Back to Journals » International Journal of Nanomedicine » Volume 12
Tumor microenvironment dual-responsive core–shell nanoparticles with hyaluronic acid-shield for efficient co-delivery of doxorubicin and plasmid DNA
Authors Wang T, Yu X, Han L, Liu T, Liu Y, Zhang N
Received 9 February 2017
Accepted for publication 5 May 2017
Published 4 July 2017 Volume 2017:12 Pages 4773—4788
DOI https://doi.org/10.2147/IJN.S134378
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Tianqi Wang, Xiaoyue Yu, Leiqiang Han, Tingxian Liu, Yongjun Liu, Na Zhang
Department of Pharmaceutics, School of Pharmaceutical Sciences, Shandong University, Jinan, Shandong Province, People’s Republic of China
Abstract: As the tumor microenvironment (TME) develops, it is critical to take the alterations of pH value, reduction and various enzymes of the TME into consideration when constructing the desirable co-delivery systems. Herein, TME pH and enzyme dual-responsive core–shell nanoparticles were prepared for the efficient co-delivery of chemotherapy drug and plasmid DNA (pDNA). A novel pH-responsive, positively charged drug loading material, doxorubicin (DOX)-4-hydrazinobenzoic acid (HBA)-polyethyleneimine (PEI) conjugate (DOX-HBA-PEI, DHP), was synthesized to fabricate positively charged polyion complex inner core DHP/DNA nanoparticles (DDN). Hyaluronic acid (HA) was an enzyme-responsive shell which could protect the core and enhance the co-delivery efficiency through CD44-mediated endocytosis. The HA-shielded pH and enzyme dual-responsive nanoparticles (HDDN) were spherical with narrow distribution. The particle size of HDDN was 148.3±3.88 nm and the zeta potential was changed to negative (-18.1±2.03 mV), which led to decreased cytotoxicity. The cumulative release of DOX from DHP at pH 5.0 (66.4%) was higher than that at pH 7.4 (30.1%), which indicated the pH sensitivity of DHP. The transfection efficiency of HDDN in 10% serum was equal to that in the absence of serum, while the transfection of DDN was significantly decreased in the presence of 10% serum. Furthermore, cellular uptake studies and co-localization assay showed that HDDN were internalized effectively through CD44-mediated endocytosis in the tumor cells. The efficient co-delivery of DOX and pEGFP was confirmed by fluorescent image taken by laser confocal microscope. It can be concluded that TME dual-responsive HA-shielded core–shell nanoparticles could be considered as a promising platform for the co-delivery of chemotherapy drug and pDNA.
Keywords: co-delivery, core–shell nanoparticles, hyaluronic acid, CD44 targeted, pH sensitive
Introduction
Cancers are complex diseases involving multiple pathways and complicated microenvironment.1 Combination therapy exerts synergistic effects and brings about more efficacious therapies than single treatment.2,3 Among them, combination of chemotherapy and gene therapy has emerged as an exciting method of treating cancers due to their complementation and the genetic regulation of cancer cell signaling by gene therapy.4,5 Combination of chemotherapy and gene therapy has the potential to overcome the multidrug resistance and reduce the dosage of chemotherapeutic drugs, which contribute to a decrease in side effects and an increase in antitumor efficiency. For example, combinations of doxorubicin (DOX) and p53 gene, paclitaxel (PTX) and VEGF-siRNA, and DOX and MDR-1-siRNA were effective in overcoming the drug resistance, reducing the side effects and exhibiting superior antitumor effect, compared with single drug treatment.6–8
Combination therapy would be more attractive with co-delivery system. The drug and gene would be loaded at a ratio and then rationally released with synchronized pharmacokinetics.9,10 So far, several vehicles, including lipoplex, polymer nanoparticles, micelles, inorganic nanoparticles and so on, have been used for the co-delivery of drug and gene.11–14 These systems are often based on the core–shell structure, which is suitable for delivering two agents. The inner core provides a carrier for the drug and gene with desirable properties, including small particle size, efficient transfection and stimuli-sensitive release behaviors. The shell outside is modified for other functions, such as enhancing the stability, improving the biocompatibility and for efficient targeting delivery.15,16 In this regard, core–shell nanoparticles are promising for the co-delivery of drug and gene.
Polyion complex (PIC), formed by the electrostatic interaction between cationic polymer and DNA, is the most simple and common vector in gene delivery.15 Cationic polymer poly(ethylenimine) (PEI) has shown great advantage in condensing genes and protecting them from digestion in serum.16 For fabrication of an inner core carrying a drug and gene, drug loading could be realized by polymer–drug conjugation.17,18 With the development of tumor microenvironment (TME), there is a dynamic process of alterations at the cellular and tissue level leading to hypoxia, acidity, release of various enzymes and decreased resting transmembrane potential on the tumor cells.1,15,16 Extra- and intracellular acidity is a primary characteristic of the TME as a result of abnormal metabolism of the tumor cells, which leads to alteration in pH value from 7.4 in the normal tissue to 6.0 in the tumor tissue or 5.0 in lysosomes.19,20 Therefore, hydrazone bond is an optimum linkage of drug and polymer with a pH-responsive drug release behavior.19–21 For this reason, the drug would be specifically released in the tumor cells, thus lessening the potential side effects.
To increase the stability of cationic core in serum and avoid erythrocyte aggregation, various electrostatic coating biomaterials including polysaccharide, polypeptide and multiblock copolymers have been deposited onto the PIC core.7,11,22 Hyaluronic acid (HA), a linear polysaccharide, is suitable for constructing core–shell nanoparticles with enzyme-sensitive, less toxic, nonimmunogenic, biodegradable and biocompatible characteristics.23 Without further modification, HA itself has the potential for the negative charge to be absorbed onto the positive core, long circulation in the blood and targeting the tumor cells through CD44 overexpressed on some tumor cells.22,24,25 Most importantly, high-molecular-weight (MW) HA is degraded into low-MW HA and tetrasaccharide by hyaluronidase-2 (Hyal2) in the tumor tissue and hyaluronidase-1 (Hyal1) in the lysosomes, which ensure disassembly of nanoparticles at the tumor site.26,27 Thus, HA-shielded core–shell nanoparticles with enzyme responsiveness would be suitable for drug and gene co-delivery.28,29
Considering efficient delivery, TME dual-responsive HA-shielded core–shell nanoparticles were fabricated for drug and gene co-delivery in this study. DOX was chosen as the model drug and pEGFP used as the reporter gene. In order to co-load the drug and gene, a pH-responsive drug loading cationic polymer was synthesized through hydrazone bond linked between DOX and 4-hydrazinobenzoic acid (HBA)-modified PEI. Thereafter, an inner core carrying DOX and gene was fabricated through the electrostatic interaction between DOX-HBA-PEI (DHP) and plasmid DNA (pDNA), which was called DHP/DNA nanoparticles (DDN). In order to shield the positive charge and increase the stability in serum, the biocompatible shell HA was coated on the surface of the core to form HA-shielded nanoparticles HA/DDN (HDDN; Scheme 1). HA shielded on pH-sensitive core was expected to improve the biocompatibility of DDN and protect pDNA from degradation to increase the transfection efficiency of HDDN in the presence of serum. The internalization of the HDDN would be increased through CD44-targeted delivery, and as such, the co-delivery of drug and gene would be realized. When at the tumor tissue, HDDN disassemble by the Hyal2 in TME and internalize through CD44-mediated endocytosis. In the lysosome, HDDN are further degraded by Hyal1 and other enzymes to expose the DDN. Because of the acidic condition in the lysosome, hydrazone bonds are broken down and DHP release DOX. With the sponge effect of PEI, DOX and gene escape from the lysosome and exert their antitumor effect.
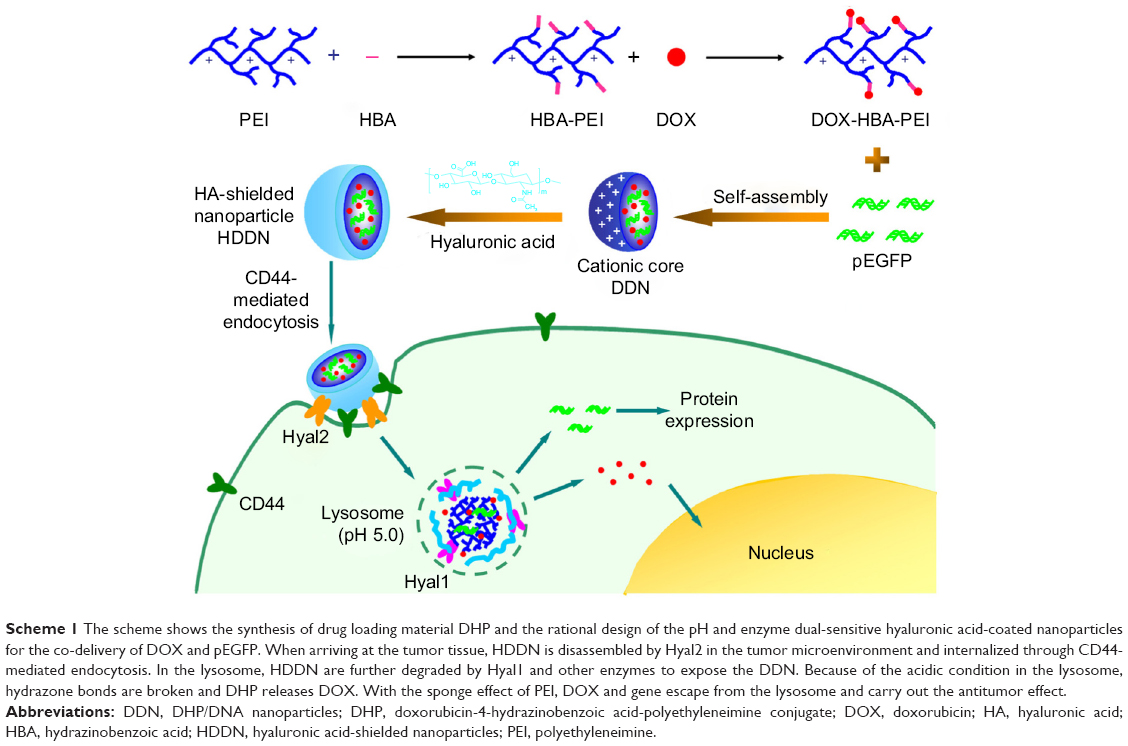 | Scheme 1 The scheme shows the synthesis of drug loading material DHP and the rational design of the pH and enzyme dual-sensitive hyaluronic acid-coated nanoparticles for the co-delivery of DOX and pEGFP. When arriving at the tumor tissue, HDDN is disassembled by Hyal2 in the tumor microenvironment and internalized through CD44-mediated endocytosis. In the lysosome, HDDN are further degraded by Hyal1 and other enzymes to expose the DDN. Because of the acidic condition in the lysosome, hydrazone bonds are broken and DHP releases DOX. With the sponge effect of PEI, DOX and gene escape from the lysosome and carry out the antitumor effect. |
In this study, the synthesis of polymer drug conjugation DHP was confirmed by proton nuclear magnetic resonance (1H NMR) and Fourier transform infrared (FTIR) spectroscopy. The pH sensitivity of DHP was determined in different pH conditions. To obtain better transfection efficiency, the ratio between polymer and gene was screened. HA-shielded nanoparticles were confirmed to be stable in serum and sensitive to hyaluronidases (Hyals) at the tumor site. The transfection of HDDN was carried out in the presence of serum. The ability of CD44-mediated intracellular uptake was detected by the fluorescence of DOX. Furthermore, co-delivery of DOX and pEGFP to the same cells was confirmed on HepG2 and B16 cells. Thus, tumor targeting and TME dual-responsive HA-shielded core–shell nanoparticles would be a potential platform for targeted co-delivery of chemotherapy drug and gene in cancer therapy.
