Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 12
Tumor necrosis factor receptor 2 as a possible marker of COPD in smokers and ex-smokers
Authors Caram LMO, Ferrari R ![]() , Nogueira DL, Oliveira MRM, Francisqueti FV, Tanni SE
, Nogueira DL, Oliveira MRM, Francisqueti FV, Tanni SE ![]() , Corrêa CR
, Corrêa CR ![]() , Godoy I
, Godoy I
Received 1 April 2017
Accepted for publication 23 May 2017
Published 7 July 2017 Volume 2017:12 Pages 2015—2021
DOI https://doi.org/10.2147/COPD.S138558
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Laura Miranda de Oliveira Caram,1 R Ferrari,1 DL Nogueira,1 MRM Oliveira,2 FV Francisqueti,2 SE Tanni,1 CR Corrêa,2 I Godoy1
1Department of Internal Medicine, 2Department of Pathology, Botucatu Medical School, UNESP – Univ Estadual Paulista, Botucatu Campus, Botucatu-São Paulo, Brazil
Introduction: Oxidative stress and systemic inflammation are higher in smokers and patients with COPD; however, markers that may help differentiate between smokers and patients with COPD have not yet been identified. We hypothesized that tumor necrosis factor-alpha receptor (TNFR) and soluble form of the receptor for advanced glycation end products (sRAGE) can be indicators of COPD in asymptomatic patients.
Patients and methods: We evaluated 32 smokers (smoking history >10 pack-years), 32 patients with mild/moderate COPD (smokers and ex-smokers), and 32 never smokers. Concentrations of C-reactive protein (CRP), interleukin (IL)-6, TNFR1 and TNFR2, advanced glycation end products (AGEs), and the sRAGE were measured in serum.
Results: There were higher CRP and AGEs concentrations in smokers and in patients with COPD (P<0.001 and P=0.01, respectively) compared to controls, without statistical difference between smokers and patients with COPD. Concentrations of sRAGE, IL-6, and TNFR1 did not differ between study groups. TNFR2 was significantly higher in patients with COPD than in smokers (P=0.004) and controls (P=0.004), and the presence of COPD (P=0.02) and CRP (P=0.001) showed a positive association with TNFR2. Positive associations for smoking (P=0.04), CRP (P=0.03), and IL-6 (P=0.03) with AGEs were also found. The interaction variable (smoking × COPD) showed a positive association with IL-6.
Conclusion: Our data suggest that TNFR2 may be a possible marker of COPD in asymptomatic smokers and ex-smokers. Although smokers and patients with early COPD presented other increased systemic inflammation markers (eg, CRP) and oxidative stress (measured by AGEs), they did not differentiate smokers from COPD.
Keywords: smoking, chronic obstructive pulmonary disease, inflammation mediators, oxidative stress
Introduction
Oxidative stress and systemic inflammation are increased in smokers and patients with COPD, are responsible for disease progression, and seem to be associated with the severity of COPD.1–3 However, whether specific inflammatory and oxidative stress blood markers are associated with systemic response to smoke or with initial stages of COPD are currently unknown.
Tobacco smoke can directly or indirectly induce the formation of advanced glycation end products (AGEs) via oxidative stress.4,5 AGEs are products of glycation and oxidation of proteins and lipids.6 Toxic glycation products are present in aqueous extracts of tobacco and can rapidly react with proteins to form AGEs or products such as methylglyoxal and glyoxal (AGE precursors), found in tobacco smoke.7 AGEs have been shown to be increased in smokers and patients with COPD.8–10
The AGEs can bind to specific receptors such as the receptor for advanced glycation end products (RAGE) – a transmembrane receptor that belongs to the immunoglobulin family (mRAGE).11,12 This interaction and mRAGE itself activate inflammatory signaling pathways such as the nuclear factor kappa B (NFκB) and can trigger proinflammatory cytokine production, leading to inflammation in various organs.11–13 In contrast, soluble forms of the receptor (sRAGE), shed from the plasma membrane via proteolytic cleavage by matrix metalloproteinases, have been shown to be protective in preventing signaling through mRAGE.14 Another mechanism of NFκB activation is through tumor necrosis factor alpha (TNF-α) receptors (TNFR); both TNFR1 and TNFR2 activation can lead to stimulation of nuclear factor NFκB in a variety of cells.15,16 Therefore, AGE, RAGE, and TNFR stimulate NFκB and can trigger the production of proinflammatory cytokines leading to inflammation in smokers and patients with COPD.11–15,17
The role of sRAGE and TNFR in smokers and patients with COPD is unclear.18–22 Some studies have shown lower sRAGE values in patients with COPD than controls, a positive association between sRAGE and forced expiratory volume in 1 second (FEV1), and a negative association between sRAGE concentrations and the presence of COPD – suggesting a protective role for sRAGE.18,19 The presence and role of TNFR1 and TNFR2 in smoking and COPD have also received little attention.21,22 A study by D’Hulst et al suggest that both receptors contribute to COPD pathogenesis, but TNFR2 seems to be more actively involved in the development of inflammation and airflow obstruction.22
Because a higher concentration of sRAGE may protect smokers from the evolution of COPD, and a higher concentration of TNFR2 seems to be associated with the development of airway obstruction, we hypothesized that serum concentrations of these mediators can be indicators of COPD in asymptomatic smokers. Therefore, our aim was to evaluate serum markers of oxidative stress (AGEs and sRAGE) and systemic inflammatory [TNFR1, TNFR2, interleukin (IL)-6, and C-reactive protein (CRP)] mediators to verify whether they could distinguish smokers from patients with COPD. To our knowledge, this is the first study evaluating the role of TNF receptors as a marker of COPD in smokers and ex-smokers.
