Back to Journals » Lung Cancer: Targets and Therapy » Volume 16
Tumor Heterogeneity and Plasticity in Small Cell Lung Cancer
Authors Duronio GN, Sage J
Received 7 June 2025
Accepted for publication 2 October 2025
Published 10 October 2025 Volume 2025:16 Pages 125—138
DOI https://doi.org/10.2147/LCTT.S511789
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sai-Hong Ou
Gina N Duronio, Julien Sage
Departments of Pediatrics and Genetics, Stanford University, Stanford, CA, USA
Correspondence: Julien Sage, 265 Campus Drive, Lokey SIM1 G2078, Stanford University, Stanford, CA, 94305, USA, Email [email protected]
Abstract: The heterogeneous nature of cell populations in human tumors is a major contributor to tumor evolution, including and perhaps most importantly in response to treatment. Here, we review current knowledge on tumor heterogeneity and cell state plasticity in small cell lung cancer (SCLC), a fast growing and highly metastatic form of lung cancer which develops rapid resistance to therapy. There is a pressing need to expand treatment options for patients with SCLC, which requires a better understanding of the mechanisms by which this disease is able to rapidly grow and evolve in response to therapy. Our current understanding points to epigenetic rather than genetic factors in defining major aspects of inter- and intra-tumoral heterogeneity in SCLC. SCLC is overall considered to be a neuroendocrine (NE) cancer type but SCLC tumors harbor a wide diversity of cancer cell states, including both NE and non-neuroendocrine (non-NE) states, defined by their mutually exclusive expression of a set of transcription factors such as ASCL1, NEUROD1, and POU2F3. The immune microenvironments of SCLC tumors also contain a great deal of heterogeneity. Here, we discuss the different SCLC cell states associated with their defining transcription factors, as well as the epigenetic mechanisms regulating the ability of SCLC cells to switch from one state to another. We further discuss how the composition of SCLC tumors and the surrounding immune cells may affect the response to chemotherapy and immunotherapy. Being able to control plasticity and heterogeneity in SCLC may in the future offer unique opportunities to improve treatment efficacy in this recalcitrant cancer.
Keywords: tumor heterogeneity, plasticity, SCLC, neuroendocrine
Introduction
Human tumors are composed of a mixture of cancer cells and non-cancer cells, including immune cells, fibroblasts, vascular cells, and nerves. Advances in single-cell technologies monitoring DNA, RNA, and proteins have demonstrated the heterogeneity of these various cancer and non-cancer cell populations. Accumulating evidence also indicates that this heterogeneity plays a key role in the evolution of tumors, including in response to therapy.1–3 A major goal of cancer research is to characterize intra-tumoral heterogeneity in depth for each type of human cancer, hoping to exploit this knowledge to develop new therapeutic strategies. In this review, we will focus our analysis of heterogeneity and plasticity in small cell lung cancer (SCLC).
SCLC is a highly aggressive neuroendocrine (NE) subtype of lung cancer which accounts for ~15% of cases worldwide. SCLC is also the deadliest lung cancer, with a median survival time of only 10–12 months and a 5-year overall survival rate of less than 15%. SCLC development has been strongly associated with heavy cigarette smoking and exposure to carcinogens. Worldwide, it is estimated that over 200,000 patients die from SCLC every year.4 One reason SCLC is difficult to treat is because SCLC tumors grow rapidly and are highly metastatic, with a large majority of patients presenting with disseminated disease by the time of diagnosis. Additionally, SCLC tumors are either intrinsically resistant to treatment or rapidly become resistant. Standard-of-care includes chemotherapy (cisplatin or carboplatin, and etoposide), radiation therapy, and immune checkpoint blockade (anti-PD-1 or anti-PD-L1). Most patients respond well initially but tumors often relapse within a few months, and second- and third-line treatment options are rarely effective.5,6 Over the past several decades, survival rates for SCLC have not kept pace with the rising rates of many other cancer types, including lung adenocarcinoma.7
Genetic alterations can contribute to how tumors become resistant to therapy. In SCLC, however, there is limited evidence that genetic evolution underlies the recalcitrant nature of these tumors. A vast majority of SCLC tumors display inactivation of the gene coding for the RB and p53 tumor suppressors.8–10 Beyond these “truncal” alterations, it is difficult to identify clear genetic subtypes in SCLC, with the possible exception of a small group of patients with RB wild-type tumors.11–13 Recent evidence indicates that upregulation of MYC expression via extrachromosomal DNA amplification during tumor progression can provide cross-resistance to chemotherapy.14 However, this mechanism may not be universal, as a genetic analysis of evolutionary trajectories of SCLC under therapy failed to identify a similar mechanism (or other recurrent genetic mechanism underlying treatment resistance).15 Based on these observations, and without excluding the key role that specific genetic alterations may have in SCLC development, metastasis, and response to therapy, the field had focused on epigenetic and transcriptional mechanisms to explain heterogeneity and plasticity in SCLC. This review largely focuses on these “non-genetic” mechanisms.
