Back to Journals » Patient Preference and Adherence » Volume 8
Two decades of subcutaneous glatiramer acetate injection: current role of the standard dose, and new high-dose low-frequency glatiramer acetate in relapsing–remitting multiple sclerosis treatment
Authors Caporro M, Disanto G, Gobbi C, Zecca C
Received 1 June 2014
Accepted for publication 2 July 2014
Published 21 August 2014 Volume 2014:8 Pages 1123—1134
DOI https://doi.org/10.2147/PPA.S68698
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Matteo Caporro, Giulio Disanto, Claudio Gobbi, Chiara Zecca
Neurocenter of Southern Switzerland, Ospedale Regionale di Lugano, Lugano, Switzerland
Abstract: Glatiramer acetate, a synthetic amino acid polymer analog of myelin basic protein, is one of the first approved drugs for the treatment of relapsing–remitting multiple sclerosis. Several clinical trials have shown consistent and sustained efficacy of glatiramer acetate 20 mg subcutaneously daily in reducing relapses and new demyelinating lesions on magnetic resonance imaging in patients with relapsing–remitting multiple sclerosis, as well as comparable efficacy to high-dose interferon beta. Some preclinical and clinical data suggest a neuroprotective role for glatiramer acetate in multiple sclerosis. Glatiramer acetate is associated with a relatively favorable side-effect profile, and importantly this was confirmed also during long-term use. Glatiramer acetate is the only multiple sclerosis treatment compound that has gained the US Food and Drug Administration pregnancy category B. All these data support its current use as a first-line treatment option for patients with clinical isolated syndrome or relapsing–remitting multiple sclerosis. More recent data have shown that high-dose glatiramer acetate (ie, 40 mg) given three times weekly is effective, safe, and well tolerated in the treatment of relapsing–remitting multiple sclerosis, prompting the approval of this dosage in the US in early 2014.This high-dose, lower-frequency glatiramer acetate might represent a new, more convenient regimen of administration, and this might enhance patients’ adherence to the treatment, crucial for optimal disease control.
Keywords: glatiramer acetate, disease modifying treatment, efficacy, safety
Introduction
Multiple sclerosis (MS) is a chronic immune-mediated disease involving the white and gray matter of the central nervous system (CNS), causing neurological dysfunction.1 It affects predominantly females, and has a prevalence varying from five to 80 per 100,000 persons worldwide.2 It is thought to be a multifactorial disease resulting from an autoimmune reaction to self-antigens in genetically predisposed individuals, and probably involving additionally several environmental factors, such as vitamin D deficiency, sun exposure, smoking, and infections. Evidence for a concomitant neurodegenerative component has been highlighted to be present already at disease onset;3 however, this prevails in the later phases of the disease.4,5
Multifocal localized inflammation of the CNS leading to demyelination, axonal damage, and astrocytosis pathologically characterizes the disease and causes impaired nerve conduction,5 leading to MS symptoms commonly involving sensory, motor, visual, balance, sphincteric, and cognitive functions, as well as fatigue.
Relapsing–remitting (RR) is the most common (80%–85%) MS subtype, characterized by flares and remissions.6,7 The first MS relapse is currently referred to as clinically isolated syndrome (CIS), corresponding to a typical clinical and paraclinical early RRMS picture that cannot however fulfill current MS diagnostic criteria.8 Approximately 60%–70% of patients with RRMS evolve to secondary progressive MS over time, and around 10% of patients can be classified as having a primary progressive or progressive relapsing course.
Though incurable, MS is currently treatable, with the aim of delaying as much as possible disability progression that may derive principally from unrecovered relapses and progressive neurological deterioration. To this end, several immunomodulating, immunosuppressive, and immunobiological agents have been developed to control inflammatory activity, prevent relapses, and possibly delay disability progression, particularly in the early phase of the disease.
Glatiramer acetate (GA; Copaxone®; Teva Pharmaceutical Industries, Petah Tikva, Israel) and beta-interferons (IFNbs) have been traditionally considered first-line treatments of RRMS, and represent the cornerstone in MS therapy.9 Until recently, these two drug types were the only immunomodulatory therapies available for the treatment of RRMS. However, these drugs are not always sufficiently efficacious to suppress inflammatory activity in all MS patients. Moreover, they may not be well tolerated due to side effects or frequent injections, which sometimes preclude adequate adherence.10 The advent of second-line drugs, such as natalizumab,11 fingolimod,12 teriflunomide,13 and dimethyl fumarate,14 as well as alemtuzumab15 in some countries, is promising both for possible higher anti-inflammatory efficacy and a more convenient way of administration (ie, either intravenous injections or oral). These advantages have, however, the price of a variable but overall less favorable safety and side-effect profile.16 Nevertheless, the approval of these new compounds changed the MS therapeutic landscape and the first-line drug-decision process in a newly diagnosed MS patient.
This paper reviews relevant data concerning the mechanism of action, efficacy, and safety of GA administered at the licensed (20 mg daily) dose, summarizes more recent data concerning the administration of GA at higher doses with lower frequency, and aims to define its current role as a treatment option in MS.
PubMed was searched for abstracts using the terms “glatiramer acetate AND multiple sclerosis” and “glatiramer acetate AND adherence”. Only articles written in English were considered, and there was no time-period restriction. The references of the resulting studies were used to identify additional articles to be included in the review (Table 1).
