")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Tumor-targeted and pH-controlled delivery of doxorubicin using gold nanorods for lung cancer therapy
Authors Amreddy N, Muralidharan R, Babu A, Mehta M, Johnson E, Zhao Y, Munshi A, Ramesh R
Received 28 July 2015
Accepted for publication 19 September 2015
Published 29 October 2015 Volume 2015:10(1) Pages 6773—6788
DOI https://doi.org/10.2147/IJN.S93237
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Narsireddy Amreddy,1,2 Ranganayaki Muralidharan,1,2 Anish Babu,1,2 Meghna Mehta,2,3 Elyse V Johnson,4 Yan D Zhao,2,5 Anupama Munshi,2,3 Rajagopal Ramesh1,2,6
1Department of Pathology, 2Stephenson Cancer Center, 3Department of Radiation Oncology University of Oklahoma Health Sciences Center, Oklahoma City, OK, USA; 4CytoViva Inc., Auburn, AL, USA; 5Department of Biostatistics and Epidemiology, 6Graduate Program in Biomedical Sciences, University of Oklahoma Health Sciences Center, Oklahoma City, OK, USA
Background: In lung cancer, the efficacy of conventional chemotherapy is limited due to poor drug accumulation in tumors and nonspecific cytotoxicity. Resolving these issues will increase therapeutic efficacy.
Methods: GNR-Dox-Tf-NPs (gold nanorod-doxorubicin-transferrin-nanoparticles) were prepared by different chemical approaches. The efficacy of these nanoparticles was carried out by cell viability in lung cancer and primary coronary artery smooth muscle cells. The receptor-mediated endocytosis studies were done with human transferrin and desferrioxamine preincubation. The GNR-Dox-Tf nanoparticles induced apoptosis, and DNA damage studies were done by Western blot, H2AX foci, and comet assay.
Results: We developed and tested a gold nanorod-based multifunctional nanoparticle system (GNR-Dox-Tf-NP) that carries Dox conjugated to a pH-sensitive linker and is targeted to the transferrin receptor overexpressed in human lung cancer (A549, HCC827) cells. GNR-Dox-Tf-NP underwent physicochemical characterization, specificity assays, tumor uptake studies, and hyperspectral imaging. Biological studies demonstrated that transferrin receptor-mediated uptake of the GNR-Dox-Tf-NP by A549 and HCC827 cells produced increased DNA damage, apoptosis, and cell killing compared with nontargeted GNR-Dox-NP. GNR-Dox-Tf-NP-mediated cytotoxicity was greater (48% A549, 46% HCC827) than GNR-Dox-NP-mediated cytotoxicity (36% A549, 39% HCC827). Further, GNR-Dox-Tf-NP markedly reduced cytotoxicity in normal human coronary artery smooth muscle cells compared with free Dox.
Conclusion: Thus, GNR-Dox-Tf nanoparticles can selectively target and deliver Dox to lung tumor cells and alleviate free Dox-mediated toxicity to normal cells.
Keywords: doxorubicin, gold, lung cancer, nanoparticles, transferrin, tumor targeting
Introduction
The utility of doxorubicin (Dox) for cancer treatment is limited by its poor accumulation in tumor tissue and cytotoxic side effects, especially irreversible cardiotoxicity, to normal tissues.1–4 Therefore, methods to increase tumor-specific drug accumulation and simultaneously reduce cytotoxicity to normal tissues will greatly improve the efficacy of Dox.5 The overexpression of receptors on the surface of tumor cells is one of several methods being tested to improve tumor-selective uptake of anticancer drugs.6,7 Studies have demonstrated that tumor cells often express cell surface receptors at high levels compared with normal cells; targeting these receptors for drug delivery produces increased drug accumulation and therapeutic efficacy.8 One such receptor is the transferrin receptor (TfR), which is overexpressed in a broad spectrum of human cancer cells, including lung cancer.9
TfR is a cell membrane-associated glycoprotein that transports iron to the cells to regulate cell growth.10 TfR has been explored as a target to deliver therapeutics into cancer cells due to its increased expression on malignant cells, accessibility on the cell surface, and constitutive endocytosis.11,12 TfR-targeted drug delivery to tumor cells can be achieved by conjugation of its natural ligand, transferrin (Tf), or monoclonal antibodies against TfR to nanoparticles (NPs) of various formulations.13,14
Gold-based NPs are promising candidates as drug carriers because they are inert, biocompatible, their surface can easily be modified, and they exhibit adequate cell penetration.15 Among the gold-based NPs, gold nanorods (GNR) have been extensively studied, due to their prolonged stability, ability to function as contrast agents, and capability of delivering drugs, DNA, small interfering ribonucleic acid, and proteins.16–19
In this study, we developed and tested a GNR-based multifunctional NP system (GNR-Dox-Tf-NP) that carries Dox conjugated to a pH-sensitive linker and is targeted to the TfR overexpressed in human lung cancer cells. The inclusion of a pH-sensitive linker to the NP facilitates the release of Dox under acidic conditions present in the endosomes/lysosomes (pH ≈4–6) of cancer cells upon internalization.20,21 We demonstrated that GNR-Dox-Tf-NP can selectively target and deliver Dox to induce cytotoxicity in lung tumor cells, with reduced toxicity to normal human coronary artery smooth muscle (HCASM) cells.
Methods
Cell lines
A549 and HCC827 non-small-cell lung cancer cells, which express different levels of TfRs, were maintained and cultured as previously described.22 The normal HCASM (PCS-100-021), the vascular cell basal medium, and the vascular smooth muscle cell growth kit were all purchased from American Type Culture Collection (ATCC). The cells were maintained in the vascular cell basal medium (ATCC PCS-100-030) that was supplemented with growth factors (smooth muscle cell growth kit; ATCC PCS-100-042 [American Type Culture Collection, Manassas, VA, USA]) per ATCC recommendation and used in this study. No ethics statement was required from the institutional review board for the use of these cell lines.
Synthesis of GNR-Dox and GNR-Dox-Tf nanoparticles
GNR-Dox and GNR-Dox-Tf NPs were synthesized as described in the Supplementary material (Figure S1). The optimum amount of Dox and Tf required for exhibiting maximum cytotoxicity was determined to be 2 μg/mL for each, respectively. The optimized GNR-Dox-NP and GNR-Dox-Tf-NP were used in all of the studies described herein.
Hyperspectral imaging
A549 cells (5×104) were seeded on coverslips on a six-well plate and were treated with GNR-Dox-NP and GNR-Dox-Tf-NP. Untreated cells served as controls. At 2 h after treatment, the cells were washed with phosphate-buffered saline (PBS). Subsequently, the cells were fixed with 1 mL of 4% paraformaldehyde for 20 min and again washed with PBS. Hyperspectral images were acquired using CytoViva hyperspectral microscopy (CytoViva Inc., Auburn, AL, USA) (Supplementary material).
Cell viability assay
A549, HCC827, and HCASM cells (0.2×106 cells/well) were seeded in six-well plates and were treated with free Dox (2 μg/mL), GNR-Dox-NP, or GNR-Dox-Tf-NP. Untreated cells served as controls. At 24 h after treatment, the cell viability was determined as previously described.23
Receptor-mediated endocytosis
A549 cells (5×104) were seeded on coverslips in two six-well plates and treated with GNR-Dox-NP and GNR-Dox-Tf-NP. Untreated cells served as controls. After NP treatment, one of the two plates was incubated at 37°C. The other plate was incubated at 4°C. After 4 h of incubation, the plates were removed, and the cells were washed with PBS, followed by treatment with 1 mL of 4% paraformaldehyde for 20 min. After fixation, the cells were again washed with PBS and counterstained with DAPI (4′6-diamidino-2-phenylindole dihydrochloride; 1:1,000) for 5 min. The cover slips were subsequently washed with PBS and were mounted on glass slides using mounting solution. Fluorescence images of cells were captured using a Nikon TiU microscope attached to a CCD (charge-coupled device) camera (Nikon Instruments Inc, New York, NY, USA) and imported into ImageJ (NIH, Bethesda, MD, USA) analysis software.
Receptor specificity studies
Receptor blocking study
A549 cells (0.2×106/well) seeded in six-well plates were either pretreated with human transferrin (HTf; 10 μM) or were untreated and were incubated at 37°C with 5% CO2 for 1 h. The cells were subsequently treated with GNR-Dox-Tf-NP in serum-free medium, and incubation continued for an additional 4 h. Then, the culture medium was replaced with fresh 5% serum-containing medium, and incubation continued for 20 h. At the end of the incubation period, the cells were subjected to cell viability assay.23,24
Receptor enhancement study
A549 cells (5×104) were seeded on coverslips and placed in a six-well plate containing 2 mL of RPMI (Roswell Park Memorial Institute) medium supplemented with 10% serum. At 24 h after incubation, the culture medium was replaced with 1 mL of serum-free medium, followed by the addition of 100 μM of desferrioxamine (DFO). Incubation continued for another 24 h. Cells that did not receive DFO served as controls. Then, GNR-Dox-Tf-NP was added to both treated and untreated cells. After 24 h of incubation, the coverslips were removed and mounted onto glass slides and were then processed for fluorescence microscopy and image capture.
In a separate set of experiments identical to those aforementioned ones, GNR-Dox-Tf-NP-treated A549 cells, in the presence and absence of DFO, were harvested. An aliquot was used for determining cell viability, while the rest of the cells were used to determine the intracellular fluorescence intensity (FI) of Dox with a Perkin Elmer Envision multiple plate reader (Perkin Elmer, Inc, Waltham, MA, USA). The FI was normalized to the cell count, and results were represented as FI per 10,000 cells.
