supports this hypothesis; however, general agreement on this point has not been reached.41,43 By taking advantage of recent advancements in imaging, including amyloid imaging technology with PET,84 higher field MRI with some potential for imaging small vessel diseases,88 and the diffusion tensor imaging method,89 it may be possible to investigate the relative contributions of AD-related pathology and ischemic changes to the increased prevalence of dementia in T2DM subjects. Furthermore, CSF biomarkers such as total tau, hyperphosphorylated tau, and the 42-amino-acid form of Aβ are now established biomarkers for AD90 and can be used to identify AD in the early or mild cognitive impairment stage of the disease with high accuracy.91,92 Identifying AD before the development of overt dementia and closely following up with the patient would make it possible to compare both the amyloid load and ischemic lesions before and after the development of dementia. Moreover, amyloid imaging and the measuring of CSF biomarkers in non-demented older people with or without IR could verify the hypothesis that insulin plays a role in the processing and deposition of Aβ. These investigations are important considering the potential future availability of disease-modifying therapeutics such as Aβ vaccination and inhibitors of Aβ secretions.
Nasal insulin or GLP-1 analogs or other medications targeting the brain insulin system may be therapies available in the future. By the intranasal method, insulin effectively bypasses the BBB and can be delivered into the brain. Clinical trials have demonstrated that intranasal insulin has some beneficial effects on cognition in patients with mild cognitive impairment and AD.93 GLP-1 analogs have been demonstrated to exert neuroprotective and aniapoptotic effects, reduce Aβ plaque accumulation, modulate long-term potentiation and synaptic plasticity, and promote the differentiation of neuronal progenitor cells. In animal models of behavior, treatment with GLP-1 receptor agonists has been demonstrated to improve measures of cognitive function, including learning and memory, as well as to reduce depressive behavior.94
It is a clinically important question whether the treatment for vascular risk factors has therapeutic potential in terms of slowing the progression of cognitive dysfunction, as neurodegenerative pathologies are not yet amenable to treatment. Vascular risk factors including T2DM and hypertension are reported to be associated with the progression of lacunae and white matter lesions in the brain.95 However, it remains to be investigated whether pharmaceutical interventions with antidiabetics and antihypertensives have protective effects against the progression of cognitive dysfunction and the development of dementia. If such beneficial effects do exist, the underlying mechanism of the therapeutic effects should also be explored. Such investigation may lead to the elucidation of the fundamental mechanism of the involvement of T2DM in the development of dementia.
With the ongoing increase in the size of the elderly population, T2DM-associated cognitive dysfunction and dementia are becoming increasingly larger problems. Developing a greater understanding of the relevant pathophysiology and establishing better therapeutic interventions are urgent needs.
Cognitive dysfunction in T2DM may start at a relatively young age; thus, starting the management at an early age may be important in terms of preventing not only dementia but also other complications.96,97
Conclusion
T2DM is associated with cognitive dysfunction and the incidence of dementia including AD. The underlying mechanism of this association should be elucidated. Such elucidation could lead to a clarification of the pathogenesis of AD and to the development of a treatment or method of prevention.98 Moreover, it is urgent that the optimal level of blood glucose control and the best combination of medications for enhancing cognitive preservation be established.
Disclosure
The author reports no conflicts of interest in this work.