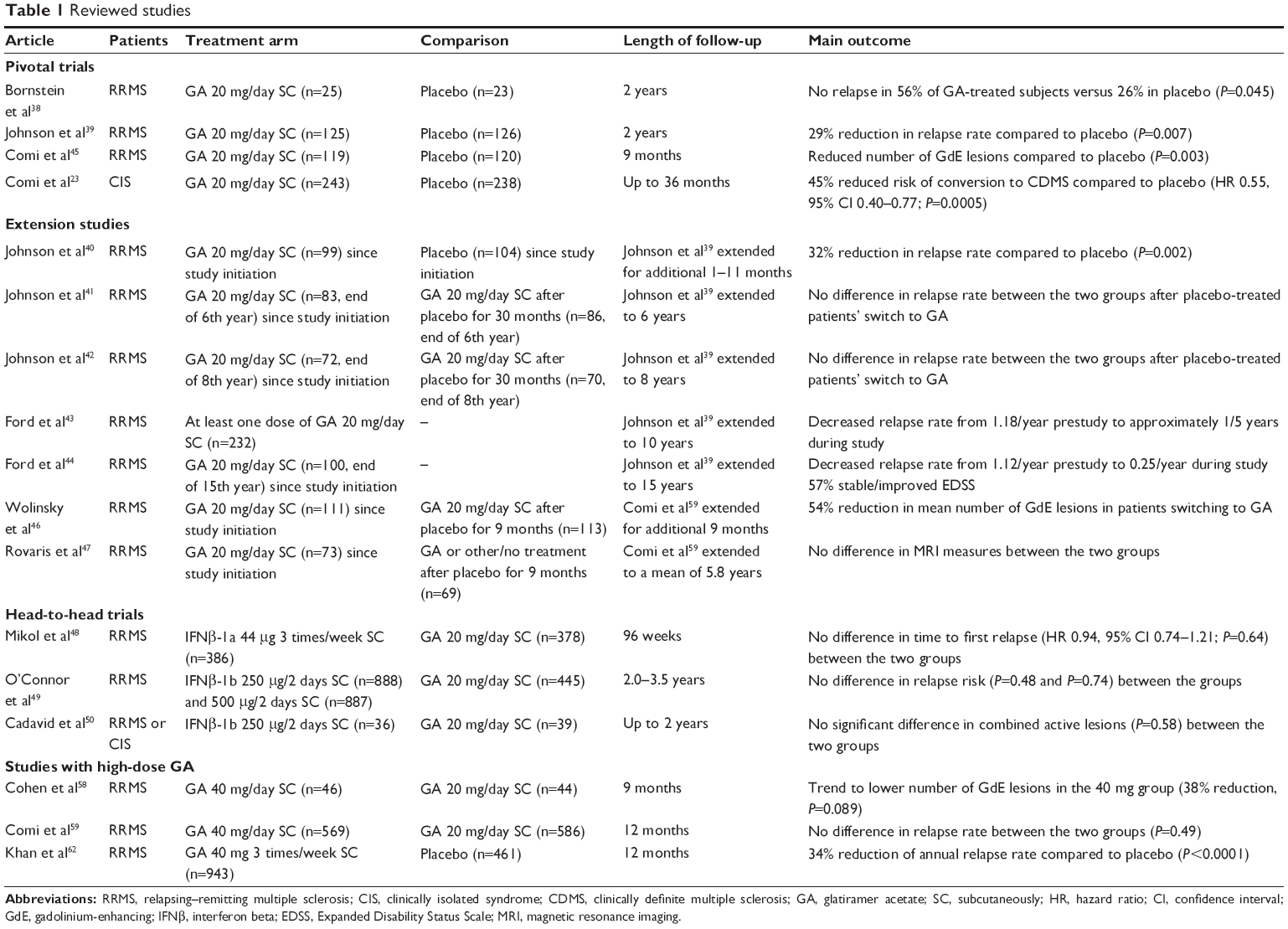 | Table 1 Reviewed studies |
Glatiramer acetate
GA (Copaxone) is a synthetic amino acid polymer analog of myelin basic protein (MBP), an antigen thought to be involved in the pathogenesis of MS.17–21 It consists of a standardized combination of four amino acids (l-alanine, l-glutamic acid, l-lysine and l-tyrosine) randomly combined to form a polymer with an average length of 40–100 amino acids.17–20
It has been empirically found to suppress autoimmune encephalomyelitis in mice,22 possibly due to a displacement of immune cells targeting native myelin components. Clinical results consistent with this rationale have also been shown in humans, leading to its licensing for MS treatment in 1997 in the US and 2000 in Europe. Initially, GA was approved as first-line treatment in RRMS at a dose of 20 mg subcutaneous (SC) injection daily. More recently, further approval was obtained for the treatment of CIS patients.23
Mechanism of action
It is believed that GA has a multifaceted mechanism of action, involving both immunomodulation and neuroprotection (Figure 1). It is basically an immunomodulator capable of modifying the immune responses that drive MS pathogenesis.17–20,24 It binds to major histocompatibility complex (MHC) class II molecules on MBP-specific antigen-presenting cells, preventing MBP itself from binding to and stimulating these cells.18,20,24 A body of preclinical and clinical data support a role of GA in inducing a T-helper (Th)-1 to Th2-cell phenotype shift. In other words, GA-reactive T cells predominantly secrete anti-inflammatory cytokines, such as IL-1, IL-4, and IL-10, characterizing Th2 regulatory cells instead of typical Th1, and proinflammatory cytokines, such as IL-2 and IL-12.18,20,24 It has to be underlined that GA per se is not able to penetrate the CNS blood–brain barrier. Its immunomodulatory function is carried out by peripheral GA-induced Th2 cells that enter the CNS, recognize myelin antigens, and are thus reactivated, ultimately reducing inflammation associated with MS.24–26 This mechanism of action is known as “bystander suppression”.27
 | Figure 1 Mechanisms of action of glatiramer acetate (GA) in multiple sclerosis. GA exhibits competitive binding at the MHC-II complex and T-cell receptor (TCR) antagonism. GA is able to displace myelin basic protein from the binding site on MHC-II molecules. Treatment with GA leads to the induction of antigen-specific TH2 T cells in the periphery (1). In addition CD8+ and CD4+CD25+ regulatory T cells are induced by GA therapy (2). The constant activation seems to have an important impact on the induction and maintenance of the regulatory/suppressive immune cells (3). Because of the daily activation, GA T cells are believed to be able to cross the blood–brain barrier (4). Inside the central nervous system, some GA-specific T cells cross-react with products of local myelin turnover presented by local antigen-presenting cells (APCs) (5). In response, anti-inflammatory cytokines are secreted, which dampen the local inflammatory process (bystander suppression) (6). Furthermore, GA-specific T cells secrete neurotrophic factors that might favor remyelination and axonal protection (7). Reprinted from Autoimmun Rev. 2007;6(7). Schrempf W, Ziemssen T. Glatiramer acetate: mechanisms of action in multiple sclerosis. 469–475. Copyright © 2007, with permission from Elsevier.78 |
In addition, several studies have suggested further effects on the immune system mediated by GA.17,24 GA induces T-regulatory cells, such as CD4+, CD8+, and CD4+CD25+ T cells, while it downregulates Th17 cells that have been associated with MS disease activity. Moreover, GA drives monocytes, dendritic cells, and microglia to preferential anti-inflammatory responses.24,25,28–34
Finally, GA seems to induce neuroprotective and/or neuroregenerative effects at the preclinical level.17,18,24,25 For instance, it increases neurotrophic factors like brain-derived neurotrophic factor, involved in neuronal and glial cell survival, and may mediate neuroprotection. There is also evidence that GA induces remyelination and enhances neurogenesis.17,18,21,24,25
The majority of patients treated with GA develop GA-reactive IgG antibodies. However, these do not appear to be related to clinical or radiological clinical course measures of efficacy.35–37
Clinical efficacy: data from clinical trials
Pivotal trials
Pivotal trials have shown consistent efficacy of GA in the treatment of RRMS patients. The first study assessing the efficacy of GA in RRMS was published more than 25 years ago.38 It was a double-blind, randomized, placebo-controlled pilot trial involving 50 RRMS patients who were treated either with daily GA 20 mg or daily placebo over 2 years. Twenty-six percent of placebo- and 56% of GA-treated patients experienced no relapses over the study period (P=0.045). Among less disabled patients (Kurtzke disability score 0–2), those taking GA improved (+1.2 Kurtzke units), while placebo-treated patients worsened (−0.5 Kurtzke units, P=0.012). In contrast, more disabled patients in both groups showed an increase in Kurtzke disability score. Limited by the small sample size, this pivotal trial provided the first clinical evidence for a role of GA in the treatment of RRMS. A number of subsequent larger multicenter trials confirmed these results.