Western blotting
A549 and HCASM cells (0.2×106/well) seeded in six-well plates were treated with GNR-Dox-NP and GNR-Dox-Tf-NP. Untreated cells served as controls. At 24 h after NP treatment, the cells were processed and subjected to Western blotting.24 Primary antibody against caspase 9 was purchased from Cell Signaling Technology, Inc., Danvers, MA, USA (1:1,000). β-actin (1:2,000; Sigma Chemicals, St Louis, MO, USA) was used as an internal loading control. Changes in the protein expression levels were quantified using Image Quant (Syngene, Cambridge, UK) software as previously described.24,25
Immunofluorescence assay
A549 cells were grown on coverslips placed in a six-well plate and treated with GNR-Dox-NP and GNR-Dox-Tf-NP. At 24 h after treatment, the cells were stained for H2AX using antihuman H2AX primary antibody (1:300; Cell Signaling) and Alexa Fluor-488-labeled secondary antibody (1:300; Invitrogen [Thermo Fisher Scientific, Waltham, MA, USA]) as previously described.26 H2AX foci were quantified by counting a minimum of 50 nuclei per treatment.
Comet assay
A549 cells (0.2×106/well) seeded in six-well plates were treated with GNR-Dox-NP and GNR-Dox-Tf-NP. At 24 h after treatment, the cells were harvested, processed, and subjected to Comet Assay (Trevigen, Gaithersburg, MD, USA) as previously described.27 The Olive tail moment was determined by screening 10 cells per field for a total of 5 fields (50 cells in total) in each sample.
Statistics
Response variables including percent cell viability, fluorescence, and foci per nuclei were compared among treatment groups using one-way analysis of variance. Multiple testing was conducted using Tukey’s adjustment for pairwise comparisons or using the Dunnett’s adjustment for comparing active treatments with the control. A P-value less than 0.05 was considered to be statistically significant. SAS 9.2 (SAS Institute Inc., Cary, NC, USA) was used for the statistical analysis.
Results
Physicochemical characterizations of GNR-Dox NPs
The surface charge, size, shape, and conjugation of Tf to GNR were determined prior to testing in biological systems. The surface charge of the NPs at each conjugation step was determined by measuring the zeta potential (Brookhaven, Holtsville, NY, USA). The zeta potential for bare GNR was +39 mV, which concurred with specifications provided by Nanopartz Inc. (A12-10-780, Loveland, CO, USA). With the addition of methoxy polyethylene glycol thiol (mPEG-SH), the surface charge was reduced to +13 mV. This was expected, as mPEG-SH, which is neutral in charge, reduces the total positive charge and is attributed to the PEG’s neutral charge. Interestingly, the addition of maleimidocapric acid hydrazine (EMCH) linker (HS-PEG-NH-NH2) conferred a zeta potential of −19 mV to the GNR system (Figure 1A). Although the amines in the EMCH linker are positively charged and are supposed to increase the net positive charge, the observed negative zeta potential could be attributed to the influence of buffer solution (PBS, pH 7.4) in which the particles were dispersed. The influence of buffer solutions on modulating the zeta potential has previously been reported.28 However, the negative charge reverted to a positive charge after the addition of Dox (+43.34 mV). The net positive charge on the final GNR-Dox-Tf-NP after adding Tf was +22.48 mV (Figure 1A).
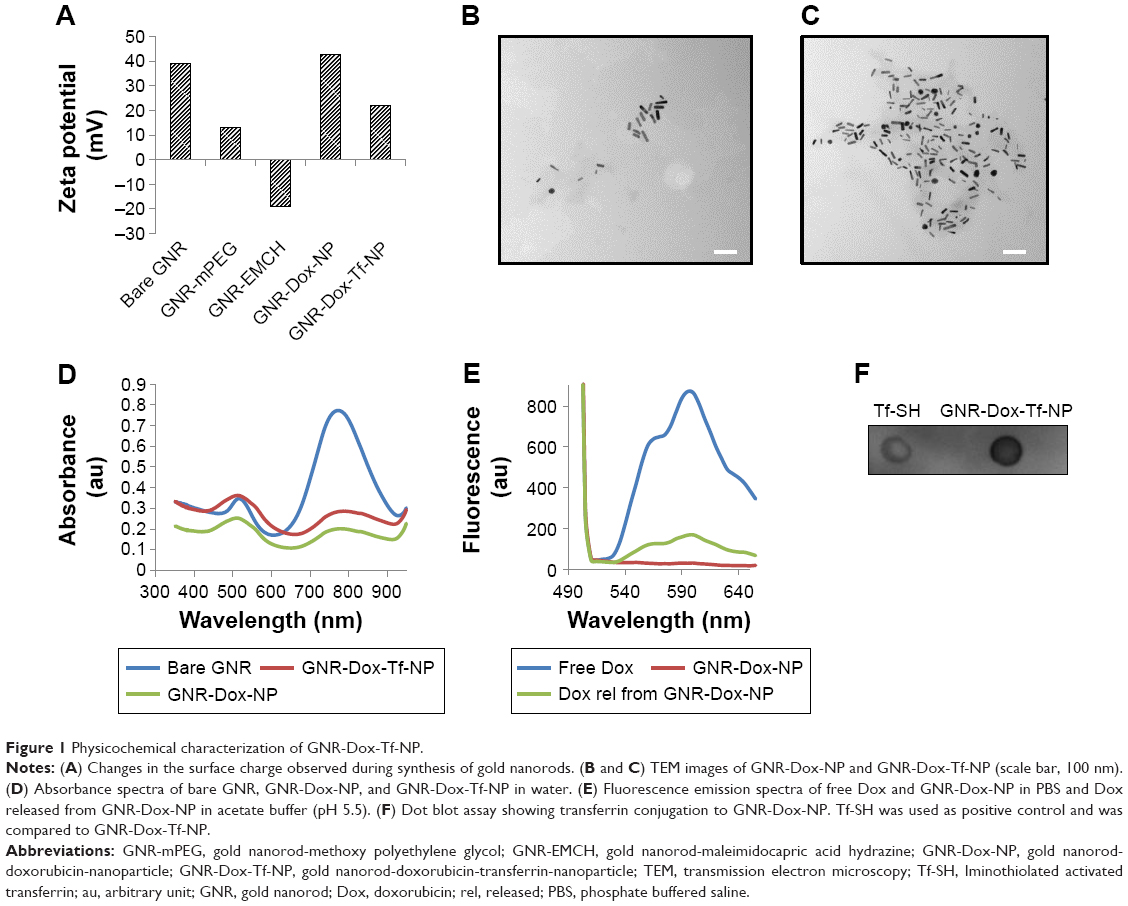 | Figure 1 Physicochemical characterization of GNR-Dox-Tf-NP. |
The size and shape of the nanorods, before and after conjugation of Dox and Tf, were measured with a transmission electron microscope (HITACHI 80 kV, HITACHI, Schaumburg, IL, USA). The average size of the bare nanorods was approximately 40 nm in length and 10 nm in width, with an aspect ratio of around 4 (Figure S2). The size and shape remained unaltered after conjugation of Dox and Tf (Figure 1B and C), suggesting that the particles were not affected by the applied conjugation chemistry.
We next determined whether conjugation of Dox and Tf affected the absorbance of GNR. The absorption spectra of GNR showed the presence of two peaks at 520 and 780 nm (Figure 1D), representing the transverse and longitudinal peaks, respectively, that remained unaffected on addition of Dox and Tf. The broad absorption peak observed at 520 nm in the GNR-Dox-NP and GNR-Dox-Tf-NP samples is attributed to interference of Dox absorbance (485 nm) with the transverse peak of the GNR.
We next determined whether the Dox attached to GNR via the acid cleavable hydrazone bond could be released under acidic pH (5.5). Fluorescence emission spectra showed that the fluorescence of free Dox at excitation/emission of 485/565 nm wavelength was quenched when conjugated to GNR (Figure 1E). However, the fluorescence was retrieved when GNR-Dox-NP was incubated in acidic pH buffer (5.5), indicating the release of Dox from GNR via acid hydrolysis.
Finally, dot blot analysis showed efficient conjugation of Tf to GNR-Dox-NP (Figure 1F).
In vitro drug release kinetic studies
To determine the rate of drug release at different pHs, GNR-Dox-NP and GNR-Dox-Tf-NP were incubated in PBS and acetate buffer solutions set at pH 7.4 and 5.5, respectively. These pH levels were chosen to mimic the pH of physiological blood pH (pH 7.4), the acidic pH of the tumor microenvironment (pH 5.8–7.6), and the pH of endosomes/lysosomes (pH 4–6). The percentage of Dox released from GNR-Dox-NP and GNR-Dox-Tf-NP in PBS and acetate buffer was estimated at different time periods using fluorescence measurements. The rate of Dox release from both GNR-Dox-NP and GNR-Dox-Tf-NP increased for the first 8 h and then plateaued, indicating sustained release of the drug over time (Figure 2A). Dox release, however, was higher at pH 5.5 than at pH 7.4 for both GNR-Dox-NP and GNR-Dox-Tf-NP. Drug release from GNR-Dox-Tf-NP was slighter higher than from GNR-Dox-NP at pH 5.5. The percentage of drug release at 24 h of incubation was 25% and 66% for GNR-Dox-Tf-NP, and 28% and 57% for GNR-Dox-NP at pH 7.4 and pH 5.5, respectively (Figure 2A). These results demonstrate that the GNR-Dox-NPs when internalized by tumor cells will efficiently release the drug, resulting in cytotoxicity.