Materials and methods
Materials
Branched PEI (MW =25 kDa) was purchased from Sigma-Aldrich (St Louis, MO, USA). HA (MW =79 kDa) was purchased from Freda (Shandong, People’s Republic of China). Doxorubicin·HCl (DOX·HCl) was purchased from Dalian Meilun Biology Technology Co. Ltd. (Dalian, People’s Republic of China). N-hydroxysuccinimide, HBA and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride were purchased from Sigma-Aldrich. Hyals were purchased from Sigma-Aldrich. The plasmid pCMV-EGFP (pEGFP-N1) carrying enhanced green fluorescent protein (EGFP) under cytomegalovirus (CMV) promoter was propagated in Escherichia coli and purified by Endo Free Plasmid Maxi Kit (Qiagen, Hilden, Germany). The purity and concentration of pDNA was then measured using NanoDrop UV–Vis Spectrophotometers (ND-2000C; Thermo Fisher Scientific, Waltham, MA, USA). Hoechst 33342 was purchased from Thermo Fisher Scientific. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was also obtained from Sigma Aldrich. GoldView was purchased from BioTeke Corporation (Beijing, People’s Republic of China). All other reagents were of commercial special grade and were used without further purification.
Cell culture
Human liver hepatocellular carcinoma cell lines (HepG2), purchased from the Chinese Academy of Sciences (Shanghai, People’s Republic of China), were kindly provided by the Institute of Immunopharmacology and Immunotherapy of Shandong University (Jinan, People’s Republic of China). Murine malignant melanoma cell lines (B16) were purchased from the Chinese Academy of Sciences (Shanghai, People’s Republic of China). All cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% (v/v) fetal bovine serum (FBS) from Sijiqing Co. Ltd. (Hangzhou, People’s Republic of China), streptomycin at 100 mg/mL and penicillin at 100 U/mL in an incubator at 37°C with 5% CO2.
Synthesis of pH-sensitive DHP conjugates
To synthesize the pH-sensitive DHP conjugates, HBA was used as a linker to conjugate the C13-ketone group of DOX to the amino group of PEI through pH-sensitive hydrazone bond. The procedure was conducted in two steps as shown in Scheme 2.
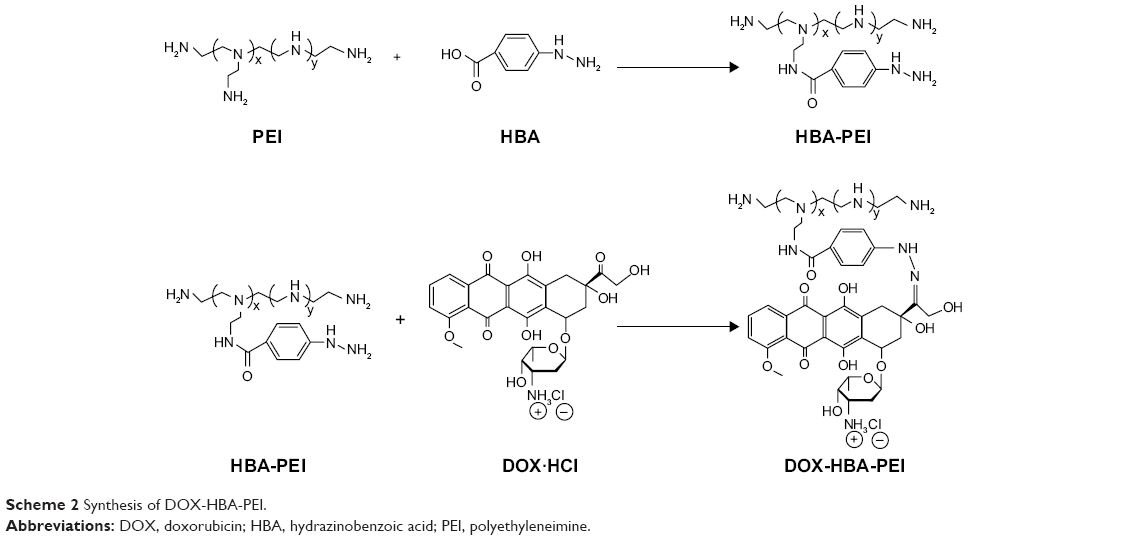 | Scheme 2 Synthesis of DOX-HBA-PEI. |
First, 2.4 μmol PEI (60 mg) and 0.19 mmol HBA (29.2 mg) were dissolved in 5 mL dimethyl sulfoxide (DMSO), and then 0.28 mmol 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (54 mg) and 0.28 mmol N-hydroxysuccinimide (32 mg) were added. The mixture was stirred for 24 h at room temperature in an atmosphere of nitrogen, after which the HBA-PEI (HP) conjugates were dialyzed against H2O to remove the unreacted HBA (molecular weight cut-off [MWCO] =8,000) and lyophilized to obtain fine powder for the next step.
When the structure of HP was confirmed, DOX was coupled to the HP conjugates. The HP conjugates (50 mg) were dissolved in conjugation buffer, and DOX (56 mg) was dissolved in DMSO. Following this, the two solutions were mixed and stirred for 24 h at room temperature. The product was dialyzed against H2O (MWCO =8,000) and freeze-dried to obtain red fine powder.
The chemical structures of HP and DHP were confirmed by 1H NMR (in D2O, 300 MHz) spectrometer and FTIR spectra. UV–Vis spectrophotometer (λ=481 nm) was used to confirm DOX conjugation and determine the drug loading capacity of DOX in the polymer by measuring the absorbance of the polymer solution at 481 nm.
In vitro pH-sensitive drug release of DOX
The release of DOX from DHP conjugates was assayed in PBS buffer of pH 7.4, 6.0 and 5.0 in a dialysis bag (MWCO =3,500).17 Thirty milligrams of dried DHP conjugates was dispersed in 5 mL of water and placed in a dialysis bag. The dialysis bag was immersed in 20 mL of release medium and incubated at 37°C. One milliliter of the samples was removed for measurement of drug concentration at desired time intervals and replaced by an equal volume of fresh media. The amount of DOX released was analyzed with a UV–Vis spectrophotometer at 481 nm. The drug release studies were performed in triplicate for each sample.
In vitro cytotoxicity of materials
Cytotoxicity of free DOX, HP and DHP conjugates was evaluated by MTT assay with HepG2 and B16 cells. HepG2 and B16 cells were seeded on 96-well plates at a cell density of 6×103 cells/well in 0.15 mL RPMI 1640 supplemented with 10% FBS. After a 24 h incubation period, the cells were incubated with various concentrations of DOX, HP and DHP (with the DOX concentration being 0.01, 0.1, 1.0, 2.5, 5.0, 10.0 and 20.0 μg/mL) in the culture medium for another 48 h. Following this, 0.02 mL MTT in PBS (5 mg/mL) was added to each well and incubated for an additional 4 h at 37°C in a 5% CO2 incubator. The MTT containing media were removed, and the formazan crystals formed by the living cells were dissolved in 0.2 mL DMSO. Cells without treatment were used as control with 100% viability. The absorbance at 570 nm was determined by a Microplate Reader (Model 680; Bio-Rad Laboratories Inc., Hercules, CA, USA). Cell viability was calculated with the formula: (Abs [sample] − Abs [blank])/(Abs [control] − Abs [blank]) ×100%.
Preparation of DDN and HDDN
To obtain HA-coated nanoparticles, the inner core DDN was first prepared. To determine the appropriate mass ratio of DHP to DNA, DHP fine powders were diluted to concentration gradients. With the conjugate solution being vortexed, an equal volume of a fixed concentration of pEGFP (80 μg/mL) was added dropwise to it. After incubating for another 30 min at room temperature, the cationic core was obtained.
The freshly prepared cationic core was dropped stepwise to concentration gradients (100, 150, 200, 250, 300, 350, 400 μg/mL) of HA solution under vortexing. HDDN were obtained after stewing for 30 min at room temperature.
