Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Tumor-targeting, pH-sensitive nanoparticles for docetaxel delivery to drug-resistant cancer cells
Authors Tran TH, Ramasamy T, Choi JY, Nguyen H ![]() , Pham T, Jeong J, Ku SK, Choi H, Yong CS, Kim JO
, Pham T, Jeong J, Ku SK, Choi H, Yong CS, Kim JO
Received 30 May 2015
Accepted for publication 8 July 2015
Published 21 August 2015 Volume 2015:10(1) Pages 5249—5262
DOI https://doi.org/10.2147/IJN.S89584
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Tuan Hiep Tran,1 Thiruganesh Ramasamy,1 Ju Yeon Choi,1 Hanh Thuy Nguyen,1 Thanh Tung Pham,1 Jee-Heon Jeong,1 Sae Kwang Ku,2 Han-Gon Choi,3 Chul Soon Yong,1 Jong Oh Kim1
1College of Pharmacy, Yeungnam University, Dae-Dong, 2College of Korean Medicine, Daegu Haany University, Gyeongsan, 3College of Pharmacy, Hanyang University, Hanyangdaehak-ro, Sangnok-gu, Ansan, South Korea
Abstract: The attachment of polyethylene glycol (PEG) increases the circulation time of drug-containing nanoparticles; however, this also negatively affects cellular uptake. To overcome this problem, unique lipid polymer hybrid (LPH) nanoparticles were developed with a pH-responsive PEG layer that detached prior to cell uptake. Docetaxel (DTX) was incorporated into the lipid core of the nanoparticles, which was then shielded with the pH-responsive block co-polymer polyethylene glycol-b-polyaspartic acid (PEG-b-PAsp) using a modified emulsion method. The optimized LPH nanoparticles were ~200 nm and had a narrow size distribution. Drug release from DTX-loaded LPH (DTX-LPH) nanoparticles was pH-sensitive, which is beneficial for tumor targeting. More importantly, DTX-LPH nanoparticles were able to effectively induce apoptosis in cancer cells. The negative surface charge and PEG shell of vehicle remarkably enhanced the blood circulation and physiological activity of DTX-LPH nanoparticles compared with that of free DTX. The nanoparticles were also found to reduce the size of tumors in tumor-bearing xenograft mice. The in vivo anticancer effect of DTX-LPH nanoparticles was further confirmed by the elevated levels of caspase-3 and poly ADP ribose polymerase found in the tumors after treatment. Thus, the results suggest that this novel LPH system could be an effective new treatment for cancer.
Keywords: docetaxel, polyaspartic acid, drug delivery systems, antitumor, pH-sensitive
Introduction
Docetaxel (DTX) is an antimitotic chemotherapy drug that promotes the assembly of tubulins into microtubulins, stabilizes microtubules, and thus inhibits cell proliferation. DTX has been approved as a first-line drug for the treatment of several cancers including those of the breast, lung, and prostate.1 The clinical application of DTX is hampered by severe side effects such as neutropenia, peripheral neuropathy, musculoskeletal toxicity, and hypersensitivity.2 DTX therapy has also been impeded by multidrug resistance (MDR), wherein cancer cells develop resistance against chemotherapeutic agents.3,4 The development of alternative, effective therapeutic approaches that overcome drug-related side effects and MDR is a high priority.
Nanoparticle-based drug delivery systems have great potential to overcome the adverse effects associated with anticancer drugs. Sufficiently small nanoparticles can take advantage of the enhanced permeability and retention effect to target cancer cells, and the attachment of polyethylene glycol (PEG) chains to the surface of nanoparticles can improve their systemic longevity.5 PEG is effective because it prevents the nanoparticles from being hindered by interactions with biomolecules; however, PEG also interferes with the interactions between nanoparticles and target cells. Therefore, additional carrier functionalities are required to overcome the fundamental limitations of PEGylated nanoparticle systems.6 Cationically charged nanoparticles interact with the anionic plasma membranes of cells and are rapidly internalized;7 however, unlike charge-neutral PEGylated nanoparticles, cationic particles are quickly adsorbed by systemic serum proteins, which “tags” them for removal by the mononuclear phagocyte system inside the liver and spleen.8 Therefore, we propose a new surface modification strategy, where the cationic surface is shielded by PEG while circulating before undergoing detachment prior to cellular uptake.
In the present study, we use pH to trigger detachment of the PEG chains from the nanoparticle. The pH in tumor tissues is far more acidic (pH =5–6) than the wider physiological environment (pH =7.4). Therefore, we prepared unique lipid polymer hybrid (LPH) nanoparticles with a cationic surface covered with a thin layer of polyethylene glycol-b-polyaspartic acid (PEG-b-PAsp). At pH 7.4, the aspartic acid moiety of the block copolymer will be negatively charged and therefore attracted to the cationic nanoparticle. However, in the tumor microenvironment (pH =5–6), the aspartic acid moiety will be neutrally charged and therefore detach from the nanoparticles and expose the positively charged lipid core. The cationic nature of the nanoparticles is expected to increase their cellular uptake and thus the intracellular concentration of DTX. To further improve the ability of the particles to overcome MDR, d-α-tocopheryl polyethylene glycol succinate (vitamin E-TPGS or simply TPGS) was also included in the nanoparticles. TPGS is expected to inhibit the ATPase section of P-glycoprotein (P-gp) and thereby overcome MDR. The pH-responsive property of the nanoparticles was confirmed by measuring zeta potentials and drug release at different pH values. The high efficacy of the DTX-loaded LPH (DTX-LPH) nanoparticles was demonstrated using in vitro assays and in vivo experiments on tumor-bearing xenograft mice.
Materials and methods
DTX was purchased from LC laboratories (Woburn, MA, USA). PEG-b-poly aspartic acid (PEG113-b-PAsp10; Mw =6,400, polydispersity index =1.20) was purchased from Alamanda Polymers, Inc. (Huntsville, AL, USA). Capryol 90 was procured from Gattefossé (Lyon, France). Didecyldimethylammonium bromide (DDAB) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) were obtained from Sigma-Aldrich Co. (St Louis, MO, USA). TPGS was supplied by Isochem (Vert-le-Petit, France). Coumarin 6 and LysoTracker Red® were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Squamous cell carcinoma (SCC-7), human breast adenocarcinoma (MCF-7), and human Caucasian breast adenocarcinoma (MDA-MB-231) cells were originally obtained from the Korean Cell Line Bank (Seoul, South Korea). All other chemicals were of reagent grade and used without further purification. The study protocols were approved by the Institutional Ethical Committee, Yeungnam University, South Korea, in accordance with national guidelines.
Preparation of DTX-LPH nanoparticles
DTX-LPH nanoparticles were prepared using a modified emulsification method. Briefly, the organic phase consisting of Capryol 90, TPGS, DDAB, and DTX was melted at 90°C to homogeneity. The aqueous phase containing PEG-b-PAsp at various concentrations was also heated to the same temperature and then mixed with the organic phase under constant homogenization (Ultra Turrax® T-25 homogenizer; IKA®-Werke, Staufen, Germany) for 3 minutes at 13,500 rpm. The coarse solution was sonicated using a high-intensity probe sonicator (Vibracell VCX130; Sonics, Newtown, CT, USA) at 90% amplitude for 5 minutes. The resulting suspension was then cooled in an ice bath. The free DTX was discarded by filtrating the obtained dispersion by using a centrifugal tube. The detailed composition of the various DTX-LPH formulations is shown in Table 1.