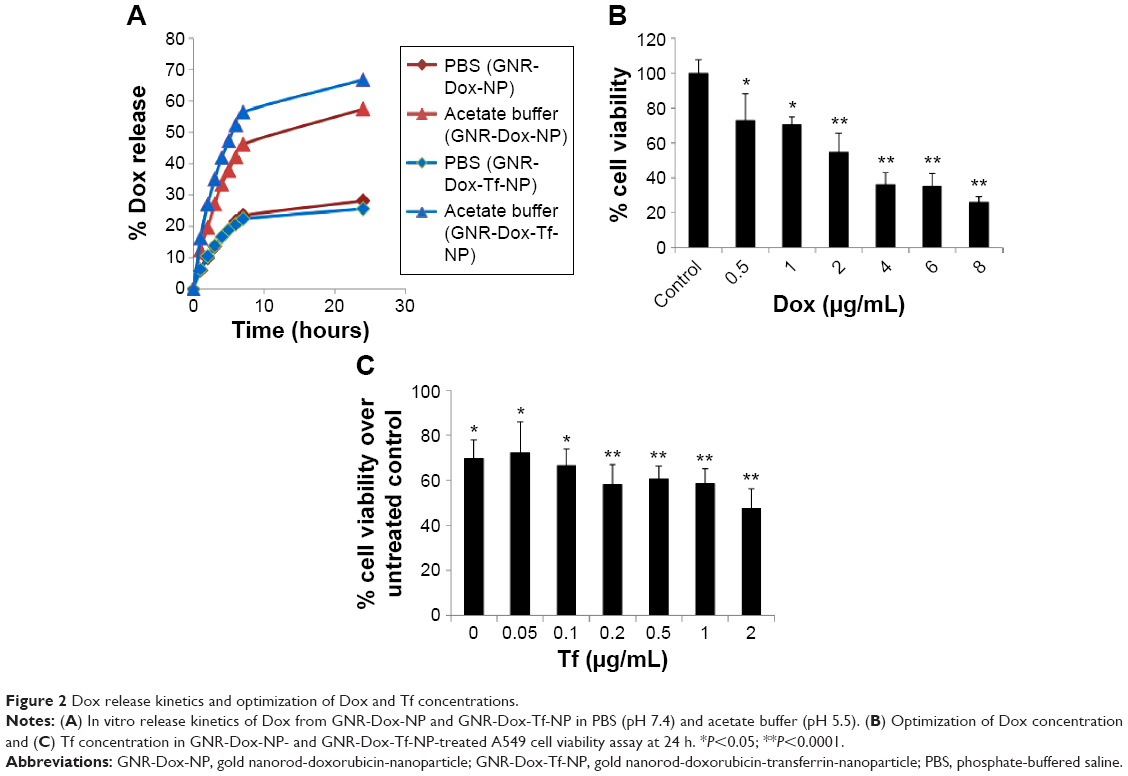 | Figure 2 Dox release kinetics and optimization of Dox and Tf concentrations. |
Optimization of Dox and Tf concentration on GNR
The Dox and Tf concentration was optimized by performing cell viability studies using A549 cells. First, varying concentrations (0.5–8 μg/mL) of Dox conjugated to GNR (GNR-Dox-NP) were tested for their cytotoxicity. A dose-dependent cytotoxicity was observed at 24 h after GNR-Dox-NP treatment, with 0.5 μg/mL of Dox showing the lowest toxicity (72% cell viability; P<0.05) and 8 μg/mL of Dox showing the highest toxicity (25% cell viability; P<0.0001; Figure 2B). The half-maximal inhibitory concentration value of GNR-Dox-NP was around 2.5 μg/mL of Dox. Based on these study results, we chose to use 2 μg/mL of Dox conjugated to GNR to produce GNR-Dox NPs.
Next, we optimized the Tf concentration to attach to GNR-Dox-NP. Different concentrations of Tf (0.05–2 μg/mL) were added to GNR-Dox-NP (1.25 μg/mL) and tested for cytotoxicity against A549 cells. A progressive reduction in cell viability was observed with increasing concentrations of Tf on GNR-Dox-NP (P<0.05–0.0001; Figure 2C). The highest percentage (71%) of viable cells was observed when treated with GNR-Dox-NP containing no Tf (P<0.05). The lowest percentage (47%) of viable cells was observed when treated with GNR-Dox-NP containing 2 μg/mL of Tf (P<0.0001; Figure 2C). Based on these results, 2 μg/mL of Tf was conjugated to GNR-Dox-NP to produce GNR-Dox-Tf-NPs.
The final concentration of Dox and Tf attached to GNR-Dox-Tf-NP and used in the study was 2 μg/mL.
GNR-Dox-Tf nanoparticles exhibit selective cytotoxicity against lung tumor cells
To determine the utility of GNR-Dox-Tf-NP as a therapeutic, we conducted cell viability and hyperspectral imaging studies using TfR-expressing A549 cells, and compared these results against those found with GNR-Dox-NP and free Dox. Hyperspectral imaging showed greater uptake of GNR-Dox-Tf nanoparticles than GNR-Dox nanoparticles in A549 cells at 2 h after treatment (Figure 3A). This result demonstrated that uptake of targeted GNR-Dox-Tf-NP is rapid and higher in A549 cells.
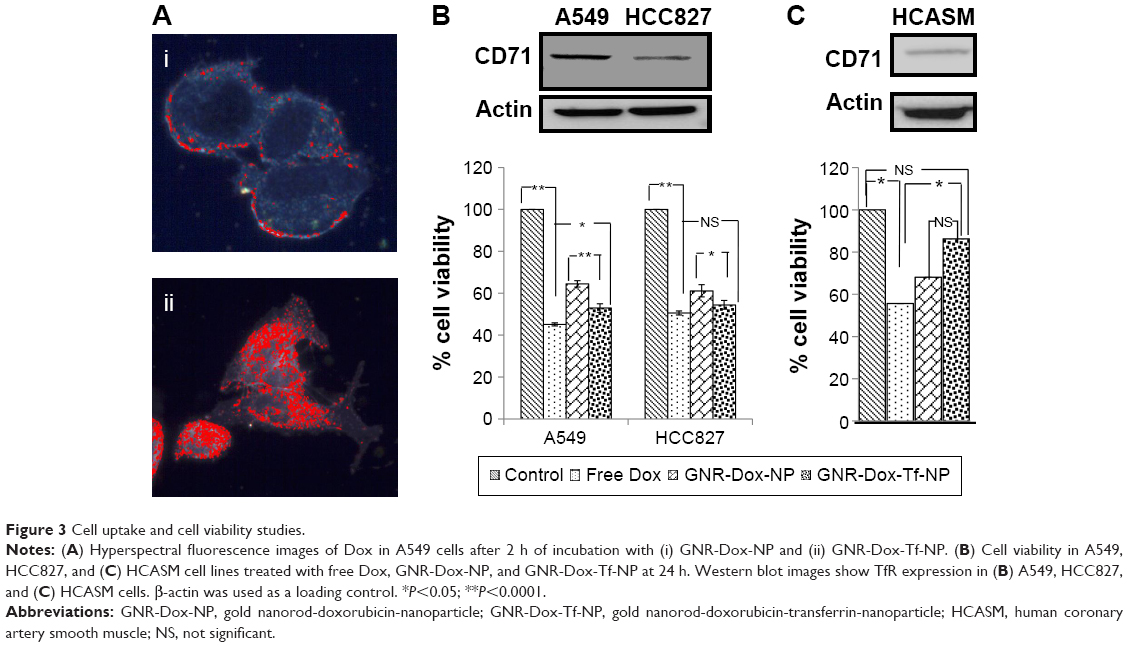 | Figure 3 Cell uptake and cell viability studies. |
A cell viability study showed that GNR-Dox-Tf-NP markedly reduced cell viability by 48% (52% viable) compared with a 36% reduction (64% viable) observed with GNR-Dox-NP treatment (P<0.0001; Figure 3B), demonstrating that GNR-Dox-Tf-NP was more effective than GNR-Dox-NP. The cytotoxicity exerted by free Dox was observed to be the highest, with viability reduced by 55% (45% viable; P<0.0001) when compared with untreated control cells. The difference in cytotoxicity between free Dox and GNR-Dox-Tf-NP could be explained by the fact that free Dox passively and rapidly diffuses into cells,29 while GNR-Dox-Tf-NP enters the cells by receptor-mediated endocytosis, followed by the controlled release of Dox from the acid-labile linker.
The possibility that the observed GNR-Dox-Tf-NP-mediated cytotoxicity was unique to the A549 cell line was excluded by testing GNR-Dox-Tf-NP against another lung cancer cell line, HCC827. Free Dox treatment reduced HCC827 viability by 50% (50% viable; P<0.0001; Figure 3B), a level akin to that observed in A549 cells. GNR-Dox-Tf-NP treatment reduced cell viability by 46% (54% viable; P<0.0001) when compared with untreated controls and was greater when compared with GNR-Dox-NP treatment, which showed a reduction in cell viability by 39% (61% viable; P<0.05; Figure 3B). There was no significant difference in the cytotoxicity activity of GNR-Dox-Tf-NP and free Dox (P>0.05). These results demonstrate the broad-spectrum cytotoxicity exhibited by GNR-Dox-Tf-NP against lung cancer cells.
The higher efficacy demonstrated by GNR-Dox-Tf-NP over GNR-Dox-NP in A549 cells (12%–14%) compared with HCC827 cells (7%–8%) is attributable to higher TfR expression in A549 cells than in HCC827 cells (upper panel, Figure 3B).