Patients and methods
From March 2013 to November 2014, 157 individuals (never smokers, smokers, and patients with mild/moderate COPD) from the Pulmonology Outpatient and Smoking Cessation units at Botucatu Medical School were evaluated, and 96 patients were included in a cross-sectional study. The other 61 patients were excluded according to exclusion criteria (Figure 1). Never smokers, active smokers (smoking history >10 pack-years), or patients with mild/moderate COPD (smoker or ex-smoker, post-bronchodilator FEV1/forced vital capacity (FVC) <0.70 and FEV1 >50%) 23 were consecutively selected. Exclusion criteria included primary diagnosis of other respiratory diseases such as asthma, restrictive disorders (tubercular sequelae, interstitial fibrosis), sleep apnea/hypopnea syndrome, or lung cancer. In addition, a primary diagnosis of unstable angina, congestive heart failure (New York Heart Association classes III or IV), or other chronic diseases, such as uncontrolled diabetes mellitus, kidney or liver failure, and cancer, were also grounds for exclusion. Patients with mild/moderate obstruction reported they were asymptomatic and not on maintenance medications and did not report exacerbation in the previous year. After initial screening, those included were evaluated over 3 days in the same week.
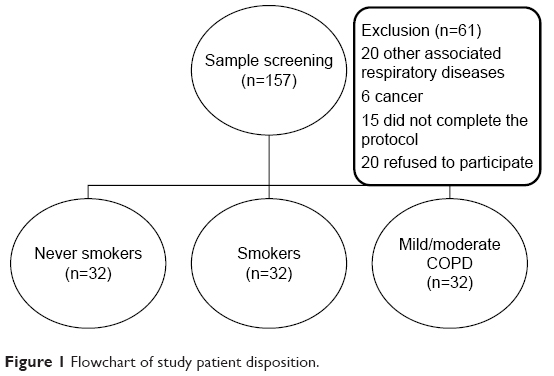 | Figure 1 Flowchart of study patient disposition. |
Sample size was calculated for a multiple linear regression effect size of 0.15, with an estimated multiple correlation squared of 0.50 and the addition of five predictors in the model (G Power 3.1.3).
Participants were made aware of the proposed study procedures and provided written informed consent for study participation. All procedures were approved by the Botucatu Medical School University Hospital Research Ethics Committee (IRB approval number 4415-2012).