References
Danaei G, Finucane MM, Lu Y, et al; Global Burden of Metabolic Risk Factors of Chronic Diseases Collaborating Group (Blood Glucose). National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 2.7 million participants. Lancet. 2011;378(9785):31–40. | ||
Abi Khalil C, Roussel R, Mohammedi K, Danchin N, Marre M. Cause-specific mortality in diabetes: recent changes in trend mortality. Eur J Prev Cardiol. 2012;19(3):374–381. | ||
McCrimmon RJ, Ryan CM, Frier BM. Diabetes and cognitive dysfunction. Lancet. 2012;379(9833):2291–2299. | ||
Stewart R, Liolitsa D. Type 2 diabetes mellitus, cognitive impairment and dementia. Diabet Med. 1999;16(2):93–112. | ||
Li L, Hölscher C. Common pathological processes in Alzheimer disease and type 2 diabetes: a review. Brain Res Rev. 2007;56(2):384–402. | ||
Ott A, Stolk RP, van Harskamp F, et al. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology. 1999;53(9):1937–1942. | ||
Arvanitakis Z, Wilson RS, Bienias JL, Evans DA, Bennett DA. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch Neurol. 2004;61(5):661–666. | ||
Peila R, Rodriguez BL, Launer LJ; Honolulu-Asia Aging Study. Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu-Asia Aging Study. Diabetes. 2002;51(4):1256–1262. | ||
Ohara T, Doi Y, Ninomiya T, et al. Glucose tolerance status and risk of dementia in the community: the Hisayama study. Neurology. 2011; 77(12):1126–1134. | ||
Cheng G, Huang C, Deng H, Wang H. Diabetes as a risk factor for dementia and mild cognitive impairment: a meta-analysis of longitudinal studies. Intern Med J. 2012;42(5):484–491. | ||
McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):263–269. | ||
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4th ed. Washington, DC: American Psychiatric Association; 2000. | ||
McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of the Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34(7):939–944. | ||
Jellinger KA. Prevalence and impact of cerebrovascular lesions in Alzheimer and lewy body diseases. Neurodegener Dis. 2010;7(1–3): 112–115. | ||
Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology. 2007;69(24):2197–2204. | ||
Lee SW, Clemenson GD, Gage FH. New neurons in an aged brain. Behav Brain Res. 2012;227(2):497–507. | ||
Lang BT, Yan Y, Dempsey RJ, Vemuganti R. Impaired neurogenesis in adult type-2 diabetic rats. Brain Res. 2009;1258:25–33. | ||
Machida M, Fujimaki S, Hidaka R, Asashima M, Kuwabara T. The insulin regulatory network in adult hippocampus and pancreatic endocrine system. Stem Cells Int. 2012;2012:959737. | ||
Farrall AJ, Wardlaw JM. Blood–brain barrier: ageing and microvascular disease – systematic review and meta-analysis. Neurobiol Aging. 2009;30(3):337–352. | ||
Kalaria RN. Cerebral vessels in ageing and Alzheimer’s disease. Pharmacol Ther. 1996;72(3):193–214. | ||
Bell RD, Zlokovic BV. Neurovascular mechanisms and blood–brain barrier disorder in Alzheimer’s disease. Acta Neuropathol. 2009;118(1): 103–113. | ||
Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci. 2007;10(11):1369–1376. | ||
Mooradian AD. Central nervous system complications of diabetes mellitus – a perspective from the blood–brain barrier. Brain Res Brain Res Rev. 1997;23(3):210–218. | ||
Kerti L, Witte AV, Winkler A, Grittner U, Rujescu D, Flöel A. Higher glucose levels associated with lower memory and reduced hippocampal microstructure. Neurology. 2013;81(20):1746–1752. | ||
Crane PK, Walker R, Hubbard RA, et al. Glucose levels and risk of dementia. N Engl J Med. 2013;369(6):540–548. | ||