Agarose gel retardation assay
To determine the DNA binding ability of DHP, agarose gel retardation assay was carried out. DDN prepared at various mass ratios (DHP to pEGFP: 30:512, 30:256, 30:128, 30:64, 30:32, 30:16, 30:8, 30:4, 30:2) were mixed with appropriate amounts of 6× loading buffer and electrophoresed on 0.8% (w/v) agarose gel containing 2 μL GoldView at 90 V for 20 min. Similarly, binding conditions for series of HDDN with various mass ratios of HA to DNA (20:8, 30:8, 40:8, 50:8, 60:8, 70:8, 80:8) were evaluated to examine whether the addition of HA led to disassembly of the complex. The location of DNA in the gel was analyzed using a UV transilluminator and a digital imaging system (Gel Doc™ XR+, Bio-Rad Laboratories Inc.).
Characterization of DDN and HDDN
The particle size distribution and the zeta potential of DDN and HDDN were measured by dynamic light scattering (DLS) using a Nano Particle Analyzer (Zetasizer Nano ZS; Malvern Instruments, Malvern, UK). Transmission electronic microscopy (TEM; JEM-1200EX; JEOL, Tokyo, Japan) was used to examine the morphology of DDN and HDDN. Samples were prepared by placing a drop of DDN or HDDN onto a copper grid and air dried, following which negative staining was performed with a drop of 2.0% aqueous solution of sodium phosphotungstate for contrast enhancement.
In vitro transfection studies of DDN and HDDN
Transfection efficiency was evaluated by flow cytometry and observed by inverted fluorescence microscope. HepG2 and B16 cells were seed on 12-well plates at 2×105 cells/well and transfected after 24 h with 80%–90% confluence. The cells were washed twice with PBS buffer before transfection. Following this, the media were removed and replaced by RPIM 1640 containing DDN or HDDN with 4 μg pEGFP per well, respectively. After transfection for 4 h at 37°C, the transfection media were removed and the cells were reincubated with 1 mL culture media for 48 h at 37°C until analysis. During the 48 h incubation, the media were exchanged once. The fluorescence of the cells was observed, after which the cells were harvested for transfection efficiency quantification. Cells transfected with PEI/DNA complexes (ratio of nitrogen to phosphorus [N/P]=10) were used as positive control and with equal amount of DNA as negative control.
To investigate the serum stability of HDDN, the experiments were carried out in the presence or absence of 10% FBS in the transfection media. After transfection for 4 h, the media were also changed by culture media.
In vitro stability of HDDN in serum, DNase I and enzyme sensitive disassembly of HDDN
The stability of HDDN in serum was characterized by the particle size of HDDN in the presence or absence of 20% serum. Serum was taken from rat and this experiment was approved by the Animal Experiment Ethics Committee of Shandong University. The experiments were carried out in compliance with the Animal Management Rules of the Ministry of Health of the People’s Republic of China (Document No 55, 2001). The particle size was measured by DLS using a Nano Particle Analyzer at different time periods (0, 15, 30, and 60 min, 1, 2, 4, and 8 h).
The stability of DDN and HDDN in DNase I was characterized by agarose gel retardation assay. After incubation with DNase I for 0.5, 1, 2, 4, 8 and 12 h, the reaction was stopped by adding 2.5 mM EDTA solution for 10 min and DNA was extracted from DDN and HDDN by 8% heparin solution. Then the sample was tested by agarose gel electrophoresis, and DNA at the same concentration was used as control.
Characteristics of HDDN were used to analyze disassembly of HDDN. In brief, HDDN were incubated with different concentrations of Hyals (0 and 120 units/mL) in 1 mL of acetate buffer (pH =4.3, 37°C). The particle size distribution and the zeta potential were measured by DLS using a Nano Particle Analyzer at different time periods (0, 15, 30, and 45 min, 1 h).
In vitro toxicity of DDN and HDDN
In vitro toxicity of DDN and HDDN in HepG2 and B16 cells was evaluated by MTT assay. HepG2 and B16 cells were seeded on 96-well plates at a cell density of 6×103 cells/well in 0.15 mL RPMI 1640 supplemented with 10% FBS. After a 24 h incubation period, the cells were incubated with various concentrations of DDN and HDDN (with the DOX concentration being 0.1, 0.5, 1.0, 2.5, 5.0 μg/mL) in the culture medium for another 48 h. Following this, 0.02 mL MTT in PBS (5 mg/mL) was added to each well and incubated for an additional 4 h at 37°C in a 5% CO2 incubator. The MTT containing media were removed, and the formazan crystals formed by the living cells were dissolved in 0.2 mL DMSO. Cells without treatment were used as control with 100% viability. The absorbance at 570 nm was determined by a Microplate Reader (Model 680, Bio-Rad Laboratories Inc.). Cell viability was calculated with the formula: (Abs [sample] − Abs [blank])/(Abs [control]− Abs [blank]) ×100%.
Cellular uptake
Cellular uptake of DDN and HDDN was visualized using an inverted fluorescence microscope (BX40; Olympus Corporation, Tokyo, Japan) and quantified by flow cytometry (BD Biosciences, San Jose, CA, USA). CD44-positive HepG2, B16 cells and CD44-negative NIH 3T3 cells were seeded in 12-well culture plates at a density of 1.5×105 cells/well. After 24 h, the culture media were replaced by fresh media containing DDN and HDDN and the cells were incubated at 37°C in 5% CO2 for 4 h. Then the cells were washed three times with cold PBS and observed by inverted fluorescence microscope. To quantify the cellular uptake of DDN and HDDN, the cells were trypsinized and resuspended in 100 μL PBS. For each sample, 5,000 events were collected and the fluorescence was detected using fluorescence-activated cell sorting. For confocal microscopy, the cells were seeded on the Coverglass-Bottom Dish. After incubation with HDDN for 0.5, 1, 2 and 4 h, the media were removed and the cells were washed with cold PBS for three times. Fluorescein isothiocyanate labeled CD44-antibody was incubated with the cells at 4°C for 2 h and then the cells were washed with cold PBS for three times. Before being observed under a laser confocal microscope, the cells were covered with antifade polyvinylpyrrolidone mounting medium.
When the internalization of HDDN was inhibited by different inhibitors, the inhibitors were added to the cells 30 min before incubation with HDDN. Chlorpromazine, genistein and cytochalasin D were used as inhibitors of clathrin-mediated pathway, caveolae-mediated pathway and macropinocytosis, respectively. The CD44-antibody, which was used to inhibit CD44-mediated pathway, was incubated with the cells for 1 h before adding HDDN to the cells.
Co-delivery of DOX and pEGFP
Co-delivery of DOX and pEGFP was visualized using a laser confocal microscope (LSM780; Carl Zeiss Meditec AG, Jena, Germany) following a similar method as transfection, as described previously. After transfection, the cells were fixed in 4% paraformaldehyde. The fixed cells were then incubated with Hoechst 33342 for 15 min at 37°C to label the nuclei and washed twice with PBS. The morphologic changes and the co-delivery of DOX and DNA were observed under a fluorescence microscope.
Statistical analysis
All studies were repeated at least three times. All data were reported as mean ± standard deviation (SD). Statistical significance was analyzed using the one-tail Student’s t-test. Comparisons between two groups were considered significant when the P-value was <0.05 (P<0.05).
Results and discussion
Synthesis of HP and DHP
The drug loading material was synthesized in two steps as shown in Scheme 2. DOX and PEI were connected through a linker containing two functional groups. The HP conjugates were first synthesized by the reaction of amino group on PEI with the carboxylic group on HBA through amidocarbonylation reactions.30 The 1H NMR spectra of HP and DNP in D2O were as shown in Figures 1A and B. The successful conjugation of HBA to PEI was confirmed by the appearance of chemical shifts at 7.0–8.0 ppm for the aromatic protons of HBA. In the FTIR spectrum of HP shown in Figure 1C, carbonyl (~1,651 cm−1) vibrations also proved this conjugation. Following this, the C-13 ketone group of DOX interacted with the hydrazine group on HP to form hydrazone bond. After purification and freeze-drying, the red powder DHP obtained indicated that DOX was successfully conjugated to the polymer. As shown in Figure 1B, the characteristic peaks of DOX at 1.25 ppm confirmed the addition of DOX. As some characteristic peaks of DOX were covered by PEI and HBA, the FITR spectrum of DOX and DHP in Figure 1D was used to verify the formation of hydrazone bond.17 Carbonyl (~1,729 cm−1) vibrations of DOX disappeared in the spectra of DHP, demonstrating that hydrazone bond was achieved and no free DOX existed in DHP.31 The drug loading capacity of DHP quantified by UV–Vis spectrophotometer was 13.90%±1.87%, which is enough for drug loading and other experiments.