Definition of SCLC Transcriptional Subtypes and States
The existence of possible SCLC subtypes came from the histopathological analysis of tumors, which identified “classic” and “variant” forms of SCLC. Early molecular studies identified expression of transcription factors (TFs) involved in neural or neuronal differentiation such as ASCL1 (Achaete-scute Family bHLH Transcription Factor 1), NEUROD1 (Neurogenic Differentiation 1), or ATOH1 (Atonal Homolog 1) in subsets of tumors.16–23 Additional studies found tumors that expressed neither ASCL1 nor NEUROD1,24 which became known as “non-neuroendocrine” (non-NE) SCLC. Later, YAP1 (yes-associated protein 1) and POU2F3 (POU domain class 2 transcription factor 3) were identified as dominant TFs in these non-NE tumors.25–28 These studies led to a first review of transcriptional subtypes that included tumors with high expression of ASCL1 (SCLC-A subtype), NEUROD1 (SCLC-N subtype), YAP1 (SCLC-Y subtype) and POU2F3 (SCLC-P subtype).29 Since then, the existence of the SCLC-Y subtype as a distinct, YAP1-driven subset of SCLC has remained controversial, as YAP1 expression has failed to define distinct sets of tumors in further histopathologic analyses of SCLC,30,31 and SCLC-Y cell lines have been reclassified as actually being derived from undifferentiated, SMARCA4-deficient lung cancers rather than true SCLC.32 Regardless, YAP1 expression remains associated with a non-NE state in SCLC and some studies have proposed the presence of YAP1 expression to be a positive prognostic marker.33 As the phenotype of A/N/P triple-negative SCLC continues to be explored, a new definition has gained popularity for a fourth “inflamed” (SCLC-I) subtype defined by distinctly high expression of immune checkpoint markers and human leukocyte antigen (HLA) genes.34 The SCLC-I subtype may include some tumors that retain some NE features (SCLC-I NE), while others are non-NE (SCLC-I non-NE),35 suggestive of a possible continuum between NE and non-NE states, with the inflammatory state as a transition state.36 Recent evidence supports the existence of a subtype defined by high levels of ATOH1,37 but more studies are needed to characterize this group.
An analysis of small-cell NE tumors (beyond SCLC) also identified a state driven by HNF4 (Hepatocyte Nuclear Factor 4) with some gastric differentiation,38 but whether SCLC-H tumors represent a new independent SCLC subtype remains to be determined. While MYC family members (c-MYC, L-MYC, and N-MYC) can be highly expressed in SCLC cells and contribute to tumor growth and chemoresistance, the expression of these TFs overlaps with the lineage-specific drivers and MYC factors are not directly used to determine specific subtypes.14,15,39–43 NFIB (Nuclear Factor 1B) is involved in both lung development and brain development and can be highly expressed in SCLC cells where it promotes neuronal features and metastasis.43–46 NFIB levels may affect response to chemotherapy.45 However, high expression of this TF can be found in both ASCL1high or NEUROD1high SCLC cells,41,44 and there is no evidence that NFIBhigh SCLC tumors represent a distinct subtype. Figure 1A represents our current view of SCLC subtypes and their notable molecular features.