To further determine the cytotoxicity of GNR-Dox-Tf toward normal cells, HCASM cells, which express low levels of TfR (upper panel, Figure 3C), were treated with GNR-Dox-Tf-NP and compared with GNR-Dox-NP and free Dox. Untreated cells served as controls. Free Dox reduced cell viability to a greater extent (55% viable; P<0.05) than did GNR-Dox-NP (68% viable) and GNR-Dox-Tf-NP (86% viable; Figure 3C) when compared with untreated control cells. GNR-Dox-Tf-NP demonstrated reduced cytotoxicity when compared with GNR-Dox-NP (16% less toxicity) and free Dox (31% less toxicity). These results demonstrate that TfR-targeted drug delivery markedly reduces Dox-induced toxicity to normal coronary artery smooth muscle cells.
GNR-Dox-Tf-NP-mediated cytotoxicity is specific and occurs via transferrin receptor
To determine whether GNR-Dox-Tf-NP-induced cytotoxicity was receptor-mediated, A549 cells were treated with GNR-Dox-Tf-NP and incubated at 37°C or at 4°C for 4 h. Receptor-mediated endocytosis is active at 37°C and is inhibited at 4°C.30 GNR-Dox-Tf-NP-treated cells incubated at 37°C demonstrated intense intracellular fluorescence that was greatly diminished in GNR-Dox-Tf-NP-treated cells incubated at 4°C (Figure 4A). These results show that GNR-Dox-Tf-NP uptake occurs via receptor-mediated endocytosis that is active at 37°C but not at 4°C.31
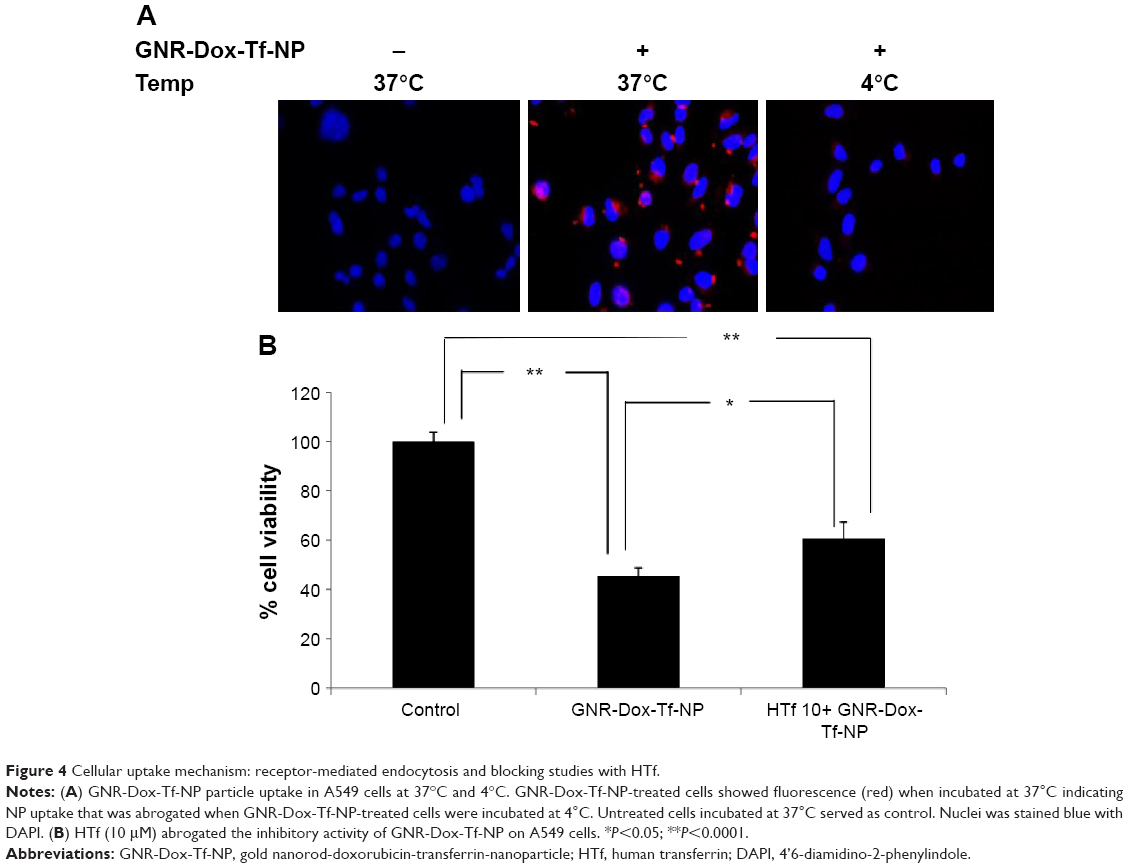 | Figure 4 Cellular uptake mechanism: receptor-mediated endocytosis and blocking studies with HTf. |
To further test receptor-mediated uptake of GNR-Dox-Tf-NP, we conducted cell viability studies in the presence of HTf, the natural ligand for TfR.32 A549 cells were pretreated with GNR-Dox-Tf-NP in the presence or absence of HTf (10 μM). Treatment with GNR-Dox-Tf-NP reduced cell viability by 55% compared with control cells receiving no treatment (P<0.05; Figure 4B). In contrast, the efficacy of GNR-Dox-Tf-NP was markedly abrogated in the presence of HTf (40% inhibition). Our results demonstrate that TfR receptor-mediated uptake of the GNR-Dox-Tf nanoparticles results in cytotoxicity.
Next, we tested the efficacy of GNR-Dox-Tf-NP in the presence of the iron-chelating ligand DFO. Incubation of cells with DFO induces TfR biosynthesis and increases TfR expression on the cell surface, thereby facilitating increased TfR-mediated particle uptake.33 Exposure of A549 cells to DFO (100 μM) prior to GNR-Dox-Tf-NP treatment increased TfR expression (Figure 5A). Fluorescence microscopy studies showed intense intracellular fluorescence in the presence of DFO (Figure 5B). Further, a 2.5 times increase in FI in DFO plus GNR-Dox-Tf treatment was observed when compared with GNR-Dox-Tf-NP treatment (P<0.0001; Figure 5C). A marked and significant reduction in cell viability in DFO plus GNR-Dox-Tf-NP treatment (22% viability) was associated with increased GNR-Dox-Tf-NP uptake when compared with the untreated control group (P<0.0001; Figure 5D). The reduction of cell viability following GNR-Dox-NP treatment, though significant (53% viability; P<0.05) when compared with untreated controls, was less than that observed in GNR-Dox-Tf-NP-treated cells. These results demonstrate that the GNR-Dox-Tf-NP-mediated cytotoxicity observed in the A549 lung cancer cell line is specific and occurs via the TfR.
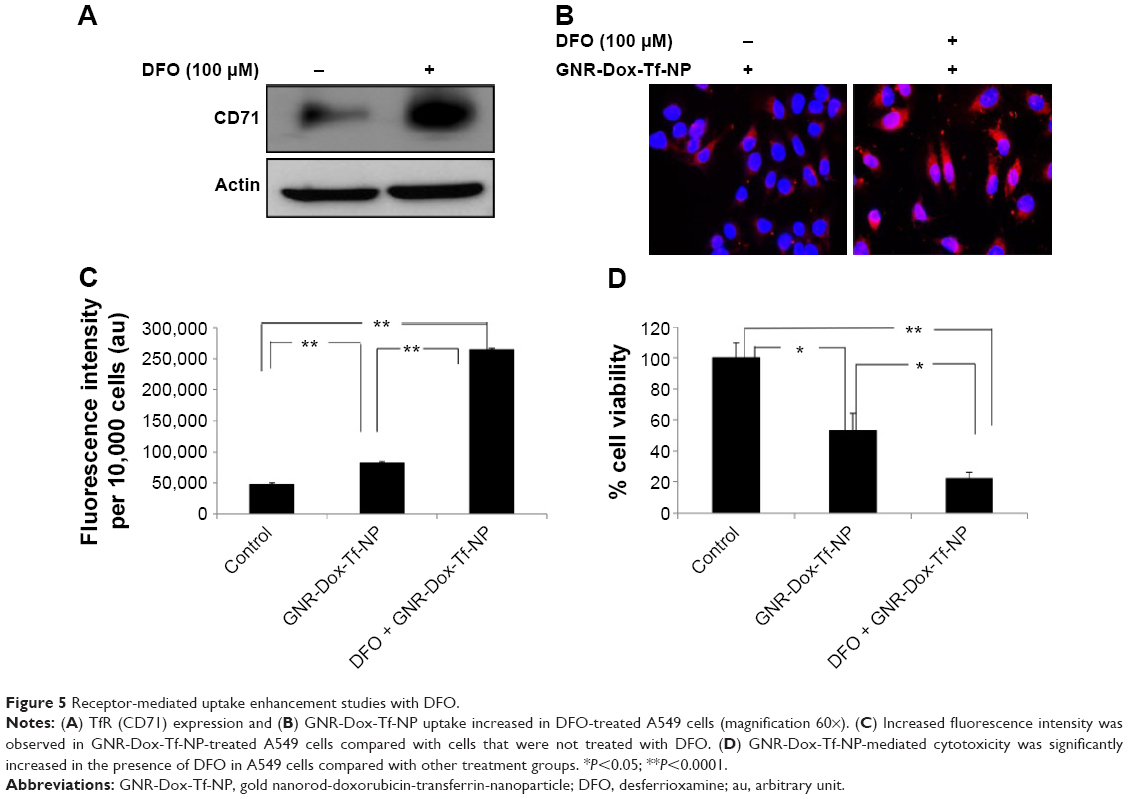 | Figure 5 Receptor-mediated uptake enhancement studies with DFO. |
GNR-Dox-Tf-NP treatment activates caspase-9 in the A549 lung cancer cell line
Dox treatment induces apoptosis in a broad spectrum of cancer cells.34 Dox has also been shown to produce cardiac tissue damage resulting in cardiotoxicity.35 We therefore investigated whether GNR-Dox-Tf-NP treatment also induced apoptosis in A549 and HCASM cells. Marked activation of caspase-9, an indicator of cells undergoing apoptosis,36 was detected in both GNR-Dox- and GNR-Dox-Tf-NP-treated A549 cells (Figure 6A). However, caspase-9 levels were significantly higher in GNR-Dox-Tf-NP-treated cells than in GNR-Dox-NP-treated cells (P<0.0001; Figure 6B). In HCASM cells, a mild increase in activated caspase-9 level was observed in GNR-Dox-Tf-NP-treated cells compared with untreated control cells (P<0.05; Figure 6C and D). In GNR-Dox-NP-treated cells, the caspase-9 level was significantly increased compared with GNR-Dox-Tf-NP and untreated controls (P<0.05; Figure 6C and D). These results demonstrate that GNR-Dox-Tf-NP induces apoptosis selectively and to greater extent in lung cancer cells, while exhibiting reduced killing toward HCASM cells.