Yamagishi S, Ueda S, Okuda S. Food-derived advanced glycation end products (AGEs): a novel therapeutic target for various disorders. Curr Pharm Des. 2007;13(27):2832–2836. | ||
Valente T, Gella A, Fernàndez-Busquets X, Unzeta M, Durany N. Immunohistochemical analysis of human brain suggests pathological synergism of Alzheimer’s disease and diabetes mellitus. Neurobiol Dis. 2010;37(1):67–76. | ||
Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8(1): 57–69. | ||
Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocr Rev. 2002;23(5):599–622. | ||
Moreira PI, Santos MS, Seiça R, Oliveira CR. Brain mitochondrial dysfunction as a link between Alzheimer’s disease and diabetes. J Neurol Sci. 2007;257(1–2):206–214. | ||
Badawi A, Klip A, Haddad P, et al. Type 2 diabetes mellitus and inflammation: Prospects for biomarkers of risk and nutritional intervention. Diabetes Metab Syndr Obes. 2010;3:173–186. | ||
Akiyama H, Barger S, Barnum S, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21(3):383–421. | ||
Zhao WQ, Townsend M. Insulin resistance and amyloidogenesis as common molecular foundation for type 2 diabetes and Alzheimer’s disease. Biochim Biophys Acta. 2009;1792(5):482–496. | ||
Watson GS, Craft S. The role of insulin resistance in the pathogenesis of Alzheimer’s disease: implications for treatment. CNS Drugs. 2003;17(1):27–45. | ||
Davis SN, Granner DK. Insulin, oral hypoglycemic agents, and the pharmacology of the endocrine pancreas. In: Hardman JG, Gilman AG, Limbird LE. Goodman and Gilman’s the Pharmacological Basis of Therapeutics. 9th ed., New York: McGraw-Hill; 1996:1487–1517. | ||
Woods SC, Seeley RJ, Baskin DG, Schwartz MW. Insulin and the blood–brain barrier. Curr Pharm Des. 2003;9(10):795–800. | ||
Havrankova J, Roth J, Brownstein M. Insulin receptors are widely distributed in the central nervous system of the rat. Nature. 1978;272(5656): 827–829. | ||
Freychet P. Insulin receptors and insulin action in the nervous system. Diab Metab Res Rev. 2000;16(6):390–392. | ||
Rivera EJ, Goldin A, Fulmer N, Tavares R, Wands JR, de la Monte SM. Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: link to brain reductions in acetylcholine. J Alzheimers Dis. 2005;8(3):247–268. | ||
Zhao L, Teter B, Morihara T, et al. Insulin-degrading enzyme as a downstream target of insulin receptor signaling cascade: implications for Alzheimer’s disease intervention. J Neurosci. 2004;24(49): 11120–11126. | ||
Ho L, Qin W, Pompl PN, et al. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer’s disease. FASEB J. 2004;18(7):902–904. | ||
Umegaki H. Pathophysiology of cognitive dysfunction in older people with type 2 diabetes: vascular changes or neurodegeneration? Age Ageing. 2010;39(1):8–10. | ||
Schubert M, Brazil DP, Burks DJ, et al. Insulin receptor substrate-2 deficiency impairs brain growth and promotes tau phosphorylation. J Neurosci. 2003;23(18):7084–7092. | ||
Talbot K, Wang HY, Kazi H, et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest. 2012;122(4):1316–1338. | ||
Bomfim TR, Forny-Germano L, Sathler LB, et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease-associated Aβ oligomers. J Clin Invest. 2012;122(4):1339–1353. | ||
Benedict C, Hallschmid M, Hatke A, et al. Intranasal insulin improves memory in humans. Psychoneuroendocrinology. 2004;29(10):1326–1334. | ||
de Zeeuw D, Parving HH, Henning RH. Microalbuminuria as an early marker for cardiovascular disease. J Am Soc Nephrol. 2006;17(8): 2100–2105. | ||
