Back to Archived Journals » Research and Reports in Biochemistry » Volume 5
Tumor necrosis factor-related apoptosis-inducing ligand-induced apoptotic pathways in cancer immunosurveillance: molecular mechanisms and prospects for therapy
Authors James B, Griffith T
Received 1 October 2014
Accepted for publication 12 November 2014
Published 22 December 2014 Volume 2015:5 Pages 1—10
DOI https://doi.org/10.2147/RRBC.S59123
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Nikolay Dokholyan
Britnie R James,1 Thomas S Griffith1–4
1Department of Urology, 2Microbiology, Immunology, and Cancer Biology Graduate Program, 3Center for Immunology, 4Masonic Cancer Center, University of Minnesota, Minneapolis, Minnesota, USA
Abstract: Since first described in 1995, tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) has generated considerable interest as a cancer therapeutic because of its ability to induce apoptosis in a range of tumor cell types while having little activity on normal cells and tissues. Since then, the vast majority of studies published on TRAIL and anti-TRAIL receptor monoclonal antibodies have focused on the tumoricidal activity of these molecules, with the intention of developing TRAIL-receptor agonists into potent cancer therapeutic agents. As promising as these agonists have proved to be in vitro and in various in vivo preclinical models, there have been a number of obstacles identified likely contributing to the underwhelming clinical trial data obtained – including a high frequency of TRAIL-resistant tumors – and reduced excitement about using TRAIL-receptor agonists as monotherapy for cancer. Consequently, it is important to understand the various mechanisms used by tumor cells to maintain TRAIL resistance and develop novel combinatorial approaches to restore TRAIL sensitivity in tumor cells. This review highlights the complexities of the TRAIL–TRAIL-receptor system, explores various methods for inducing TRAIL-induced death of tumor cells, and discusses some of the mechanisms that regulate tumor resistance to TRAIL and the way in which this resistance can be countered.
Keywords: TRAIL, TRAIL-R, TRAIL-receptor agonists, TRAIL resistance, cell death, tumor
Introduction
The process of cell death plays an essential natural physiological role in removing cells of the body that are produced in excess, diseased/damaged, or have completed a defined function. In the broadest sense, there are two means by which cell death can occur that are defined by classical biochemical and morphological criteria.1–3 One mechanism of cell death, “necrosis”, is typically induced by injury or a traumatic event, leading to the inability to maintain cell-membrane integrity and eventually the violent rupture of the cell(s).4 Necrosis generally leads to uncontrolled release of cellular components that can initiate an inflammatory response when it develops in vivo. “Apoptosis”, the other means by which cell death can proceed, is a more “civil” means of death characterized by numerous cellular changes that limit the release of cellular components with inflammatory potential.3
Apoptosis is an essential process in a number of basic physiological events and is regulated by many intracellular and extracellular cues.5,6 Tight control of this process is, obviously, critical to the survival of the organism, as evidenced by the studies from Hengartner and Horvitz describing the detailed apoptotic cell death that occurs in the nematode Caenorhabditis elegans.7,8 Failure of the apoptotic process has been implicated in certain autoimmune diseases (eg, autoimmune lymphoproliferative syndrome [ALPS]),9,10 but cancer is probably the best-studied disease state linked to a defect in the apoptotic cell death process.6 The identification and development of novel therapeutic agents that target the apoptotic pathway, through direct induction or counteracting resistance mechanisms, in tumor cells has become a focus of many laboratories around the world. Although there are many mechanisms that can ultimately result in cellular apoptosis, some of the best characterized are those induced through an active, instructive process mediated by the death receptors (DRs) of the tumor necrosis factor (TNF) receptor superfamily and their respective death ligands.11 These DRs are characterized by cysteine-rich extracellular domains,12,13 and a cytoplasmic sequence (termed the “death domain” [DD]) that serves as the aggregation point for the proteins that initiate the apoptotic signaling machinery.13–15 A variety of immunological functions are under the control of the corresponding death ligands in the TNF superfamily, with TNF and Fas ligand (FasL) being two of the most studied. Activation-induced cell death of lymphocytes, autoimmunity, maintenance of immune privilege, and tumor immunosurveillance are highly dependent on the proper expression and function of the TNF/TNF receptor and FasL/Fas pathways.16–20 However, interest in developing TNF and FasL as cancer therapeutics decreased significantly with reports describing life-threatening toxicity associated with the systemic administration of TNF and FasL (or anti-Fas monoclonal antibody [mAb]).21,22
Another member of the TNF superfamily capable of inducing apoptosis is tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), which was first identified after a screen of an expressed sequence tag database using a conserved sequence within a number of TNF family members.23 A similar approach was used by Pitti et al, who designated their discovery “Apo2 ligand” (Apo2L) due to its close homology to the Fas/apolipoprotein (Apo) 1 ligand.24 As with other TNF superfamily members, multimeric (or cross-linked) TRAIL is the most biologically active form, rather than monomeric.23 Early studies investigating the biology of TRAIL revealed two characteristics that were unique compared with other TNF superfamily members. First, TRAIL messenger RNA (mRNA) is constitutively expressed throughout the body.23 In contrast, the expression of other TNF superfamily members is tightly regulated and often transient. It has been hypothesized that TRAIL expression on cells within the immune system is critical in the immune system’s ability to eliminate nascent tumors, and there is increasing evidence supporting the role of TRAIL-induced death and/or immune cell activation in the efficacy of immunosurveillance of tumors. Studies using TRAIL-deficient mice and TRAIL-neutralizing antibodies have confirmed a role for TRAIL-induced death in controlling the progression of tumors and contributing to antitumor immunity.25–28 Second, unlike TNF and FasL, TRAIL preferentially induces apoptosis in transformed cells, while demonstrating little-to-no cytotoxic activity against normal cells and tissues.23 A number of subsequent reports have confirmed that malignant cells are the primary targets of TRAIL in vivo.29–33 Cells undergoing TRAIL-induced death exhibit many of the same canonical hallmarks of apoptosis induced by TNF or FasL, including DNA fragmentation, rapid expression of phosphatidylserine on the cell membrane, and caspase-mediated cleavage of multiple intracellular proteins.23,24,34,35
Because of the ability to preferentially induce tumor cell apoptosis, a number of recombinant TRAIL isoforms have been tested in preclinical studies using human tumor xenografts or immunocompetent mouse tumor models. Systemic administration of large amounts of recombinant TRAIL protein is one means by which the TRAIL receptors on cancer cells can be engaged to activate the apoptotic cell death machinery extrinsically.29,32 Identification of the cell-surface receptors that bind TRAIL and signal for apoptosis led to the development of a variety of mAb-based therapies.35–39 In addition to exploiting this pathway via recombinant proteins, others have developed gene-transfer therapies using viral vectors that encode for TRAIL protein.40–43 These methods can allow for the prolonged expression of TRAIL and with additional modifications to the vectors they can be targeted to specific sites of interest. In this review, we summarize the biology of TRAIL and the TRAIL receptors, strategies for targeting the TRAIL-induced death pathway, and preclinical/clinical studies aimed at exploiting this pathway for the treatment of cancers.