The first large Phase III double-blind, placebo-controlled study included 251 RRMS subjects 18–45 years old, with an Expanded Disability Status Scale (EDSS) score of 0–5.0, a history of at least two relapses in the 2 years prior to study entry, and a disease duration of at least 1 year. Participants were randomized to receive GA or placebo by daily SC injection for 2 years, with a reduction of 29% in the annualized relapse rate (ARR; primary end point) in favor of the GA group (0.59 versus 0.84, respectively; P=0.0007).39 Among secondary clinical outcomes, median time to first relapse from baseline and the proportion of relapse-free patients over 2 years showed a trend favoring GA over placebo (287 versus 198 days, P=0.097; 33.6% versus 27.0%, P=0.098; respectively). Overall, parameters of disability change also favored GA over placebo (EDSS change from baseline −0.05 versus 0.21, P=0.023), though the proportion of patients who were free from disability progression was similar between groups (78.4% versus 75.4%, not significant). The main limitation of this trial was the absence of magnetic resonance imaging (MRI) monitoring.
A pivotal GA study by Johnson et al was followed by a prospective, open-label study replicating the benefits of early versus delayed GA at 3, 6 and 8 years.41,42 Further 10-year extension data were obtained from 47% of the original cohort,43 and showed that continuous GA treatment led to more than 80% decline in relapse rate (from 1.18 relapses/year prestudy to one relapse/5 years), with no significant disability progression, evaluated using the EDSS score. Recently, data concerning 15-year extension were published.44 Of the initially randomized subjects, 43% were still on GA treatment, and had received only this immunomodulator during the disease course. Of those, two-thirds had not reached secondary progression, 57% had stable or improved EDSS, and 82% of patients could still walk. The comparator cohort that had interrupted GA obtained fairly good results as well; however, the mean disease duration in these patients was much shorter (13 versus 22 years).
The clinical efficacy of GA was replicated in a European/Canadian trial45 involving 239 RRMS subjects, and extended results toward a benefit on MRI disease activity. Main inclusion criteria were age between 18 and 50 years, a disease duration of at least 1 year, an EDSS score up to 5.0, and documented disease activity (at least one relapse in the preceding 2 years, and at least one gadolinium-enhanced [GdE] lesion on their screening brain MRI). Patients were randomized to either daily injections of GA 20 mg or placebo and treated for 9 months, and were followed with monthly brain MRIs. GA-treated patients showed a significant reduction in total GdE lesions (primary end point −10.8 versus −18.0, P=0.003), number and volume of new T2 lesions, and brain-atrophy progression, as well as clinical efficacy measured by reduction of mean relapse rate. Interestingly, the treatment effect of GA on the mean number of GdE lesions per patient per month, as well as mean number of relapses per patient, consistently appeared only from month 6 after GA start. However, the short duration of the study prevented assessment of treatment effects on disability progression, especially in light of the delayed onset of GA action.
In the 9-month, open-label phase of the European/Canadian study45 involving 94% of the original cohort, the effect of GA treatment was sustained: a 54% reduction in the mean number of GdE lesions for those switching from placebo to GA and a further 24.6% reduction for those remaining on GA were observed.46 A 5.8-year extension phase47 involving 63.4% of the original cohort showed that 66% of the patients were still on GA and had the highest relapse-free period, compared either to IFN switchers or to untreated patients (3.5 versus 1.3 versus 2.9, respectively). No significant differences for any MRI parameters were found at 5 years between originally GA- or placebo-treated subjects. However, the proportion of patients not requiring walking aids was lower in the first group (P=0.034), suggesting that an earlier initiation of GA might have a favorable impact on long-term disease evolution.
In conclusion, between the late 1980s and early 2000s, the results of three pivotal trials were published, which consistently assessed the efficacy of GA in the treatment of patients with RRMS, showing an approximately 30% reduction in relapse rate and consistent benefits on MRI for surrogates of disease activity compared to placebo. Open-label extension studies consistently showed a sustained efficacy of GA up to 15 years in a subgroup of patients participating in pivotal trials, though these were limited by several factors, including absence of a placebo arm and positive selection of responders.
CIS trial
GA has been recently labeled for CIS based on the favorable results of the PreCISe (Evaluate Early Glatiramer Acetate Treatment in Delaying Conversion to Clinically Definite Multiple Sclerosis of Subjects Presenting with Clinically Isolated Syndrome) trial.23 This involved 481 subjects presenting with a monofocal CIS and two or more T2 brain lesions (≥6 mm), that were randomly assigned to either SC GA 20 mg per day or placebo. A significant delay in conversion to clinically definite MS (722 versus 336 days in the treatment versus placebo groups, P=0.0005), as well as consistent benefits on radiological parameters (number/volume of new T2, number of new T1 GdE and T1 hypointense lesions) were observed after approximately 2.4 years of treatment. The study included only a restricted subgroup of CIS subjects, and did not provide information concerning the impact of GA on disability progression. In conclusion, the PreCISe trial provided substantial information in favor of the effectiveness of GA in the treatment of early forms of MS.
Head-to-head trials
Three head-to-head trials48–50 assessed the efficacy of GA compared to high-dose IFNs. REGARD48 (REbif vs Glatiramer Acetate in Relapsing MS Disease) was a randomized, open-label trial comparing SC IFNβ-1a 44 μg three times per week (Rebif®; EMD Millipore, Billerica, MA, USA) to daily SC GA 20 mg in 764 RRMS patients. Main inclusion criteria were 18–60 years of age, EDSS score between 0 and 5.5, and at least one relapse in the year prior to study entry. After 96 weeks, there were no differences between the two treatment groups in the time to relapse (primary outcome, P=0.64) or in the number or volume of active T2 lesion load on MRI. However, the IFN group showed significantly fewer GdE lesions, and the GA-group had less pronounced brain-volume loss (1.073% versus 1.24%, P=0.018). The main limitations of the study were a lack of patient blinding to treatment (although the assessor was blinded) and a low on-study relapse rate, which could have prevented the capturing of differences between the two active compounds.
BEYOND (Betaferon Efficacy Yielding Outcomes of a New Dose) was a large randomized trial49 involving 2,244 RRMS patients randomized to either 250 μg SC or 500 IFNβ-1b (Betaseron®; Bayer, Leverkusen, Germany) every other day or daily 20 mg GA SC over a minimum of 2 years. Main inclusion criteria were 18–55 years of age, EDSS score between 0 and 5.0, and at least one relapse in the year prior to study entry. Results demonstrated no statistically significant differences in the ARR (0.33, 0.36, and 0.34, respectively), disability progression measured by EDSS, the majority of MRI-monitoring parameters (GdE lesions, T1 lesions, normalized brain volume) among groups. Partial blinding was a major limitation.