de Bresser J, Reijmer YD, van den Berg E, et al; Utrecht Diabetic Encephalopathy Study Group. Microvascular determinants of cognitive decline and brain volume change in elderly patients with type 2 diabetes. Dement Geriatr Cogn Disord. 2010;30(5):381–386. | ||
Kawamura T, Umemura T, Umegaki H, et al. Effect of renal impairment on cognitive function during a 3-year follow-up in elderly patients with type 2 diabetes: association with microinflammation. J Diabetes Investig. Epub 2014 Feb 4. | ||
Umegaki H, Iimuro S, Shinozaki T, et al; Japanese Elderly Diabetes Intervention Trial Study Group. Risk factors associated with cognitive decline in the elderly with type 2 diabetes: baseline data analysis of the Japanese Elderly Diabetes Intervention Trial. Geriatr Gerontol Int. 2012;12 Suppl 1:103–109. | ||
Umemura T, Kawamura T, Umegaki H, et al. Association of chronic kidney disease and cerebral small vessel disease with cognitive impairment in elderly patients with type 2 diabetes. Dement Geriatr Cogn Dis Extra. 2013;3(1):212–222. | ||
Dalkara T, Gursoy-Ozdemir Y, Yemisci M. Brain microvascular pericytes in health and disease. Acta Neuropathol. 2011;122(1):1–9. | ||
Weller RO, Massey A, Kuo YM, Roher AE. Cerebral amyloid angiopathy: accumulation of A beta in interstitial fluid drainage pathways in Alzheimer’s disease. Ann N Y Acad Sci. 2000;903:110–117. | ||
van Harten B, de Leeuw FE, Weinstein HC, Scheltens P, Biessels GJ. Brain imaging in patients with diabetes: a systematic review. Diabetes Care. 2006;29(11):2539–2548. | ||
Manschot SM, Brands AM, van der Grond J, et al; Utrecht Diabetic Encephalopathy Study Group. Brain magnetic resonance imaging correlates of impaired cognition in patients with type 2 diabetes. Diabetes. 2006;55(4):1106–1113. | ||
Korf ES, van Straaten EC, de Leeuw FE, et al; LADIS Study Group. Diabetes mellitus, hypertension and medial temporal lobe atrophy: the LADIS study. Diabet Med. 2007;24(2):166–171. | ||
Last D, Alsop DC, Abduljalil AM, et al. Global and regional effects of type 2 diabetes on brain tissue volumes and cerebral vasoreactivity. Diabetes Care. 2007;30(5):1193–1199. | ||
Gold SM, Dziobek I, Sweat V, et al. Hippocampal damage and memory impairments as possible early brain complications of type 2 diabetes. Diabetologia. 2007;50(4):711–719. | ||
Bruehl H, Wolf OT, Sweat V, Tirsi A, Richardson S, Convit A. Modifiers of cognitive function and brain structure in middle-aged and elderly individuals with type 2 diabetes mellitus. Brain Res. 2009;1280: 186–194. | ||
Burns JM, Donnelly JE, Anderson HS, et al. Peripheral insulin and brain structure in early Alzheimer disease. Neurology. 2007;69(11): 1094–1104. | ||
Burns JM, Honea RA, Vidoni ED, Hutfles LJ, Brooks WM, Swerdlow RH. Insulin is differentially related to cognitive decline and atrophy in Alzheimer’s disease and aging. Biochim Biophys Acta. 2012;1822(3): 333–339. | ||
den Heijer T, Vermeer SE, van Dijk EJ, et al. Type 2 diabetes and atrophy of medial temporal lobe structures on brain MRI. Diabetologia. 2003;46(12):1604–1610. | ||
Willette AA, Xu G, Johnson SC, et al. Insulin resistance, brain atrophy, and cognitive performance in late middle-aged adults. Diabetes Care. 2013;36(2):443–449. | ||
Moran C, Phan TG, Chen J, et al. Brain atrophy in type 2 diabetes: regional distribution and influence on cognition. Diabetes Care. 2013; 36(12):4036–4042. | ||
Baker LD, Cross DJ, Minoshima S, Belongia D, Watson GS, Craft S. Insulin resistance and Alzheimer-like reductions in regional cerebral glucose metabolism for cognitively normal adults with prediabetes or early type 2 diabetes. Arch Neurol. 2011;68(1):51–57. | ||