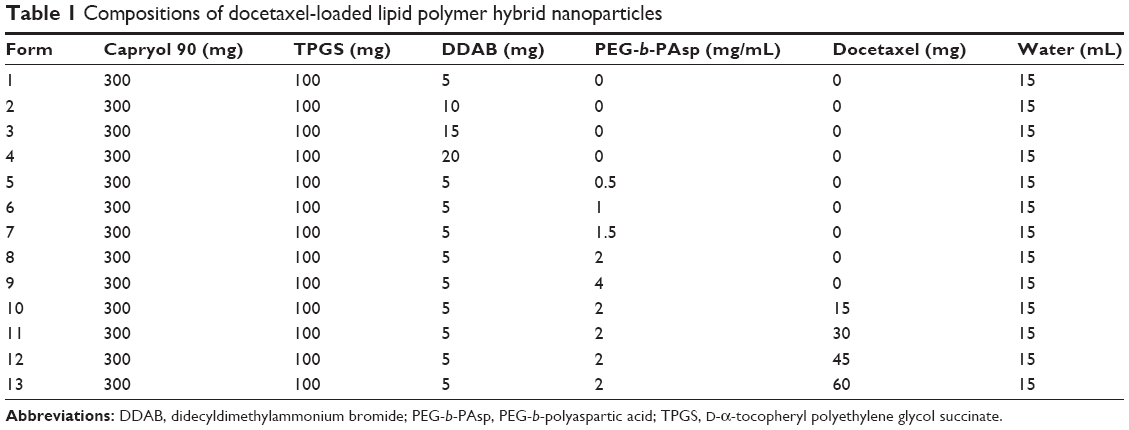 | Table 1 Compositions of docetaxel-loaded lipid polymer hybrid nanoparticles |
Freeze-drying process was carried out using mannitol (5%, w/v) as a cryoprotectant. The resulting nanoparticle dispersions were placed into glass bottles and prefrozen at −80°C for 24 hours. After this time, the samples were freeze-dried using a lyophilizer (FDA5518; Ilshin, South Korea) at a temperature of −25°C for 24 hours.
Physical characterization
Drug loading content
The drug loading efficiency (LE) and drug loading capacity (LC) of DTX-LPH nanoparticles were determined indirectly by measuring the amount of free drug in the dispersion medium using a Amicon centrifugal tube (molecular weight cut-off =10,000 Da; Merck Millipore, Billerica, MA, USA). In detail, 1 mL of the DTX-LPH solution was added into an Amicon centrifugal tube. After centrifugation at 5,000 rpm for 15 minutes, the concentration of unbound DTX in the filtrates was determined by high-performance liquid chromatography (HPLC). The LE% and LC% were calculated using the following equations:9
|
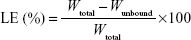
|
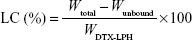
where Wtotal, Wunbound, and WDTX-LPH are the total amount of DTX added, unbound DTX, and DTX-LPH, respectively. The amount of DTX was quantified using HPLC (Hitachi Ltd., Tokyo, Japan) with a UV/Vis detector (Model L-2420) set at a wavelength of 232 nm. The column was an Inertsil C8 column (GL Science, 3.5 μm, 15 cm ×0.46 cm) with a flow rate of 1.0 mL/min, using a mobile phase consisting of acetonitrile and phosphate buffer (pH =3) at a volume ratio of 60/40.10
Measurement of size, size distribution, and zeta potential
The particle size, size distribution, and zeta potential of DTX-LPH nanoparticles were measured using dynamic light scattering (DLS; Zetasizer Nano ZS; Malvern Instruments, Malvern, UK). Measurements were conducted at a scattering angle of θ =90° to the incident beam at 25°C.11 Zeta potential was examined using the Smoluchowski equation. All measurements were determined using the manufacturer’s software (NanoDTS, version 6.34). The samples were diluted ten times using distilled water and homogenously dispersed before measurement. All experiments were performed at least three times.
The optimized nanoparticles were incubated in acidic conditions (acetate-buffered saline [ABS], pH =5.0) in a ratio of 1:10 to assess the effect of an acidic environment on zeta potential. After incubation for 1 hour and 2 hours at 37°C, samples were obtained and measured as mentioned earlier.
Morphological studies
The shape and surface morphology of DTX-LPH nanoparticles was observed using transmission electron microscopy (H-7100; Hitachi) at an accelerating voltage of 100 kV. Briefly, a drop of formulation was placed on a carbon-coated copper grid (300 mesh) and allowed to settle for 5 minutes. Excess dispersion was removed, and 2% phosphotungstic acid solution was added as a negative staining agent. The samples were air-dried under infrared radiation for 10 minutes. In addition, the shape and size of the DTX-LPH nanoparticles were examined using atomic force microscopy. The samples were scanned at a rate of 100 mV/s in the range 50 μm ×50 μm using Nanoscope IIIa (Veeco, Plainview, NY, USA).
Physical properties
The X-ray diffraction patterns of the samples were obtained with an X-ray diffractometer (X’Pert PRO MPD; PANalytical, Almelo, the Netherlands) using Cu Kα radiation with a voltage of 40 kV and a current of 30 mA with a scan step size of 0.02. The measurement was conducted thrice, and the representative pattern was shown.
In vitro drug release
The drug release profiles were characterized in different pH media (7.4 and 5.0) using the dialysis method. Briefly, a suspension of DTX-LPH nanoparticles (1 mL) was transferred to a dialysis bag (molecular weight cut-off =3.5 KDa). The dialysis bag was placed in phosphate-buffered saline (PBS) or ABS (35 mL) at 37°C inside a shaking water bath (HST–205 SW; Hanbaek ST Co., Seoul, South Korea) with continuous shaking at 100 rpm. At a predesignated time, the solution (0.5 mL) was removed and replaced with an equal volume of fresh buffer. The amount of DTX released into the solution was measured using HPLC as described in the section “Drug loading content.” Release experiments were performed in triplicates.
In vitro cell studies
In vitro cytotoxicity assay
SCC-7, MCF-7, and MDA-MB-231 cells were cultured at 37°C in 5% CO2 atmosphere in RPMI 1640 medium containing 10% fetal bovine serum, penicillin (100 U/mL), and streptomycin (0.1 mg/mL). The in vitro cytotoxicity was evaluated as reported previously.12,13 Briefly, the cells were seeded at a density of 1×104 cells/well in 96-well culture plates. After 24 hours of incubation to allow cell adhesion, the cells were treated with free DTX, DTX-LPH, or blank LPH at concentrations ranging from 0.1 μg/mL to 50 μg/mL followed by incubation for an additional 24 hours. Untreated cells were used as a control. After 24 hours and 48 hours incubation, MTT solution (100 μL; 1.25 mg/mL) was added to each of the 96 wells, followed by incubation in the dark for 3–4 hours. Finally, DMSO was added to dissolve the formazan crystals, and the absorbance was read at 570 nm using a microplate reader (Multiskan EX; Thermo Fisher Scientific). Cell viability was calculated using the following formula (OD, optical density):
|
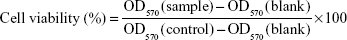
Flow cytometry
All flow cytometry analyses were carried out using a FACSVerse flow cytometer (BD Biosciences, San Jose, CA, USA). For cellular uptake study, cells were seeded in six-well plates with a density of 2×105 cells/well in RPMI 1640 (2.0 mL), incubated for 24 hours, and then treated with C6-LPH at a concentration of 2 μg/mL for 45 minutes and 90 minutes. Untreated cells were used as a control. The harvested cells were suspended in PBS and analyzed using flow cytometry.14 An Annexin V FITC kit (BD Biosciences) was used to measure apoptosis.15 SCC-7 and MCF-7 cells were treated with fresh DMEM, free DTX, and DTX-LPH at an equivalent concentration of 10 μg/mL, and MDA-MB-231 cells were treated with a concentration of 25 μg/mL for 24 hours; thereafter, apoptosis was measured according to the manufacturer’s instructions.
Confocal laser scanning microscopy
Cells were seeded on coverslips in 12-well plates at a density of 1×105 cells/well. For cellular uptake studies, cells were stained with Coumarin 6-labeled DTX-LPH and incubated for 45 minutes and 90 minutes. LysoTracker Red was then added, the cells were fixed, and then observed under a fluorescence microscope. The nuclear morphologies of the cells after different treatments were observed using Hoechst 33342 staining. Cells were treated with DTX or DTX-LPH nanoparticles at a concentration of 10 μg/mL. After 24 hours, cells were fixed and stained with Hoechst 33342 (10 μg/mL) for 8 minutes. Nuclei morphology was observed using a laser scanning confocal microscope (TCS SP2; Leica Microsystems, Wetzlar, Germany).2,16
Western blotting analysis
Cells were seeded and incubated in a 12-well plate for 24 hours in an automated incubator. Cells were treated with free DTX and DTX-LPH and further incubated for 24 hours. After harvesting, cells were lysed, treated with proteinase inhibitors, and stored for 40 minutes in ice. After centrifugation at 13,000 rpm for 20 minutes at 4°C, the supernatant was collected and the protein concentration was determined using a BCA Protein Assay Kit. The total protein was separated on a 12% Bis-Tris polyacrylamide gel (at 150 mA for 120 minutes) and transferred to polyvinylidene fluoride membranes. Following incubation with 5% nonfat milk powder in PBS buffer containing Tween-20 (PBST; pH 7.2), membranes were incubated in 5% nonfat milk powder in PBST with p-53, p-27, and p-21 antibody (Santa Cruz Biotechnology, Inc., Dallas, TX, USA) overnight. Samples were subsequently incubated with the appropriate secondary antibody for 1 hour and visualized using enhanced chemiluminescence. The gels were photographed, and UVP gel analysis was used to evaluate protein levels.17
In vivo pharmacokinetic study
The pharmacokinetic study was carried out in male Sprague Dawley rats weighing 250±10 g. The animals were quarantined in an animal house maintained at 25°C±2°C and 50%–60% relative humidity and were fasted for 12 hours prior to the experiments. The study protocols were approved by the Institutional Animal Ethical Committee, Yeungnam University, South Korea, in accordance with national guidelines.