TRAIL and TRAIL receptors
Within the course of several months, a number of publications reported the identification of multiple cellular receptors with the ability to bind to either soluble or membrane-bound TRAIL. Four membrane-bound human TRAIL receptors were identified: DR4,44 DR5/ tumor necrosis factor-related apoptosis-inducing ligand receptor (TRAIL-R) 2/tumor necrosis factor-related apoptosis-inducing ligand receptor inducer of cell killing 2 (TRICK2)/KILLER,45–52 TRAIL-R3/decoy receptor (DcR) 1/tumor necrosis factor-related apoptosis-inducing ligand receptor without an intracellular domain (TRID)/lymphocyte inhibitor of TRAIL (LIT),45,46,49,50,53,54 and TRAIL-R4/DcR2/tumor necrosis factor-related apoptosis-inducing ligand receptor with a truncated death domain (TRUNDD)55–57 (we refer to these receptors as TRAIL-R1, -R2, -R3, and -R4, respectively; Figure 1). The TRAIL receptors are all expressed in a cell surface form and (with the exception of TRAIL-R3) are constitutively expressed in a wide variety of cells and tissues. Both TRAIL-R1 and -R2 contain DDs in the intracellular portions of the molecules, and ligation of either of these receptors is capable of inducing apoptosis.44–52 TRAIL-R3 is expressed as a glycosylphosphatidylinositol (GPI)-linked cell-surface protein with no known signaling properties.45,46,49,50,53,54 TRAIL-R4 contains only a partial DD and ligation of this receptor does not lead to apoptosis.55–57 Interestingly, the genes encoding these four receptors are all highly homologous (ranging from 54% to 70% identical) and map to a cluster in human chromosome 8p21-23, suggesting that they arose by gene duplication in the recent evolutionary past. It is believed that the DcRs can thus “compete” for binding of TRAIL, and reduce the apoptotic potential of TRAIL.46 In mice, genes for multiple TRAIL-receptor homologs have been identified, but only one receptor (KILLER/DR5) has been characterized at the protein level.58,59
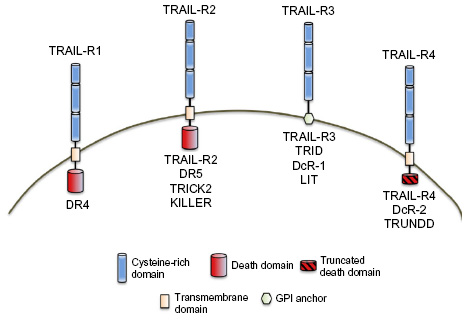 | Figure 1 Human tumor necrosis factor-related apoptosis-inducing ligand-receptor (TRAIL-R) structure and nomenclature. Death domains designate receptors (TRAIL-R1 and -R2) capable of inducing apoptosis. TRAIL-R3 and TRAIL-R4 are incapable of inducing apoptosis due to the lack of a functional intracellular death domain. |
Like other TNF family member proteins, TRAIL monomers form bell-shaped trimers as a result of head-to-tail interactions.60 TRAIL trimerization, like that seen for other TNF family members, corresponds to superior biologic activity over that of monomeric or dimeric versions of TRAIL.23 TRAIL-R1 (in humans) or -R2 ltrimerization activates the extrinsic apoptotic pathway61 (Figure 2), one of the two main pathways in which apoptotic death results from the systematic disassembly of the cell. Once trimerized, TRAIL-R1 or -R2 serves as the aggregation point for a multimeric protein structure called the “death-inducing signaling complex” (DISC) that is comprised of the ligated TRAIL-R1 or -R2, Fas-associated death-domain protein (FADD), and procaspases 8 and 10.62,63 Autocatalytic cleavage of procaspase 8 into its active form is followed by the cleavage and activation of caspases 3, 6, and 7, which are the “effector caspases” responsible for the proteolytic cleavage of multiple protein targets necessary for the maintenance of general cellular integrity (such as caspase-activated deoxyribonuclease [CAD], and poly(ADP-ribose)polymerase [PARP]).63 Active caspase 8 then amplifies the apoptotic signal by cleaving the pro-apoptotic B-cell leukemia/lymphoma 2 (Bcl-2) family protein Bcl-2 homology (BH) 3 interacting-domain death agonist (Bid), part of the BH3-only group of proteins that potently regulate apoptotic cell death,64 thereby simultaneously triggering the intrinsic apoptotic pathway that leads to a number of changes at the mitochondrial level that are controlled by interactions of pro- and anti-apoptotic proteins.65,66 The loss of mitochondrial membrane potential permits the escape of cytochrome c (Cyt-c) and second mitochondria-derived activator of caspases/direct inhibitor of apoptosis protein binding protein with low isoelectric point (Smac/DIABLO) into the cytosol. Cytosolic Cyt-c combines with adenosine triphosphate (ATP) and apoptotic peptidase-activating factor 1 (APAF-1) to form the apoptosome, which activates caspase 9.67,68 Smac/DIABLO blocks the function of caspase inhibitors such as X-linked inhibitor of apoptosis protein (XIAP).69,70 Thus, a stronger apoptotic signaling is generated after engagement of the intrinsic apoptotic pathway.