Schrijvers EM, Witteman JC, Sijbrands EJ, Hofman A, Koudstaal PJ, Breteler MM. Insulin metabolism and the risk of Alzheimer disease: the Rotterdam Study. Neurology. 2010;75(22):1982–1987. | ||
Matsuzaki T, Sasaki K, Tanizaki Y, et al. Insulin resistance is associated with the pathology of Alzheimer disease: the Hisayama study. Neurology. 2010;75(9):764–770. | ||
Beeri MS, Silverman JM, Davis KL, et al. Type 2 diabetes is negatively associated with Alzheimer’s disease neuropathology. J Gerontol A Biol Sci Med Sci. 2005;60(4):471–475. | ||
Arvanitakis Z, Schneider JA, Wilson RS, et al. Diabetes is related to cerebral infarction but not to AD pathology in older persons. Neurology. 2006;67(11):1960–1965. | ||
Thambisetty M, Jeffrey Metter E, Yang A, et al. Glucose intolerance, insulin resistance, and pathological features of Alzheimer disease in the Baltimore Longitudinal Study of Aging. JAMA Neurol. 2013;70(9):1167–1172. | ||
Tomita N, Furukawa K, Okamura N, et al. Brain accumulation of amyloid β protein visualized by positron emission tomography and BF-227 in Alzheimer’s disease patients with or without diabetes mellitus. Geriatr Gerontol Int. 2013;13(1):215–221. | ||
Strittmatter WJ, Roses AD. Apolipoprotein E and Alzheimer disease. Proc Natl Acad Sci U S A. 1995;92(11):4725–4727. | ||
Dore GA, Elias MF, Robbins MA, Elias PK, Nagy Z. Presence of the APOE epsilon4 allele modifies the relationship between type 2 diabetes and cognitive performance: the Maine-Syracuse Study. Diabetologia. 2009;52(12):2551–2560. | ||
Irie F, Fitzpatrick AL, Lopez OL, et al. Enhanced risk for Alzheimer disease in persons with type 2 diabetes and APOE epsilon4: the Cardiovascular Health Study Cognition Study. Arch Neurol. 2008;65(1):89–93. | ||
Luitse MJ, Biessels GJ, Rutten GE, Kappelle LJ. Diabetes, hyperglycaemia, and acute ischaemic stroke. Lancet Neurol. 2012;11(3):261–271. | ||
Reijmer YD, Leemans A, Brundel M, Kappelle LJ, Biessels GJ; Utrecht Vascular Cognitive Impairment Study Group. Disruption of the cerebral white matter network is related to slowing of information processing speed in patients with type 2 diabetes. Diabetes. 2013;62(6):2112–2115. | ||
Reijmer YD, Brundel M, de Bresser J, Kappelle LJ, Leemans A, Biessels GJ; Utrecht Vascular Cognitive Impairment Study Group. Microstructural white matter abnormalities and cognitive functioning in type 2 diabetes: a diffusion tensor imaging study. Diabetes Care. 2013;36(1):137–144. | ||
Cukierman-Yaffe T, Gerstein HC, Williamson JD, et al; Action to Control Cardiovascular Risk in Diabetes-Memory in Diabetes (ACCORD-MIND) Investigators. Relationship between baseline glycemic control and cognitive function in individuals with type 2 diabetes and other cardiovascular risk factors: the action to control cardiovascular risk in diabetes-memory in diabetes (ACCORD-MIND) trial. Diabetes Care. 2009;32(2):221–226. | ||
Maggi S, Limongi F, Noale M, et al; ILSA Study Group. Diabetes as a risk factor for cognitive decline in older patients. Dement Geriatr Cogn Disord. 2009;27(1):24–33. | ||
Peters R, Beckett N, Forette F, et al; HYVET investigators. Incident dementia and blood pressure lowering in the Hypertension in the Very Elderly Trial cognitive function assessment (HYVET-COG): a double-blind, placebo controlled trial. Lancet Neurol. 2008;7(8):683–689. | ||