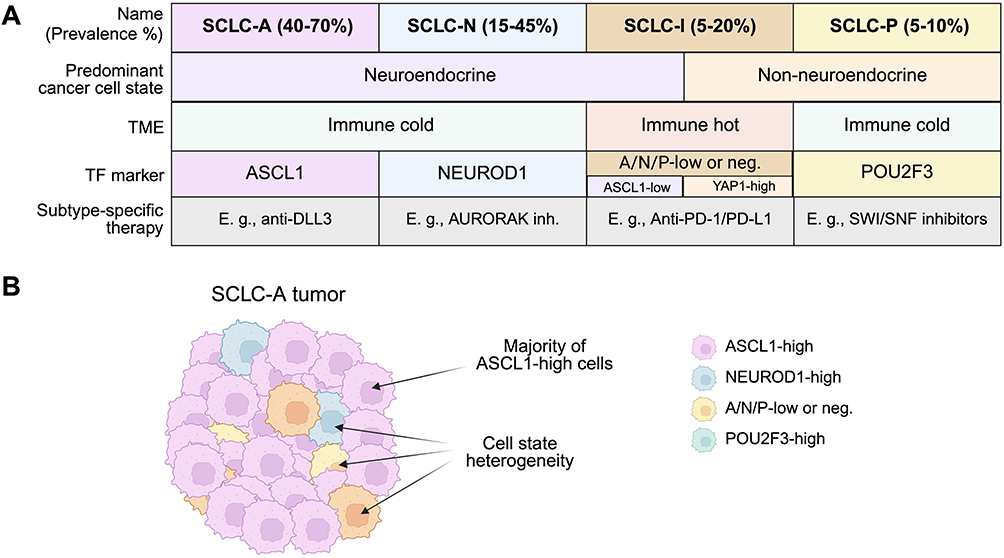 |
Figure 1 SCLC subtypes and states. (A) Current definitions, notable and shared features, and prevalence of the major SCLC subtypes. SCLC is broadly grouped into NE and non-NE disease, of which the majority are considered to be immune-cold in regard to immune cell infiltrate and MHC I expression. Subtypes are defined by dominant expression of transcription factors (TFs) or by lack of transcription factor expression in the case of some inflamed tumors. Though not yet used for clinical decision making, subtype-specific characteristics may prime SCLC cases to be more amenable to distinct therapeutic strategies. Note that the possible ATOH1 subtype, which has been less characterized, is not shown. (B) Graphical depiction of a heterogeneous SCLC-A subtype tumor harboring a majority of ASCL1high SCLC cells and various TF-drive cell-states. The presence of majority ASCL1high cells defines the tumor as SCLC-A, although cells lacking ASCL1 expression in lieu of NEUROD1, POU2F3, or triple-negative phenotypes may also be present within the tumor at large. Created in BioRender. Duronio, G. (2025) https://BioRender.com/ybftwp2. |
To summarize, the four TF-subtypes are currently the major framework through which we categorize inter-tumoral heterogeneity in SCLC.36,47,48 However, each tumor displays intra-tumoral heterogeneity, and populations of cancer cells defined by the different TFs can be present in the same tumor: in this context, we refer to the TF-definitions as distinct cancer cell states. A tumor in which the majority of cancer cells are in a specific state (eg, ASCL1high) belongs to the subtype associated with this state (ie, SCLC-A) (Figure 1B). This distinction between tumor subtypes versus cell states is notable as multiple TF-driven cell states may be present in the same patient. Additionally, the primary tumor and metastases may belong to different subtypes, as changes in the composition of cancer cell states may occur during progression, and different metastases in the same patient might also belong to distinct subtypes.
Given this as well as emerging evidence that subtype switching may occur after therapy,34,49 there is great interest in understanding the molecular mechanisms driving each state with hopes of translating this to a clinical advantage. However, it is notable that to date no correlations between patient demographic features such as sex, gender, age, or extent of smoking history and molecular subtype have been found.50 Continuing to analyze and classify patient tissue samples is an important effort for the field to pursue in order to enhance our understanding of how the subtypes can translate into meaningful clinical utility.
Emerging Evidence of the Clinical Significance of SCLC Subtypes
While there has been no baseline difference in overall survival found amongst SCLC subtypes,50 recent evidence indicates that our new understanding of SCLC heterogeneity may drive the implementation of subtype-specific treatment protocols. Our goal here is not to provide an exhaustive list of therapies being developed against SCLC, as done in other recent reviews,36,47,48,51 but to provide some salient examples.