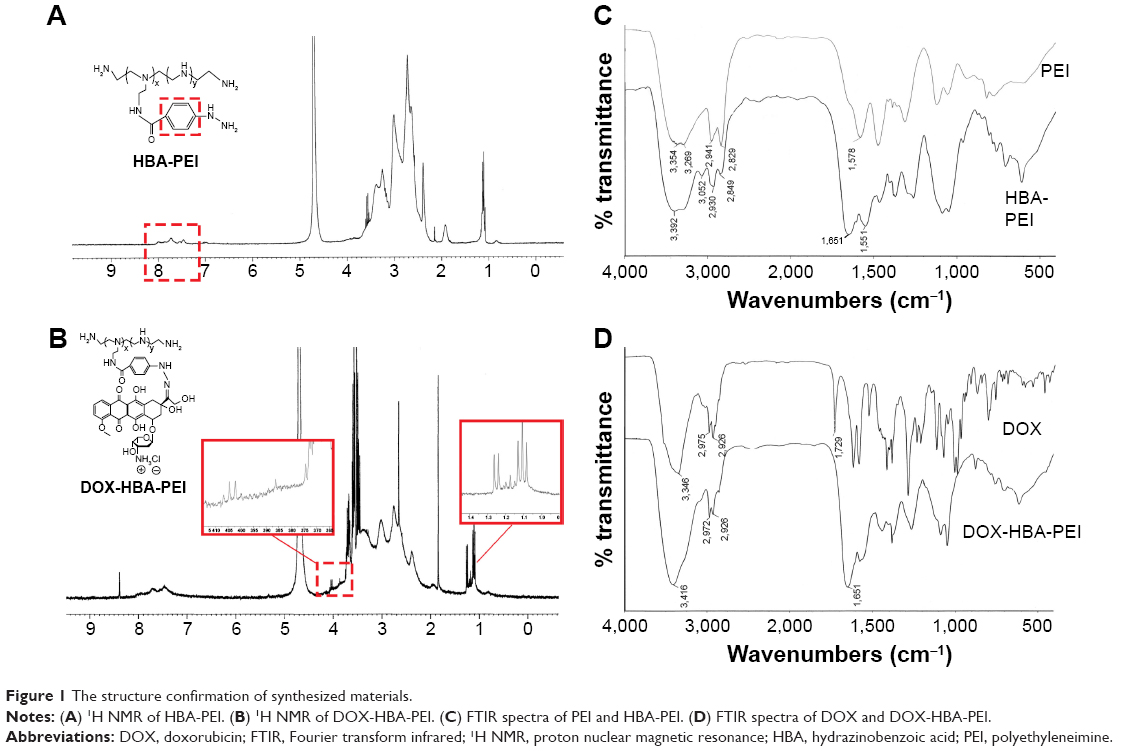 | Figure 1 The structure confirmation of synthesized materials. |
In vitro pH-sensitive drug release of DOX
In vitro release studies were performed at three pH levels for mimicking different conditions including physiologic conditions (pH 7.4), tumor sites (pH 6.0) and lysosomes in the tumor cells (pH 5.0).32 As shown in Figure 2, the cumulative release was significantly lower at pH 7.4 than at pH 5.0. At pH 7.4, the cumulative release of DOX was only 30.1% after 48 h. With the pH value lowered to 5.0, the cumulative release of DOX reached 66.4%, which revealed that the hydrazone bond of the polymer was broken and more drugs were exposed. The release of DOX from DHP exhibited pH-sensitive behaviors due to the acid-cleavable hydrazone bond between DOX and HBA. When the conjugates arrived at the lysosomes of tumor cells, lower pH value triggered the breakage of the hydrazone bond and the release of DOX. This pH-triggered release profile could prevent leakage of DOX in the blood circulation and increase the accumulation of DOX in the tumor site.33
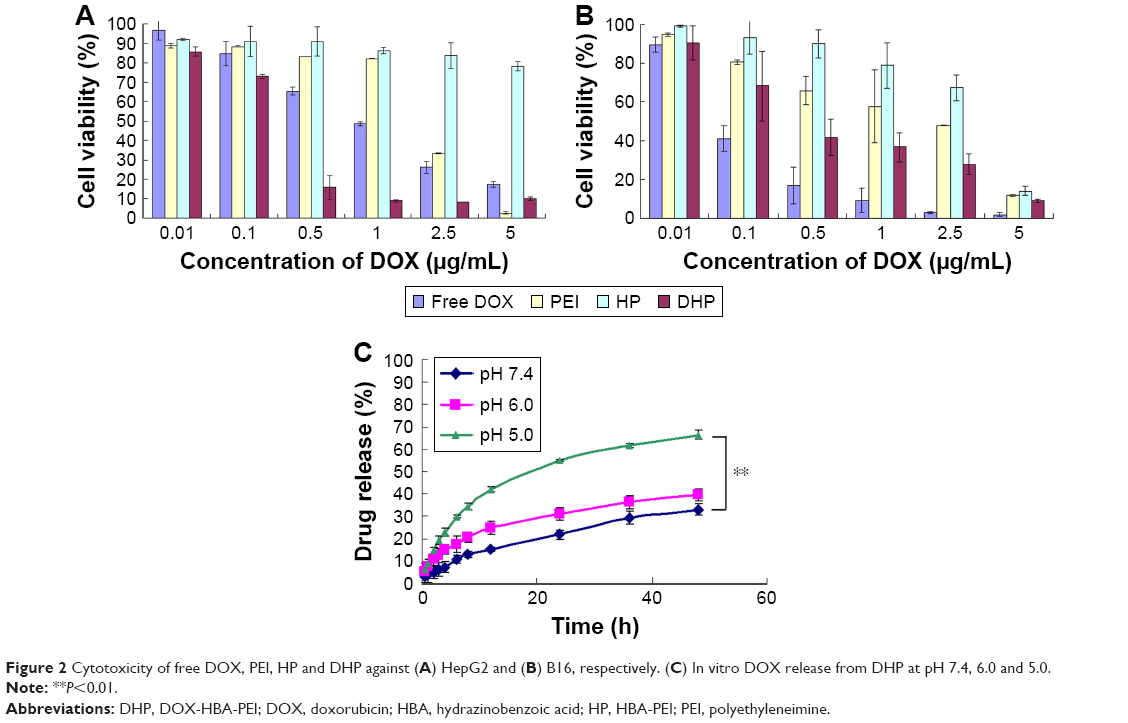 | Figure 2 Cytotoxicity of free DOX, PEI, HP and DHP against (A) HepG2 and (B) B16, respectively. (C) In vitro DOX release from DHP at pH 7.4, 6.0 and 5.0. |
In vitro cytotoxicity of multifunctional materials
The in vitro cytotoxicity of DOX, PEI, HP and DHP against HepG2 and B16 cell lines was studied using the MTT assay after 48 h of incubation, respectively. The concentration of DOX, PEI, HP and DHP was equal to the concentration of DOX in DHP. With the same concentration of PEI, the cell viability of HP was >70% compared with <40% for PEI (Figure 2A and B). The IC50 of PEI and HP was 0.46±0.10 and 1.44±0.15 μg/mL, respectively, in HepG2 cells and 0.37±0.09 and 1.79±0.71 μg/mL, respectively, in B16 cells. This result shows that HP presents good biocompatibility through modified amino groups on PEI.34 However, the cell viability of HP at 5 μg/mL was only 14.07%±2.25% in B16 cells. The low cell viability of HP at high concentration may be due to the excessive free amino group on PEI.35 When DOX was conjugated to HP, the drug loading material DHP had the potential of antitumor efficacy and exhibited a dose-dependent cytotoxic effect in HepG2 and B16 cells. The IC50 of DHP was 0.12±0.01 and 0.36±0.25 μg/mL in HepG2 and B16 cells, respectively. In HepG2 cells, DHP was more toxic than DOX, of which the IC50 was 0.99±0.31 μg/mL. On the contrary, the IC50 of DOX in B16 cells was 0.11±0.02 μg/mL, which indicates that DOX was more toxic than DHP. This opposite result obtained in different cells was due to the respective chemosensitivity and drug resistance to DOX.36 In spite of this, DOX and DHP showed no significant difference in cell viability in HepG2 and B16 cells, indicating that DHP, as a drug loading material, exhibited obvious antitumor efficacy in both cell lines.
Preparation and characterization of DDN
The inner core-DDN was a PIC formed by electrostatic interaction between the cationic polymers DHP and pEGFP. Considering the easy destruction of nucleic acid, pEGFP was dropped into DHP solution to avoid the destruction of pEGFP. With different mass ratios of DHP to DNA from 30:512 to 30:2, agarose gel retardation assay was carried out to determine the DNA binding ability of DHP.
As shown in Figure 3A, free DNA could not be observed only when the mass ratio was from 30:16 to 30:2, reflecting that DNA could be condensed completely. This data indicated that modification of PEI by drug loading did not influence the binding ability of DHP. It could be used as a cationic polymer for pEGFP delivery. DDN at different mass ratios of DHP to DNA were performed and characterized. The particle size and the zeta potential both increased with the mass ratio becoming larger (Table 1) due to the augmentation of concentration of DHP with more amino groups. The particle size was 65–90 nm and the zeta potential of DDN was around 20 mV. TEM photo further demonstrated the formation of DDN. Figure 3B and C shows the morphology of DDN (30:4) wherein it is found to have a kind of analogous spherical shape with no conglutination.
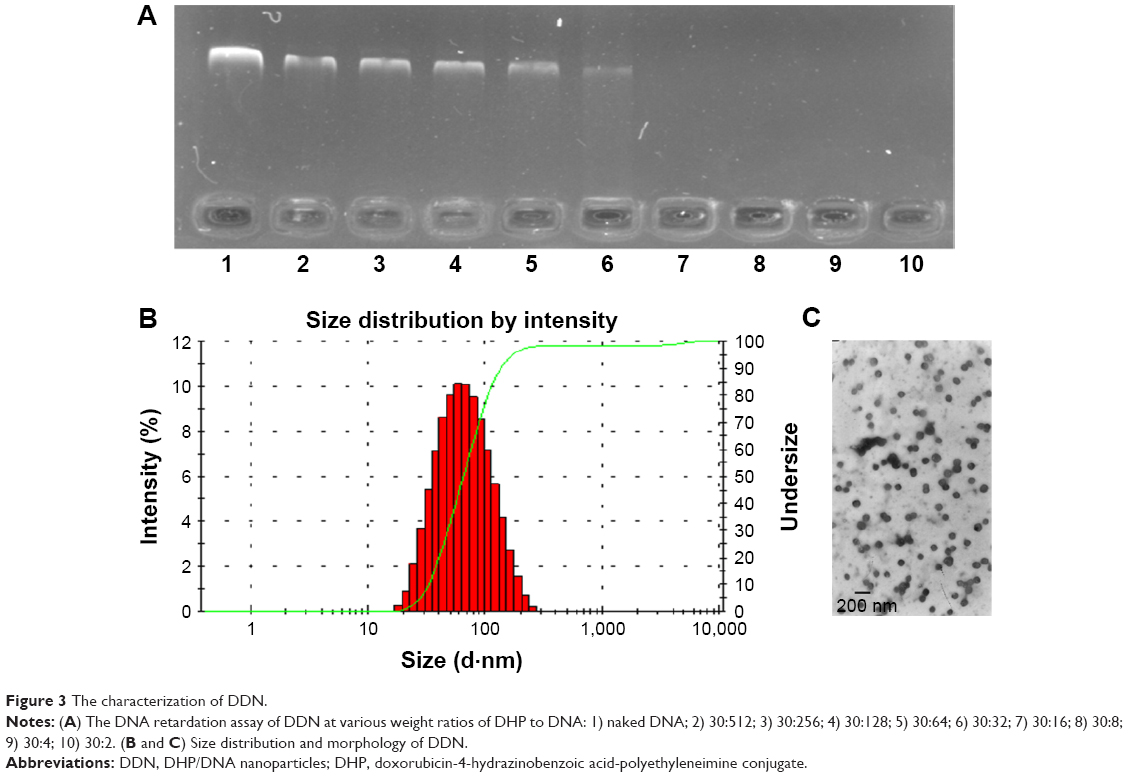 | Figure 3 The characterization of DDN. |
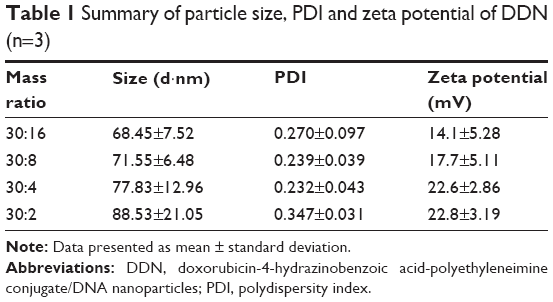 | Table 1 Summary of particle size, PDI and zeta potential of DDN (n=3) |
In vitro transfection of DDN
Enough transfection efficiency was required for gene delivery.37 Therefore, it is important to consider the transfection efficiency of DDN when making sure the prescription of inner core.