The rats were divided into two groups with four rats in each group. Two groups of rats received free DTX or DTX-LPH by intravenous (IV) administration. For IV injection of free DTX, DTX was dissolved in PEG200/water (50:50) and administered at a dose of 10 mg/kg. Lyophilized DTX-LPH was re-dispersed in physiological saline at an appropriate concentration before injection. Blood samples (300 μL) were collected from the right femoral artery at predetermined times (0.25 hour, 0.5 hour, 1 hour, 2 hours, 3 hours, 4 hours, 6 hours, 8 hours, 12 hours, and 24 hours) after administration of the treatment. The samples were collected in heparin-containing tubes (100 IU/mL) and then immediately centrifuged (Eppendorf, Hauppauge, NY, USA) at 14,000 rpm for 10 minutes. The plasma supernatant was collected and stored at -20°C until further analysis.18 For extraction of DTX and for precipitation of unwanted protein, plasma (150 μL) was mixed with acetonitrile (150 μL) for 15 minutes. The samples were then centrifuged at 13,000 rpm for 10 minutes, and supernatant (20 μL) was injected into the HPLC system for DTX measurement, as described in the section “Drug loading content.”
The pharmacokinetic profiles of free DTX and DTX-LPH were calculated using the Win-NonLin pharmacokinetic software (v4.0, Pharsight Corporation, St Louis, MO, USA). The area under the curve of plasma drug concentration against time from time zero to infinity (AUC0–∞) and the half-life of elimination (t1/2) were both measured. Mean residence time (MRT) was obtained by summation of the central and tissue compartments.
Establishment of in vivo antitumor activity
In vivo antitumor effect
The in vivo antitumor efficacy of DTX-LPH was evaluated in SCC-7 cell-bearing Balb/c nude mice. SCC-7 cells (2×106) were injected subcutaneously into the right flanks of nude mice resulting in cancer xenografts. When the tumor volumes reached approximately 100 mm3, the antitumor study was started. Mice were randomly divided into three groups (n=7): group 1 for PBS (control), group 2 for DTX suspension, and group 3 for DTX-LPH (10 mg/kg based on DTX concentration). Treatments were administered once every 3 days for a total of three administrations. Tumor volume and mouse weight were measured at the scheduled time points. Tumor volume was calculated using the equation V(L × W2)/2, where L represents the longest diameter, and W represents the shortest diameter perpendicular to length. At the end of the experiment, the animals were sacrificed and the tumor masses harvested, weighed, photographed, and stored in formalin 10% for further studies.19 The protocols for the animal studies were approved by the Institutional Animal Ethical Committee, Yeungnam University, South Korea.
Histological characterization
SCC7 tumor cell xenograft masses were separated from surrounding tissues and fixed in 10% neutral buffered formalin, crossly trimmed, and embedded in paraffin. After this, samples were serially sectioned (3–4 μm), stained with hematoxylin and eosin (H&E), and then the histopathological profiles of each sample were observed under an optical microscope (Nikkon Corporation, Tokyo, Japan). Additionally, the tumor cell volumes and the intact tumor cell occupied regions (%/mm2 of tumor mass) were calculated using a computer-based automated image analyzer (iSolution FL ver 9.1; IMT i-solution Inc., Vancouver, Quebec, Canada).
Immunohistochemistry
Changes in the immunoreactivity against the apoptotic markers, caspase-3, and poly ADP ribose polymerase (PARP) were observed in tumor masses using purified primary antibodies with an avidin–biotin–peroxidase complex (ABC) and peroxidase substrate kit (Vector Labs, Burlingame, CA, USA).20,21 Briefly, endogenous peroxidase activity was blocked by incubating in methanol and 0.3% H2O2 for 30 minutes, and nonspecific binding of immunoglobulin was blocked with normal horse serum blocking solution for 1 hour in humidity chamber after heat-induced (95°C–100°C) epitope retrieval in 10 mM citrate buffer (pH =6.0).22 Primary antisera were treated overnight at 4°C in a humidity chamber and then incubated with biotinylated universal secondary antibody and ABC reagents for 1 hour at room temperature in a humidity chamber. Finally, sections were made to react with a peroxidase substrate kit for 3 minutes at room temperature. All sections were rinsed three times in 0.01 M PBS between each step. Samples were regarded as testing positive for apoptosis if covered by 20% or more of each marker of apoptosis (caspase-3 and PARP). The percentage regions occupied by caspase-3- and PARP-positive cells located in tumor mass were measured by automated image analyzer (%/mm2 of tumor mass).
Statistical analysis
Statistical analysis was performed using Student’s t-test for pairs of groups and one-way ANOVA for multiple groups. All results were expressed as the mean ± standard deviation of the mean. A value of P<0.05 was considered to be significant.
Results and discussion
Preparation of DTX-LPH nanoparticles
The DTX-LPH nanoparticles were prepared using the emulsification method and subsequently coated with polymer. Figure 1 shows the parameters for the various formulations. We coated the positively charged nanoparticles with PEG-b-PAsp in order to prolong plasma circulation and minimize carrier-mediated toxicity. The deposition of the polyelectrolyte layer on the LPH surface was confirmed by zeta potential experiments that showed charge reversal from highly positive (+41.4±1.3 mV) to negative (-22.6±0.3 mV) values when the polymer layer was added. The optimized DTX-LPH formulation consisted of Capryol 90 (300 mg), TPGS (100 mg), DDAB (5 mg), and DTX (30 mg). This formulation had a particle size of 199.2±15.9 nm with a narrow size distribution (PD =0.277±0.031). DTX-LPH showed high entrapment efficiency (91.4%) as well as LC (6.9%), indicating suitability for therapeutic applications. As shown in Figure 1, when added to acidic medium, the negative charge gradually became positive due to the protonation behavior of the aspartic acid residues of PEG-b-PAsp.