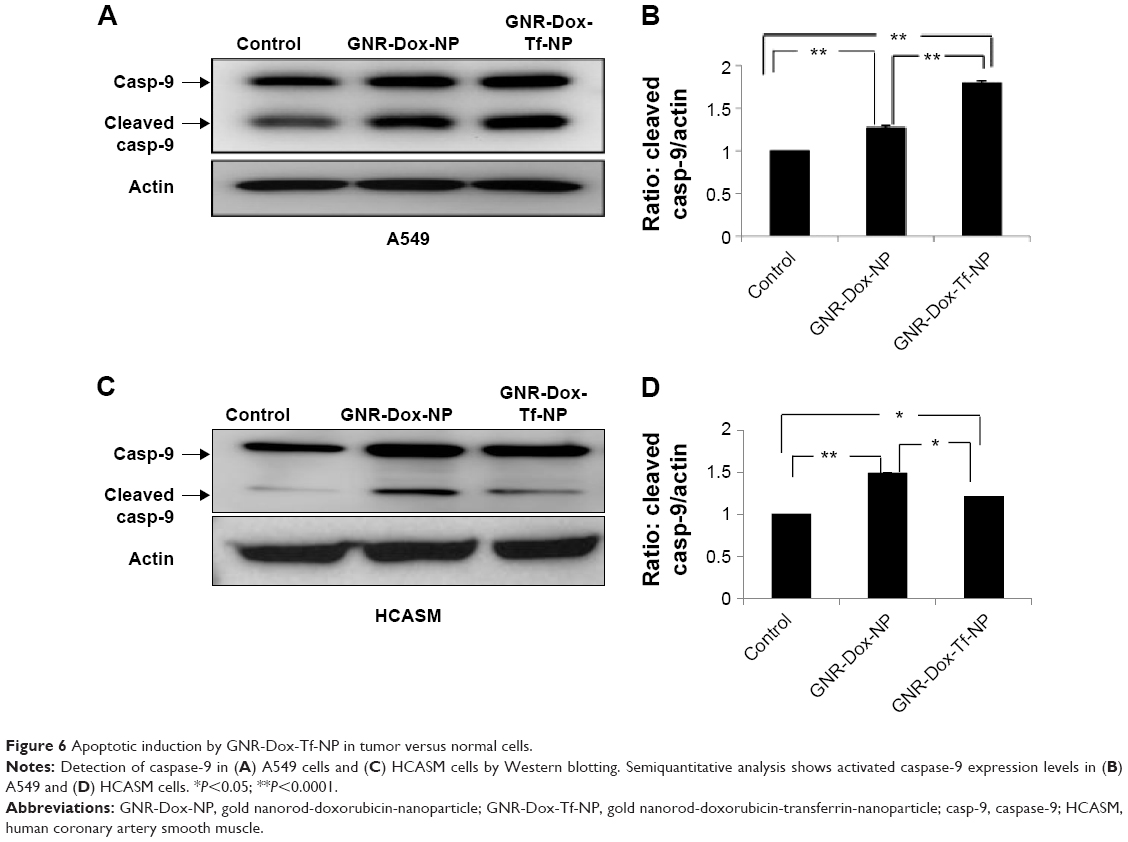 | Figure 6 Apoptotic induction by GNR-Dox-Tf-NP in tumor versus normal cells. |
GNR-Dox-Tf-NP treatment induces DNA damage in the A549 lung cancer cell line
Dox treatment produces DNA damage by inducing DNA double-strand breaks.37 Therefore, in this study, we looked for DNA damage in GNR-Dox-Tf-NP-treated cells by staining for H2AX foci38 and conducting a comet assay.27,39 A significant increase in H2AX foci was observed in both GNR-Dox-NP- and GNR-Dox-Tf-NP-treated cells compared with untreated control cells (P<0.0001; Figure 7A). However, the number of H2AX foci in GNR-Dox-Tf-NP-treated cells was significantly higher than that in GNR-Dox-NP-treated cells (P<0.05; Figure 7A).
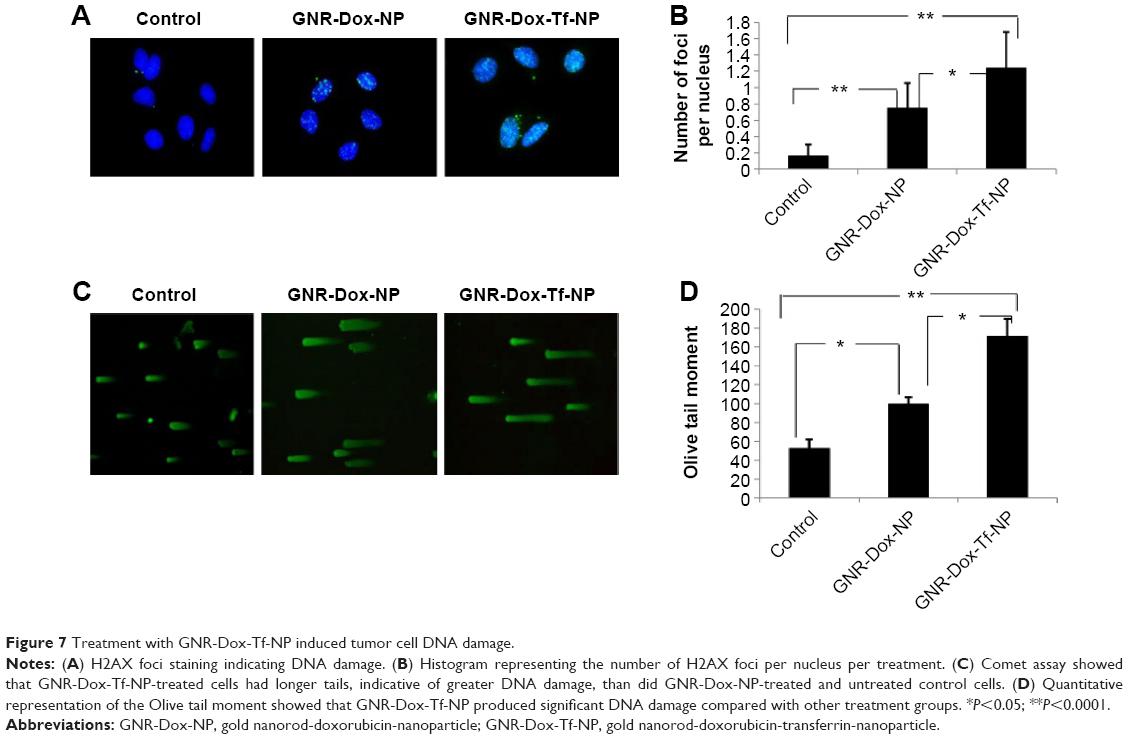 | Figure 7 Treatment with GNR-Dox-Tf-NP induced tumor cell DNA damage. |
Comet assay also demonstrated that GNR-Dox-Tf-NP-treated cells produced greater DNA damage than did GNR-Dox-NP-treated and -untreated control cells, as indicated by the intense and long tail (P<0.05; Figure 7B). GNR-Dox-NP treatment also resulted in more DNA damage than was observed in untreated control cells (P<0.05). These results demonstrate that GNR-Dox-Tf-NPs induce a greater magnitude of DNA damage than the control groups resulting in cell death.
Discussion
The goal of this study was to develop a strategy for increased delivery of Dox to tumor cells and to circumvent the nonspecific toxicity to normal cells. To achieve this goal, we have developed and tested a multifunctional TfR-targeted GNR drug delivery system that efficiently carries and releases Dox under acid-labile conditions (GNR-Dox-Tf-NP), such as the acidic environment present in the intracellular endosomal compartment and under hypoxic conditions.40 Physicochemical studies revealed that the GNR-Dox-Tf nanoparticles were positively charged and approximately 40 nm in size. Further, the nanoparticles exhibited desirable drug release kinetics under acidic pH (5.5) supporting their testing in biological systems.