Some of the PEI-based delivery systems have been reported in the previous studies.38,39 In this study, we prepared DDN and the PEI/DNA complexes (N/P =10) that were chosen as the positive control. The obtained transfection efficiencies of the two gene delivery systems were all similar in comparison with the reported systems. As shown in Figure 4, the transfection efficiency of PEI/DNA complexes (N/P =10) was 24.03%±0.47% and 39.34%±0.86% in B16 and HepG2 cell lines, similar to that of PEI/DNA complexes reported previously. Taranejoo and Wang chose PEI/DNA (N/P =10) as the positive control; the transfection of PEI/DNA complexes (N/P =10) was around 20% in HEK293 cells and 16.38%±1.20% in HepG2 cells.38,39 The transfection efficiency of DDN at 30:4 in B16 and HepG2 cells was 25.03%±1.83% and 36.65%±6.63%, respectively, which was similar to that of the PEI-modified DNA delivery systems reported previously. Taranejoo prepared GCS-ss-PEI/DNA and the transfection of GCS-ss-PEI/DNA (N/P =20) was 28.6%±2.8% in HEK293 cells.38 The transfection efficiency of OTMCS-PEI-R13-h/DNA (N/P=10) prepared by Lv was around 20%.39 These researches proved that modification of PEI did not affect the transfection ability of PEI. Thus, the DDN obtained in this study could be used as gene and drug co-delivery carriers. Compared with other mass ratios, the transfection efficiency at 30:4 was significantly higher in both HepG2 and B16 cell lines. The fluorescence image also showed more green fluorescence at 30:4 in agreement with quantification. DDN at 30:4 was determined as the optimal formulation and it would be used in the following experiments for preparing HDDN.
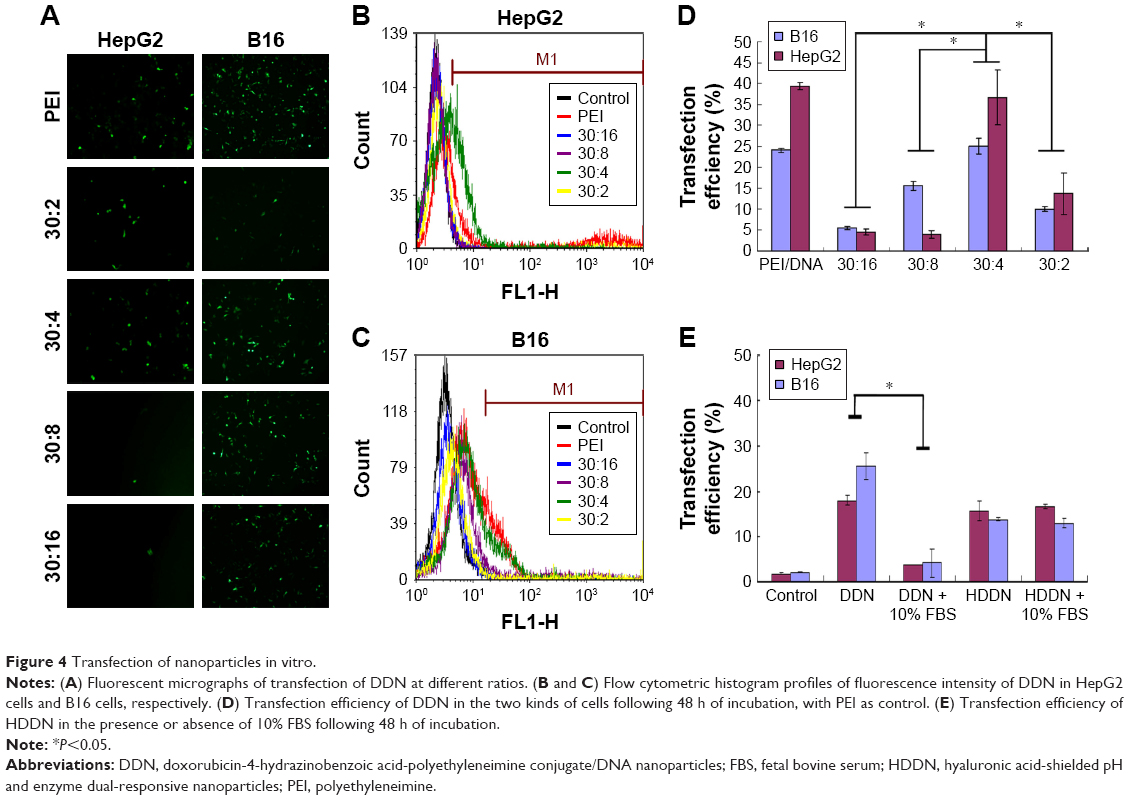 | Figure 4 Transfection of nanoparticles in vitro. |
Preparation and characterization of HDDN
The procedure of shielding positive charge was carried out in many ways, such as electrostatic interaction and PEGylation, chemical modification.12,40 In this study, the biodegradable material HA with a negative charge was chosen as a shell to shield the positive charge of DDN. The formation of both DDN and HDDN was through electrostatic interaction. Therefore, it must be confirmed that the addition of HA would not induce disassembly of the inner core.
Agarose gel retardation assay indicated that HA and DNA at all mass ratios would not influence the structure of the inner core (Figure 5A). In this occasion, the mass ratio of HA to DNA from 20:8 to 80:8 was chosen for further characterization. However, when the mass ratios were 20:8 and 30:8, HA dispersed in the solution was unable to be absorbed on the DDN to form rounding nanoparticles observed under TEM. The particle size, polydispersity index, and zeta potential of other mass ratios are shown in Table 2.
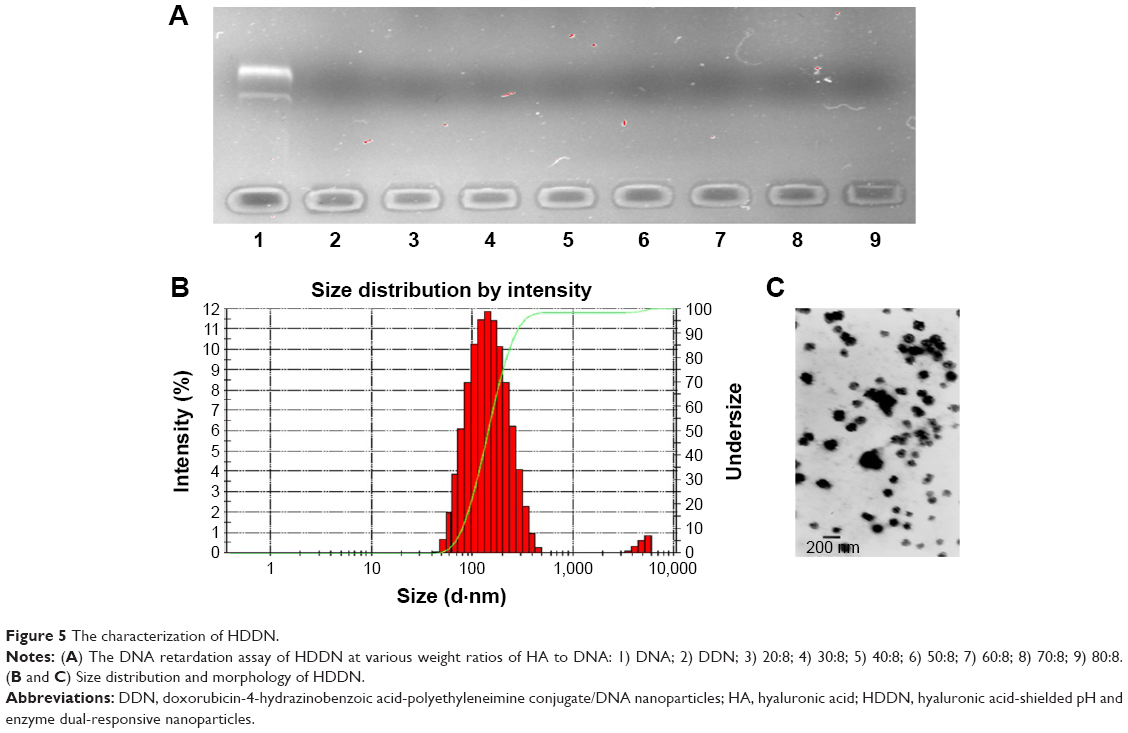 | Figure 5 The characterization of HDDN. |
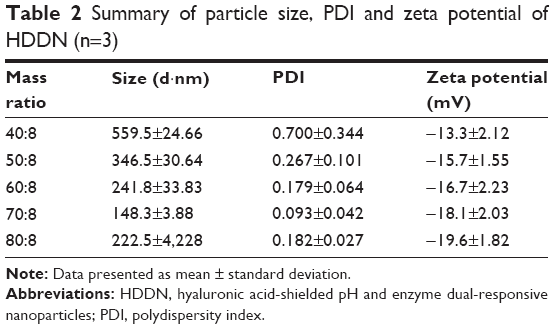 | Table 2 Summary of particle size, PDI and zeta potential of HDDN (n=3) |
With an increase in mass ratio, the particle size decreased at first and increased later. This is related to the concentration of HA in HDDN. With more and more HA being added, the interaction between HA and DDN was more and more strong. When the interaction becomes maximum, more HA would not decrease the size through interaction and, on the contrary, increase the size because of its large MW. Hence, HDDN at 70:8 had the least size (148.3±3.88 nm), polydispersity index (0.093±0.042) and proper zeta potential (−18.1±2.03 mV), which is stable and suitable for enhanced permeability and retention effect and cellular uptake.41 TEM images showed round morphology of HDDN at the optimal prescription with few agglomerations because of the viscoelasticity of HA as a biomacromolecule (Figure 5B and C).