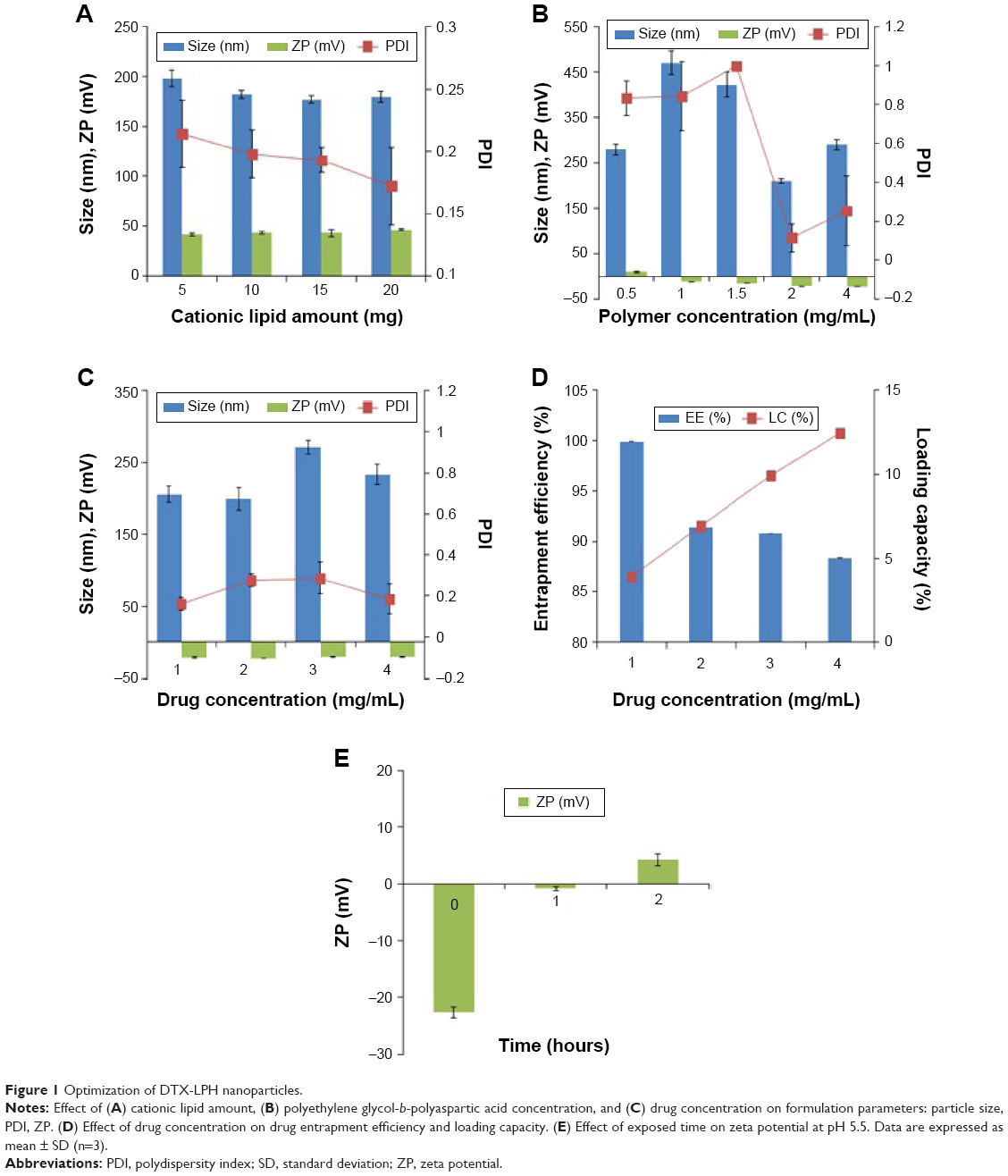 | Figure 1 Optimization of DTX-LPH nanoparticles. |
The morphology of DTX-LPH is shown in Figure 2A and B. Transmission electron microscopy images revealed a nanosized, spherical-shaped, compact core-shell structure with a size consistent with the DLS data. A similar result was observed using atomic force microscopy, which also provided the 3D shape of the nanoparticles. X-ray diffraction analysis was performed to evaluate the physical status of drug and formulation (Figure 2C). The diffractogram for free DTX showed numerous sharp, intense peaks reflecting its high crystallinity. The lack of a characteristic peak in blank LPH and DTX-LPH indicates that the drug is in the amorphous form or molecularly dispersed after encapsulation.10
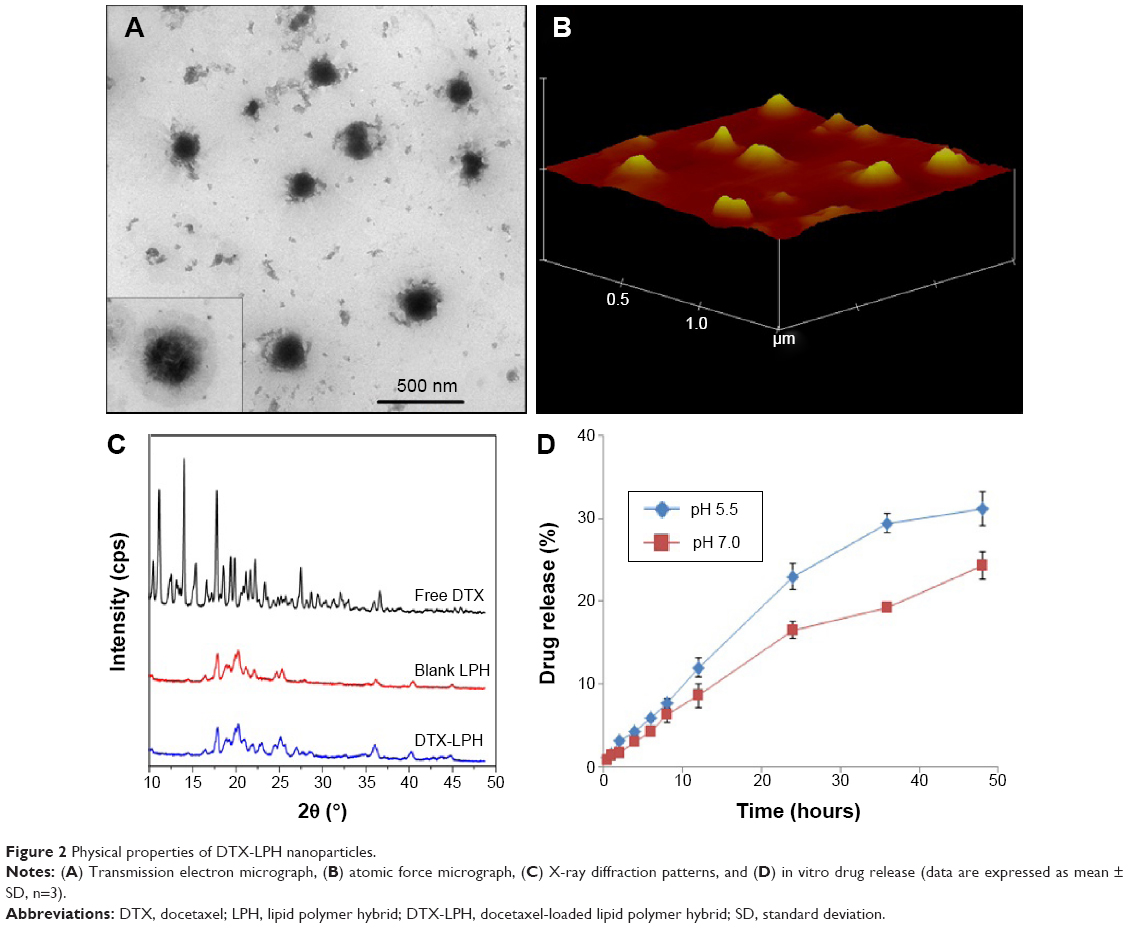 | Figure 2 Physical properties of DTX-LPH nanoparticles. |
The release profiles of DTX from DTX-LPH were evaluated in PBS (pH =7.4) and ABS (pH =5.0) (Figure 2D). In both release media, DTX-LPH exhibited a biphasic release pattern, with 20% of the drug released within the first 24 hours, followed by a sustained release pattern over the next 24 hours. As expected, the drug release was higher at pH 5.0 than at pH 7.4. The enhanced drug release at acidic pH conditions might be attributed to the pH-responsive nature of PEG-b-PAsp polymers. This phenomenon is beneficial for drug delivery because the acidic extracellular and intracellular environments of tumors would be expected to accelerate drug release.23
In vitro cell studies
To ascertain the potential of nanoparticles for intracellular drug delivery, Coumarin 6-loaded LPH (C6-LPH) was exposed to three different cells (SCC-7, MCF-7, and MDA-MB-231). The time-dependent cellular uptake of nanoparticles was analyzed using (fluorescence-activated cell sorting [FACS] and CLSM (Figure 3). In SCC-7 and MCF-7 cells, C6-LPH clearly underwent time-dependent cellular uptake as the fluorescence intensity was significantly higher after 90 minutes of incubation than after 45 minutes (Figure 3B). In addition, confocal microscopy was used to examine the mechanism of cellular uptake. LPH penetrated into cells mainly via endocytosis, which led to the delivery of the transported cargo to lysosomes.24 After 45 minutes of incubation, only a small number of complexes entered cells (less fluorescence intensity) and proceeded into lysosomes, but no complexes entered the nuclei (data not shown). However, a dramatic increase in cellular uptake was observed after 90 minutes of incubation as evidenced by an increase in fluorescence intensity (Figure 3A). The high cellular uptake of LPH in all cancer cells was attributed to the electrostatic interactions between the positively charged nanoparticles and the negatively charged cellular membrane. Generally, the tumor extracellular environment is slightly acidic (pH =6.8), and the pH of the bloodstream is ~7.4. Therefore, the surface charge of LPH will change to positive in the tumor extracellular fluid, which will enhance the uptake of the nanoparticles in the cell. In addition, the nanometric size of the particles (~200 nm) might contribute to their cellular internalization. From the result, it can be expected that negatively charged LPH will minimize the undesirable systemic side effects and facilitate the accumulation of the nanoparticles in the tumors. A similar pH influence on zeta potential has been reported previously in the literature.25–27 As a result, the use of LPH nanoparticles may dramatically enhance the concentration of the drug in cells, thereby improving drug cytotoxicity and more effectively overcoming MDR in cancer cells (Figure 4). In all cell lines studied, DTX exhibited a time- and dose-dependent cytotoxic effect. As expected, DTX-LPH nanoparticles were also more effective than free DTX in killing tumor cells, especially in drug-resistant MDA-MB-231 cells. Interestingly, in the case of MDA-MB-231 cells, free DTX did not induce appreciable cytotoxicity at doses up to 50 μg/mL even after 48 hours of incubation. The limited cytotoxicity of free DTX could be attributed to P-glycoprotein–mediated efflux pumps, which can dramatically reduce the intracellular drug concentration and thus limit the cytotoxicity of free drugs in tumors. In the case of DTX-LPH, enhanced cellular DTX retention, intracellular acid-responsive drug release, and minimization of drug loss due to efflux resulted in enhanced cytotoxicity.28 Blank LPH did not cause any significant cytotoxicity in any cell lines across all concentrations tested (up to 50 μg/mL).29 DTX disrupts microtubule formation resulting in cell death via the apoptosis pathway.30 Therefore, the cytotoxic effect of different formulations was further confirmed by Hoechst staining. As shown in Figure 5A, chromatin condensation and apoptotic body formation of nuclei were clearly evident in the DTX and DTX-LPH treatment group, while control cells were homogenous and smooth. These data further suggested that DTX-LPH more effectively induced apoptosis than free DTX.