Both GNR-Dox-Tf-NP-treated A549 and HCC827 lung cancer cell lines showed a significant reduction in cell viability compared with GNR-Dox-NP-treated cells and untreated control groups. However, A549 cells, which express higher levels of TfR than HCC827 cells, were more sensitive to GNR-Dox-Tf-NP treatment, reflecting a receptor dosage effect. HCASM cells, unlike tumor cells, expressed low levels of TfR and were relatively less sensitive to GNR-Dox-Tf-NP treatment when compared with free Dox treatment. These results demonstrated the utility of receptor-targeted drug delivery, minimizing toxicity to normal coronary artery smooth muscle cells while exhibiting potent anticancer activity against tumor cells. Another important observation made in this study was that although we have used a relatively high concentration of Dox (equivalent to 3.4 μM) in the GNR-Dox-NP system, the observed toxicity in the HCASM cells was significantly reduced when compared to free Dox. This is in contrast to the reports that demonstrated that normal cells including cardiomyocytes when exposed to very low concentrations of free Dox (0.1 μM) exhibited toxicity that increased with increasing drug concentration.35,41,42 Thus, our study results demonstrate that higher Dox concentration can be delivered to tumor cells using the GNR-Dox-Tf-NP system while sparing normal cells. Furthermore, the GNR-Dox-Tf-NP treatment is likely to produce reduced cardiotoxicity, while efficiently killing tumor cells, a feature that is preferred for Dox-based cancer treatment. While our in vitro study results are promising, it is to be realized that further improvements are to be made to the GNR-Dox-Tf-NP system for increasing tumor cytotoxicity. Further, the safety of the GNR-Dox-Tf-NP is to be determined in in vivo studies prior to advancing to clinical testing.
The fact that the observed GNR-Dox-Tf-NP toxicity in cancer cells was receptor-mediated was determined by conducting specificity studies. GNR-Dox-Tf nanoparticles uptake by the lung cancer cells occurred via receptor-mediated endocytosis, as particle uptake by the cells was observed when incubated at 37°C but not when incubated at 4°C. This observation concurred with previous studies that showed that receptor-mediated endocytosis is active at 37°C and reduced at 4°C.31 The reduction in endocytic activity at a lower temperature occurs because the receptors are not active to bind with their ligands.31 Further, receptor specificity was demonstrated by using an excess of HTf, a natural ligand for TfR. The cytotoxicity of GNR-Dox-Tf in the presence of HTf was greatly mitigated (Figure 8). In contrast, increasing the TfR expression in A549 cells using the iron chelator DFO resulted in a significant increase in GNR-Dox-Tf particle uptake and in enhanced cytotoxicity (Figure 8). The increase in TfR expression after DFO treatment is attributed to its ability to interrupt the normal intracellular iron balance.33 Our study results thus demonstrated that the GNR-Dox-Tf nanoparticles are receptor-specific and enter cells via endocytosis to exert their anticancer activity.
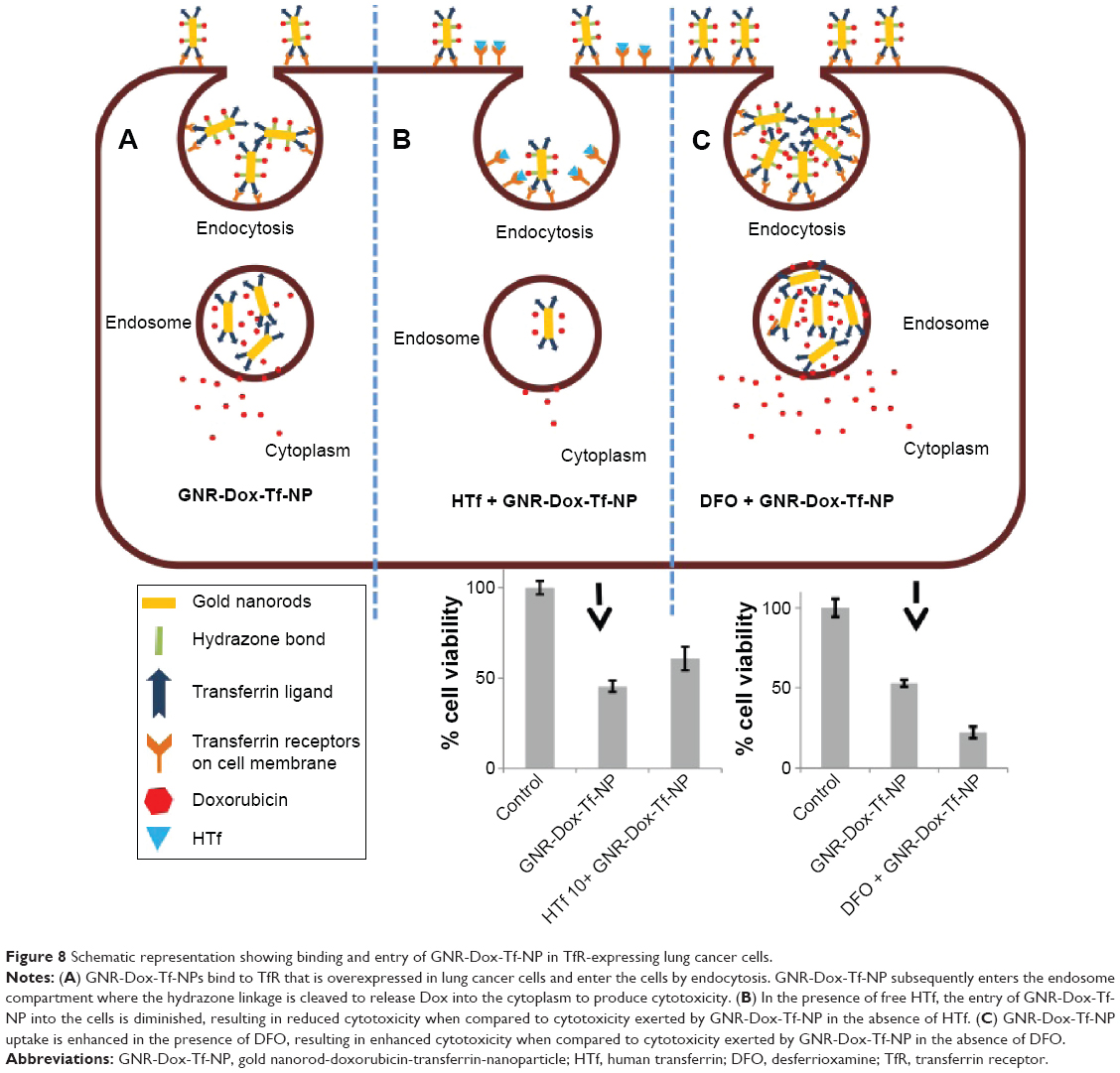 | Figure 8 Schematic representation showing binding and entry of GNR-Dox-Tf-NP in TfR-expressing lung cancer cells. |
Molecular studies showed that GNR-Dox-Tf exerted its anticancer activity in A549 cells by activating caspase-9, a molecular indicator of cells undergoing apoptosis.34,36 Although caspase-9 activation was observed in HCASM cells, the level of activation was less than that observed in A549 cells, suggesting that tumor cells were more sensitive to GNR-Dox-Tf-NP therapy. Further, GNR-Dox-Tf-NP-treated A549 cancer cells showed a significant increase in both H2AX foci and DNA laddering when compared with GNR-Dox-NP and untreated control cells, indicating that the cells underwent DNA damage and double-strand breaks.37,38
Conclusion
In this study, we have successfully fabricated targeted GNR-Dox-Tf-NP with pH-sensitive drug release capabilities for the treatment of lung cancer. Our findings demonstrate that GNR-Dox-Tf-NP selectively targets Tf overexpressing lung cancer cells and facilitates their cellular entry via receptor-mediated endocytosis (Figure 8). The targeting and pH-responsive drug release properties of GNR-Dox-Tf-NP enhanced the effectiveness of the therapeutic agent in cancer cells by inducing apoptosis and DNA damage. Importantly, GNR-Dox-Tf-NP exhibited reduced toxicity in normal coronary artery smooth muscle cells. This study demonstrates that GNR-Dox-Tf-NP will selectively kill tumor cells with reduced cardiotoxicity and serves as a strong foundation for our future in vivo study.
Acknowledgments
The authors appreciate the assistance received from the cancer functional genomics core and electron microscope core facility at the University of Oklahoma and Oklahoma Medical Research Foundation, respectively. The authors thank Kathy Kyler at the office of Vice President of Research, OUHSC, for editorial assistance. This study was supported in part by a grant received from the National Cancer Institute (R01 CA167516-03), an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences (P20 GM103639) of the National Institutes of Health, and by the Jim and Christy Everest Endowed Chair in Cancer Developmental Therapeutics.
Author contributions
NA, RM, AB, MM, and EVJ conducted the studies and collected data; YDZ performed statistical analysis; NA, RM, AB, MM, EVJ, YDZ, AM, and RR conceived and designed the studies; NA, AB, AM, and RR wrote the manuscript. NA, RM, AB, MM, EVJ, YDZ, AM, and RR critically analyzed and interpreted the data; NA, RM, AB, MM, EVJ, YDZ, AM, and RR critically reviewed, provided suggestions, and edited the manuscript. RR supervised the project.
Disclosure
RR is an Oklahoma TSET research scholar and holds the Jim and Christy Everest Endowed Chair in Cancer Developmental Therapeutics. The authors report no other conflicts of interest in this work.