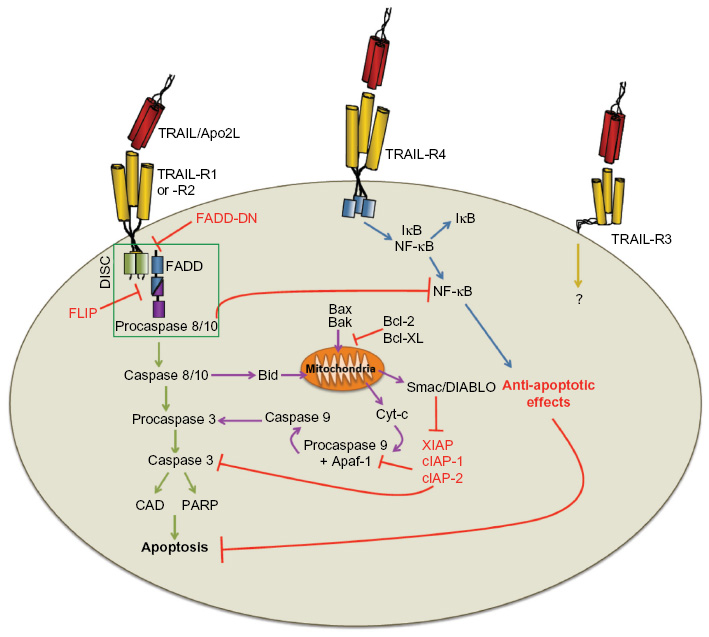 | Figure 2 Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced signaling pathways. |
Strategies for targeting the TRAIL-induced death pathway in cancer: preclinical/clinical studies
Targeting the TRAIL pathway to induce the death of malignant cells can be accomplished in a number of ways – exogenous deliveries of recombinant tumor necrosis factor-related apoptosis-inducing ligand (rTRAIL) protein or agonistic antibodies specific for TRAIL-R1 or -R2. The external carboxy-terminal region of TRAIL contains the receptor-binding domain and can be cleaved to yield a biologically active form of soluble TRAIL.71 Studies using rTRAIL in vivo have demonstrated low toxicity to normal cells while inducing tumor cell death in mouse models of cancer.29,32 However, subsequent studies using human cells suggested that tagged forms of the rTRAIL protein could in fact induce apoptosis in normal, primary human hepatocytes, similar to other TNF family member proteins.72 Studies of non-tagged forms of rTRAIL have shown that they are not toxic to normal human cells, while still inducing the death of human xenograft tumors in vivo.73,74 To date, there has been one rTRAIL compound developed for clinical application, dulanermin, which is comprised of the homology TNF domain within the extracellular domain of human TRAIL.29,75 In a dose-escalation study, dulanermin was found to be well tolerated as a single agent and resulted in an antitumor activity (partial response) in one cohort of patients and stable disease in another.75 Dulanermin has also been used in a number of clinical trials in which it was found to be nontoxic and well tolerated when combined with standard-of-care treatments.76,77 Unfortunately these combination studies have not indicated the definitive superiority of rTRAIL combination therapy over standard-of-care treatment alone. rTRAIL delivery is advantageous due to the fact that it can bind both TRAIL-R1 or -R2 resulting in a greater apoptotic signal; however rTRAIL can also bind to DcRs, which would result in no apoptotic event at all. Future clinical trials will be required to provide further evidence for the efficacy of rTRAIL-related therapies for cancer.
In addition to rTRAIL therapy, agonistic mAbs to TRAIL-R1 and -R2 have become attractive methods for inducing the TRAIL-apoptotic pathway in cancer cells. mAbs provide specific activation of the receptors that induce death, as opposed to rTRAIL protein, which has the potential to also bind non-active DcRs. mAbs also allow for a longer half-life in vivo compared with rTRAIL. A multitude of preclinical studies have been conducted to address the efficacy of mAbs for antitumor activity. The agonistic anti-mouse TRAIL-R-specific mAb MD5-1 has shown promising antitumor results in mouse models of cancer and is well tolerated. Takeda et al demonstrated the antitumor activity of MD5-1 in a syngeneic tumor model with immunocompetent mice.78 This study found that MD5-1 could inhibit the growth of TRAIL-sensitive tumors, but more importantly demonstrated the ability of MD5-1 to induce a tumor-specific immune response that subsequently eradicated TRAIL-resistant variants. These studies further elucidated the role for Fc receptor (FcR)-bearing immune cells cross-linking the Fc region of MD5-1 to mediate tumor cell lysis. Previous researchers had shown the necessity of cross-linking MD5-1 in vitro, but this was the first study to describe the role of FcR-expressing innate immune cells (eg, macrophage and dendritic cells) in MD5-1-mediated lysis in vivo.79
This observation was profound, as it led to the understanding of how MD5-1 mAb treatment induced an antitumor immune response capable of eradicating otherwise TRAIL-resistant cells. MD5-1-mediated tumor cell lysis results from cross-linking innate immune cells that express FcR with apoptotic tumor cells coated by MD5-1. The cross-linking event activates the immune cells, as well as induces the recruitment of other FcR-expressing cells to the tumor microenvironment – increasing the number of effector cells present. The activated FcR-expressing immune cells can endocytose the apoptotic tumor cell and cross-present tumor antigens to T-cells leading to the development of a tumor-specific immune response.78,79 This FcR-dependent phenomenon has also been documented using drozitumab, a human anti-TRAIL-R1 mAb, and other TNF family member mAbs.80,81 These preclinical studies have set the groundwork for mAbs to TRAIL-R1 or -R2 to move into clinical trials for cancer.
Multiple agonistic TRAIL-receptor-specific mAbs have been or are currently in human clinical trials: the anti-TRAIL-R1 mAb mapatumumab and anti-TRAIL-R2 mAbs conatumumab, lexatumumab, tigatuzumab, and drozitumab.82 As monotherapies, all of these mAbs have shown little-to-no adverse events in Phase I and II clinical trials testing for toxicity.36,37,83,84 For many of the mAbs, responses were sporadic and few, though stable disease was demonstrated in a population of patients in many of the trials suggesting an antitumor response. Further preclinical and clinical trials combining the various mAbs with other treatments have shown some promising results, suggesting additive and/or synergistic roles for mAbs with current, standard therapies.85–87 Most recently, a preclinical study from Tuthill et al demonstrated the synergistic relationship between the human anti-TRAIL-R2 mAb conatumumab and human rTRAIL dulanermin to kill primary ovarian cancer cells.88 They revealed that conatumumab binds a different epitope within TRAIL-R2 than dulanermin, allowing the concomitant binding of both reagents resulting in enhanced cross-linking and apoptosis-inducing capacity. Further, the combination therapy enhanced DISC formation and subsequent caspase 8 activation in the cancer cells.
In summary, TRAIL-receptor agonists have proven to be well tolerated and have yielded antitumor activity to some degree in patients (ie, stable disease or partial responses), though additional trials will need to be done to achieve significant activity with TRAIL-based therapies for cancer.