The BECOME (Betaseron vs Copaxone in Multiple Sclerosis with Triple-Dose Gadolinium and 3-Tesla MRI Endpoints) study50 compared primarily radiological efficacy of SC IFNβ-1b 250 μg every other day and daily SC 20 mg GA over 2 years in 75 RRMS and CIS patients. Main inclusion criteria were 18–55 years of age and EDSS score <5.5. The study was performed using a 3 T MRI machine and administering triple-dose Gd combined with delayed imaging, which is known to more than double lesion detection compared to standard techniques. Treatment arms showed similar results in the number of combined active lesions (overall GdE plus new nonenhancing fluid-attenuated inversion-recovery lesions) per patient per scan at year 1, obtained by monthly brain MRI monitoring. The main limitations of this study were the relatively small sample size, lack of patient blinding, and the monocentric design.
It has to be underlined that in all these head-to-head trials, the on-study relapse rate was much lower than the rates reported in the pivotal trials (0.29–0.34 in head-to-head studies versus 0.59 in the Johnson et al trial),39 mainly reflecting a change in study populations over two decades. Increasing treatment options and changes in MS diagnostic criteria probably account for this phenomenon.
Among the novel therapeutic compounds for MS, only the Phase III CONFIRM (Comparator and an Oral Fumarate in Relapsing–Remitting Multiple Sclerosis) trial51 testing BG-12 included a treatment arm with GA monotherapy. This was a multicenter, placebo-controlled, randomized trial involving 1,417 RRMS patients from 18 to 55 years of age, having an EDSS score up to 5.0, who had experienced at least one clinical relapse in the past year or showed one GdE brain lesion at baseline. Enrolled subjects were randomized either to BG-12 240 mg twice a day, BG-12 240 mg three times a day, GA 20 mg SC daily, or placebo, and were treated for 2 years. Compared to placebo, relapse risk was reduced by 29%, 34%, and 45% by GA (P=0.01), twice-daily (P=0.002) and three-times-daily (P<0.001) BG-12, respectively (primary end point). Secondary end points, including disability progression and several radiological surrogates of disease activity, showed consistent results. Although the study was not powered to test the superiority or inferiority of BG-12 versus GA, both BG-12 doses were associated with results numerically similar or superior to GA across all study end points. A post hoc analysis with direct comparisons indicated a trend of superiority of BG-12 over GA (ARR – twice-daily BG-12 versus GA, P=0.10, thrice-daily BG-12 versus GA, P=0.02; new or enlarging T2 lesions – twice-daily BG-12 versus GA, P=0.007, thrice-daily BG-12 versus GA, P=0.002). The limitations of this study were the partial blinding to treatment assignment, the late modification of the trial design with the inclusion of an additional comparative arm with GA required by healthy authorities, and the insufficient power for assessing direct efficacy comparisons between GA and BG-12.
In conclusion, three head-to-head trials showed comparable efficacy between GA and high-dose IFNβs in the treatment of RRMS, both from a clinical and radiological perspective. Notably, some results suggest that GA better protects against brain-volume loss, while high dose IFNβs are associated with fewer GdE lesions.
Evidence for neuroprotection from clinical trials
Besides preventing new MS-lesion formation, treatment with GA was also found to reduce the accumulation of permanent black holes, namely persistently hypointense T1 lesions, which are indicative of irreversible tissue damage and correlate with disability.20 Indeed, compared to placebo, the proportion of new T1 lesions that evolved into chronic black holes in RRMS patients participating in the European/Canadian trial52 was statistically significantly lower (18.9% versus 26.3%, P=0.04). However, it should be stressed that under GA treatment, a similar (BEYOND trial, IFNβ 21.6% versus GA 23.5%; P>0.20)53 or smaller (BECOME trial, IFNβ 9.8% versus GA 15.2%; P=0.20)54 proportion of acute black holes turned into persistent black holes compared to IFNβ.
MS is characterized by a more rapid rate of brain-volume decrease compared to the general population, which argues in favor of a mechanism of neurodegeneration behind the disease. In an MRI subgroup analysis of the pivotal trial by Johnson et al,39 GA significantly reduced the loss of brain volume compared to placebo (mean annual brain-volume change: −0.6% and −1.8%, respectively; P=0.0078).55 This finding was, however, not confirmed by the European/Canadian trial.45 Notably, the REGARD study48 showed that GA better protects from brain-volume loss, while high-dose IFNβ is associated with fewer GdE lesions, which might support a neuroprotective property of GA partially independent of anti-inflammatory activity.
The integrity of axons in MS can be assessed in vivo by assessing levels of N-acetylaspartate, a neuronal marker compound.56 A spectroscopy analysis of a subgroup of 34 subjects participating in the Phase III PreCISe trial23 showed that treatment with GA was associated with an improvement, whereas placebo was associated with a decline in brain neuroaxonal integrity, as indicated by an increase and decrease in the ratio between N-acetylaspartate and creatine, respectively.56
In conclusion, several clinical studies suggest a possible neuroprotective effect of GA in MS. However, it has not yet been clarified if this effect is peculiar to GA or a general consequence of any efficacious anti-inflammatory compound.
High-dose GA and different regimens
GA is currently approved as a 20 mg daily SC injection. However, some experimental data have suggested that higher doses were associated with greater efficacy.57
High-dose GA
A Phase II clinical trial was performed to evaluate differences in efficacy, safety, and tolerability between 40 mg and 20 mg daily doses of SC GA in 38 RRMS patients for 9 months.58 The primary efficacy end point was the total number of GdE lesions on MRI at 7, 8, and 9 months of follow-up. Results showed a trend for increased efficacy in the higher compared to the lower dose (number of GdE lesions 3.62 versus 2.26, respectively, rate ratio 0.62; P=0.09). The decrease in GdE lesions occurred earlier in the 40 mg group (from month 3) than in the 20 mg group. Safety profiles were comparable between the two doses, with a mild increase of local injection-site manifestations with the 40 mg dose. The main limitation of this trial was the small sample size.
A more recent Phase III, randomized, double-blind, parallel-group trial compared daily administration of SC 40 mg or 20 mg GA over 1 year.59 This study was conducted in 136 sites across 20 different countries worldwide. A total of 1,262 patients were screened and 1,155 (91.5%) randomized to receive single prefilled daily shots of either 20 or 40 mg GA for 12 months. The primary efficacy outcome was the relapse rate in the first year, while secondary outcomes included MRI efficacy parameters, such as the cumulative number of GdE lesions, the number of new T2 lesions at 12 months, and brain-volume changes. At 1 year, no differences were found in the primary end point (mean number of relapses 0.28 versus 0.27 for 20 mg and 40 mg doses, respectively; mean annualized number of relapses 0.33 versus 0.35, respectively). The decrease in mean number of GdE lesions at 3 months compared to baseline (frequent MRI cohort, n=234) was more evident in the 40 mg group (37.6% reduction, P=0.012) than in the 20 mg one (21.9% reduction, P=0.172). The percentage reduction in GdE lesions was consistently higher in the 40 mg than in the 20 mg group at 6, 9, and 12 months, results supported by similar findings in the entire study population at 12 months (0.68 in the 20 mg group versus 0.54 in the 40 mg group, −21%). The mean number of new T2 lesions and percentage brain-volume changes at month 12 were similar in both groups.