References
Gerber DE. Targeted therapies: a new generation of cancer treatments. Am Fam Physician. 2008;77(3):311–319. | ||
Griffin AM, Butow PN, Coates AS, et al. On the receiving end V: patient perceptions of the side effects of cancer chemotherapy in 1993. Ann Oncol. 1996;7(2):189–195. | ||
Muller I, Jenner A, Bruchelt G, Niethammer D, Halliwell B. Effect of concentration on the cytotoxic mechanism of doxorubicin-apoptosis and oxidative DNA damage. Biochem Biophys Res Commun. 1997;230(2):254–257. | ||
Buzdar AU, Marcus C, Smith TL, Blumenschein GR. Early and delayed clinical cardiotoxicity of doxorubicin. Cancer. 1985;55(12):2761–2765. | ||
Arap W, Pasqualini R, Ruoslahti E. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science. 1998;279(5349):377–380. | ||
Bareford LM, Swaan PW. Endocytic mechanisms for targeted drug delivery. Adv Drug Deliv Rev. 2007;59(8):748–758. | ||
Weaver M, Laske DW. Transferrin receptor ligand-targeted toxin conjugate (Tf-CRM107) for therapy of malignant gliomas. J Neurooncol. 2003;65(1):3–13. | ||
Kukowska-Latallo JF, Candido KA, Cao Z, et al. Nanoparticle targeting of anticancer drug improves therapeutic response in animal model of human epithelial cancer. Cancer Res. 2005;65(12):5317–5324. | ||
Calzolari A, Oliviero I, Deaglio S, et al. Transferrin receptor 2 is frequently expressed in human cancer cell lines. Blood Cell Mol Dis. 2007;39(1):82–91. | ||
Kawabata H, Yang R, Hirama T, et al. Molecular cloning of transferrin receptor 2. A new member of the transferrin receptor-like family. J Biol Chem. 1999;274(30):20826–20832. | ||
Qian ZM, Li H, Sun H, Ho K. Targeted drug delivery via the transferrin receptor-mediated endocytosis pathway. Pharmacol Rev. 2002;54(4):561–587. | ||
Daniels TR, Delgado T, Helguera G, Penichet ML. The transferrin receptor part II: targeted delivery of therapeutic agents into cancer cells. Clin Immunol. 2006;121(2):159–176. | ||
Dixit S, Novak T, Miller K, Zhu Y, Kenney ME, Broome AM. Transferrin receptor-targeted theranostic gold nanoparticles for photosensitizer delivery in brain tumors. Nanoscale. 2015;7(5):1782–1790. | ||
Li JL, Wang L, Liu XY, et al. In vitro cancer cell imaging and therapy using transferrin-conjugated gold nanoparticles. Cancer Lett. 2009;274(2):319–326. | ||
Wang F, Wang YC, Dou S, Xiong MH, Sun TM, Wang J. Doxorubicin-tethered responsive gold nanoparticles facilitate intracellular drug delivery for overcoming multidrug resistance in cancer cells. ACS Nano. 2011;5(5):3679–3692. | ||
Wijaya A, Schaffer SB, Pallares IG, Hamad-Schifferli K. Selective release of multiple DNA oligonucleotides from gold nanorods. ACS Nano. 2009;3(1):80–86. | ||
Salem AK, Searson PC, Leong KW. Multifunctional nanorods for gene delivery. Nat Mater. 2003;2(10):668–671. | ||
Huang HC, Barua S, Kay DB, Rege K. Simultaneous enhancement of photothermal stability and gene delivery efficacy of gold nanorods using polyelectrolytes. ACS Nano. 2009;3(10):2941–2952. | ||
Chanda N, Shukla R, Katti KV, Kannan R. Gastrin releasing protein receptor specific gold nanorods: breast and prostate tumor avid nanovectors for molecular imaging. Nano Lett. 2009;9(5):1798–1805. | ||
Huschka R, Zuloaga J, Knight MW, Brown LV, Nordlander P, Halas NJ. Light-induced release of DNA from gold nanoparticles: nanoshells and nanorods. J Am Chem Soc. 2011;133(31):12247–12255. | ||
Sun Z, Ni W, Yang Z, Kou X, Li L, Wang J. pH-controlled reversible assembly and disassembly of gold nanorods. Small. 2008;4(9):1287–1292. | ||
Ramesh R, Ito I, Gopalan B, Saito Y, Mhashilkar AM, Chada S. Ectopic production of MDA-7/IL-24 inhibits invasion and migration of human lung cancer cells. Mol Ther. 2004;9(4):510–518. | ||
Inoue S, Hartman A, Branch CD, et al. mda-7 in combination with bevacizumab treatment produces a synergistic and complete inhibitory effect on lung tumor xenograft. Mol Ther. 2007;15(2):287–294. | ||
Panneerselvam J, Shanker M, Jin J, et al. Phosphorylation of interleukin (IL)-24 is required for mediating its anti-cancer activity. Oncotarget. 2015;6(18):16271–16286. | ||
Panneerselvam J, Jin J, Shanker M, et al. IL-24 inhibits lung cancer cell migration and invasion by disrupting the SDF-1/CXCR4 Signaling Axis. PLoS One. 2015;10(3):e0122439. | ||
Kuroda S, Tam J, Roth JA, Sokolov K, Ramesh R. EGFR-targeted plasmonic magnetic nanoparticles suppress lung tumor growth by abrogating G2/M cell-cycle arrest and inducing DNA damage. Int J Nanomedicine. 2014;9:3825–3839. | ||
Tanaka T, Munshi A, Brooks C, Liu J, Hobbs ML, Meyn RE. Gefitinib radiosensitizes non-small cell lung cancer cells by suppressing cellular DNA repair capacity. Clin Cancer Res. 2008;14(4):1266–1273. | ||
Cugia F, Monduzzi M, Ninhamb BW, Salis A. Interplay of ion specificity, pH and buffers: insights from electrophoretic mobility and pH measurements of lysozyme solutions. RSC Adv. 2013;3(17):5882–5888. | ||
El-Kareh AW, Secomb TW. Two-mechanism peak concentration model for cellular pharmacodynamics of doxorubicin. Neoplasia. 2005;7(7):705–713. | ||
Schafer K, Braun HA, Isenberg C. Effect of menthol on cold receptor activity. Analysis of receptor processes. J Gen Physiol. 1986;88(6):757–776. | ||
Moore A, Josephson L, Bhorade RM, Basilion JP, Weissleder R. Human transferrin receptor gene as a marker gene for MR imaging. Radiology. 2001;221(1):244–250. | ||
Lebro JA, Bennett MJ, Vaughn DE, et al. Crystal structure of the hemochromatosis protein HFE and characterization of its interaction with transferrin receptor. Cell. 1998;93(1):111–123. | ||
Mattia E, Rao K, Shapiro DS, Sussman HH, Klausner RD. Biosynthetic regulation of the human transferrin receptor by desferrioxamine in K562 cells. J Biol Chem. 1984;259(5):2689–2692. | ||
Casares N, Pequignot MO, Tesniere A, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med. 2005;202(12):1691–1701. | ||
Maejima Y, Adachi S, Ito H, Hirao K, Isobe M. Induction of premature senescence in cardiomyocytes by doxorubicin as a novel mechanism of myocardial damage. Aging Cell. 2008;7(2):125–136. | ||
Wang IK, Lin-Shiau SY, Lin JK. Induction of apoptosis by apigenin and related flavonoids through cytochrome c release and activation of Caspase-9 and Caspase-3 in Leukaemia HL-60 Cells. Eur J Cancer. 1999;35(10):1517–1525. | ||
Soleimani R, Heytens E, Darzynkiewicz Z, Oktay K. Mechanisms of chemotherapy-induced human ovarian aging: double strand DNA breaks and microvascular compromise. Aging (Albany NY). 2011;3(8):782–793. | ||
Sedelnikova OA, William MB. GammaH2AX in cancer cells: a potential biomarker for cancer diagnostics, prediction and recurrence. Cell Cycle. 2006;5(24):2909–2913. | ||
Olive PL, Banáth JP. The comet assay: a method to measure DNA damage in individual cells. Nat Protoc. 2006;1(1):23–29. | ||
Duan X, Xiao J, Yin Q, et al. Smart pH-sensitive and temporal-controlled polymeric micelles for effective combination therapy of doxorubicin and disulfiram. ACS Nano. 2013;7(7):5858–5869. | ||
Wang L, Ma W, Markovich R, Chen JW, Wang PH. Regulation of cardiomyocyte apoptotic signaling by insulin-like growth factor I. Circ Res. 1998;83(5):516–522. | ||
Sawyer DB, Fukazawa R, Arstall MA, Kelly RA. Daunorubicin-induced apoptosis in rat cardiac myocytes is inhibited by dexrazoxane. Circ Res. 1999;84(3):257–265. |
Supplementary materials
Materials
HS-PEG-OMe (6,000 Da), HS-PEG-SH (3,400 Da), doxorubicin (Dox), transferrin (Tf), iminothiolane, desferrioxamine, and ethylenediaminetetraacetic acid (EDTA) were all purchased from Sigma Chemicals. Gold nanorods (GNR) were procured from Nanopartz Inc. Maleimidocapric acid hydrazine (EMCH, Thermo Scientific), SephadexG-25 (PD10, GE, Erie, PA, USA), bovine serum albumin (BSA; KSE Scientific, Durham, NC, USA), CD71 mouse monoclonal primary antibody (Cell Signaling, Danvers, MA, USA), RPMI medium (Roswell Park Memorial Institute) (Cellgro, Manassas, VA, USA), trypan blue (Lonza, MD, USA), and polyvinylidene fluoride-membrane (Millipore, Billerica, MA, USA) were all purchased from commercial vendors.
Functionalization of gold nanorods
Presynthesized cetyltrimethylammonium bromide (CTAB) stabilized GNRs were purchased from Nanopartz Inc. and conjugated with HS-PEG-OMe (mPEG-SH, methoxy polyethylene glycol thiol) and HS-PEG-EMCH (HS-PEG-NH-NH2). Briefly, 10 mL of (1.3 nM) GNR was mixed with 500 μL of 1 mM mPEG-SH, and the mixture was stirred for 5 h at room temperature. Then, the unbound mPEG-SH and CTAB molecules were removed by centrifugation at 13,500 rpm for 15 min, and the pellet was dispersed in 5 mL of milliQ water and labeled as GNR-S-mPEG.