Mechanisms regulating resistance to TRAIL-induced apoptosis
One of the biggest questions regarding the biology of TRAIL has been what determines its selectivity for tumor cells. The molecular mechanisms underlying the profound differential sensitivities of normal and cancerous cells have been a subject of intense interest. An early hypothesis put forth to explain this difference was based on the observations that neither TRAIL-R3 nor -R4 was capable of activating the apoptotic process. Additionally ligation of TRAIL-R4 can trigger the nuclear factor of kappa B (NF-κB) pathway leading to anti-apoptotic effects and protection of the cell from TRAIL-R1 and -R2-induced death.55 Thus, it seemed possible that these receptors might act as “decoys” and functionally sequester TRAIL from the death-inducing TRAIL-R1 and -R2. Indeed, some early results were consistent with this hypothesis, including the observation that overexpression of TRAIL-R3 in TRAIL-sensitive target cells conferred resistance.46,49 In addition, treatment of endothelial cells with phospholipase C (to “strip” GPI-linked TRAIL-R3) in the presence of cycloheximide (to prevent re-expression of this molecule) was reported to result in the conversion of these cells from TRAIL resistant to sensitive.46 Similarly, overexpression of TRAIL-R4 in TRAIL-sensitive cells has also been shown to inhibit TRAIL-induced apoptosis.55–57 As attractive as the “decoy hypothesis” was as an explanation for the resistance of normal cells to the effects of TRAIL, it suffered from a variety of flaws. First, although TRAIL-R4 mRNA is detected in a wide variety of human tissues, significant levels of TRAIL-R3 mRNA are only seen in peripheral blood, spleen, lung, and skeletal muscle (lower levels have been reported in other tissues upon extended exposures of Northern blots).46,49,50,53,54 Second, in order for either TRAIL-R3 or -R4 to effectively act as “decoy” receptors they either need to be expressed at much higher levels or have markedly higher affinities than the two “death-inducing” receptors (TRAIL-R4 and -R2). Not only are the levels of expression of TRAIL-R3 and -R4 not significantly greater than -R1 and -R2 (indeed, as noted, the expression pattern of -R3 seems to be much more restricted than those of -R1 and -R2), but the affinities of the four TRAIL receptors are actually quite similar.50,53,55
If susceptibility to the apoptosis-inducing effects of TRAIL is not controlled by “decoy” receptors, how is it controlled? An initial clue to this mystery was provided by the observation that a number of tumor cells that are normally resistant to TRAIL are rendered susceptible by treatment with either actinomycin D or cycloheximide,34 suggesting that resistance of many tumors is mediated by a labile but constitutively produced protein(s) that interfere(s) with activation of the intracellular signaling process(es) that ultimately results in apoptosis. One potential candidate for such an intracellular inhibitor of apoptosis is the inhibitor of caspase 8 activation, cellular FLICE-inhibitory protein (cFLIP).89 Further investigation demonstrated that TRAIL-resistant tumor cells expressed high levels cFLIP. In contrast, TRAIL-sensitive tumor cells contained either no detectable cFLIP, or very low levels of this protein.34 Upon treatment of tumor target cells with actinomycin D the intracellular levels of cFLIP were found to rapidly decrease, and the sensitivity of the target cells to TRAIL commensurately increased. Expression or non-expression of cFLIP does not appear to be the sole regulator of sensitivity to TRAIL, as additional anti-apoptotic molecules, such as anti-apoptotic members of the Bcl-2 family of proteins, inhibitors of apoptosis proteins (IAPS), Akt, and Toso, have also been suggested to regulate TRAIL-receptor signaling.34,90–93
Finally, posttranslational modifications have been suggested to participate in the regulation of tumor cell sensitivity to TRAIL, as there are data suggesting a link between DR O-glycosylation mediated by polypeptide N-acetylgalactosaminyltransferase 14 (GALNT14) and TRAIL sensitivity.94 This relationship has only been observed in human non-small-cell lung carcinoma, pancreatic cancer, and melanoma cell lines, so it is unknown to what extent GALNT14-mediated O-glycosylation contributes to the sensitivity in a broader panel of tumor cells. Moreover, the presence of TRAIL DRs in lipid rafts within the cell membrane constitutes another essential mechanism for efficient signaling.95–97 There has even been investigation into the relationship between tumor cell sensitivity to TRAIL and cell-cycle progression,98,99 as G0-, G1-, or G2-arrested tumor cells were more sensitive to TRAIL than non-arrested cells.
Countering resistance to TRAIL-based therapeutic agents
Because of these resistance mechanisms, identifying combinations with current therapies capable of sensitizing tumors to TRAIL-induced apoptosis has become an important topic of research. Synergistic effects of current cancer therapies in combination with TRAIL agonists have previously been reported in the literature. These potentiating effects are commonly due to expression regulation of proteins that play important roles in the TRAIL-induced death pathway, and it is likely that these combinations can make TRAIL-receptor agonists a feasible approach for treating cancer.
Of the drugs that currently exist for the treatment of cancer, histone deacetylase inhibitors (HDACi) are attractive candidates for combination therapy with TRAIL-receptor agonists. HDACi epigenetically alter gene expression as a result of increased histone acetylation. HDACi have been shown to not only increase the cell-surface expression of TRAIL-R1 and -R2, but also to increase the signaling efficiency upon ligation of the receptors.95,100–102 Furthermore, HDACi can alter the expression of pro- and anti-apoptotic proteins that regulate the TRAIL-induced death pathway. Increased expression/activation of caspase 8, Bid, and Bax by HDACi has been observed.103,104 HDACi-induced TRAIL sensitization can also occur through the downregulation of anti-apoptotic Bcl-2 family proteins and cFLIP,105,106 known inhibitors of the TRAIL-apoptotic pathway. Proteasome inhibitors have also been investigated as possible combinatorial agents. Prolonged proteasome inhibition results in increased expression and accumulation of pro-apoptotic proteins, decreased cFLIP, and induction of cell-cycle arrest – all of which can increase TRAIL sensitivity.107–109 Proteasome inhibitors, such as bortezomib, can also increase TRAIL-receptor agonist sensitivity of cancer cells through upregulation of TRAIL-R1/-R2.110,111 Aside from HDACi and proteasome inhibitors, a broad range of standard chemotherapeutics have also demonstrated similar results to increase sensitivity in combination with TRAIL-receptor agonists.85–87,112
Chemotherapeutics are efficient at killing tumor cells; however their ability (as single agents) to induce long-lasting immunity against tumors can be limited and drug-type dependent.113–116 Therefore, a growing area of research is the combination of TRAIL-receptor agonists with cytokine therapy or immunomodulators to help elicit antitumor immunity. Interferons (IFNs) are a pleiotropic family of cytokines that directly act on and inhibit tumor cell function. For these reasons IFNs have been used to treat over 14 types of malignancies and have the longest documented use in oncology.117,118 In contrast to simply killing tumor cells, IFNs play a crucial role in the induction of antitumor immunity,119–121 and can modulate the death-inducing signaling pathway.122,123 Preclinical studies combining IFN-γ with rTRAIL or anti-TRAIL-R1/-R2 mAb resulted in increased tumor cell death and inhibition of tumor outgrowth in xenografted mice.124 Immunomodulators, such as mAb against immune-stimulating or immune-suppressive cellular receptors,125 provide another combinatorial option for optimizing DR-agonist efficacy. One such option is the implementation of “trimAb”, a combination of anti-TRAIL-R2 mAb with T-cell activating mAb against cluster of differentiation (CD) 40 and CD137.38 The concomitant activation of T-cells has become an attractive approach to augment the therapeutic effect of anti-TRAIL-R2 mAb.126,127 Because anti-TRAIL-R2 mAb can activate innate immune cells via FcR cross-linking, the addition of T-cell activating mAb is thought to potentiate the induction of antitumor immunity. Mechanistically, trimAb works to kill the tumor cells (via anti-TRAIL-R2) resulting in the release of tumor antigens that can be effectively recognized by the immune system (with help from anti-CD40 and CD137 stimulation) to mount a robust antitumor response.128 Preclinical models using trimAb have demonstrated the ability to elicit tumor-specific T-cell responses capable of eradicating established tumors, even those that were otherwise resistant to TRAIL-induced death.38
Conclusion and perspectives
Though all of these combinatorial treatments are promising, one must remember that while these drugs can increase cancer cell sensitivity to TRAIL-receptor-agonist-induced death, they may also sensitize normal cells as well. It will be important to carefully assess the off-target effects of combinatorial therapies. However, evidence from preclinical and even clinical studies clearly demonstrates the potential for TRAIL-based therapeutics in the treatment of cancers. The pitfalls of these therapies have been outlined, and the strategies to overcome these shortcomings are being rigorously investigated. Novel/altered TRAIL DR agonists with increased sensitivity, selection, and potency are needed and are currently being developed. Even if TRAIL-based monotherapies are suboptimal, combination therapies are still a promising avenue, especially those that include immunomodulators, given that anti-TRAIL-R1 and -R2 mAbs naturally induce an immune response via FcR cross-linking.