The tolerability profile was comparable between the two dose groups, and was similar to that observed in previous GA 20 mg trials. This first Phase III trial on GA with a 20 versus 40 mg daily dose comparison suggested no gain in efficacy with higher GA doses over a 12-month period. Nevertheless, the beneficial effect of GA 40 mg on MRI activity appeared to start earlier than in the 20 mg dose regimen.45 The main limitation of the study was the short duration.
In conclusion, these two trials did not demonstrate superior efficacy of daily double-dose GA compared to the approved 20 mg GA, thus excluding this as a possible strategy to increase GA efficacy in MS treatment. Importantly, these two trials highlighted that GA 40 mg administration was feasible, well tolerated, and associated with a side-effect profile similar to the approved 20 mg GA, and raised the idea to redistribute weekly 20 mg GA doses, reducing the number of injections to improve treatment adherence.
Low-frequency GA
Two small randomized exploratory studies tested different administration regimens of GA 20 mg.60,61 Khan et al60 compared daily administration of GA 20 mg to GA 20 mg every other day in 30 treatment-naïve RRMS patients. After 2 years, no differences in relapse rate, disability progression, change in T2 brain lesion volume, or GdE lesions between the two groups were reported. In another study, Khan et al61 compared continued daily GA 20 mg to a switch to GA 20 mg twice a week in 48 RRMS patients that had been treated with GA 20 mg daily for at least 1 year. After 2 years, relapse rate, EDSS scores, and several MRI parameters (T2 lesions, GdE lesions, percentage of brain-volume change) were similar in the two groups. Notably, the overall tolerability profile was considerably higher in the group of patients receiving GA less frequently.60,61 The limitation of these trials was the small sample size not allowing definitive conclusions.
Low-frequency high-dose GA
The GALA (Glatiramer Acetate Low-frequency Administration) study group62 designed a randomized, placebo-controlled, parallel-group, Phase III trial aimed at evaluating the efficacy and safety of a less frequent GA-administration regimen in a larger sample size. This treatment schedule might provide a convenient frequency of injections while maintaining a similar weekly dose as the approved daily 20 mg regimen. This study included 142 sites across 17 countries, and enrolled a total of 1,404 RRMS patients. Out of these patients, 943 were randomized to GA 40 mg three times weekly and 461 to placebo three times weekly for 12 months.
An open-label phase in which all patients were invited to continue treatment with the same 40 mg GA thrice-weekly regimen is ongoing. A total of 1,219 patients were evaluable at 12 months (86.8%). Patients receiving GA 40 mg three times weekly demonstrated a significant reduction (34%) in ARR, which represented the primary end point of the study (mean ARR 0.331 and 0.505 in GA and placebo, respectively; P<0.0001).
In addition, the GA-treated group showed a longer time to relapse and a greater proportion of relapse-free patients. MRI parameters were also evaluated as secondary end points, and GA-treated patients showed a significant reduction at both 6 and 12 months in the cumulative number of GdE lesions and cumulative number of new or newly enlarging T2 lesions compared to placebo. No significant differences were found in terms of brain volume.
The tolerability profile was consistent with that observed in trials testing the currently used 20 mg daily formulation. A major limitation of the trial was the absence of a third arm comparing the efficacy of GA 40 mg three times a week to GA 20 mg daily.
The ARR reduction observed in the GALA study (34%) is roughly in line with that reported in the pivotal trial (29%);39 however, populations in the two trials differed in baseline characteristics, reflecting more advanced and severe MS disease in the pivotal trial compared to the GALA trial. This is confirmed by the substantial difference in the absolute ARR reported in the two trials (placebo group – pivotal 0.84, GALA 0.505; GA group – pivotal 0.59, GALA 0.331), making indirect efficacy comparison unreliable.
In summary, the GALA study proved the efficacy of GA 40 mg three times weekly in reducing the ARR and MRI activity in RRMS patients over a 12-month period, with a safety profile that was comparable to that of the approved 20 mg daily dose. These results suggest that GA 40 mg three times weekly represents a valid alternative administration regimen for patients with poor needle tolerance who prefer a less frequent injection schedule. This treatment schedule was approved in the US in early 2014.
Clinical safety
GA’s safety profile in RRMS has been studied and confirmed by long-term studies up to 22 years and an experience with this drug of more than 1 million patient-years.44
Side-effect profile: GA 20 mg daily
Side effects with GA are usually mild and well tolerated. Injection-site reactions (ISRs) have been reported as the most commonly complained-of adverse event, reported in around 80% of patients.63 ISRs consist of pain, itching, swelling, and redness, and are isolated or randomly associated. Some strategies have been proposed to reduce the incidence and severity of ISRs, such as warm compresses applied to the injection site 5 minutes before self-injection, which has been demonstrated to be effective.64
Around 10% of GA-treated MS patients complain about an immediate postinjection reaction, which includes symptoms of flushing, chest pain, palpitations, anxiety, dyspnea, tachycardia, throat constriction, and urticaria.65 Immediate postinjection-reaction symptoms are usually transient and self-limited, requiring no specific treatment, but can be frightening when first experienced and surely influence adherence.
Around 2% of patients participating in GA-related clinical trials experienced localized lipoatrophy or other skin abnormalities,66 which might be disfiguring and sometimes become permanent. There is no known treatment for lipoatrophy, which has to be prevented by instructing patients in proper injection technique and advice to change injection sites daily.
Side-effect profile: alternative treatment regimens
Both daily and thrice-weekly GA 40 mg showed a side-effect profile comparable to low doses. In the GALA study,62 GA-treated patients experienced local adverse events, such as ISRs (35.2% versus 5% with placebo), 99.9% of which were of mild or moderate intensity. These ISRs were most commonly represented by erythema, pruritus, or pain. The frequency of serious adverse events was similar between GA and placebo groups (approximately 4.5% in each group). These severe episodes led to treatment discontinuation in 3.1% of GA- and 1.3% of placebo-treated patients.