Umegaki H, Kawamura T, Mogi N, Umemura T, Kanai A, Sano T. Glucose control levels, ischaemic brain lesions, and hyperinsulinaemia were associated with cognitive dysfunction in diabetic elderly. Age Ageing. 2008;37(4):458–461. | ||
Launer LJ, Miller ME, Williamson JD, et al; ACCORD MIND investigators. Effects of intensive glucose lowering on brain structure and function in people with type 2 diabetes (ACCORD MIND): a randomised open-label substudy. Lancet Neurol. 2011;10(11):969–977. | ||
Luchsinger JA, Palmas W, Teresi JA, et al. Improved diabetes control in the elderly delays global cognitive decline. J Nutr Health Aging. 2011;15(6):445–449. | ||
Ikonomovic MD, Klunk WE, Abrahamson EE, et al. Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer’s disease. Brain. 2008;131(Pt 6):1630–1645. | ||
van den Berg E, Reijmer YD, de Bresser J, Kessels RP, Kappelle LJ, Biessels GJ, Utrecht Diabetic Encephalopathy Study Group. A 4 year follow-up study of cognitive functioning in patients with type 2 diabetes mellitus. Diabetologia. 2010;53(1):58-65. | ||
Shorr RI, de Rekeneire N, Resnick HE, et al. Glycemia and cognitive function in older adults using glucose-lowering drugs. J Nutr Health Aging. 2006;10(4):297–301. | ||
Whitmer RA, Karter AJ, Yaffe K, Quesenberry CP, Selby JV. Hypoglycemic episodes and risk of dementia in older patients with type 2 diabetes mellitus. JAMA. 2009;301(15):1565–1572. | ||
Novak V, Abduljalil AM, Novak P, Robitaille PM. High-resolution ultrahigh-field MRI of stroke. Magn Reson Imaging. 2005;23(4): 539–548. | ||
Kodl CT, Franc DT, Rao JP, et al. Diffusion tensor imaging identifies deficits in white matter microstructure in subjects with type 1 diabetes that correlate with reduced neurocognitive function. Diabetes. 2008;57(11):3083–3089. | ||
Zetterberg LO, Wahlund K, Blennow. Cerebrospinal fluid markers for prediction of Alzheimer’s disease. Neurosci Lett. 2003;352(1):67–69. | ||
Hansson H, Zetterberg P, Buchhave E, Londos E, Blennow K, Minthon L. Association between CSF biomarkers and incipient Alzheimer’s disease in patients with mild cognitive impairment: a follow-up study. Lancet Neurol. 2006;5(3):228–234. | ||
Ewers M, Buerger K, Teipel SJ, et al. Multicenter assessment of CSF-phosphorylated tau for the prediction of conversion of MCI. Neurology. 2007;69(24):2205–2212. | ||
Schiöth HB, Craft S, Brooks SJ, Frey WH, Benedict C. Brain insulin signaling and Alzheimer’s disease: current evidence and future directions. Mol Neurobiol. 2012;46(1):4–10. | ||
McIntyre RS, Powell AM, Kaidanovich-Beilin O, et al. The neuroprotective effects of GLP-1: possible treatments for cognitive deficits in individuals with mood disorders. Behav Brain Res. 2013;237:164–171. | ||
Gouw AA, van der Flier WM, Fazekas F, et al; LADIS Study Group. Progression of white matter hyperintensities and incidence of new lacunes over a 3-year period: the Leukoaraiosis and Disability study. Stroke. 2008;39(5):1414–1420. | ||
van Eersel ME, Joosten H, Gansevoort RT, Dullaart RP, Slaets JP, Izaks GJ. The interaction of age and type 2 diabetes on executive function and memory in persons aged 35 years or older. PLoS One. 2013; 8(12):e82991. | ||
Joosten H, van Eersel ME, Gansevoort RT, Bilo HJ, Slaets JP, Izaks GJ. Cardiovascular risk profile and cognitive function in young, middle-aged, and elderly subjects. Stroke. 2013;44(6):1543–1549. | ||
Tang J, Pei Y, Zhou G. When aging-onset diabetes is coming across with Alzheimer disease: comparable pathogenesis and therapy. Exp Gerontol. 2013;48(8):744–750. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.