Acknowledgments
The authors thank Dr David H Lynch for helpful discussions. This work was supported by National Institutes of Health Grant CA109446 (TSG) and a Careers in Immunology Fellowship from the American Association of Immunologists (TSG and BRJ).
Disclosure
The authors report no conflicts of interest in this work.
References
Cohen JJ. Apoptosis. Immunol Today. 1993;14(3):126–130. | |
Wyllie AH, Kerr JF, Currie AR. Cell death: the significance of apoptosis. Int Rev Cytol. 1980;68:251–306. | |
Vaux DL, Strasser A. The molecular biology of apoptosis. Proc Natl Acad Sci U S A. 1996;93(6):2239–2244. | |
Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26(4):239–257. | |
Steller H. Mechanisms and genes of cellular suicide. Science. 1995;267(5203):1445–1449. | |
Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267(5203):1456–1462. | |
Hengartner MO, Horvitz HR. The ins and outs of programmed cell death during C. elegans development. Philos Trans R Soc Lond B Biol Sci. 1994;345(1313):243–246. | |
Hengartner MO, Horvitz HR. Programmed cell death in Caenorhabditis elegans. Curr Opin Genet Dev. 1994;4(4):581–586. | |
Jackson CE, Puck JM. Autoimmune lymphoproliferative syndrome, a disorder of apoptosis. Curr Opin Pediatr. 1999;11(6):521–527. | |
Siegel RM, Fleisher TA. The role of Fas and related death receptors in autoimmune and other disease states. J Allergy Clin Immunol. 1999;103(5 Pt 1):729–738. | |
Li J, Yin Q, Wu H. Structural basis of signal transduction in the TNF receptor superfamily. Adv Immunol. 2013;119:135–153. | |
Armitage RJ. Tumor necrosis factor receptor superfamily members and their ligands. Curr Opin Immunol. 1994;6(3):407–413. | |
Smith CA, Farrah T, Goodwin RG. The TNF receptor superfamily of cellular and viral proteins: activation, costimulation, and death. Cell. 1994;76(6):959–962. | |
Itoh N, Nagata S. A novel protein domain required for apoptosis. Mutational analysis of human Fas antigen. J Biol Chem. 1993;268(15):10932–10937. | |
Tartaglia LA, Ayres TM, Wong GH, Goeddel DV. A novel domain within the 55 kd TNF receptor signals cell death. Cell. 1993;74(5):845–853. | |
Alderson MR, Tough TW, Davis-Smith T, et al. Fas ligand mediates activation-induced cell death in human T lymphocytes. J Exp Med. 1995;181(1):71–77. | |
Cerami A, Beutler B. The role of cachectin/TNF in endotoxic shock and cachexia. Immunol Today. 1988;9(1):28–31. | |
Griffith TS, Brunner T, Fletcher SM, Green DR, Ferguson TA. Fas ligand-induced apoptosis as a mechanism of immune privilege. Science. 1995;270(5239):1189–1192. | |
Hahne M, Rimoldi D, Schröter M, et al. Melanoma cell expression of Fas (Apo-1/CD95) ligand: implications for tumor immune escape. Science. 1996;274(5291):1363–1366. | |
Zheng L, Fisher G, Miller RE, Peschon J, Lynch DH, Lenardo MJ. Induction of apoptosis in mature T cells by tumour necrosis factor. Nature. 1995;377(6547):348–351. | |
Ogasawara J, Watanabe-Fukunaga R, Adachi M, et al. Lethal effect of the anti-Fas antibody in mice. Nature. 1993;364(6440):806–809. | |
Havell EA, Fiers W, North RJ. The antitumor function of tumor necrosis factor (TNF), I. Therapeutic action of TNF against an established murine sarcoma is indirect, immunologically dependent, and limited by severe toxicity. J Exp Med. 1988;167(3):1067–1085. | |
Wiley SR, Schooley K, Smolak PJ, et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity. 1995;3(6):673–682. | |
Pitti RM, Marsters SA, Ruppert S, Donahue CJ, Moore A, Ashkenazi A. Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J Biol Chem. 1996;271(22):12687–12690. | |
Cretney E, Takeda K, Yagita H, Glaccum M, Peschon JJ, Smyth MJ. Increased susceptibility to tumor initiation and metastasis in TNF-related apoptosis-inducing ligand-deficient mice. J Immunol. 2002;168(3):1356–1361. | |
Seki N, Hayakawa Y, Brooks AD, et al. Tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis is an important endogenous mechanism for resistance to liver metastases in murine renal cancer. Cancer Res. 2003;63(1):207–213. | |
Takeda K, Smyth MJ, Cretney E, et al. Critical role for tumor necrosis factor-related apoptosis-inducing ligand in immune surveillance against tumor development. J Exp Med. 2002;195(2):161–169. | |
Takeda K, Smyth MJ, Cretney E, et al. Involvement of tumor necrosis factor-related apoptosis-inducing ligand in NK cell-mediated and IFN-gamma-dependent suppression of subcutaneous tumor growth. Cell Immunol. 2001;214(2):194–200. | |
Ashkenazi A, Pai RC, Fong S, et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest. 1999;104(2):155–162. | |
Huang X, Lin T, Gu J, et al. Combined TRAIL and Bax gene therapy prolonged survival in mice with ovarian cancer xenograft. Gene Ther. 2002;9(20):1379–1386. | |
Lin T, Zhang L, Davis J, et al. Combination of TRAIL gene therapy and chemotherapy enhances antitumor and antimetastasis effects in chemosensitive and chemoresistant breast cancers. Mol Ther. 2003;8(3):441–448. | |
Walczak H, Miller RE, Ariail K, et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat Med. 1999;5(2):157–163. | |
Yang F, Shi P, Xi X, et al. Recombinant adenoviruses expressing TRAIL demonstrate antitumor effects on non-small cell lung cancer (NSCLC). Med Oncol. 2006;23(2):191–204. | |
Griffith TS, Chin WA, Jackson GC, Lynch DH, Kubin MZ. Intracellular regulation of TRAIL-induced apoptosis in human melanoma cells. J Immunol. 1998;161(6):2833–2840. | |
Griffith TS, Rauch CT, Smolak PJ, et al. Functional analysis of TRAIL receptors using monoclonal antibodies. J Immunol. 1999;162(5):2597–2605. | |