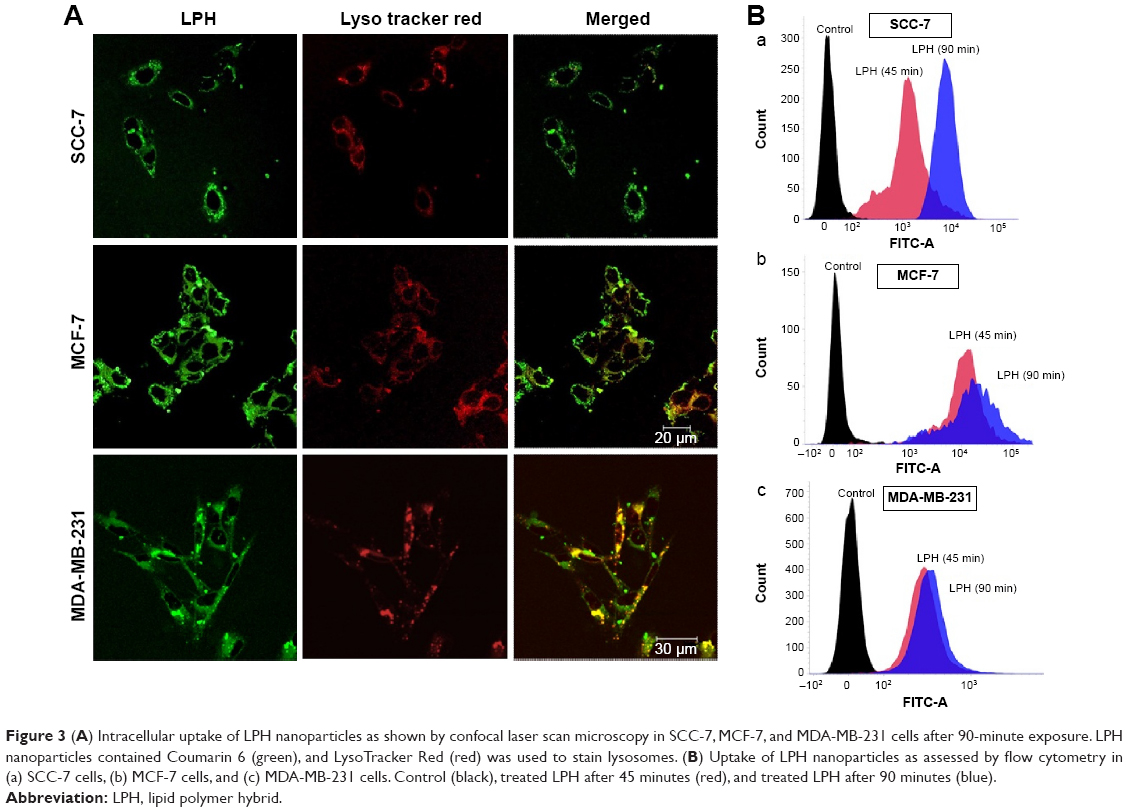 | Figure 3 (A) Intracellular uptake of LPH nanoparticles as shown by confocal laser scan microscopy in SCC-7, MCF-7, and MDA-MB-231 cells after 90-minute exposure. LPH nanoparticles contained Coumarin 6 (green), and LysoTracker Red (red) was used to stain lysosomes. (B) Uptake of LPH nanoparticles as assessed by flow cytometry in (a) SCC-7 cells, (b) MCF-7 cells, and (c) MDA-MB-231 cells. Control (black), treated LPH after 45 minutes (red), and treated LPH after 90 minutes (blue). |
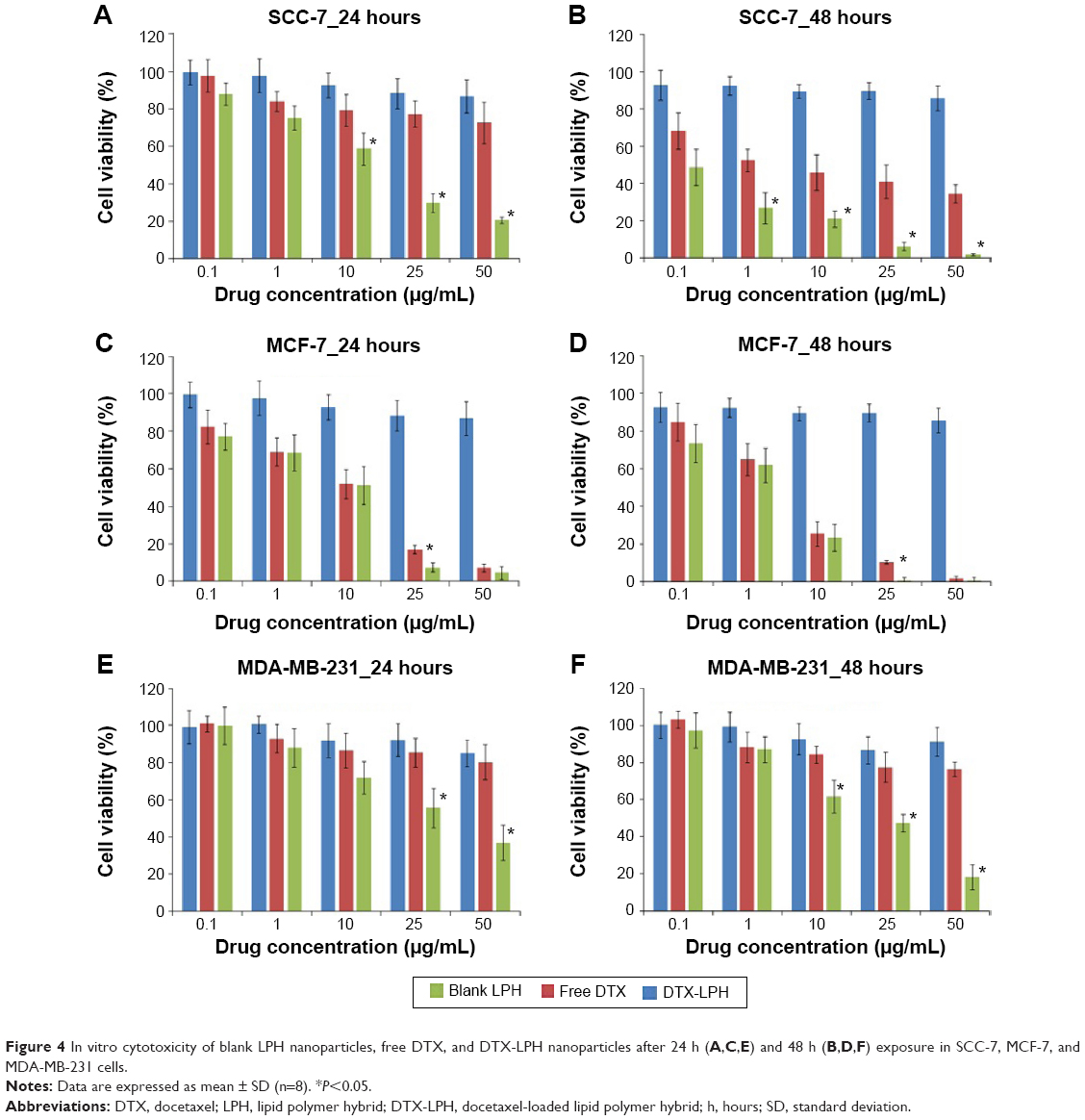 | Figure 4 In vitro cytotoxicity of blank LPH nanoparticles, free DTX, and DTX-LPH nanoparticles after 24 h (A,C,E) and 48 h (B,D,F) exposure in SCC-7, MCF-7, and MDA-MB-231 cells. |
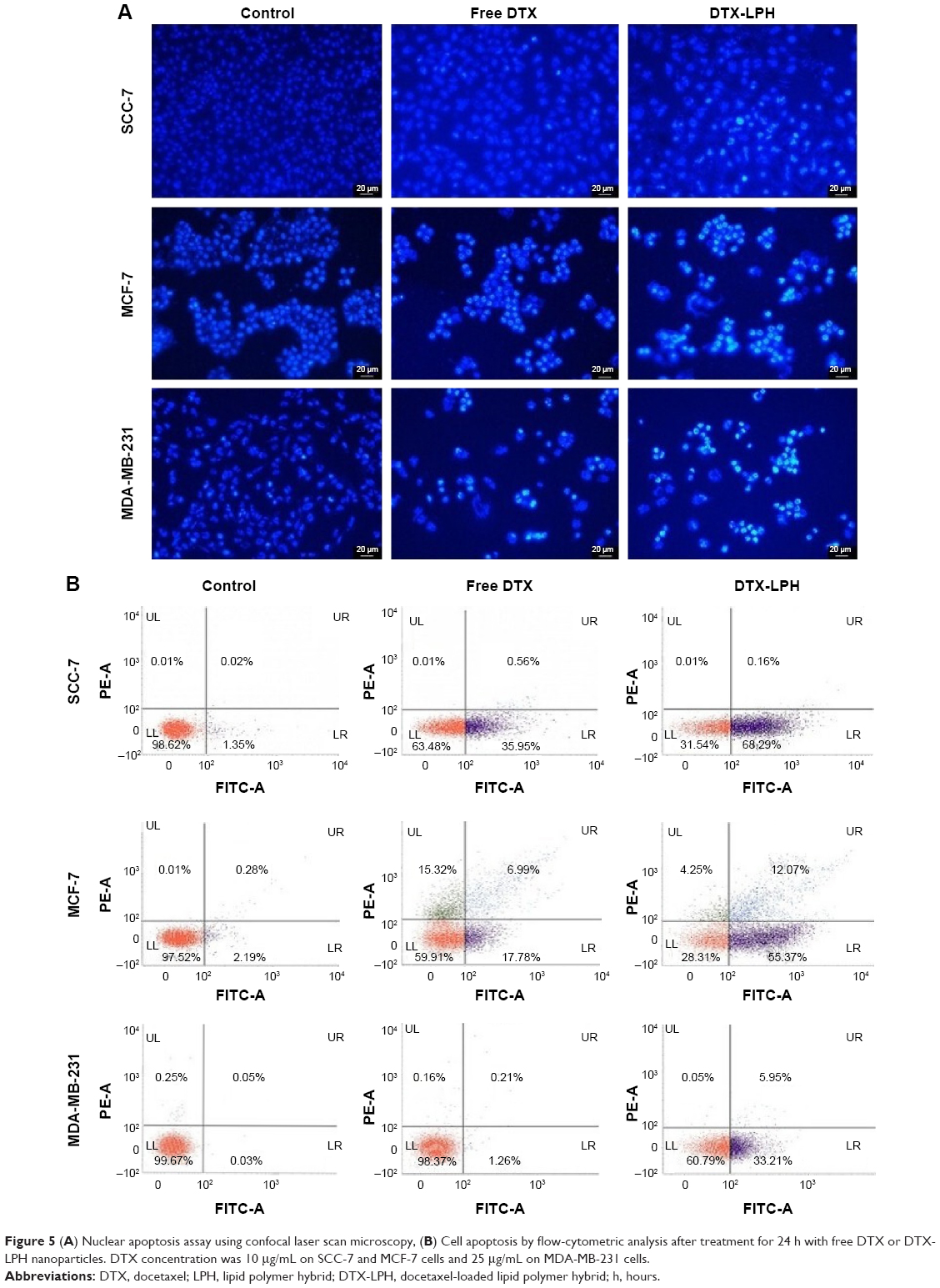 | Figure 5 (A) Nuclear apoptosis assay using confocal laser scan microscopy, (B) Cell apoptosis by flow-cytometric analysis after treatment for 24 h with free DTX or DTX-LPH nanoparticles. DTX concentration was 10 μg/mL on SCC-7 and MCF-7 cells and 25 μg/mL on MDA-MB-231 cells. |
Annexin V/propidium iodide (PI) staining-based apoptosis assays were performed in order to quantitate the different phase of apoptosis. The lower-right quadrant in the chart (Annexin V-positive, PI-negative) indicates the percentage of cells undergoing early apoptosis, while the upper-right quadrant is a symbol of late apoptosis. Early apoptosis is identified by plasma membrane reorganization and is detected by Annexin V-FITC, whereas later stage apoptosis is shown by DNA damage and tests positive for both Annexin V and PI. As shown in Figure 5B, DTX-LPH exhibited a greater proportion of early and late apoptosis compared with the free drug, while free DTX showed limited effectiveness. Following DTX-LPH treatment in SCC7 cancer cells, 68.29% of cells were in the early apoptosis stage compared with only 35.95% following free DTX treatment. The superior anticancer effect of DTX-LPH was also more evident in MDA-MB-231 cells. Following DTX-LPH treatment, 33.21% of cells were in the early apoptosis phase compared with only 1.26% following treatment with free DTX. This finding was consistent with the results of the qualitative apoptosis assays.