Pulmonary function, pulse oximetry, smoking status, and dyspnea score
Pre and post-bronchodilator spirometry were performed using a KOKO spirometer (Ferrari KOKO Louisville, CO 80027, USA) according to criteria set by the American Thoracic Society (ATS).24 FEV1 values were expressed in liters and as percentages of FVC and reference values.25 Pulse oximetry (SpO2) was assessed using an Onyx oximeter (Model 9500 Oximeter; Nonin Medical Inc., Minneapolis, MN, USA) while the patients were breathing room air. Smoking history and current smoking status were investigated and complemented by assessing nicotine dependence intensity (Fargeström Test).26 Confirmation of smoking status was performed by measuring carbon monoxide (CO) in exhaled air by a standardized technique (Micro CO Meter, ©Cardinal Health, England, UK). A value of exhaled CO >6.0 ppm was considered to indicate active smoking.27,28 Dyspnea was evaluated by the Borg Dyspnoea Scale Score.29
Nutritional assessment
Body weight and height were measured by the Filizola® scale, and the body mass index ([BMI] = weight [kg]/height [m2]) was calculated. Body composition was evaluated by bioelectrical impedance (BIA 101; RJL Systems, Detroit, MI, USA) according to the European Society for Parenteral and Enteral Nutrition guidelines.30 Fat-free mass (FFM, kg) was calculated using a group-specific regression equation developed by Kyle et al.30 The FFM index (FFMI = FFM/height2) was also calculated. Lean body mass depletion was defined as an FFMI <15 kg/m2 for women and <16 kg/m2 for men.31
Blood sampling and analysis of oxidative stress and systemic inflammation: AGEs, sRAGE, IL-6, CRP, and TNFR1 and TNFR2
Fasting peripheral blood was collected (08.00 hours) and serum stored at −80°C until analysis. IL-6, TNFR1, TNFR2, AGEs, and sRAGE (lower detection limit of 0.70, 0.16, 0.2, 0.5, and 1.23 pg/mL, respectively) were measured in duplicate by high-sensitivity commercial kits using enzyme-linked immunosorbent assay (ELISA) according to manufacturer’s instructions (BioSource International Inc., CA, USA). AGEs represent a class of covalently modified proteins generated by oxidative and non-oxidative pathways, involving sugars or their degradation products. Further, AGEs exert their damaging and pro-inflammatory effects by activating signaling cascades via specific receptors named RAGE.32 Soluble RAGE (sRAGE), a free form of RAGE, acts as a suppressor of RAGE activation and signaling, because it plays its role as a decoy for RAGE ligands in order to prevent signal transduction and to avoid the effects of the RAGE-binding interaction. Readings were obtained in a Spectra Max 190 microplate spectrophotometer (Molecular Devices®, Sunnyvale, CA, USA). CRP was measured in duplicate by high-sensitivity particle-enhanced immunonephelometry (Cardio-Phase, Dade Behring Marburg GmbH, Marburg, USA), with a lower detection limit of 0.007 mg/L. All measurements were performed after the final evaluation using kits with the same lot number to avoid measurement bias.
Statistical analysis
Descriptive statistics were used to describe the features of all participants. Mean ± SD or medians and interquartile range (25%–75%) were used depending on data distribution. Categorical variables were expressed as percentages. Chi-square was used to compare the values of categorical variables.
Analysis of variance (ANOVA), followed by the Tukey or Kruskal–Wallis test, and further followed by Dunn’s test was used to compare demographic and general characteristics between smokers, patients with mild/moderate COPD, and never smokers.
Multiple linear regression analysis was used to assess associations between COPD (absence =0, presence =1), smoking status (absence =0, presence =1), interaction variable (smoking × COPD), exercise capacity, health status, body composition, systemic inflammatory status (CRP, IL-6, TNFR1, and TNFR2), and oxidative stress (AGEs and sRAGE) in the three study groups. Setting variables in the model were: sex and age. Collinearity was prevented by deleting a variable showing correlation. The level of significance was set at 5%. All analyses were performing using IBM SPSS Statistics 22 and Sigma Plot 11.0.
Results
Table 1 shows the general characteristics of the 96 subjects enrolled in the study and the comparison between never smokers, smokers, and patients with mild/moderate COPD. Patients with COPD presented reduced lean body mass index; however, the proportion of depleted and non-depleted subjects did not differ between groups. In addition, 44% of patients with COPD were active smokers. Smoking history was higher in the COPD group compared to smokers and never smokers. Spirometry variables were lower in patients with COPD versus smokers as well as in smokers versus never smokers.