First, inhibitors of CDK4/6 (Cyclin-dependent kinase 4/6) have shown efficacy in RB wild-type breast cancer,52 and their possible efficacy in the ~6% of SCLC cases that are RB wild-type has led to a recent ongoing clinical trial (clinical trial NCT04010357).13 The analysis of gene expression levels shows that CDK4 is more highly expressed in SCLC cells compared to CDK6, and it will be interesting in the future to test CDK4-selective inhibitors, which may have fewer side effects and possibly the same efficacy.53 CDK2/4/6 inhibitors may also be worth testing in the SCLC context, where CDK2 activity is suspected to be high.54 Notably, CDK4/6 inhibitors may also be used in RB mutant SCLC, where they can induce transient cell cycle arrest in lymphocytes and reduce myelosuppression upon chemoradiotherapy.55 This approach provides an example where the genetics of SCLC may inform specific treatment.
Second, the atypical NOTCH ligand DLL3 (Delta-like 3) has become a target of great interest in SCLC (and other NE tumors) due to its specific and upregulated cell surface expression in cancer cells.56 While several approaches are likely to prove successful, the bispecific T-cell engager (BiTE) Tarlatamab recently received accelerated FDA approval for the treatment of SCLC.57,58 Notably, the DLL3 gene is a target of ASCL1,23,59,60 indicating that SCLC-A tumors may benefit the most from strategies targeting DLL3.
Third, cells in the SCLC-P state are highly dependent on POU2F3 expression.28 A genetic screen recently identified genes whose products are essential for the expression of POU2F3 in SCLC cells, including members of the SWI/SNF (Switch/Sucrose Non-Fermentable) chromatin remodeling complex (eg, SMARCD1 or BRD9).61 The clinical development of small molecule inhibitors and degraders of SWI/SNF components (eg, clinical trials NCT04879017 and NCT04891757) suggests an avenue for the treatment of SCLC-P tumors.
Fourth, the SCLC-I subtype has been associated with better responses to T-cell-based immunotherapies.35,36 Despite a high tumor mutation burden in SCLC tumors,8 the beneficial effects of ICIs remain limited to only ~15% of patients.62–66 One explanation for the relative lack of efficacy of T-cell immune checkpoint inhibitors in SCLC is the low expression of MHC I (major histocompatibility class I) molecules on the surface of SCLC cells and the subsequent lack of tumor antigen presentation.67–70 However, when stratified by subtype patients with NE SCLC-I demonstrated the longest median progression-free survival of 5.47 months as well as the largest improvement in median overall survival between atezolizumab plus chemotherapy treated and chemotherapy-only treated controls, at 16.37 and 8.64 months, respectively.35 It is possible that SCLC cells in inflamed tumors are exposed to interferon gamma (IFNγ) and other cytokines that may help remodel the tumor microenvironment and induce the expression of MHC I molecules (see below). However, this does not explain the disparity in the survival benefit that atezolizumab treatment conferred to NE SCLC-I but not non-NE SCLC-I, indicating that all of the mechanisms that sensitize SCLC to immunotherapy are yet to be discovered.
As subtype-specific therapies begin to be implemented, a key question in the future will be how resistance evolves and what therapy should be used in cases of subtype-switching. For example, a patient with an SCLC-A tumors being treated with Tarlatamab may experience the emergence of resistant tumors where most SCLC cells have low levels of ASCL1 (and thus DLL3).71 However, if these resistant tumors are of the SCLC-I subtype, T-cell based immunotherapies may be used as a next treatment step. This type of staged, evolution-responsive approach will require a more refined characterization of cancer cell states, close monitoring of SCLC cell populations under therapy, and the development of additional cell state-specific therapies.