Forero-Torres A, Shah J, Wood T, et al. Phase I trial of weekly tigatuzumab, an agonistic humanized monoclonal antibody targeting death receptor 5 (DR5). Cancer Biother Radiopharm. 2010;25(1):13–19. | |
Rosevear HM, Lightfoot AJ, Griffith TS. Conatumumab, a fully human mAb against death receptor 5 for the treatment of cancer. Curr Opin Investig Drugs. 2010;11(6):688–698. | |
Uno T, Takeda K, Kojima Y, et al. Eradication of established tumors in mice by a combination antibody-based therapy. Nat Med. 2006;12(6):693–698. | |
Holoch PA, Griffith TS. TNF-related apoptosis-inducing ligand (TRAIL):a new path to anti-cancer therapies. Eur J Pharmacol. 2009;625(1–3):63–72. | |
Griffith TS, Anderson RD, Davidson BL, Williams RD, Ratliff TL. Adenoviral-mediated transfer of the TNF-related apoptosis-inducing ligand/Apo-2 ligand gene induces tumor cell apoptosis. J Immunol. 2000;165(5):2886–2894. | |
Griffith TS, Kawakita M, Tian J, et al. Inhibition of murine prostate tumor growth and activation of immunoregulatory cells with recombinant canarypox viruses. J Natl Cancer Inst. 2001;93(13):998–1007. | |
James BR, Anderson KG, Brincks EL, et al. CpG-mediated modulation of MDSC contributes to the efficacy of Ad5-TRAIL therapy against renal cell carcinoma. Cancer Immunol Immunother. 2014;63(11):1213–1227. | |
James BR, Brincks EL, Kucaba TA, Boon L, Griffith TS. Effective TRAIL-based immunotherapy requires both plasmacytoid and CD8a DC. Cancer Immunol Immunother. 2014;63(7):685–697. | |
Pan G, O’Rourke K, Chinnaiyan AM, et al. The receptor for the cytotoxic ligand TRAIL. Science. 1997;276(5309):111–113. | |
Pan G, Ni J, Wei YF, Yu G, Gentz R, Dixit VM. An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science. 1997;277(5327):815–818. | |
Sheridan JP, Marsters SA, Pitti RM, et al. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science. 1997;277(5327):818–821. | |
Walczak H, Degli-Esposti MA, Johnson RS, et al. TRAIL-R2: a novel apoptosis-mediating receptor for TRAIL. EMBO J. 1997;16(17):5386–5397. | |
Chaudhary PM, Eby M, Jasmin A, Bookwalter A, Murray J, Hood L. Death receptor 5, a new member of the TNFR family, and DR4 induce FADD-dependent apoptosis and activate the NF-kappaB pathway. Immunity. 1997;7(6):821–830. | |
MacFarlane M, Ahmad M, Srinivasula SM, Fernandes-Alnemri T, Cohen GM, Alnemri ES. Identification and molecular cloning of two novel receptors for the cytotoxic ligand TRAIL. J Biol Chem. 1997;272(41):25417–25420. | |
Schneider P, Bodmer JL, Thome M, Hofmann K, Holler N, Tschopp J. Characterization of two receptors for TRAIL. FEBS Lett. 1997;416(3):329–334. | |
Screaton GR, Mongkolsapaya J, Xu XN, Cowper AE, McMichael AJ, Bell JI. TRICK2, a new alternatively spliced receptor that transduces the cytotoxic signal from TRAIL. Curr Biol. 1997;7(9):693–696. | |
Wu GS, Burns TF, McDonald ER 3rd, et al. KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat Genet. 1997;17(2):141–143. | |
Degli-Esposti MA, Smolak PJ, Walczak H, et al. Cloning and characterization of TRAIL-R3, a novel member of the emerging TRAIL receptor family. J Exp Med. 1997;186:1165–1170. | |
Mongkolsapaya J, Cowper AE, Xu XN, et al. Lymphocyte inhibitor of TRAIL (TNF-related apoptosis-inducing ligand):a new receptor protecting lymphocytes from the death ligand TRAIL. J Immunol. 1998;160(1):3–6. | |
Degli-Esposti MA, Dougall WC, Smolak PJ, Waugh JY, Smith CA, Goodwin RG. The novel receptor TRAIL-R4 induces NF-kappaB and protects against TRAIL-mediated apoptosis, yet retains an incomplete death domain. Immunity. 1997;7(6):813–820. | |
Marsters SA, Sheridan JP, Pitti RM, et al. A novel receptor for Apo2L/TRAIL contains a truncated death domain. Curr Biol. 1997;7(12):1003–1006. | |
Pan G, Ni J, Yu G, Wei YF, Dixit VM. TRUNDD, a new member of the TRAIL receptor family that antagonizes TRAIL signalling. FEBS Lett. 1998;424(1–2):41–45. | |
Schneider P, Olson D, Tardivel A, et al. Identification of a new murine tumor necrosis factor receptor locus that contains two novel murine receptors for tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). J Biol Chem. 2003;278(7):5444–5454. | |
Wu GS, Burns TF, Zhan Y, Alnemri ES, El-Deiry WS. Molecular cloning and functional analysis of the mouse homologue of the KILLER/DR5 tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) death receptor. Cancer Res. 1999;59(12):2770–2775. | |
Cha SS, Kim MS, Choi YH, et al. 2.8 A resolution crystal structure of human TRAIL, a cytokine with selective antitumor activity. Immunity. 1999;11(2):253–261. | |
Testa U. Apoptotic mechanisms in the control of erythropoiesis. Leukemia. 2004;18(7):1176–1199. | |
Sprick MR, Weigand MA, Rieser E, et al. FADD/MORT1 and caspase-8 are recruited to TRAIL receptors 1 and 2 and are essential for apoptosis mediated by TRAIL receptor 2. Immunity. 2000;12(6):599–609. | |
Kischkel FC, Lawrence DA, Chuntharapai A, Schow P, Kim KJ, Ashkenazi A. Apo2L/TRAIL-dependent recruitment of endogenous FADD and caspase-8 to death receptors 4 and 5. Immunity. 2000;12(6):611–620. | |
Kelekar A, Thompson CB. Bcl-2-family proteins: the role of the BH3 domain in apoptosis. Trends Cell Biol. 1998;8(8):324–330. | |
Wei MC, Zong WX, Cheng EH, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292(5517):727–730. | |
Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94(4):491–501. | |
Li P, Nijhawan D, Budihardjo I, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91(4):479–489. | |
Schulze-Osthoff K, Ferrari D, Los M, Wesselborg S, Peter ME. Apoptosis signaling by death receptors. Eur J Biochem. 1998;254(3):439–459. | |
Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102(1):33–42. | |