Stability of HDDN in serum, DNase I and Hyals
As HA was sensitive to Hyals in the tumor tissue and lysosomes, disassembly of HDDN was confirmed in vitro (Figure 6A). On incubation at 37°C, the particle size and the zeta potential of HDDN rapidly increased at 15 min and kept still up to 1 h. The increase of zeta potential and particle size was caused by the degradation of HA and the monosaccharide taken off from HDDN. HA was degraded into low-MW HA and monosaccharide. Low-MW HA was still on the DDN and the monosaccharide was taken off from HDDN. The collapse of HDDN in a short time proved that HDDN was susceptible to Hyal-1, which is in accordance with another report.22 These results indicated the potential of enzyme-triggered release and disassembly of HDDN.
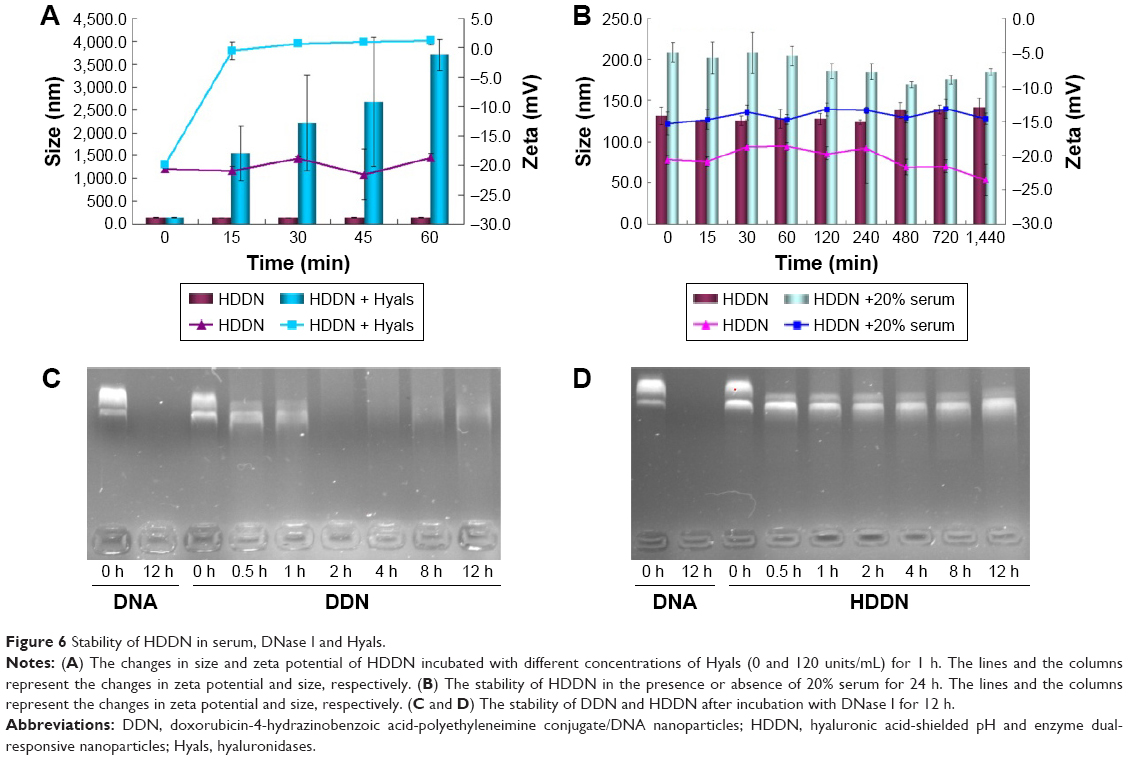 | Figure 6 Stability of HDDN in serum, DNase I and Hyals. |
Since genes are sensitive to the presence of serum, the stability of HDDN in serum was determined. The particle size of HDDN at 37°C in the presence or absence of serum was measured to represent the stability of HDDN, as shown in Figure 6B. The particle size of HDDN was increased at the moment of adding serum. As the time went on, the particle size did not increase, which indicated that HDDN was stable in the presence of serum. The in vitro transfection efficiency of DDN and HDDN also proved the stability of HDDN. In the presence of 10% FBS, the transfection efficiency of HDDN was 15.76%±0.58% and 13.73%±1.07% in HepG2 and B16 cells, respectively, which is similar to that of HA-based core–shell nanoparticles reported previously.42 Badwaik et al prepared HA-Ad:CD-PEI+:pDNA for efficient gene delivery. The HA-Ad was modified on the CD-PEI/DNA to reduce the positive charge of the cationic gene complex. The transfection efficiency of HA-Ad:CD-PEI+ :pDNA (N/P =5) was around 20% in Hela cells.42 There was no significant difference in the transfection efficiency between the groups with the presence of 10% FBS and the absence of 10% FBS. However, there was a significant difference in the transfection of DDN in the presence or absence of 10% FBS, as shown in Figure 4E. Since DDN had a positive charge, they were able to bind the negative protein in the serum and influence the stability of gene. HA coated on DDN supported negative charge and prevented the gene from contacting the serum. These results indicate that HA has the potential for protecting the gene from degradation in the serum.
Furthermore, HA coated on DDN had the potential to protect the gene from being degraded by DNase I, which was important for gene delivery. As shown in Figure 6C, pEGFP could be easily degraded by DNase I. After being loaded in the carrier, the plasmid was not sensitive to DNase I. After incubation with DNase I, pEGFP was degraded at 2 h in DDN, while it was stable for 12 h in HDDN (Figure 6D). This result indicated that HA coated outside was favorable to increase the stability of gene. HDDN was a stable carrier for gene delivery.
In vitro toxicity of DDN and HDDN
The cytotoxicity of DDN and HDDN should be taken into consideration to determine the efficiency of nanoparticles. As shown in Figure 7, the cell viabilities of DDN were lower than HHDN in both cell lines. In HepG2 cells, the IC50 values of HDDN and DDN were 13.88±1.77 and 7.33±1.85 μg/mL, respectively. The IC50 values of DDN and HDDN were 1.33±0.30 and 3.68±0.56 μg/mL, respectively, in B16 cells, which exhibited antitumor effect in vitro. The higher toxicity of DDN was caused by the positive charge on the surface of DDN, which destroyed the cell membrane through aggregation on the cell surfaces.
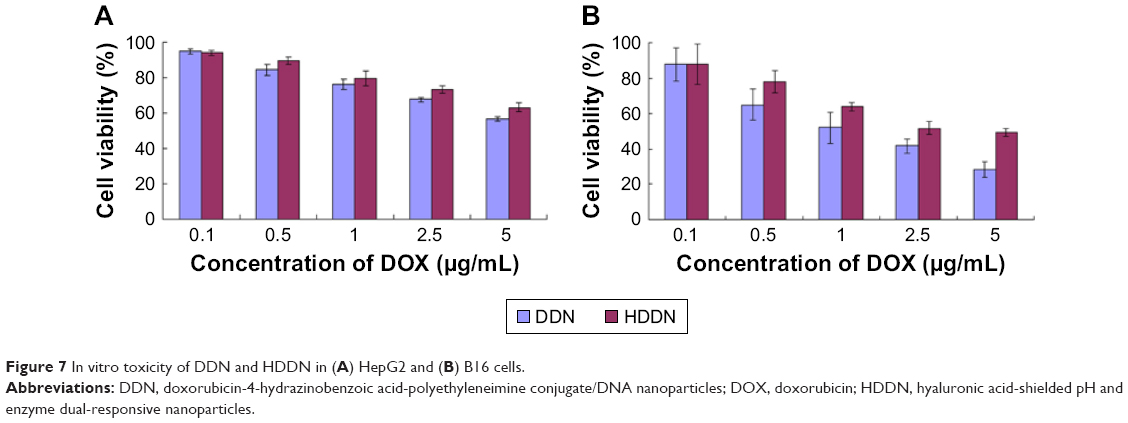 | Figure 7 In vitro toxicity of DDN and HDDN in (A) HepG2 and (B) B16 cells. |
Cellular uptake
HA-modified nanoparticles were internalized through CD44-mediated endocytosis because of the specific affinity of HA to CD44. As shown in Figure 8A, the red fluoresences were observed in both HepG2 and B16 cell lines in HDDN. As illustrated in Figure 8B, flow cytometry was used for the quantitative assay of the cellular uptake of HDDN in CD44-positive HepG2 and B16 cell lines and CD44-negative NIH 3T3 cell lines. Both DDN and HDDN could be internalized efficiently after 4 h of incubation in three CD44-positive cell lines, ensuring the entry of NPs into tumor cells and the release of DOX. However, the fluorescence images presented in Figure 8A show that red fluorescence did not exist in nuclear region after incubation with DDN and HDDN. This result indicates that DOX did not enter the nuclear region, which is in accordance with the release behaviors of DHP at 4 h in this study.