Influence of the Cell Type of Origin on SCLC Heterogeneity
A question in the field that has long gone unsolved is whether there is one definitive cell type of origin for SCLC, and if so, which cell type this is. Although most SCLC tumors contain a majority of distinctly NE cells, pulmonary neuroendocrine cells (PNECs) are quite rare in the lung. The scarcity of these cells brings it into question whether they are likely to be the main cell of origin or whether SCLC transforms from other epithelial cell types. Genetically engineered mouse models (GEMMs) have been instrumental in addressing this question.72,73 While PNECs, club cells, basal cells, or even alveolar type II cells may all be competent to initiate SCLC under the right genetic context, recent work indicates that basal cells, with their stem cell potential in the lung epithelium, may serve as a common cell of origin.74 Beyond the identification of cell types from which SCLC may originate, these studies have shown different patterns of heterogeneity that depend on the cell type of origin in fully developed tumors. In the RPR2 mouse model (inactivation of RB, p53, and the RB-related protein RBL2 in the lungs using the Cre/Lox system), tumors initiated from a viral vector with expression of the Cre recombinase under the PNEC-specific promoter of the CALCA gene (coding for CGRP) were less heterogeneous (fewer non-NE cells) than tumors initiated with Cre driven from the broadly-expressed CMV promoter.75 In addition, tumors initiated in PNECs metastasized mostly using an NFIB-independent mechanism, different from what had been described for tumors using CMV-Cre.44,75 While this work did not address the question of cell type of origin in the CMV-promoter model, it provided the first definitive evidence that SCLC derived from a particular cell of origin has distinct features and independent mechanisms of progression. Importantly, SCLC tumors initiated from basal cells in the MYC-driven SCLC GEMM (known as the RPM model) closely recapitulate the heterogeneity of human tumors, including POU2F3-positive cells not seen before in other mouse models,74 further illustrating how the cell type of origin can influence tumor heterogeneity.
The changes in tumor heterogeneity observed in these mouse models dependent on the identity of the cell type of origin have been early pointers to the critical role of epigenetic mechanisms in regulating cell state transitions in SCLC.
Evidence for Cellular Plasticity in SCLC
As our understanding of heterogeneity within SCLC tumors has grown, so has our understanding of the mechanisms allowing SCLC cells to switch from one state to another. Tracing the lineage of cells in human tumors is extremely challenging, and nearly all studies investigating plasticity in SCLC have relied on cell models and genetically engineered mice.
It has long been known that SCLC cells in culture have different morphologies, growing either attached to the substratum or in suspension, and these phenotypes have been linked for many years to the classic and variant phenotypes of SCLC with classic SCLC cells growing in floating aggregates and variant SCLC cells often growing more attached to the plate;76,77 in general, non-NE SCLC cells tend to grow more adherent to the plate. Investigators working on mouse and human SCLC cells have often observed cell populations with various ratios of “floaters” and “stickers” in different culture conditions, suggestive of plasticity between the two states. The quantification of these two states, together with a molecular analysis of NE markers can be used to investigate how different pathways may affect cell state switching in SCLC.
The investigation of these distinct SCLC states in vitro has led to the understanding that in addition to the distinction between the NE and non-NE subtypes of SCLC disease, the most fundamental division of cell states when considering the intra-tumoral heterogeneity of SCLC is between the NE and non-NE cells present within single tumors that derive from a common precursor.18 Together with the analysis of mouse models (xenografts and GEMMs), these studies have uncovered mechanisms promoting the transition from a NE state to a non-NE state within SCLC. For example, activation of the MAPK signaling pathway by different means has been shown to promote a non-NE state to arise from NE SCLC cells.18,78,79 Activation of Notch signaling similarly promotes the NE-to-non-NE transition.8,80–82 Mechanistically, key targets of the Notch pathway in the reprogramming towards a non-NE state include HES1 and REST, which together block the induction of NE programs by ASCL1 and directly inhibit NE programs.83,84 Of interest in SCLC, Notch pathway activity may also be activated downstream of YAP1.82,85 REST may also be induced in NE SCLC cells by RUNX2 independent of Notch signaling.86 Notably, Notch signaling within SCLC tumors serves both pro- and anti-tumorigenic functions: Notch signaling can inhibit SCLC growth by promoting the transition of NE cells to a slower growing non-NE state, but non-NE cells are often more chemoresistant than NE cells and can promote the survival of NE cells and their metastases.45,82,87,88 Despite this dual role, SCLC tends to select for loss-of-function mutations to the Notch pathway receptors,8 and knockout of Notch1 or Notch2 in the RPR2 model accelerates SCLC development.86 In addition, knockout of RBPJ, an essential factor for Notch signaling transcriptional output, does not prevent tumor development in the RPR2 model,85 suggesting that the tumor suppressive effect of Notch signaling is its dominant function, at least under normal tumor growth conditions.