In another vial, 1 mL of 5 mM HS-PEG-SH solution was mixed with 1 mL of 5 mM maleimidocapric acid hydrazine in 2 mL of 0.1 M sodium phosphate containing EDTA (0.1 M; pH 7.15) solution. The mixture was stirred for 12 h at room temperature. The conjugated HS-PEG-EMCH (HS-PEG-NH-NH2) solution was purified by dialysis against milliQ water using a 3,500 Da dialysis bag.
From the purified HS-PEG-EMCH solution, 1.5 mL (1 mM) of the solution was added to GNR-S-mPEG solution and stirred for 6 h at room temperature. Then, the solution was centrifuged to remove unbound and nonspecific attached molecules, and was dispersed in 5 mL of phosphate-buffered saline (PBS) and labeled as GNR particles.
Conjugation of doxorubicin onto the functionalized GNRs
Doxorubicin (Dox; 600 μg) was added to the GNR particles (EMCH-PEG-S-GNR-S-mPEG) dispersed in 5 mL of PBS solution, and the mixture was stirred for 48 h at 37°C. The unbound Dox molecules were removed by centrifugation, and the resulting pellet was dispersed in 2 mL of PBS solution. The bound Dox concentration was estimated using absorbance spectroscopy by measuring optical density at 485 nm. Hereafter, this compound is referred to as GNR-Dox-NP.
Transferrin activation and conjugation onto the functionalized GNRs
Activation of Tf was achieved by mixing 13 mg of Tf (166 nmol) with 0.5 mg of iminothiolane (3.6 μmol) in 0.1 M sodium phosphate/EDTA (pH 8.0) solution, stirred for 4 h at 4°C to produce Tf-SH (iminothiolated Tf). Tf-SH was subsequently purified by size exclusion chromatography using a Sephadex G-25 (PD10) column. The purified Tf-SH was next incubated with GNR-Dox NP for 4 h at room temperature to produce GNR-Dox-Tf-NP. Conjugation of Tf to GNR-Dox-NP was determined by dot blot analysis.
Evaluation of the pH-controlled drug release studies
GNR-Dox-NP and GNR-Dox-Tf-NP were equally dispersed in two different buffer solutions, pH 5.5 acetate buffer (0.1 M) and 7.4 PBS (0.1 M). These two solutions were incubated at 37°C with mild stirring. At intervals of 1 h for 8 h, the solutions were centrifuged, and an aliquot of the supernatant was collected and an equal amount of buffers was added at each time point of sample collection. The collected samples were measured for fluorescence at 560/485 nm emission and excitation wavelengths, respectively. The percentage of Dox released in each buffer solution was calculated by cumulative fluorescence values and compared with free Dox.
Dot blot analysis
For determining efficient conjugation of Tf to GNR-Dox-NP, dot blot analysis was performed. Briefly, 3 μL of GNR-Dox-Tf-NP and Tf-SH were spotted onto a nitrocellulose membrane and allowed to dry for 20 min at room temperature. The membrane was then soaked in 5% BSA dissolved in TBS-T (10 mM Tris, pH 7.5, containing 0.15 M NaCl, 0.1% Tween 20) for 1 h at room temperature, followed by incubation with CD71 mouse monoclonal primary antibody (1:200 dilution with BSA) for 30 min at room temperature. Then, the membrane was washed three times with TBS-T for 5 min, followed by incubation with the appropriate secondary antimouse antibody (1:1,000) for 40 min at room temperature. The membrane was then washed with TBS-T three more times, incubated with a chemiluminescence reagent (Thermo Scientific) for 1 min, and blots developed by exposing the membrane to an autoradiographic film (Thermo Scientific).
Optimization of Dox and Tf concentration
A549 cells (0.2×106/well) suspended in RPMI media containing 10% serum were seeded in six-well tissue culture plates and incubated at 37°C. After 24 h, the culture medium was aspirated and replaced with 1 mL of serum-free medium. Incubation continued for 1.30 h to activate the receptors. Then, the cells were treated with GNR-Dox-NP carrying different concentrations (0.5, 1, 2, 4, 6, and 8 μg/mL) of Dox. After 4 h of Dox treatment, the culture medium was replaced with media containing 5% serum. Incubation continued for an additional 20 h. After the incubation period, the cells were washed twice with PBS and harvested. The number of viable cells was determined by trypan blue assay as previously described. The number of viable cells was calculated and expressed as a percentage. Cells that were not treated with Dox served as controls. Based on these results, we chose 2 μg/mL of Dox for all of the studies described herein.
To optimize the Tf concentration, A549 cells were seeded in six-well plates as described above for Dox optimization. We added GNR-Dox-Tf-NP with fixed Dox concentration (1.25 μg/mL) and varying Tf concentrations (0.05, 0.1, 0.2, 0.5, 1, and 2 μg/mL) to the cells. At 24 h after treatment, the cells were washed, and trypsinized, and cell viability was determined. From the results, 2 μg/mL of Tf was identified as the optimal concentration for the studies described herein.
Hyperspectral imaging
Hyperspectral data was acquired using CytoViva’s hyperspectral imaging system (CytoViva Inc., Auburn, AL, USA). The system contains a high signal-to-noise dark-field illuminator and hyperspectral imaging components integrated on a research-grade microscope. Using a motorized stage, hyperspectral data were collected in the 400–1,000 nm range for every pixel in the image one row at a time, utilizing the “pushbroom” approach. These data were compiled and presented as a datacube containing spectral and spatial data.
To identify the GNRs conjugated with Dox and Tf and Dox alone, a particle filter was run on all images to gather spectra corresponding to each substance to create reference spectral libraries. Particle filter collected spectra based on a set intensity threshold. Once the spectra were collected, they were filtered against the negative control image to eliminate any false positives. The remaining spectra comprised the spectral libraries representing GNR-Dox-NP, GNR-Dox-Tf-NP, and Dox alone.
The spectral libraries were then used to spectrally identify and spatially map GNR-Dox-NP, GNR-Dox-Tf-NP, and Dox in the 2 hour images of the cells containing each compound. Using the spectral angle mapper algorithm, the reference library was compared to each spectrum in the respective 2 hour images. Any pixel in the image whose spectrum matched a spectrum in the library was false-colored red.
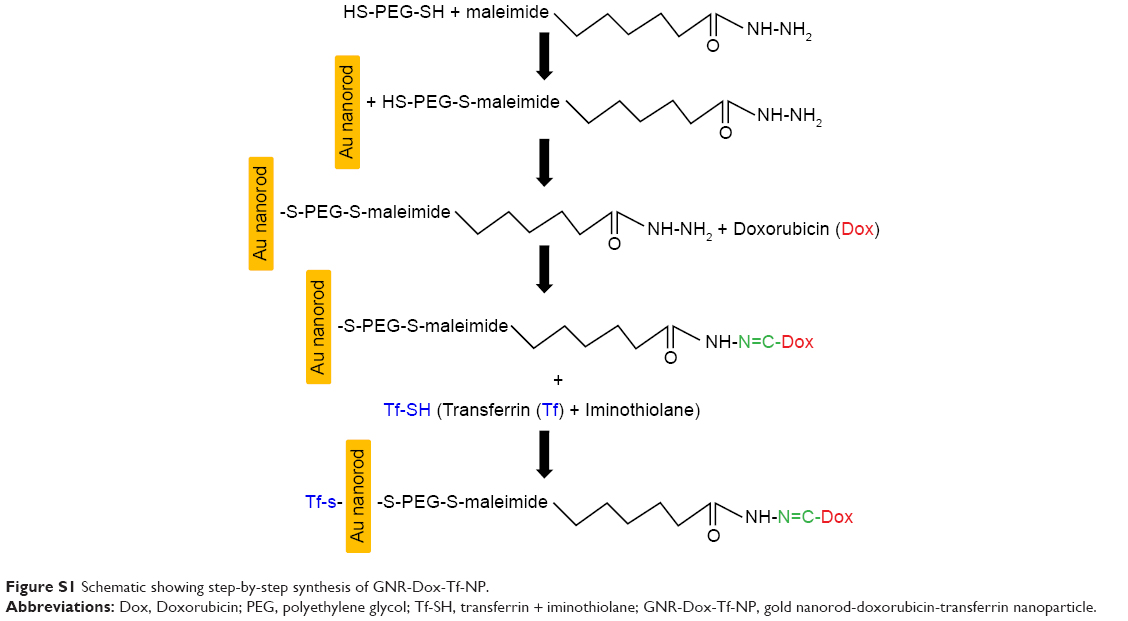 | Figure S1 Schematic showing step-by-step synthesis of GNR-Dox-Tf-NP. |
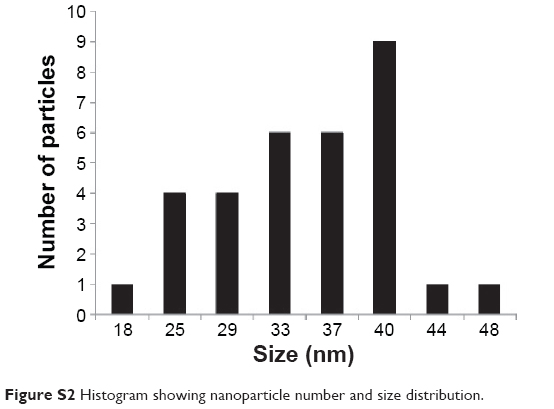 | Figure S2 Histogram showing nanoparticle number and size distribution. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.