Verhagen AM, Ekert PG, Pakusch M, et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102(1):43–53. | |
Mariani SM, Krammer PH. Differential regulation of TRAIL and CD95 ligand in transformed cells of the T and B lymphocyte lineage. Eur J Immunol. 1998;28(3):973–982. | |
Jo M, Kim TH, Seol DW, et al. Apoptosis induced in normal human hepatocytes by tumor necrosis factor-related apoptosis-inducing ligand. Nat Med. 2000;6(5):564–567. | |
Hao C, Song JH, Hsi B, et al. TRAIL inhibits tumor growth but is nontoxic to human hepatocytes in chimeric mice. Cancer Res. 2004;64(23):8502–8506. | |
Lawrence D, Shahrokh Z, Marsters S, et al. Differential hepatocyte toxicity of recombinant Apo2L/TRAIL versions. Nat Med. 2001;7(4):383–385. | |
Herbst RS, Eckhardt SG, Kurzrock R, et al. Phase I dose-escalation study of recombinant human Apo2L/TRAIL, a dual proapoptotic receptor agonist, in patients with advanced cancer. J Clin Oncol. 2010;28(17):2839–2846. | |
Sun S, Li Z, Sun L, et al. Results on efficacy and safety of cancer treatment with or without tumor necrosis factor-related apoptosis-inducing ligand-related agents: A meta-analysis. Mol Clin Oncol. 2014;2(3):440–448. | |
Soria JC, Márk Z, Zatloukal P, et al. Randomized phase II study of dulanermin in combination with paclitaxel, carboplatin, and bevacizumab in advanced non-small-cell lung cancer. J Clin Oncol. 2011;29(33):4442–4451. | |
Takeda K, Yamaguchi N, Akiba H, et al. Induction of tumor-specific T cell immunity by anti-DR5 antibody therapy. J Exp Med. 2004;199(4):437–448. | |
Haynes NM, Hawkins ED, Li M, et al. CD11c+ dendritic cells and B cells contribute to the tumoricidal activity of anti-DR5 antibody therapy in established tumors. J Immunol. 2010;185(1):532–541. | |
White AL, Chan HT, French RR, et al. FcγRIIB controls the potency of agonistic anti-TNFR mAbs. Cancer Immunol Immunother. 2013;62(5):941–948. | |
Wilson NS, Yang B, Yang A, et al. An Fcγ receptor-dependent mechanism drives antibody-mediated target-receptor signaling in cancer cells. Cancer Cell. 2011;19(1):101–113. | |
Lemke J, von Karstedt S, Zinngrebe J, Walczak H. Getting TRAIL back on track for cancer therapy. Cell Death Differ. 2014;21(9):1350–1364. | |
Tolcher AW, Mita M, Meropol NJ, et al. Phase I pharmacokinetic and biologic correlative study of mapatumumab, a fully human monoclonal antibody with agonist activity to tumor necrosis factor-related apoptosis-inducing ligand receptor-1. J Clin Oncol. 2007;25(11):1390–1395. | |
Plummer R, Attard G, Pacey S, et al. Phase 1 and pharmacokinetic study of lexatumumab in patients with advanced cancers. Clin Cancer Res. 2007;13(20):6187–6194. | |
Mom CH, Verweij J, Oldenhuis CN, et al. Mapatumumab, a fully human agonistic monoclonal antibody that targets TRAIL-R1, in combination with gemcitabine and cisplatin: a phase I study. Clin Cancer Res. 2009;15(17):5584–5590. | |
Cohn AL, Tabernero J, Maurel J, et al. A randomized, placebo-controlled phase 2 study of ganitumab or conatumumab in combination with FOLFIRI for second-line treatment of mutant KRAS metastatic colorectal cancer. Ann Oncol. 2013;24(7):1777–1785. | |
Rajeshkumar NV, Rasheed ZA, García-García E, et al. A combination of DR5 agonistic monoclonal antibody with gemcitabine targets pancreatic cancer stem cells and results in long-term disease control in human pancreatic cancer model. Mol Cancer Ther. 2010;9(9):2582–2592. | |
Tuthill MH, Montinaro A, Zinngrebe J, et al. TRAIL-R2-specific antibodies and recombinant TRAIL can synergise to kill cancer cells. Oncogene. Epub June 9, 2014. | |
Irmler M, Thome M, Hahne M, et al. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388(6638):190–195. | |
Deng Y, Lin Y, Wu X. TRAIL-induced apoptosis requires Bax-dependent mitochondrial release of Smac/DIABLO. Genes Dev. 2002;16(1):33–45. | |
Fulda S, Meyer E, Debatin KM. Inhibition of TRAIL-induced apoptosis by Bcl-2 overexpression. Oncogene. 2002;21(15):2283–2294. | |
Griffith TS, Fialkov JM, Scott DL, et al. Induction and regulation of tumor necrosis factor-related apoptosis-inducing ligand/Apo-2 ligand-mediated apoptosis in renal cell carcinoma. Cancer Res. 2002;62(11):3093–3099. | |
Ng CP, Bonavida B. X-linked inhibitor of apoptosis (XIAP) blocks Apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis of prostate cancer cells in the presence of mitochondrial activation: sensitization by overexpression of second mitochondria-derived activator of caspase/direct IAP-binding protein with low pl (Smac/DIABLO). Mol Cancer Ther. 2002;1(12):1051–1058. | |
Wagner KW, Punnoose EA, Januario T, et al. Death-receptor O-glycosylation controls tumor-cell sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nat Med. 2007;13(9):1070–1077. | |
VanOosten RL, Moore JM, Ludwig AT, Griffith TS. Depsipeptide (FR901228) enhances the cytotoxic activity of TRAIL by redistributing TRAIL receptor to membrane lipid rafts. Mol Ther. 2005;11(4):542–552. | |
Delmas D, Rébé C, Micheau O, et al. Redistribution of CD95, DR4 and DR5 in rafts accounts for the synergistic toxicity of resveratrol and death receptor ligands in colon carcinoma cells. Oncogene. 2004;23(55):8979–8986. | |
Ouyang W, Yang C, Zhang S, et al. Absence of death receptor translocation into lipid rafts in acquired TRAIL-resistant NSCLC cells. Int J Oncol. 2013;42(2):699–711. | |
Ehrhardt H, Wachter F, Grunert M, Jeremias I. Cell cycle-arrested tumor cells exhibit increased sensitivity towards TRAIL-induced apoptosis. Cell Death Dis. 2013;4:e661. | |
Jin Z, Dicker DT, El-Deiry WS. Enhanced sensitivity of G1 arrested human cancer cells suggests a novel therapeutic strategy using a combination of simvastatin and TRAIL. Cell Cycle. 2002;1(1):82–89. | |