More granular than the histopathologic and transcriptomic classification of individual SCLC samples and cell lines into NE and non-NE, there exists functional evidence for plasticity within the various cell and tumor TF-subtypes described to date. In addition to identifying and characterizing the molecular subtypes of SCLC, this finding has sparked great interest in understanding whether the subtypes can interconvert, or whether they perhaps represent an evolutionary process as SCLC progresses rather than arising as distinct clonal entities. Indeed, a thorough, temporal analysis of SCLC evolution in the RPM model revealed that SCLC cells can evolve from an ASCL1high to a NEUROD1high cell state, eventually progressing into a YAP1high state.89 Correspondingly, these transitions were also marked by a reduction of NE factor expression and an overall phenotypic switch towards a non-NE state, which was again associated with increased Notch signaling downstream of MYC.89 In this study, MYC binding directly correlates with increased chromatin accessibility at key Notch pathway components including Hes1, thus confirming one directly mediated genetic mechanism for the phenotypic switch.89 The same group reinforced these findings with an evolutionary analysis of transcriptomic data to demonstrate that SCLC cell states exist on a continuum, rather than as distinct, static entities.90 This finding has been validated by independent computational analyses on extensive single-cell transcriptomic data of SCLC, which agreeingly found that tumors are composed of cell states which can interconvert between one another.91 Known mechanisms for subtype interconversion in SCLC are diagrammed in Figure 2. A recent study interrogating the mechanisms of SCLC-A to SCLC-N subtype plasticity specifically investigated what unique gene programs are present in SCLC-A/N double-positive disease, hypothesizing that this might represent an intermediate state along the transition. While this study did not find any significantly activated gene programs that were unique to the A/N state, it uncovered new mechanisms for micro-RNA mediated suppression of NFIB, a key mediator of SCLC metastasis, specifically in NEUROD1high SCLC.92
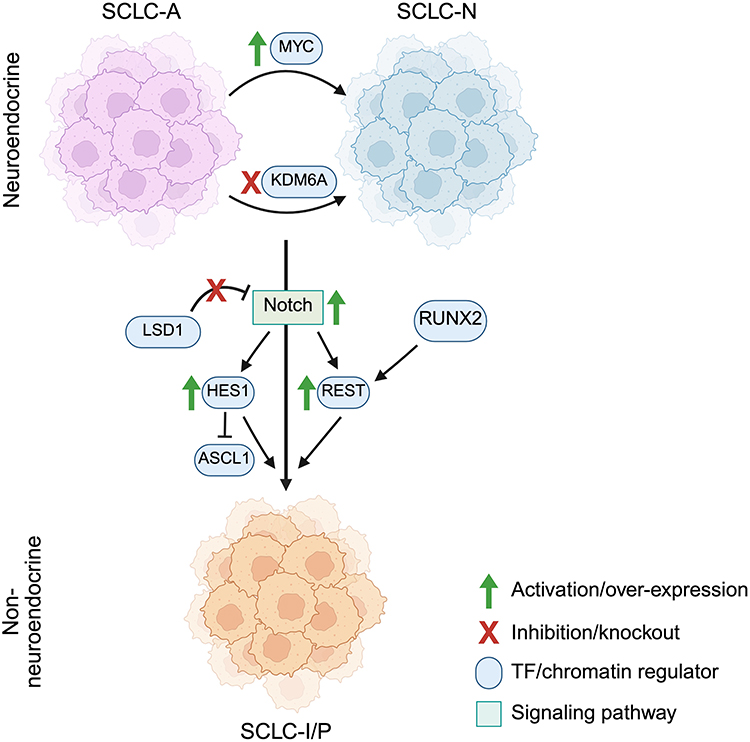 |
Figure 2 Mechanisms for cell state and TF subtype interconversion in SCLC. The major NE types of SCLC, SCLC-A and SCLC-N, can interconvert between one another, for example by the activation or silencing of additional genes, MYC and KDM6A respectively. A major determinant of NE versus non-NE cell state in SCLC tumors is the input of Notch signaling, whose activation promotes a non-NE cell state transition. This transition is mediated intracellularly by activation of downstream Notch pathway components HES1 and REST, though REST activity can be independently triggered through RUNX2. HES1 can inhibit ASCL1 activity and thus prevent the activation of NE gene programs. LSD1 is normally an inhibitor of the expression of NOTCH. Inhibition of the histone lysine demethylase LSD1 relieves the repression of the Notch pathway, thus promoting the NE to non-NE transition. The major subtypes of non-NE SCLC are SCLC-I and SCLC-P, though no specific determinants of their interconversion are currently known. Created in BioRender. Duronio, G. (2025) https://BioRender.com/xhtoivp. |
Notably, vast heterogeneity coupled with cell state plasticity has been observed even in circulating tumor cells (CTCs) sampled from patients with SCLC by liquid biopsy. By performing highly sensitive genomic analyses of CTCs, it has been confirmed that even rare cell phenotypes shared truncal genetic alterations with other cell states, indicating they arose from one another and reinforcing the extensive plasticity possible for SCLC.93 The existence of such plasticity and heterogeneity even amongst small samples of CTCs exemplifies how central these qualities are to SCLC and its pathogenesis.