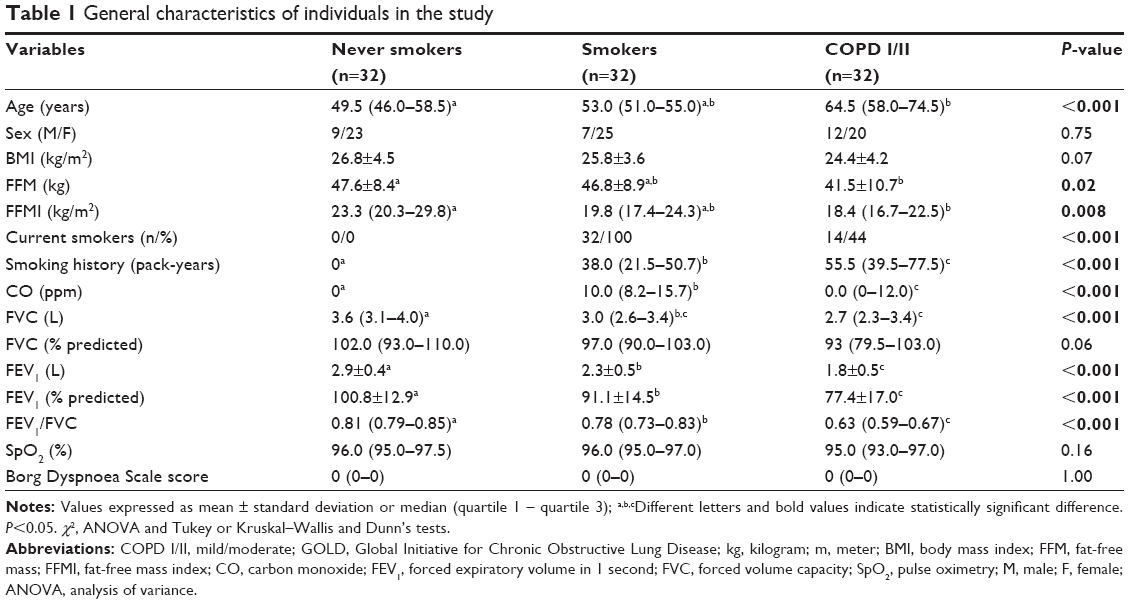 | Table 1 General characteristics of individuals in the study |
Figure 2 shows AGEs concentrations in the three groups. Smokers (P<0.001) and patients with COPD (P<0.001) showed higher values than never smokers. There was no statistically significant difference between smokers and patients with COPD. sRAGE concentrations did not differ between groups (P=0.92).
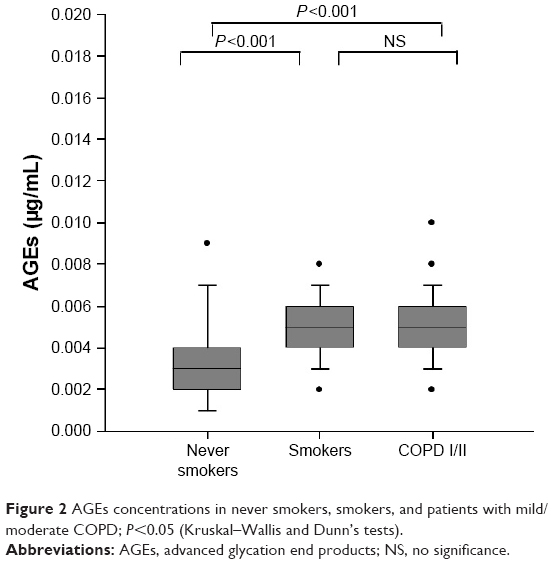 | Figure 2 AGEs concentrations in never smokers, smokers, and patients with mild/moderate COPD; P<0.05 (Kruskal–Wallis and Dunn’s tests). |
Systemic inflammatory status assessed by TNFR1 concentrations did not differ between groups [never smokers: 119.2 (104.3–139.5) pg/mL; smokers: 122.8 (103.7–136.7) pg/mL; COPD I/II: 134.0 (115.7–210.3) pg/mL, P=0.07]; however, patients with COPD showed higher TNFR2 values than smokers and never smokers (Figure 3).
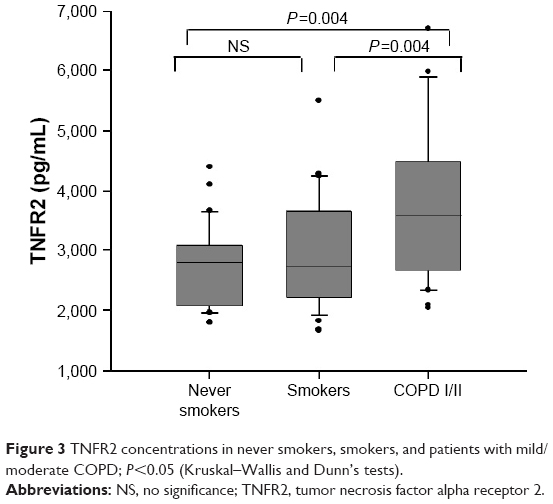 | Figure 3 TNFR2 concentrations in never smokers, smokers, and patients with mild/moderate COPD; P<0.05 (Kruskal–Wallis and Dunn’s tests). |
Smokers and patients with COPD presented higher CRP values than never smokers. However, there was no statistically significant difference between smokers and patients with COPD (Figure 4). Concentrations of IL-6 did not differ between groups [never smokers: 1.2 (0.9–3.4) pg/mL; smokers: 1.9 (1.5–3.6) pg/mL; COPD I/II: 2.8 (1.3–4.7) pg/mL, P=0.07].