Next, we explored the effects of free DTX and DTX-LPH on downstream apoptosis-related protein signaling pathways such as p53, p27, and p21 in MDA-MB-231 cancer cells. The results showed that both DTX and DTX-LPH increased the expression of p53, p27, and p21 (Figure 6). The activation of p53-mediated promotion of apoptosis in tumor cells is an important mechanism of antitumor drugs. We saw that DTX-induced p53 protein expression and DTX-LPH had the strongest effect. The free drug and DTX-LPH also affected induction of the cyclin-dependent kinase inhibitors p21 and p27, which are well known to regulate cell cycle progression during the G1 and S phases.31,32 Enhancements of p53, p27, and p21 expressions suggest that the drug inhibits cancer propagation and induces cell cycle arrest because of its ability to regulate cell apoptosis and cell cycle-related genes.33,34
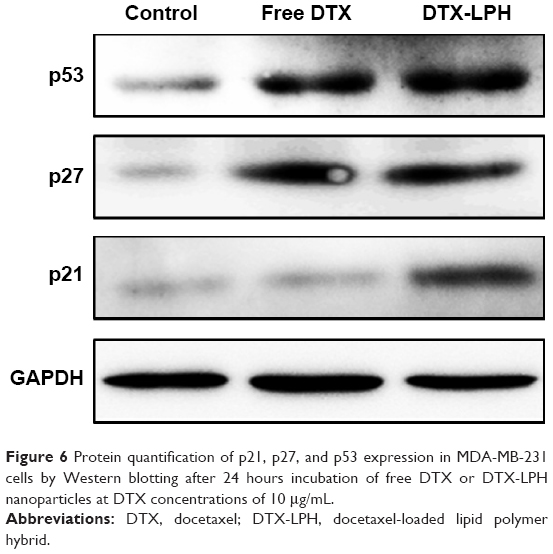 | Figure 6 Protein quantification of p21, p27, and p53 expression in MDA-MB-231 cells by Western blotting after 24 hours incubation of free DTX or DTX-LPH nanoparticles at DTX concentrations of 10 μg/mL. |
Pharmacokinetics study
As shown in Figure 7 and Table 2, the plasma concentration of DTX was markedly higher for the entirety of the studied period when delivered using LPH than when delivered alone. Free DTX readily disappeared from the blood stream within 6 hours of drug administration. The corresponding pharmacokinetic parameters also revealed the superior systemic performance of DTX-LPH. For instance, DTX-LPH showed 3.4-fold higher AUC0–∞ than free DTX. LPH significantly increased the half-life (t1/2~4.39) of DTX compared with the free drug (t1/2~1.09). The prolonged blood circulation profile of LPH was mainly attributed to the shielding effect of PEG, the excellent stability of the carrier/drug formulation in blood circulation, and the negative surface charge of the nanoparticles.35,36 Additionally, nanosized particles might be able to evade the macrophage system in the systemic blood circulation.