Long-term safety and drug interactions
After decades of continuous use, no cumulative toxicities or late-emerging adverse events have been reported. No drug interactions are known for GA, and due to the absence of immunosuppressive effects, no increase of opportunistic infections or malignancies are associated with its use.39–42,44
Pregnancy and fertility
GA is the only disease-modifying therapy with a US Food and Drug Administration (FDA) pregnancy category B rating, indicating no fetal risks in animal studies. However, due to the lack of adequate and well-controlled studies in human pregnancies, GA is currently advised to be stopped prior to an anticipated pregnancy or when pregnancy is recognized.67 Postmarketing surveillance data on GA-treated women showed a miscarriage rate of 17%,68 comparable to the 10%–15% reported for the general population in the US.69 No apparent increased risks of adverse fetal or pregnancy outcomes were reported.68 Further published open-label observational studies on GA exposure during pregnancy (beyond the first trimester, in some cases throughout the entire pregnancy period) seem to confirm preclinical studies, and suggest that GA is not associated with teratogenicity or a higher rate of miscarriage.70–72 Lastly, GA has no reported effects on fertility, as shown in a multigeneration reproduction and fertility study in rats.73
In conclusion, GA has an excellent safety profile both in the short and long (up to 22 years) term, and is the only MS immunomodulant therapy classified as FDA B category for pregnancy risk. It has no drug-to-drug interactions relevant to clinical practice. Its adverse-event profile includes mainly skin-related side effects and systemic reactions in a minority of patients.
Adherence to GA treatment
The lack of adherence over the long term represents one of the main causes for a suboptimal response to MS treatments.74 GA is an MS drug requiring a higher frequency of self-injections compared to other MS treatments, and adherence has been shown to be inversely proportional to the number of injections.75 Main barriers to proper adherence to GA include needle phobia and self-injection anxiety.74
At the 15-year follow-up of the pivotal study by Johnson et al44 43% of the initially randomized subjects were still on GA treatment. With regard to a lower-frequency administration regimen, data in the head-to-head BEYOND study49 showed a similar proportion of dropouts in the GA group (16%) as the other two treatment groups receiving IFNβ-1b 250 μg (12%) or 500 μg (18%), despite the higher frequency of SC injection in the first group. Similar proportions were observed in the BECOME50 (GA 30% versus IFNβ-1a 20%) and REGARD48 (GA 14% versus IFNβ-1a 21%) trials. In an open-label observational study on a German cohort of 308 MS patients, GA showed a significantly lower discontinuation rate compared to IFNβs during 6 and 24 months of observation (8.9% versus 32.9%, respectively), due to the lower rate of systemic adverse events.76 On the other hand, a recent meta-analysis77 investigated adherence rates to disease-modifying treatments in MS: data for GA showed slightly lower adherence rates compared to IFNβ-1a intramuscularly once per week, IFNβ1-b SC every other day, and IFNβ1-a SC three times per week (56.8%, 69.4%, 63.8%, and 58.4% respectively).
In conclusion, adherence to standard-regimen GA treatment (20 mg daily) may be challenging, mainly due to the higher number of injections and slightly lower adverse avents than other injected drugs. Reinforcing the importance to administer the treatment regularly is a key task for treating neurologists. Together with a good tolerability profile in the long term, newer and lower-frequency treatment regimens might improve patient adherence to therapy.
Conclusion and future
Decades after its approval, GA is still a well-established treatment option for RRMS and CIS. It is associated with good and sustained clinical efficacy, with approximately 30% reduction of the ARR, which is comparable to high-dose IFNs, and an optimal safety profile, even in the long term. All these data support its current use as a first-line treatment option for CIS and RRMS patients. GA could be electively considered as a treatment option particularly in some clinical conditions, such as polymorbid and/or polytreated patients, as it has no relevant drug-to-drug interactions, female patients planning a pregnancy (FDA pregnancy category B), and patients suffering from relevant fatigue, which can potentially be worsened by IFNβ side effects.
The main barrier to GA treatment is daily injections and consequent ISRs, with the risk of reduced adherence and therefore efficacy. More recent data have shown that high-dose GA (ie, 40 mg) given three times weekly is safe and well tolerated. Provided that similar efficacy between the two is reported, this new alternative administration regimen might largely substitute the standard one, with net advantage in terms of tolerability.
Acknowledgment
We thank Liliane Petrini (PhD) for writing assistance.
Author contributions
MC contributed to the drafting of the manuscript. GD contributed to the drafting of the manuscript and the preparation of the table. All authors made substantial contributions, critically revised the manuscript, and approve the final version.
Disclosure
CG has received personal compensation from Teva, Merck Serono, Biogen Idec, Bayer Schering, Novartis, and Genzyme. CZ has received personal compensation from Teva, Merck Serono, Biogen Idec, Bayer Schering, Novartis, and Genzyme. The other authors report no conflicts of interest in this work.
References
Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372(9648): 1502–1517. | ||
World Health Organization. Atlas: Multiple Sclerosis Resources in the World 2008. Geneva: WHO; 2008. Available from: http://whqlibdoc.who.int/publications/2008/9789241563758_eng.pdf. Accessed May 12, 2014. | ||
Ellwardt E, Zipp F. Molecular mechanisms linking neuroinflammation and neurodegeneration in MS. Exp Neurol. Epub 2014 Feb 14. | ||
Nylander A, Hafler DA. Multiple sclerosis. J Clin Invest. 2012;122(4): 1180–1188. | ||
Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. N Engl J Med. 2000;343(13):938–952. | ||
Stüve O, Oksenberg J. Multiple sclerosis overview. 2010. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20301492. Accessed July 3, 2014. | ||
Scott LJ. Glatiramer acetate: a review of its use in patients with relapsing–remitting multiple sclerosis and in delaying the onset of clinically definite multiple sclerosis. CNS Drugs. 2013;27(11):971–988. | ||
Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69(2):292–302. | ||
Marta M, Giovannoni G. Disease modifying drugs in multiple sclerosis: mechanisms of action and new drugs in the horizon. CNS Neurol Disord Drug Targets. 2012;11(5):610–623. | ||