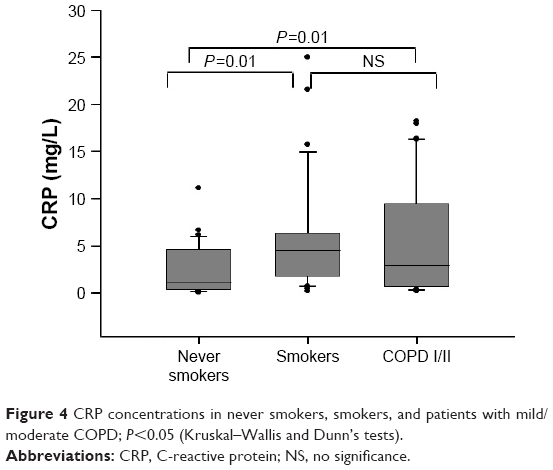 | Figure 4 CRP concentrations in never smokers, smokers, and patients with mild/moderate COPD; P<0.05 (Kruskal–Wallis and Dunn’s tests). |
In the analysis of predictors of oxidative stress and systemic inflammation, smoking presented a positive association with AGEs and CRP, whereas the presence of COPD and FEV1% predicted values were associated with TNFR2; moreover, there was an association between CRP and AGEs, TNFR1 and -2, and also between AGEs and IL-6 (Table 2). We included the interaction variable (smoking × COPD) in all models (data not shown); however, the interaction variable showed association only with IL-6 (Table 2).
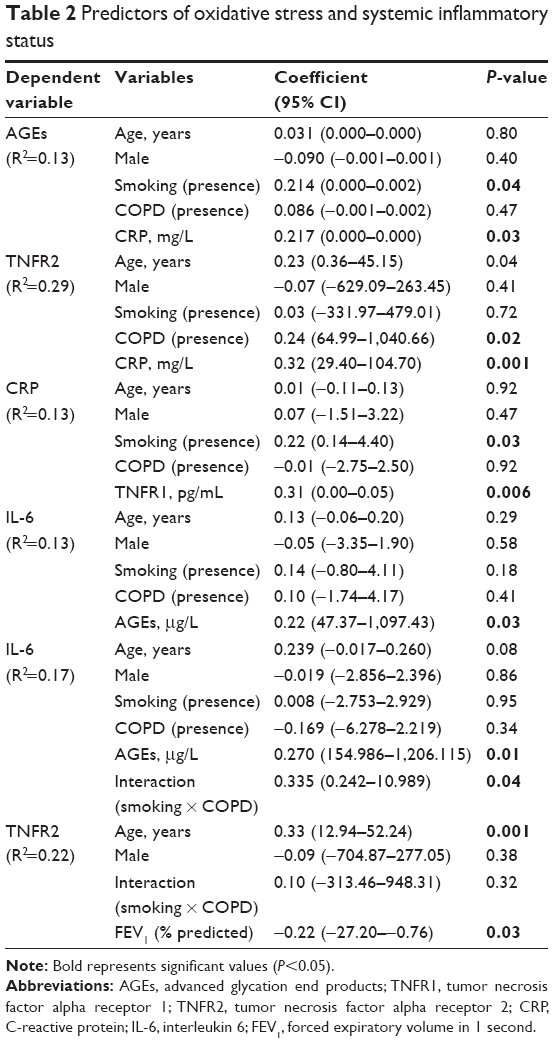 | Table 2 Predictors of oxidative stress and systemic inflammatory status |
Discussion
The main result of this study indicates that TNFR2 may be a possible marker of COPD in asymptomatic smokers and ex-smokers because its concentration is different between smokers and asymptomatic patients with COPD. Elevated oxidative stress measured by AGEs, and systemic inflammation measured by CRP and IL-6, did not differ between patients with COPD and smokers. sRAGEs that may protect smokers from COPD did not differ between study groups.