VanOosten RL, Earel JK, Griffith TS. Enhancement of Ad5-TRAIL cytotoxicity against renal cell carcinoma with histone deacetylase inhibitors. Cancer Gene Ther. 2006;13(6):628–632. | |
VanOosten RL, Earel JK Jr, Griffith TS. Histone deacetylase inhibitors enhance Ad5-TRAIL killing of TRAIL-resistant prostate tumor cells through increased caspase-2 activity. Apoptosis. 2007;12(3):561–571. | |
VanOosten RL, Moore JM, Karacay B, Griffith TS. Histone deacetylase inhibitors modulate renal cell carcinoma sensitivity to TRAIL/Apo-2L-induced apoptosis by enhancing TRAIL-R2 expression. Cancer Biol Ther. 2005;4(10):1104–1112. | |
Häcker S, Dittrich A, Mohr A, et al. Histone deacetylase inhibitors cooperate with IFN-gamma to restore caspase-8 expression and overcome TRAIL resistance in cancers with silencing of caspase-8. Oncogene. 2009;28(35):3097–3110. | |
Rosato RR, Almenara JA, Dai Y, Grant S. Simultaneous activation of the intrinsic and extrinsic pathways by histone deacetylase (HDAC) inhibitors and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) synergistically induces mitochondrial damage and apoptosis in human leukemia cells. Mol Cancer Ther. 2003;2(12):1273–1284. | |
Watanabe K, Okamoto K, Yonehara S. Sensitization of osteosarcoma cells to death receptor-mediated apoptosis by HDAC inhibitors through downregulation of cellular FLIP. Cell Death Diff. 2005;12(1):10–18. | |
Gillespie S, Borrow J, Zhang XD, Hersey P. Bim plays a crucial role in synergistic induction of apoptosis by the histone deacetylase inhibitor SBHA and TRAIL in melanoma cells. Apoptosis. 2006;11(12):2251–2265. | |
Bonvini P, Zorzi E, Basso G, Rosolen A. Bortezomib-mediated 26S proteasome inhibition causes cell-cycle arrest and induces apoptosis in CD-30+ anaplastic large cell lymphoma. Leukemia. 2007;21(4):838–842. | |
Seki N, Toh U, Sayers TJ, et al. Bortezomib sensitizes human esophageal squamous cell carcinoma cells to TRAIL-mediated apoptosis via activation of both extrinsic and intrinsic apoptosis pathways. Mol Cancer Ther. 2010;9(6):1842–1851. | |
Shanker A, Brooks AD, Tristan CA, et al. Treating metastatic solid tumors with bortezomib and a tumor necrosis factor-related apoptosis-inducing ligand receptor agonist antibody. J Natl Cancer Inst. 2008;100(9):649–662. | |
Smith MR, Jin F, Joshi I. Bortezomib sensitizes non-Hodgkin’s lymphoma cells to apoptosis induced by antibodies to tumor necrosis factor related apoptosis-inducing ligand (TRAIL) receptors TRAIL-R1 and TRAIL-R2. Clin Cancer Res. 2007;13(18 Pt 2):5528s–5534s. | |
Voortman J, Resende TP, Abou El Hassan MA, Giaccone G, Kruyt FA. TRAIL therapy in non-small cell lung cancer cells: sensitization to death receptor-mediated apoptosis by proteasome inhibitor bortezomib. Mol Cancer Ther. 2007;6(7):2103–2112. | |
Newsom-Davis T, Prieske S, Walczak H. Is TRAIL the holy grail of cancer therapy? Apoptosis. 2009;14(4):607–623. | |
Zitvogel L, Apetoh L, Ghiringhelli F, André F, Tesniere A, Kroemer G. The anticancer immune response: indispensable for therapeutic success? J Clin Invest. 2008;118(6):1991–2001. | |
Zitvogel L, Kepp O, Kroemer G. Immune parameters affecting the efficacy of chemotherapeutic regimens. Nat Rev Clin Oncol. 2011;8(3):151–160. | |
Obeid M, Tesniere A, Ghiringhelli F, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13(1):54–61. | |
Casares N, Pequignot MO, Tesniere A, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med. 2005;202(12):1691–1701. | |
Ferrantini M, Capone I, Belardelli F. Interferon-alpha and cancer: mechanisms of action and new perspectives of clinical use. Biochimie. 2007;89(6–7):884–893. | |
Vilcek J. Fifty years of interferon research: aiming at a moving target. Immunity. 2006;25(3):343–348. | |
Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol. 2006;6(11):836–848. | |
Fuertes MB, Kacha AK, Kline J, et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med. 2011;208(10):2005–2016. | |
Swann JB, Hayakawa Y, Zerafa N, et al. Type I IFN contributes to NK cell homeostasis, activation, and antitumor function. J Immunol. 2007;178(12):7540–7549. | |
Varela N, Munoz-Pinedo C, Ruiz-Ruiz C, Robledo G, Pedroso M, López-Rivas A. Interferon-gamma sensitizes human myeloid leukemia cells to death receptor-mediated apoptosis by a pleiotropic mechanism. J Biol Chem. 2001;276(21):17779–17787. | |
Kayagaki N, Yamaguchi N, Nakayama M, Eto H, Okumura K, Yagita H. Type I interferons (IFNs) regulate tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) expression on human T cells: A novel mechanism for the antitumor effects of type I IFNs. J Exp Med. 1999;189(9):1451–1460. | |
Merchant MS, Yang X, Melchionda F, et al. Interferon gamma enhances the effectiveness of tumor necrosis factor-related apoptosis-inducing ligand receptor agonists in a xenograft model of Ewing’s sarcoma. Cancer Res. 2004;64(22):8349–8356. | |
Mitsui J, Nishikawa H, Muraoka D, et al. Two distinct mechanisms of augmented antitumor activity by modulation of immunostimulatory/inhibitory signals. Clin Cancer Res. 2010;16(10):2781–2791. | |
Takeda K, Kojima Y, Uno T, et al. Combination therapy of established tumors by antibodies targeting immune activating and suppressing molecules. J immunol. 2010;184(10):5493–5501. | |
Westwood JA, Darcy PK, Guru PM, et al. Three agonist antibodies in combination with high-dose IL-2 eradicate orthotopic kidney cancer in mice. J Transl Med. 2010;8:42. | |
Takeda K, Okumura K, Smyth MJ. Combination antibody-based cancer immunotherapy. Cancer Sci. 2007;98(9):1297–1302. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.