Halpern R, Agarwal S, Dembek C, Borton L, Lopez-Bresnahan M. Comparison of adherence and persistence among multiple sclerosis patients treated with disease-modifying therapies: a retrospective administrative claims analysis. Patient Prefer Adherence. 2011;5:73–84. | ||
Polman CH, O’Connor PW, Havrdova E, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354(9):899–910. | ||
Kappos L, Radue EW, O’Connor P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010; 362(5):387–401. | ||
O’Connor P, Wolinsky JS, Confavreux C, et al. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med. 2011; 365(14):1293–1303. | ||
Gold R, Kappos L, Arnold DL, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. 2012;367(12):1098–1107. | ||
Cohen JA, Coles AJ, Arnold DL, et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing–remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet. 2012; 380(9856):1819–1828. | ||
Wingerchuk DM, Carter JL. Multiple sclerosis: current and emerging disease-modifying therapies and treatment strategies. Mayo Clin Proc. 2014;89(2):225–240. | ||
Johnson KP. Risks vs benefits of glatiramer acetate: a changing perspective as new therapies emerge for multiple sclerosis. Ther Clin Risk Manag. 2010;6:153–172. | ||
Lalive PH, Neuhaus O, Benkhoucha M, et al. Glatiramer acetate in the treatment of multiple sclerosis: emerging concepts regarding its mechanism of action. CNS Drugs. 2011;25(5):401–414. | ||
Jalilian B, Einarsson HB, Vorup-Jensen T. Glatiramer acetate in treatment of multiple sclerosis: a toolbox of random co-polymers for targeting inflammatory mechanisms of both the innate and adaptive immune system? Int J Mol Sci. 2012;13(11):14579–14605. | ||
Blanchette F, Neuhaus O. Glatiramer acetate: evidence for a dual mechanism of action. J Neurol. 2008;255 Suppl 1:26–36. | ||
Arnon R, Aharoni R. Neurogenesis and neuroprotection in the CNS-fundamental elements in the effect of glatiramer acetate on treatment of autoimmune neurological disorders. Mol Neurobiol. 2007; 36(3):245–253. | ||
Teitelbaum D, Fridkis-Hareli M, Arnon R, Sela M. Copolymer 1 inhibits chronic relapsing experimental allergic encephalomyelitis induced by proteolipid protein (PLP) peptides in mice and interferes with PLP-specific T cell responses. J Neuroimmunol. 1996;64(2):209–217. | ||
Comi G, Martinelli V, Rodegher M, et al. Effect of glatiramer acetate on conversion to clinically definite multiple sclerosis in patients with clinically isolated syndrome (PreCISe study): a randomised, double-blind, placebo-controlled trial. Lancet. 2009;374(9700):1503–1511. | ||
Aharoni R. The mechanism of action of glatiramer acetate in multiple sclerosis and beyond. Autoimmun Rev. 2013;12(5):543–553. | ||
Carter NJ, Keating GM. Glatiramer acetate: a review of its use in relapsing–remitting multiple sclerosis and in delaying the onset of clinically definite multiple sclerosis. Drugs. 2010;70(12):1545–1577. | ||
Simpson D, Noble S, Perry C. Glatiramer acetate: a review of its use in relapsing–remitting multiple sclerosis. CNS Drugs. 2002;16(12):825–850. Review. | ||
Yong VW. Differential mechanisms of action of interferon-beta and glatiramer acetate in MS. Neurology. 2002;59(6):802–808. | ||
Chiarini M, Sottini A, Ghidini C, et al. Renewal of the T-cell compartment in multiple sclerosis patients treated with glatiramer acetate. Mult Scler. 2010;16(2):218–227. | ||
Ratchford JN, Endres CJ, Hammoud DA, et al. Decreased microglial activation in MS patients treated with glatiramer acetate. J Neurol. 2012;259(6):1199–1205. | ||
Sellebjerg F, Hesse D, Limborg S, et al. Dendritic cell, monocyte and T cell activation and response to glatiramer acetate in multiple sclerosis. Mult Scler. 2013;19(2):179–187. | ||
Pul R, Morbiducci F, Škuljec J, et al. Glatiramer acetate increases phagocytic activity of human monocytes in vitro and in multiple sclerosis patients. PLoS One. 2012;7(12):e51867. | ||
Burger D, Molnarfi N, Weber MS, et al. Glatiramer acetate increases IL-1 receptor antagonist but decreases T cell-induced IL-1β in human monocytes and multiple sclerosis. Proc Natl Acad Sci U S A. 2009;106(11):4355–4359. | ||
Stasiolek M, Bayas A, Kruse N, et al. Impaired maturation and altered regulatory function of plasmacytoid dendritic cells in multiple sclerosis. Brain. 2006;129(Pt 5):1293–1305. | ||
Høglund RA, Holmøy T, Harbo HF, Maghazachi AA. A one year follow-up study of natural killer and dendritic cells activities in multiple sclerosis patients receiving glatiramer acetate (GA). PLoS One. 2013; 8(4):e62237. | ||
Teva Neuroscience. Copaxone (glatiramer acetate) solution for subcutaneous injection [prescribing information]. Overland Park (KS): Teva Neuroscience; 2012. Available from: http://copaxone.com/pdfs/PrescribingInformation.aspx. Accessed May 12, 2014. | ||
Sellebjerg F, Hedegaard CJ, Krakauer M, et al. Glatiramer acetate antibodies, gene expression and disease activity in multiple sclerosis. Mult Scler. 2012;18(3):305–313. | ||
Teitelbaum D, Brenner T, Abramsky O, Aharoni R, Sela M, Arnon R. Antibodies to glatiramer acetate do not interfere with its biological functions and therapeutic efficacy. Mult Scler. 2003;9(6):592–599. | ||
Bornstein MB, Miller A, Slagle S, et al. A pilot trial of Cop 1 in exacerbating-remitting multiple sclerosis. N Engl J Med. 1987;317(7): 408–414. | ||
Johnson KP, Brooks BR, Cohen JA, et al. Copolymer 1 reduces relapse rate and improves disability in relapsing–remitting multiple sclerosis: results of a phase III multicenter, double-blind placebo-controlled trial. The Copolymer 1 Multiple Sclerosis Study Group. Neurology. 1995; 45(7):1268–1276. | ||
Johnson KP, Brooks BR, Cohen JA et al. Extended use of glatiramer acetate (Copaxone) is well tolerated and maintains its clinical effect on multiple sclerosis relapse rate and degree of disability. Copolymer 1 Multiple Sclerosis Study Group. Neurology. 1998;50(3):701–708. | ||
Johnson KP, Brooks BR, Ford CC, et al. Glatiramer acetate (Copaxone): comparison of continuous versus delayed therapy in a six-year organized multiple sclerosis trial. Mult Scler. 2003;9(6):585–591. | ||
Johnson KP, Ford CC, Lisak RP, Wolinsky JS. Glatiramer acetate (Copaxone): neurologic consequence of delaying glatiramer acetate therapy for multiple sclerosis: 8-year data. Acta Neurol Scand. 2005;111(1):42–47. | ||
Ford CC, Johnson KP, Lisak RP, et al. A prospective open-label study of glatiramer acetate: over a decade of continuous use in multiple sclerosis patients. Mult Scler. 2006;12(3):309–320. | ||
Ford C, Goodman AD, Johnson K, et al. Continuous long-term immunomodulatory therapy in relapsing multiple sclerosis: results from the 15-year analysis of the US prospective open-label study of glatiramer acetate. Mult Scler. 2010;16(3):342–350. | ||