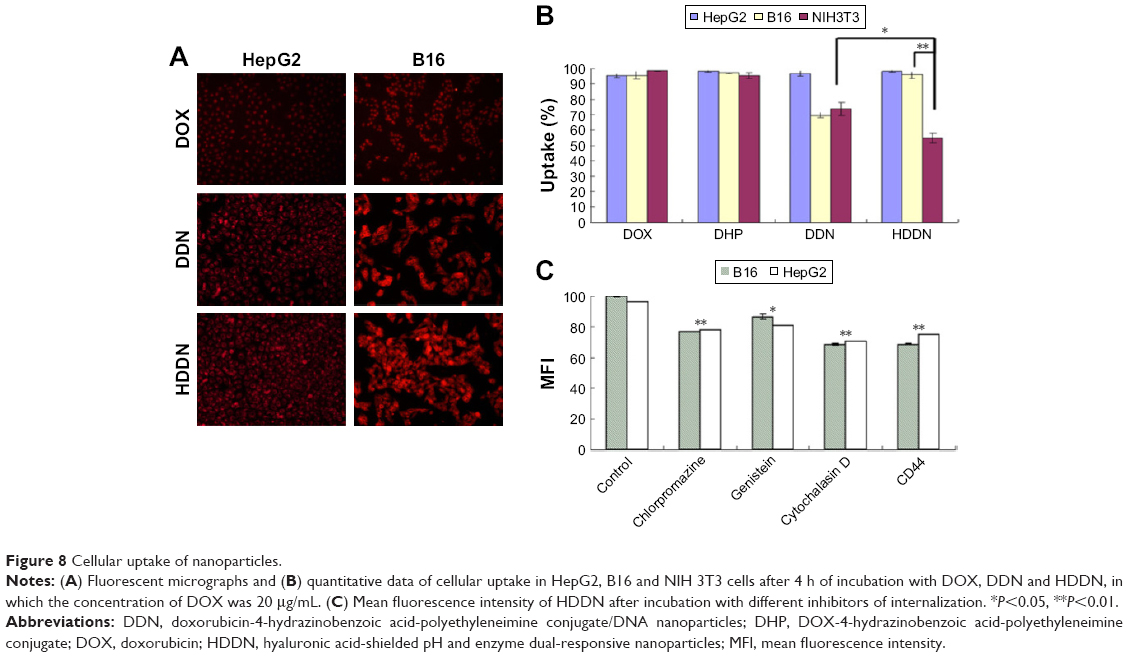 | Figure 8 Cellular uptake of nanoparticles. |
The mechanism of internalization was carried out as shown in Figure 8C and Figure 9. In CD44-positive cells, the cellular uptake of DDN through the interaction between the positive charge on DDN and the negative charge on the cell membrane was equal to that of HDDN through CD44-mediated endocytosis.22 But in CD44-negative cells, the cellular uptake of HDDN was lower than that of DDN. When CD44 was blocked by CD44-antibody, the mean fluorescence intensity of HDDN decreased by 20%. As shown in Figure 9, CD44 was localized on the membrane at 0.5 h on HepG2 cells and B16 cells, which are CD44-positive cells lines. As HDDN was internalized, CD44 and HDDN were co-localized on the membrane at 1 h and in the cell at 2 h. After incubation with HDDN for 4 h, CD44 and HDDN were separated and CD44 recycled back to membrane. These results demonstrated that HDDN with HA outside had the potential to target CD44 and was internalized through CD44-mediated endocytosis to increase the accumulation in tumor cells.
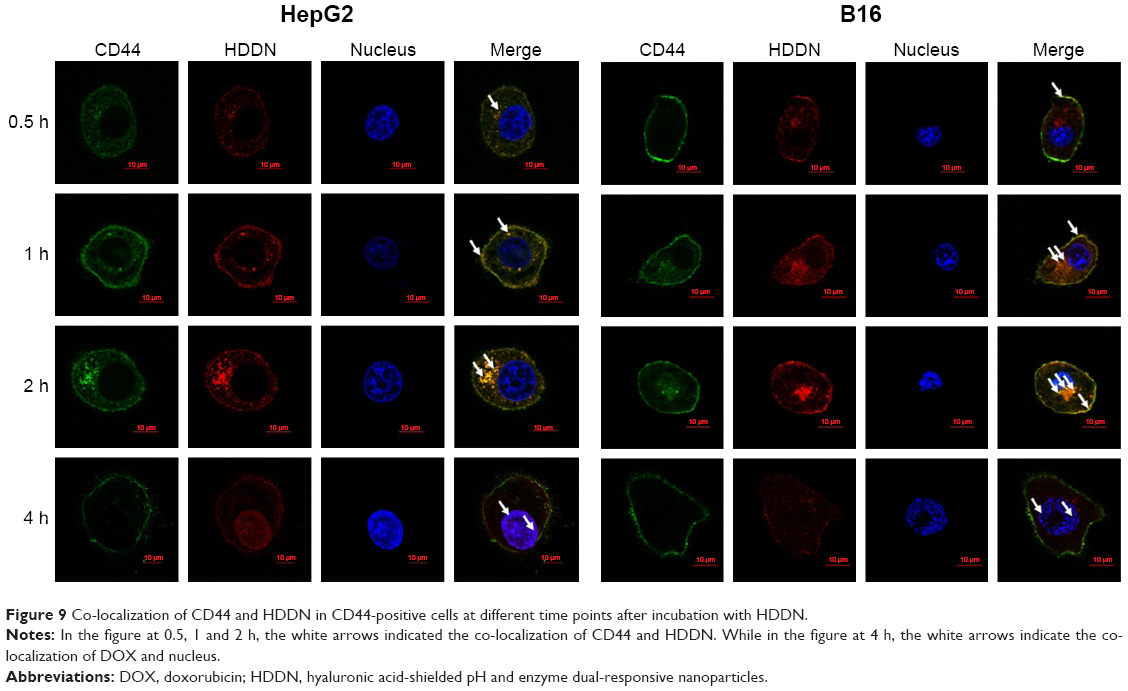 | Figure 9 Co-localization of CD44 and HDDN in CD44-positive cells at different time points after incubation with HDDN. |
Even though CD44 was inhibited by CD44-antibody, HDDN was still internalized into the cells. In order to clarify the endocytosis mechanism of HDDN, three inhibitors of different pathways were added before incubation with HDDN. As shown in Figure 8C, the uptake of HDDN was inhibited by chlorpromazine and cytochalasin D in CD44-positive cells, which represented that HDDN was internalized through clathrin-mediated pathway and macropinocytosis. In a similar way, although HDDN failed to enter NIH 3T3 cells through receptor-mediated endocytosis, there was still 54.77%±3.27% NIH 3T3 cells with HDDN inside. These results demonstrated that HA was a CD44-targeted ligand and HDDN was a favorable nanocarrier for DOX and gene delivery.
Co-delivery of drug and gene
In order to identify the co-delivery ability of HDDN, we used laser confocal microscope to investigate the fluorescence of pEGFP and DOX in HepG2 cells and B16 cells. As shown in Figure 10, red fluorescence of DOX was observed in both HepG2 cells and B16 cells, indicating that HDDN was successfully delivered into these cells. Overlapping of red and blue fluorescence generates violet stains in the nucleus of the cells, indicating that the DOX was released from HDDN and entered into nucleus for causing DNA damage. At the same time, some bright green fluorescent expressions were also observed in both cells. Overlapping of red and green fluorescent generates yellow stains in the merged image. This result directly demonstrated that DOX and pEGFP were co-delivered into the same cells by HDDN. The HA-coated nanoparticles were found to be an ideal carrier for DOX and pEGFP delivery and cell transfection. They provide a proposal for combining DOX and gene and achieving synergistic effect in cancer therapy.
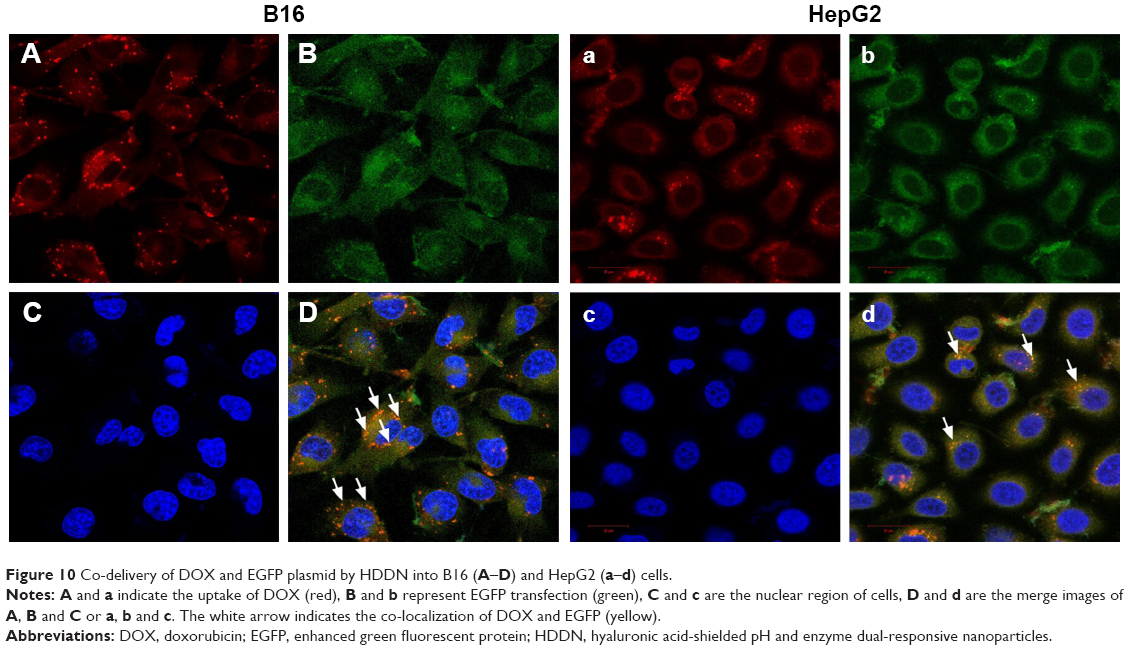 | Figure 10 Co-delivery of DOX and EGFP plasmid by HDDN into B16 (A–D) and HepG2 (a–d) cells. |
Conclusion
In this study, we developed TME dual-responsive HA-shielded core–shell nanoparticles for the co-delivery of DOX and pEGFP. DHP, a drug loading material, was successfully synthesized with high loading efficiency, pH-sensitive release behaviors and antitumor effect. The cationic core DDN prepared through electrostatic interaction showed spherical or ellipsoidal shape, uniform particle size distribution, positive zeta potential and high transfection efficiency. HA was chosen as an enzyme-responsive shell for shielding the positive charge and targeting CD44 on the tumor cells. HDDN was stable in serum and the gene was protected from being degraded due to the HA shell. In vitro transfection and cellular uptake indicated that HDDN showed negative charge, high transfection efficiency in the presence of serum and active targeting capability after HA modification. Efficient co-delivery of DOX and pEGFP was proved. Based on these results, it was concluded that TME dual-responsive HA-shield core–shell nanoparticles could be considered as a promising platform for the efficient co-delivery of DOX and gene by a simplified process.