Patients with COPD showed increased TNFR2 concentrations in comparison to smokers and never smokers, and a positive association between COPD presence and TNFR2 was found. No previous study with a similar sample and design was identified in the literature. However, rats with TNFR1 and TNFR2 deficiency,22 after 6 months of exposure to tobacco, showed increased concentrations of both receptors; however, the relative increase in TNFR2 was higher than that of TNFR1. Vernooy et al showed higher TNFR2 concentrations in moderate COPD (FEV1% predicted =55±14) compared to healthy smokers (FEV1% predicted =98±16); however, they did not show any correlation between the degree of airflow limitation and TNFR2 concentration.33 The actual involvement of TNF receptors in the pathogenesis of COPD remains unknown and the mechanism that defines the dominant effect is still unclear. D’Hulst et al suggest that both TNFRs contribute to the pathogenesis of COPD, but that TNFR2 is the most active receptor in the development of inflammation and emphysema.22
Although most of the biological effects induced by TNF-α have been attributed to TNFR1, TNFR2 has been reported to stimulate TNF-α-induced T-cell proliferation.34 It appears that TNFR2 induces activation of NFκB signaling, differentially, under specific inflammatory conditions with assistance from some co-factors.15 According to D’Hulst et al, observations of reduced T-lymphocyte activation in the lungs of mice lacking TNFR2 and high lymphocyte numbers in bronchoalveolar lavage fluid of TNFR1 knockout (KO) mice exclusively expressing TNFR2 then further underline the importance of the R2 receptor in T-cell proliferation.22 NFκB activation can also be induced by TNFR2 that can be activated by TNFα to further activate NFκB.15 Therefore, the present data confirm the importance of this marker in COPD pathogenesis and that, although both TNFRs are involved in long-term cigarette smoke-induced pulmonary inflammation and emphysema, the contribution of TNFR2 is most prominent.
The increase of AGEs in patients with COPD is consistent with results from previous studies,9,10 and we did not find differences in sRAGE concentration between groups. The Tesra study, evaluating only patients with COPD, did not identify differences in sRAGE concentrations between COPD II and III; however, a negative association was identified between sRAGE and emphysema after adjustment for spirometry and demographic variables. According to the data from the Tesra study, a lower sRAGE concentration may be associated with emphysema, regardless of disease stage.35 We can speculate that an sRAGE deficiency is involved in the pathogenesis of emphysema and could possibly provide targets for new treatments.36 However, Smith et al showed that circulating sRAGE is lower in severe and very severe COPD [FEV1% predicted =37 (23–49)] and their concentrations present a correlation to the degree of airflow limitation.19 Patients with mild/moderate COPD, evaluated in the present study, may have associated lower intensity emphysema and inflammation, and our data suggest that sRAGE does not differentiate smokers from patients with COPD. However, large-scale studies are needed to determine whether sRAGE is a marker of COPD or is involved in the pathogenesis of emphysema.
Smokers and patients with COPD showed higher CRP concentrations than never smokers. Our data are consistent with recent studies.21,37–39 Although IL-6 concentrations did not differ between groups, we identify a positive association between AGEs and IL-6/CRP, which is in agreement with studies showing the effects of AGEs on cytokine production and suggests a dose-dependent association between AGEs and concentrations of IL-6 and CRP.40,41 We found a positive association between the interaction “smoking and COPD” with IL-6 concentrations. This result has not been previously described, although a higher systemic concentration of IL-6 in smokers and COPD is well established.42–44
Our study has some potential limitations. A recent study44 showed that symptoms are more prevalent in current or ex-smokers with preserved pulmonary function than in healthy controls who had never smoked. The authors showed respiratory symptoms in 50% of current or ex-smokers with preserved pulmonary function evaluated by the COPD Assessment Test (CAT).45 We did not measure respiratory symptoms using the CAT; however, we evaluated dyspnea by the Borg Dyspnoea Scale score, and no difference was found between groups. In addition, our conclusion is based on a cross-sectional study and indicates the need for long-term follow-up studies of inflammatory mediators in smokers to confirm the present findings.
Conclusion
Smokers and patients with early COPD present with increased systemic inflammation, measured by TNFR2 and CRP, and oxidative stress, measured by AGEs. TNFR2 was the only indicator of inflammation that differed between smokers and patients with COPD. The increase in TNFR2 suggests greater inflammation intensity in individuals with COPD. Therefore, we suggest that TNFR2 is more sensitive in identifying increased inflammation in the early stages of COPD, and could be a possible marker of COPD in asymptomatic smokers and ex-smokers.
Acknowledgments
This study received financial support from Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP; Foundation for the Support of Research in São Paulo State; grant no: 2012/22321-0). Laura Miranda de Oliveira Caram was a recipient of a scholarship grant from CAPES.
Disclosure
The authors report no conflicts of interests in this work.