Comi G, Filippi M, Wolinsky JS, et al. European/Canadian multicenter, double-blind, randomized, placebo-controlled study of the effects of glatiramer acetate on magnetic resonance imaging-measured disease activity and burden in patients with relapsing multiple sclerosis. Ann Neurol. 2001;49(3):290–297. | ||
Wolinsky JS, Comi G, Filippi M, et al. Copaxone’s effect on MRI-monitored disease in relapsing MS is reproducible and sustained. Neurology. 2002;59(8):1284–1286. | ||
Rovaris M, Comi G, Rocca MA, et al. Long-term follow-up of patients treated with glatiramer acetate: a multicentre, multinational extension of the European/Canadian double-blind, placebo-controlled, MRI-monitored trial. Mult Scler. 2007;13(4):502–508. | ||
Mikol DD, Barkhof F, Chang P, et al. Comparison of subcutaneous interferon beta-1a with glatiramer acetate in patients with relapsing multiple sclerosis (the REbif vs Glatiramer Acetate in Relapsing MS Disease [REGARD] study): a multicentre, randomised, parallel, open-label trial. Lancet Neurol. 2008;7(10):903–914. | ||
O’Connor P, Filippi M, Arnason B, et al. 250 microg or 500 microg interferon beta-1b versus 20 mg glatiramer acetate in relapsing–remitting multiple sclerosis: a prospective, randomised, multicentre study. Lancet Neurol. 2009;8(10):889–897. | ||
Cadavid D, Wolansky LJ, Skurnick J, et al. Efficacy of treatment of MS with IFNβ-1b or glatiramer acetate by monthly brain MRI in the BECOME study. Neurology. 2009;72(23):1976–1983. | ||
Fox RJ, Miller DH, Phillips JT, et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med. 2012;367(12):1087–1097. | ||
Filippi M, Rovaris M, Rocca MA, Sormani MP, Wolinsky JS, Comi G. Glatiramer acetate reduces the proportion of new MS lesions evolving into “black holes”. Neurology. 2001;57(4):731–733. | ||
Cadavid D, Cheriyan J, Skurnick J, Lincoln JA, Wolansky LJ, Cook SD. New acute and chronic black holes in patients with multiple sclerosis randomised to interferon beta-1b or glatiramer acetate. J Neurol Neurosurg Psychiatry. 2009;80(12):1337–1343. | ||
Filippi M, Rocca MA, Camesasca F, et al. Interferon β-1b and glatiramer acetate effects on permanent black hole evolution. Neurology. 2011;76(14):1222–1228. | ||
Ge Y, Grossman RI, Udupa JK, et al. Glatiramer acetate (Copaxone) treatment in relapsing–remitting MS: quantitative MR assessment. Neurology. 2000;54(4):813–817. | ||
Arnold DL, Narayanan S, Antel S. Neuroprotection with glatiramer acetate: evidence from the PreCISe trial. J Neurol. 2013;260(7): 1901–1906. | ||
Teitelbaum D, Arnon R, Sela M. Immunomodulation of experimental autoimmune encephalomyelitis by oral administration of copolymer 1. Proc Natl Acad Sci U S A. 1999;96(7):3842–3847. | ||
Cohen JA, Rovaris M, Goodman AD, Ladkani D, Wynn D, Filippi M. Randomized, double-blind, dose-comparison study of glatiramer acetate in relapsing–remitting MS. Neurology. 2007;68(12):939–944. | ||
Comi G, Cohen JA, Arnold DL, Wynn D, Filippi M. Phase III dose-comparison study of glatiramer acetate for multiple sclerosis. Ann Neurol. 2011;69(1):75–82. | ||
Khan O, Caon C, Zak I, et al. Randomized, prospective, rater-blinded, four-year, pilot study to compare the effect of daily versus every-other-day glatiramer acetate 20 mg subcutaneous injections in relapsing–remitting multiple sclerosis. Mult Scler. 2008;14 Suppl 1:S296. | ||
Khan O, Perumal J, Caon C, et al. Glatiramer acetate 20 mg subcutaneous twice-weekly versus daily injections: results of a pilot, prospective, randomized, and rater-blinded clinical and MRI 2-year study in relapsing–remitting multiple sclerosis. Mult Scler. 2009;15 Suppl 2:S249–S250. | ||
Khan O, Rieckmann P, Boyko A, Selmaj K, Zivadinov R. Three times weekly glatiramer acetate in relapsing–remitting multiple sclerosis. Ann Neurol. 2013;73(6):705–713. | ||
Boster A, Bartoszek MP, O’Connell C, Pitt D, Racke M. Efficacy, safety, and cost-effectiveness of glatiramer acetate in the treatment of relapsing–remitting multiple sclerosis. Ther Adv Neurol Disord. 2011; 4(5):319–332. | ||
Jolly H, Simpson K, Bishop B, et al. Impact of warm compresses on local injection-site reactions with self-administered glatiramer acetate. J Neurosci Nurs. 2008;40(4):232–239. | ||
Teva Pharmaceuticals. Copaxone [package insert]. Kfar Saba, Israel: Teva Pharmaceuticals; 2007. | ||
Teva Neuroscience. Copaxone [package insert]. Kansas City (MO): Teva Neuroscience; 2009. | ||
Houtchens MK, Kolb CM. Multiple sclerosis and pregnancy: therapeutic considerations. J Neurol. 2013;260(5):1202–1214. | ||
Coyle P, Johnson K, Pardo L. Pregnancy outcomes in patients with multiple sclerosis treated with glatiramer acetate (Copaxone). Poster presented at: 19th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS); September 17–20, 2003; Milan, Italy. | ||
Ventura SJ, Curtin SC, Abma JC, Henshaw SK. Estimated pregnancy rates and rates of pregnancy outcomes for the United States, 1990–2008. Nat Vital Stat Rep. 2012;60(7):1–21. | ||
Fragoso YD, Finkelsztejn A, Kaimen-Maciel DR, et al. Long-term use of glatiramer acetate by 11 pregnant women with multiple sclerosis: a retrospective, multicentre case series. CNS Drugs. 2010;24(11):969–976. | ||
Salminen HJ, Leggett H, Boggild M. Glatiramer acetate exposure in pregnancy: preliminary safety and birth outcomes. J Neurol. 2010; 257(12):2020–2023. | ||
Hellwig K, Gold R. Glatiramer acetate and interferon-beta throughout gestation and postpartum in women with multiple sclerosis. J Neurol. 2011;258(3):502–503. | ||
Hale TW. Medications and Mothers’ Milk. 14th ed. Plano (TX): Hale; 2010. | ||
Costello K, Kennedy P, Scanzillo J. Recognizing nonadherence in patients with multiple sclerosis and maintaining treatment adherence in the long term. Medscape J Med. 2008;10(9):225. | ||
Devonshire V, Lapierre Y, Macdonell R, et al. The Global Adherence Project (GAP) – a multicenter observational study on adherence to disease-modifying therapies in patients suffering from relapsing–remitting multiple sclerosis. Poster presented at: 22nd Congress of the European Committee for Treatment and Research in Multiple Sclerosis; September 27–30, 2006; Madrid, Spain. | ||
Haas J, Firzlaff M. Twenty-four-month comparison of immunomodulatory treatments – a retrospective open label study in 308 RRMS patients treated with beta interferons or glatiramer acetate (Copaxone). Eur J Neurol. 2005;12(6):425–431. | ||
Menzin J, Caon C, Nichols C, White LA, Friedman M, Pill MW. Narrative review of the literature on adherence to disease-modifying therapies among patients with multiple sclerosis. J Manag Care Pharm. 2013,19(1 Suppl A):S24–S40. | ||
Schrempf W, Ziemssen T. Glatiramer acetate: mechanisms of action in multiple sclerosis. Autoimmun Rev. 2007;6(7):469–475. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.