Acknowledgments
This work was supported by The Fundamental Research Funds of Shandong University (No 2014QY004). Special thanks to Dr Livesey Olerile for his support in English grammar corrections.
Disclosure
The authors report no conflicts of interest in this work.
References
Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19(11):1423–1437. | ||
Sharma RA, Plummer R, Stock JK, et al. Clinical development of new drug-radiotherapy combinations. Nat Rev Clin Oncol. 2016;13(10):627–642. | ||
Xu X, Ho W, Zhang X, Bertrand N, Farokhzad O. Cancer nanomedicine: from targeted delivery to combination therapy. Trends Mol Med. 2015;21(4):223–232. | ||
Teo PY, Cheng W, Hedrick JL, Yang YY. Co-delivery of drugs and plasmid DNA for cancer therapy. Adv Drug Deliv Rev. 2016;98:41–63. | ||
Li J, Wang Y, Zhu Y, Oupicky D. Recent advances in delivery of drug-nucleic acid combinations for cancer treatment. J Control Release. 2013;172(2):589–600. | ||
Ganjavi H, Gee M, Narendran A, et al. Adenovirus-mediated p53 gene therapy in osteosarcoma cell lines: sensitization to cisplatin and doxorubicin. Cancer Gene Ther. 2006;13(4):415–419. | ||
Feng Q, Yu MZ, Wang JC, et al. Synergistic inhibition of breast cancer by co-delivery of VEGF siRNA and paclitaxel via vapreotide-modified core-shell nanoparticles. Biomaterials. 2014;35(18):5028–5038. | ||
Xiong XB, Lavasanifar A. Traceable multifunctional micellar nanocarriers for cancer-targeted co-delivery of MDR-1 siRNA and doxorubicin. ACS Nano. 2011;5(6):5202–5213. | ||
Kemp JA, Shim MS, Heo CY, Kwon YJ. “Combo” nanomedicine: co-delivery of multi-modal therapeutics for efficient, targeted, and safe cancer therapy. Adv Drug Deliv Rev. 2016;98:3–18. | ||
Torchilin VP. Multifunctional, stimuli-sensitive nanoparticulate systems for drug delivery. Nat Rev Drug Discov. 2014;13(11):813–827. | ||
Yao Y, Su Z, Liang Y, Zhang N. pH-Sensitive carboxymethyl chitosan-modified cationic liposomes for sorafenib and siRNA co-delivery. Int J Nanomedicine. 2015;10:6185–6197. | ||
Liu T, Wang M, Wang T, Yao Y, Zhang N. Co-delivery of doxorubicin and siRNA by a simplified platform with oligodeoxynucleotides as a drug carrier. Colloids Surf B Biointerfaces. 2015;126:531–540. | ||
Yin T, Wang L, Yin L, Zhou J, Huo M. Co-delivery of hydrophobic paclitaxel and hydrophilic AURKA specific siRNA by redox-sensitive micelles for effective treatment of breast cancer. Biomaterials. 2015;61:10–25. | ||
Meng H, Liong M, Xia T, et al. Engineered design of mesoporous silica nanoparticles to deliver doxorubicin and P-glycoprotein siRNA to overcome drug resistance in a cancer cell line. ACS Nano. 2010;4(8):4539–4550. | ||
Huo H, Gao Y, Wang Y, et al. Polyion complex micelles composed of pegylated polyasparthydrazide derivatives for siRNA delivery to the brain. J Colloid Interface Sci. 2015;447:8–15. | ||
Patnaik S, Gupta KC. Novel polyethylenimine-derived nanoparticles for in vivo gene delivery. Expert Opin Drug Deliv. 2013;10(2):215–228. | ||
Liu C, Liu F, Feng L, Li M, Zhang J, Zhang N. The targeted co-delivery of DNA and doxorubicin to tumor cells via multifunctional PEI-PEG based nanoparticles. Biomaterials. 2013;34(10):2547–2564. | ||
Liu F, Feng L, Zhang L, Zhang X, Zhang N. Synthesis, characterization and antitumor evaluation of CMCS-DTX conjugates as novel delivery platform for docetaxel. Int J Pharm. 2013;451(1–2):41–49. | ||
Webb BA, Chimenti M, Jacobson MP, Barber DL. Dysregulated pH: a perfect storm for cancer progression. Nat Rev Cancer. 2011;11(9):671–677. | ||
Jhaveri A, Deshpande P, Torchilin V. Stimuli-sensitive nanopreparations for combination cancer therapy. J Control Release. 2014;190:352–370. | ||
Wojtkowiak JW, Verduzco D, Schramm KJ, Gillies RJ. Drug resistance and cellular adaptation to tumor acidic pH microenvironment. Mol Pharm. 2011;8(6):2032–2038. | ||
Choi KY, Yoon HY, Kim JH, et al. Smart nanocarrier based on PEGylated hyaluronic acid for cancer therapy. ACS Nano. 2011;5(11):8591–8599. | ||
Chen B, Miller RJ, Dhal PK. Hyaluronic acid-based drug conjugates: state-of-the-art and perspectives. J Biomed Nanotechnol. 2014;10(1):4–16. | ||
Yang XY, Li YX, Li M, Zhang L, Feng LX, Zhang N. Hyaluronic acid-coated nanostructured lipid carriers for targeting paclitaxel to cancer. Cancer Lett. 2013;334(2):338–345. | ||
Choi KY, Saravanakumar G, Park JH, Park K. Hyaluronic acid-based nanocarriers for intracellular targeting: interfacial interactions with proteins in cancer. Colloids Surf B Biointerfaces. 2012;99:82–94. | ||
Veiman KL, Kunnapuu K, Lehto T, et al. PEG shielded MMP sensitive CPPs for efficient and tumor specific gene delivery in vivo. J Control Release. 2015;209:238–247. | ||
Lokeshwar VB, Selzer MG. Hyalurondiase: both a tumor promoter and suppressor. Semin Cancer Biol. 2008;18(4):281–287. | ||
Lee CS, Park W, Park SJ, Na K. Endolysosomal environment-responsive photodynamic nanocarrier to enhance cytosolic drug delivery via photosensitizer-mediated membrane disruption. Biomaterials. 2013;34(36):9227–9236. | ||
Kim SW, Oh KT, Youn YS, Lee ES. Hyaluronated nanoparticles with pH- and enzyme-responsive drug release properties. Colloids Surf B Biointerfaces. 2014;116:359–364. | ||
Shi J, Liu Y, Wang L, et al. A tumoral acidic pH-responsive drug delivery system based on a novel photosensitizer (fullerene) for in vitro and in vivo chemo-photodynamic therapy. Acta Biomater. 2014;10(3):1280–1291. | ||
Shi J, Zhang H, Wang L, et al. PEI-derivatized fullerene drug delivery using folate as a homing device targeting to tumor. Biomaterials. 2013;34(1):251–261. | ||
Taddei ML, Giannoni E, Comito G, Chiarugi P. Microenvironment and tumor cell plasticity: an easy way out. Cancer Lett. 2013;341(1):80–96. | ||
Wang L, Ren KF, Wang HB, Wang Y, Ji J. pH-sensitive controlled release of doxorubicin from polyelectrolyte multilayers. Colloids Surf B Biointerfaces. 2015;125:127–133. | ||
Yamada H, Loretz B, Lehr CM. Design of starch-graft-PEI polymers: an effective and biodegradable gene delivery platform. Biomacromolecules. 2014;15(5):1753–1761. | ||
Jager M, Schubert S, Ochrimenko S, Fischer D, Schubert US. Branched and linear poly(ethylene imine)-based conjugates: synthetic modification, characterization, and application. Chem Soc Rev. 2012;41(13):4755–4767. | ||
Iyer AK, Singh A, Ganta S, Amiji MM. Role of integrated cancer nanomedicine in overcoming drug resistance. Adv Drug Deliv Rev. 2013;65(13–14):1784–1802. | ||
Pack DW, Hoffman AS, Pun S, Stayton PS. Design and development of polymers for gene delivery. Nat Rev Drug Discov. 2005;4(7):581–593. | ||
Taranejoo S, Chandrasekaran R, Cheng W, Hourigan K. Bioreducible PEI-functionalized glycol chitosan: a novel gene vector with reduced cytotoxicity and improved transfection efficiency. Carbohydr Polym. 2016;153:160–168. | ||
Lv H1, Zhu Q1, Liu K, et al. Coupling of a bifunctional peptide R13 to OTMCS-PEI copolymer as a gene vector increases transfection efficiency and tumor targeting. Int J Nanomedicine. 2014;9:1311–1322. | ||
Tian H, Guo Z, Lin L, et al. pH-responsive zwitterionic copolypeptides as charge conversional shielding system for gene carriers. J Control Release. 2014;174:117–125. | ||
Errico A. New technology: nanotechnology targets cancer cells. Nat Rev Clin Oncol. 2013;10(12):667. | ||
Badwaik V, Liu L, Gunasekera D, et al. Mechanistic insight into receptor-mediated delivery of cationic-β-cyclodextrin: hyaluronic acid-adamantamethamidyl host: guest pDNA nanoparticles to CD44(+) cells. Mol Pharm. 2016;13(3):1176–1184. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.