References
Lee W, Thomas PS. Oxidative stress in COPD and its measurement through exhaled breath condensate. Clin Transl Sci. 2009;2(2):150–155. | ||
Lopez-Campos JL, Calero-Acuña C, Lopez-Ramirez C, et al. Implications of the inflammatory response for the identification of biomarkers of chronic obstructive pulmonary disease. Biomark Med. 2016;10(2):109–122. | ||
Holguin F. Oxidative stress in airway diseases. Ann Am Thorac Soc. 2013;10(Suppl):S150–S157. | ||
Brownlee M. Advanced protein glycosylation in diabetes and aging. Annu Rev Med. 1995;46:223–234. | ||
Yamagishi S, Matsui T, Nakamura K. Possible involvement of tobacco-derived advanced glycation end products (AGEs) in an increased risk for developing cancers and cardiovascular disease in former smokers. Med Hypotheses. 2008;71(2):259–261. | ||
Ahmad S, Khan MS, Akhter F, et al. Glycoxidation of biological macromolecules: a critical approach to halt the menace of glycation. Glycobiology. 2014;24(11):979–990. | ||
Cerami C, Founds H, Nicholl I, et al. Tobacco smoke is a source of toxic reactive glycation products. Proc Natl Acad Sci U S A. 1997;94(25):13915–13920. | ||
Boschetto P, Campo I, Stendardo M, et al. Plasma sRAGE and N-(carboxymethyl) lysine in patients with CHF and/or COPD. Eur J Clin Invest. 2013;43(6):562–569. | ||
Hoonhorst SJ, Lo Tam Loi AT, Hartman JE, et al. Advanced glycation end products in the skin are enhanced in COPD. Metabolism. 2014;63(9):1149–1156. | ||
Wu L, Ma L, Nicholson LF, Black PN. Advanced glycation end products and its receptor (RAGE) are increased in patients with COPD. Respir Med. 2011;105(3):329–336. | ||
Neeper M, Schmidt AM, Brett J, et al. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J Biol Chem. 1992;267(21):14998–15004. | ||
Schmidt AM, Yan SD, Yan SF, Stern DM. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Invest. 2001;108(7):949–955. | ||
Sukkar MB, Ullah MA, Gan WJ, et al. RAGE: a new frontier in chronic airways disease. Br J Pharmacol. 2012;167(6):1161–1176. | ||
Yonchuk JG, Silverman EK, Bowler RP, et al. Circulating soluble receptor for advanced glycation end products (sRAGE) as a biomarker of emphysema and the RAGE axis in the lung. Am J Respir Crit Care Med. 2015;192(7):785–792. | ||
Yang S, Wang Y, Mei K, et al. Tumor necrosis factor receptor 2 (TNFR2) interleukin-17 receptor D (IL-17RD) heteromerization reveals a novel mechanism for NF-κB activation. J Biol Chem. 2015;290(2):861–871. | ||
Ait-Ali D, Turquier V, Tanguy Y, et al. Tumor necrosis factor (TNF)-alpha persistently activates nuclear factor-kappaB signaling through the type 2 TNF receptor in chromaffin cells: implications for long-term regulation of neuropeptide gene expression in inflammation. Endocrinology. 2008;149(6):2840–2852. | ||
Barnes PJ. Cellular and molecular mechanisms of chronic obstructive pulmonary disease. Clin Chest Med. 2014;35(1):71–86. | ||
Miniati M, Monti S, Basta G, Cocci F, Fornai E, Bottai M. Soluble receptor for advanced glycation end products in COPD: relationship with emphysema and chronic cor pulmonale: a case-control study. Respir Res. 2011;12:37. | ||
Smith DJ, Yerkovich ST, Towers MA, Carroll ML, Thomas R, Upham JW. Reduced soluble receptor for advanced glycation end-products in COPD. Eur Respir J. 2011;37(3):516–522. | ||
Iwamoto H, Gao J, Koskela J, et al. Differences in plasma and sputum biomarkers between COPD and COPD-asthma overlap. Eur Respir J. 2014;43(2):421–429. | ||
Ji J, von Schéele I, Bergström J, et al. Compartment differences of inflammatory activity in chronic obstructive pulmonary disease. Respir Res. 2014;15:104. | ||
D’hulst AI, Bracke KR, Maes T, et al. Role of tumour necrosis factor-alpha receptor p75 in cigarette smoke-induced pulmonary inflammation and emphysema. Eur Respir J. 2006;28(1):102–112. | ||
Vogelmeier CF, Criner GJ, Martinez FJ, et al. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease 2017 Report: GOLD Executive Summary. Eur Respir J. 2017;49(3):pii:1700214. | ||
Standardization of spirometry – 1987 update. Statement of the American Thoracic Society. Am Rev Respir Dis. 1987;136(5):1285–1298. | ||
Pereira C, Barreto S, Simões J, Pereira F, Gerstler J, Nakatani J. Valores de referência para a espirometria em uma amostra da populaçäo brasileira adulta. J Bras Pneumol. 1992;18:10–22. | ||