 | Figure 7 Plasma concentration-time profile of DTX after intravenous administration at a dose of 10 mg/kg of free DTX (□), or DTX-LPH nanoparticles (◼). |
 | Table 2 Pharmacokinetic parameters of DTX in rats after intravenous administration of free DTX and DTX-LPH nanoparticles at a dose of 10 mg/kg |
In vivo antitumor efficacy
The in vivo antitumor efficacy of DTX-LPH was examined in SCC-7 tumor cell-bearing xenograft mice (Figure 8A). As shown, free DTX had limited influence on tumor growth, while DTX-LPH most effectively reduced tumor volume. This enhanced antitumor effect of DTX-LPH was attributed to multiple factors including sustained drug release and prolonged circulation in the bloodstream, which resulted in higher accumulation of drug in the tumor via enhanced permeability and retention effect. Importantly, the P-gp inhibitory property of TPGS might enhance the absorption of drugs by effectively reducing P-gp-mediated drug removal. The toxicity of the formulations was assessed by measuring the change in the body weight (Figure 8B). DTX-LPH did not reduce the body weight of experimental mice, whereas mice treated with free DTX underwent a weight loss of ~20% indicating a severe drug-induced toxicity. This observation indicated that the drug-related toxicity could be overcome when the drugs were encapsulated into LPH nanoparticles.19
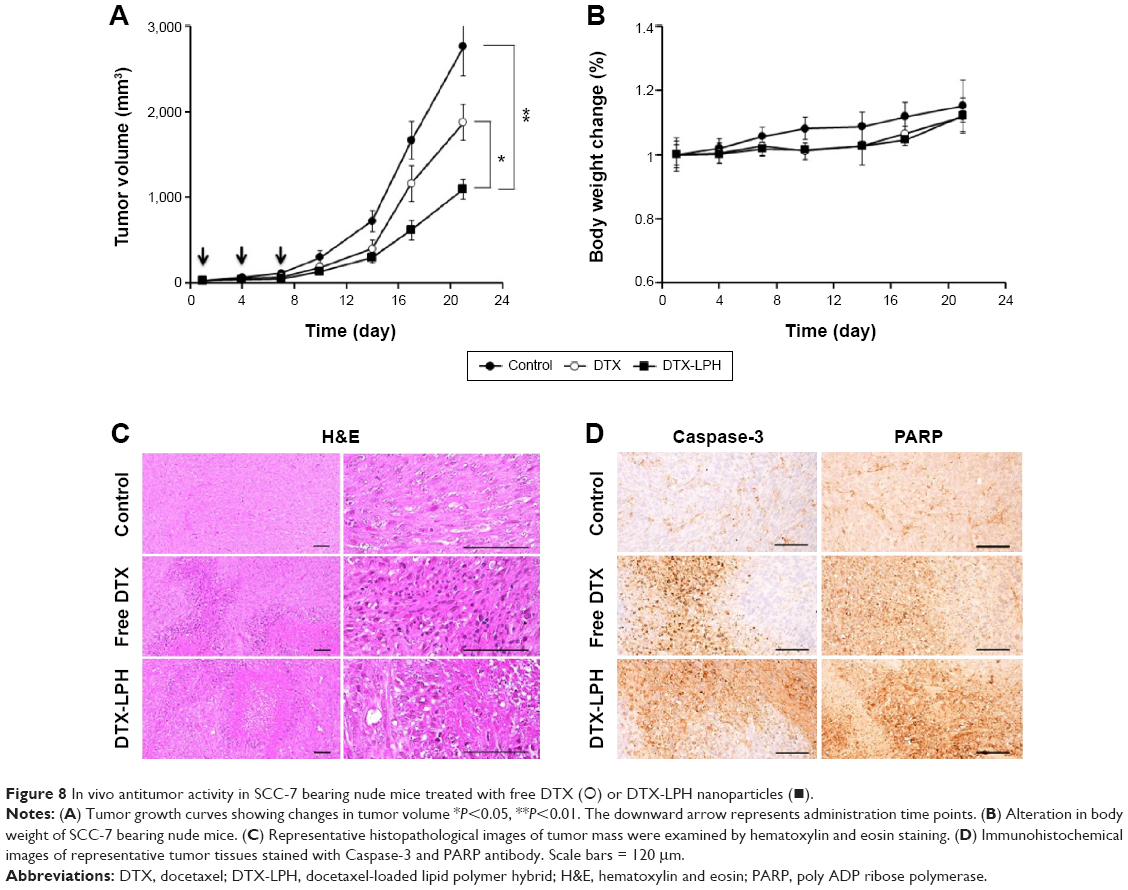 | Figure 8 In vivo antitumor activity in SCC-7 bearing nude mice treated with free DTX (○) or DTX-LPH nanoparticles (◼). |
The results of histomorphometrical analysis are shown in Table 3, and the representative histological-immunohistochemical profiles of tumor masses are shown in Figure 8C and D. The tumor cell volumes in DTX- and DTX-LPH-treated tumor masses were reduced by 35.2% and 62.3%, respectively. The percentage of caspase-3-immunolabeled cells in DTX- and DTX-LPH-treated tumor masses increased by 214.5% and 412.6%, and the percentage of PARP-positive cells increased by 212.6% and 371.7%, compared with untreated control, respectively. Significant decreases in tumor cell volume and increases in caspase-3 and PARP-positive cells were observed after both DTX and DTX-LPH treatments; however, the DTX-LTH treatment resulted in greater changes.
 | Table 3 Histomorphometrical analysis of tumor masses taken from xenograft athymic nude mice |
Conclusion
In summary, hybrid nanoparticles successfully improved the chemotherapeutic potency of DTX. The LPH nanoparticles had a pH-responsive drug release pattern that was beneficial for tumor targeting. More importantly, the DTX-LPH showed great ability to induce apoptosis and kill cancer cells. The enhanced efficacy of encapsulated DTX could be explained by its long blood circulation, which maintained a sufficiently high drug concentration for effective therapy.37 In addition, the improved intracellular uptake due to the charge-reversal characteristics allowed the drug to reach the cytoplasm.38 Moreover, the enhanced drug release in acidic condition along with the presence of TPGS in formulation could inhibit the activity of P-gp and reduce P-gp–mediated drug resistance.39 The histological-immunohistochemical profile was consistent with the results of the in vitro cytotoxicity assays and the apoptotic studies carried out using FACS, CLSM, and Western blotting. DTX-LPH significantly reduced the size of tumors in SCC7 cancer-bearing animals, and its effectiveness was further confirmed by the elevated levels of caspase-3 and PARP in tumor mass. Overall, it was demonstrated that LPH nanoparticles could provide a new platform for treating cancer.
Acknowledgments
This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (No 2015R1A2A2A01004118, 2015R1A2A2A04004806).
Disclosure
The authors report no conflicts of interest in this work.
References
Wang Z, Wu Y, Zeng X, et al. Antitumor efficiency of D-α-tocopheryl polyethylene glycol 1000 succinate-b-poly (ε-caprolactone-ran-lactide) nanoparticle-based delivery of docetaxel in mice bearing cervical cancer. J Biomed Nanotechnol. 2014;10:1509–1519. | ||
Zhao S, Tan S, Guo Y, et al. pH-sensitive docetaxel-loaded d-α-tocopheryl polyethylene glycol succinate – poly (β-amino ester) copolymer nanoparticles for overcoming multidrug resistance. Biomacromolecules. 2013;14:2636–2646. | ||
Pradhan R, Poudel BK, Ramasamy T, Choi H-G, Yong CS, Kim JO. Docetaxel-loaded polylactic acid-co-glycolic acid nanoparticles: formulation, physicochemical characterization and cytotoxicity studies. J Nanosci Nanotechnol. 2013;13:5948–5956. | ||
Seo YG, Kim D-W, Yeo WH, et al. Docetaxel-loaded thermosensitive and bioadhesive nanomicelles as a rectal drug delivery system for enhanced chemotherapeutic effect. Pharm Res. 2013;30:1860–1870. | ||
Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2:751–760. | ||
Guo Q, Li X, Yang Y, et al. Enhanced 4T1 breast carcinoma anticancer activity by co-delivery of doxorubicin and curcumin with core – shell drug-carrier based on heparin modified poly (L-lactide) grafted polyethylenimine cationic nanoparticles. J Biomed Nanotechnol. 2014;10:227–237. | ||
Yim H, Park S-J, Bae YH, Na K. Biodegradable cationic nanoparticles loaded with an anticancer drug for deep penetration of heterogeneous tumours. Biomaterials. 2013;34:7674–7682. | ||
Albanese A, Tang PS, Chan WC. The effect of nanoparticle size, shape, and surface chemistry on biological systems. Annu Rev Biomed Eng. 2012;14:1–16. | ||
Tran TH, Ramasamy T, Truong DH, et al. Development of vorinostat-loaded solid lipid nanoparticles to enhance pharmacokinetics and efficacy against multidrug-resistant cancer cells. Pharm Res. 2014;31:1978–1988. | ||
Seo YG, Kim DH, Ramasamy T, et al. Development of docetaxel-loaded solid self-nanoemulsifying drug delivery system (SNEDDS) for enhanced chemotherapeutic effect. Int J Pharm. 2013;452:412–420. | ||
Poudel BK, Pradhan R, Gupta B, Choi JY, Yong CS, Kim JO. Preparation and characterization of alginate gel core-lipid nanocapsules for co-delivery of hydrophilic and hydrophobic anti-cancer drugs. J Pharm Invest. 2014;44:485–491. | ||
Nguyen HT, Tran TH, Kim JO, Yong CS, Nguyen CN. Enhancing the in vitro anti-cancer efficacy of artesunate by loading into poly-d, l-lactide-co-glycolide (PLGA) nanoparticles. Arch Pharm Res. 2015;38:716–724. | ||
Ramasamy T, Tran TH, Choi JY, et al. Layer-by-layer coated lipid – polymer hybrid nanoparticles designed for use in anticancer drug delivery. Carbohydr Polym. 2014;102:653–661. | ||
Li M, Tang Z, Sun H, Ding J, Song W, Chen X. pH and reduction dual-responsive nanogel cross-linked by quaternization reaction for enhanced cellular internalization and intracellular drug delivery. Polym Chem. 2013;4:1199–1207. | ||
Yu B, Tang C, Yin C. Enhanced antitumor efficacy of folate modified amphiphilic nanoparticles through co-delivery of chemotherapeutic drugs and genes. Biomaterials. 2014;35:6369–6378. | ||
Lim H, Noh J, Kim Y, et al. Acid-degradable cationic poly (ketal amidoamine) for enhanced RNA interference in vitro and in vivo. Biomacromolecules. 2013;14:240–247. | ||
Palma G, Conte C, Barbieri A, et al. Antitumor activity of PEGylated biodegradable nanoparticles for sustained release of docetaxel in triple-negative breast cancer. Int J Pharm. 2014;473:55–63. | ||
Tran TH, Choi JY, Ramasamy T, et al. Hyaluronic acid-coated solid lipid nanoparticles for targeted delivery of vorinostat to CD44 overexpressing cancer cells. Carbohydr Polym. 2014;114:407–415. | ||
Ramasamy T, Kim JH, Choi JY, et al. pH sensitive polyelectrolyte complex micelles for highly effective combination chemotherapy. J Mater Chem B. 2014;2:6324–6333. | ||
Barrett KL, Willingham JM, Garvin AJ, Willingham MC. Advances in cytochemical methods for detection of apoptosis. J Histochem Cytochem. 2001;49:821–832. | ||
Smyth PG, Berman SA, Bursztajn S. Markers of apoptosis: methods for elucidating the mechanism of apoptotic cell death from the nervous system. Biotechniques. 2002;32:648–665. | ||
Shi S-R, Chaiwun B, Young L, Cote R, Taylor C. Antigen retrieval technique utilizing citrate buffer or urea solution for immunohistochemical demonstration of androgen receptor in formalin-fixed paraffin sections. J Histochem Cytochem. 1993;41:1599–1604. | ||
Zhou T, Xiao C, Fan J, et al. A nanogel of on-site tunable pH-response for efficient anticancer drug delivery. Acta Biomater. 2013;9:4546–4557. | ||
Mandal B, Bhattacharjee H, Mittal N, et al. Core – shell-type lipid – polymer hybrid nanoparticles as a drug delivery platform. Nanomedicine. 2013;9:474–491. | ||
Huang Y, Tang Z, Zhang X, et al. pH-triggered charge-reversal polypeptide nanoparticles for cisplatin delivery: preparation and in vitro evaluation. Biomacromolecules. 2013;14:2023–2032. | ||
Xu P, Van Kirk EA, Zhan Y, Murdoch WJ, Radosz M, Shen Y. Targeted charge-reversal nanoparticles for nuclear drug delivery. Angew Chem Int Ed. 2007;46:4999–5002. | ||
Meng F, Zhong Y, Cheng R, Deng C, Zhong Z. pH-sensitive polymeric nanoparticles for tumor-targeting doxorubicin delivery: concept and recent advances. Nanomedicine (Lond). 2014;9:487–499. | ||
Youm I, Agrahari V, Murowchick JB, Youan BB. Uptake and cytotoxicity of docetaxel-loaded hyaluronic acid-grafted oily core nanocapsules in MDA-MB 231 cancer cells. Pharm Res. 2014;31:2439–2452. | ||
Tran TH, Ramasamy T, Cho HJ, et al. Formulation and optimization of raloxifene-loaded solid lipid nanoparticles to enhance oral bioavailability. J Nanosci Nanotechnol. 2014;14:4820–4831. | ||
Bae WK, Park MS, Lee JH, et al. Docetaxel-loaded thermoresponsive conjugated linoleic acid-incorporated poloxamer hydrogel for the suppression of peritoneal metastasis of gastric cancer. Biomaterials. 2013;34:1433–1441. | ||
Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer. 2008;8:253–267. | ||
Gartel AL, Radhakrishnan SK. Lost in transcription: p21 repression, mechanisms, and consequences. Cancer Res. 2005;65:3980–3985. | ||
Brown I, Shalli K, McDonald SL, et al. Reduced expression of p27 is a novel mechanism of docetaxel resistance in breast cancer cells. Breast Cancer Res. 2004;6:R601–R607. | ||
Katayose Y, Kim M, Rakkar AN, Li Z, Cowan KH, Seth P. Promoting apoptosis: a novel activity associated with the cyclin-dependent kinase inhibitor p27. Cancer Res. 1997;57:5441–5445. | ||
Lv S, Li M, Tang Z, et al. Doxorubicin-loaded amphiphilic polypeptide-based nanoparticles as an efficient drug delivery system for cancer therapy. Acta Biomater. 2013;9:9330–9342. | ||
Okada H. Targeted siRNA therapy using cytoplasm-responsive nanocarriers and cell-penetrating peptides. J Pharm Invest. 2014;44:505–516. | ||
Ramasamy T, Haidar ZS, Tran TH, et al. Layer-by-layer assembly of liposomal nanoparticles with PEGylated polyelectrolytes enhances systemic delivery of multiple anticancer drugs. Acta Biomater. 2014;10:5116–5127. | ||
Torrecilla D, Lozano MV, Lallana E, et al. Anti-tumor efficacy of chitosan-g-poly(ethylene glycol) nanocapsules containing docetaxel: Anti-TMEFF-2 functionalized nanocapsules vs non-functionalized nanocapsules. Eur J Pharm Biopharm. 2013;83:330–337. | ||
Zhang Z, Tan S, Feng S-S. Vitamin E TPGS as a molecular biomaterial for drug delivery. Biomaterials. 2012;33:4889–4906. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.