Reichert J, Araújo AJ, Gonçalves CM, et al; Sociedade Brasileira de Pneumologia e Tisiologia. Diretrizes para cessação do tabagismo [Smoking cessation guidelines – 2008.]. J Bras Pneumol. 2008;34(10):845–880. Portuguese. | ||
Middleton ET, Morice AH. Breath carbon monoxide as an indication of smoking habit. Chest. 2000;117(3):758–763. | ||
Santos U, Gannam S, Abe J, Esteves P, Filho M, Wakassa T. Emprego da determinação de monóxido de carbono no ar exalado para a detecção do consumo de tabaco [Use of carbon monoxide in the exhaled air for the detection of tobacco consumption]. J Bras Pneumol. 2001;27:231–236. Portuguese. | ||
Borg. Escala CR10 de Borg. In: Borg G. Escala de Borg para dor e esforço percebido [Borg’s Perceived Exertion and Pain Scales]. 1 ed. São Paulo 2000. | ||
Kyle UG, Bosaeus I, De Lorenzo AD, et al; Composition of the ESPEN Working Group. Bioelectrical impedance analysis – part I: review of principles and methods. Clin Nutr. 2004;23(5):1226–1243. | ||
Schols AM, Broekhuizen R, Weling-Scheepers CA, Wouters EF. Body composition and mortality in chronic obstructive pulmonary disease. Am J Clin Nutr. 2005;82(1):53–59. | ||
Vistoli G, De Maddis D, Cipak A, Zarkovic N, Carini M, Aldini G. Advanced glycoxidation and lipoxidation end products (AGEs and ALEs): an overview of their mechanisms of formation. Free Radic Res. 2013;47 Suppl 1:3–27. | ||
Vernooy JH, Küçükaycan M, Jacobs JA, et al. Local and systemic inflammation in patients with chronic obstructive pulmonary disease: soluble tumor necrosis factor receptors are increased in sputum. Am J Respir Crit Care Med. 2002;166(9):1218–1224. | ||
Tartaglia LA, Goeddel DV, Reynolds C, et al. Stimulation of human T-cell proliferation by specific activation of the 75-kDa tumor necrosis factor receptor. J Immunol. 1993;151(9):4637–4641. | ||
Cheng DT, Kim DK, Cockayne DA, et al; TESRA and ECLIPSE Investigators. Systemic soluble receptor for advanced glycation endproducts is a biomarker of emphysema and associated with AGER genetic variants in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;188(8):948–957. | ||
Carolan BJ, Hughes G, Morrow J, et al. The association of plasma biomarkers with computed tomography-assessed emphysema phenotypes. Respir Res. 2014;15:127. | ||
Agustí A, Edwards LD, Rennard SI, et al; Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) Investigators. Persistent systemic inflammation is associated with poor clinical outcomes in COPD: a novel phenotype. PLoS One. 2012;7(5):e37483. | ||
Serapinas D, Narbekovas A, Juskevicius J, Sakalauskas R. Systemic inflammation in COPD in relation to smoking status. Multidiscip Respir Med. 2011;6(4):214–219. | ||
Tonstad S, Cowan JL. C-reactive protein as a predictor of disease in smokers and former smokers: a review. Int J Clin Pract. 2009;63(11):1634–1641. | ||
Hein GE, Köhler M, Oelzner P, Stein G, Franke S. The advanced glycation end product pentosidine correlates to IL-6 and other relevant inflammatory markers in rheumatoid arthritis. Rheumatol Int. 2005;26(2):137–141. | ||
Chen YJ, Sheu ML, Tsai KS, Yang RS, Liu SH. Advanced glycation end products induce peroxisome proliferator-activated receptor γ down-regulation-related inflammatory signals in human chondrocytes via Toll-like receptor-4 and receptor for advanced glycation end products. PLoS One. 2013;8(6):e66611. | ||
Sunyer J, Forastiere F, Pekkanen J, et al. Interaction between smoking and the interleukin-6 gene affects systemic levels of inflammatory biomarkers. Nicotine Tob Res. 2009;11(11):1347–1353. | ||
Caram LM, Amaral RA, Ferrari R, et al. Serum vitamin A and inflammatory markers in individuals with and without chronic obstructive pulmonary disease. Mediators Inflamm. 2015;2015:862086. | ||
Tanni SE, Pelegrino NR, Angeleli AY, Correa C, Godoy I. Smoking status and tumor necrosis factor-alpha mediated systemic inflammation in COPD patients. J Inflamm (Lond). 2010;7:29. | ||
Woodruff PG, Barr RG, Bleecker E, et al; SPIROMICS Research Group. Clinical significance of symptoms in smokers with preserved pulmonary function. N Engl J Med. 2016;374(19):1811–1821. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.