Epigenetic Regulators of Cellular Plasticity in SCLC
Given that the SCLC molecular subtypes appear not to be regulated at the level of genetic variation but rather epigenetically/transcriptionally, it follows that epigenetic regulators could play a large role in regulating the SCLC cell state. The aforementioned study which defined bidirectional cell state transitions in SCLC found that the interconversion of these cell states and their balance within SCLC models could be altered by drugs that target epigenetic modulators.91 One of the earliest examples of the importance of epigenetic regulators to SCLC was the finding that inhibition of lysine-specific demethylase 1 (LSD1) led to a strong reduction in the proliferation of SCLC cell lines.94 Later studies sought to elucidate the mechanism of LSD1 activity in SCLC, and found that it is a central regulator of NE cell state, and its inhibition led to an activation of the Notch pathway which in turn suppressed NE marker gene expression and promoted a transition to a non-NE phenotype, as discussed above.
It has been well established that the Notch pathway is crucial for determining the balance of NE and non-NE cell states within tumors, but how exactly the more granular TF-subtypes are regulated has only just begun to be elucidated. One example of specific subtype states being modulated by a single epigenetic regulator was the discovery that the transition of SCLC-A into SCLC-N can be induced by inactivation of the H3K27 histone demethylase KDM6A.95 By performing a CRISPR-Cas9 screen in SCLC GEMMs, the authors found that KDM6A expression promotes ASCL1high SCLC, and that its inactivation allows for expression of NEUROD1 as well as co-expression of these factors within single SCLC samples.95 The authors demonstrated through chromatin analyses (ATAC-seq) that loss of KDM6A function leads to an increase in chromatin accessibility at the Neurod1 gene locus that is accompanied by NEUROD1 protein expression. The ability of SCLC to transition between two of the four major subtypes following inactivation of a single gene poses interesting questions regarding the utility of employing subtype-specific treatments to fight a disease where these states can be so readily interconverted.
Another such example of direct epigenetic regulation of subtype maintenance is the recent finding that SCLC-P, as discussed above, relies on activity of the SWI/SNF complex to maintain POU2F3 expression which drives the oncogenic program of this subtype.61 Importantly, the authors demonstrated that pharmacologic disruption of the enzymatic activity of the complex attenuated growth of SCLC-P xenografts, demonstrating the essentiality of POU2F3 expression in this subtype and reinforcing the importance of employing cell state-specific therapeutics for improving SCLC treatment.
A third pertinent example is the role of the pioneer TFs FOXA1/2 in driving NFIB-independent metastasis. Given that SCLC metastasis happens rapidly and early in disease progression, with greater than 70% of patients presenting with metastases at the time of diagnosis, it is crucial to understand all the mechanisms by which SCLC spreads throughout the body. Outside of the established NFIB-driven mechanism for SCLC metastasis, in the context of NFIB-knockout, FOXA1/2 expression is likely to drive this process instead, highlighting how crucial epigenetic-driven plasticity is in conferring SCLC with its most aggressive features.
Heterogeneity and Plasticity in Disease Progression and Response to Treatment
Given the propensity of SCLC for developing therapy resistance, there has been great interest in understanding how the heterogeneity of SCLC tumors changes in response to treatment. Deriving xenografts from CTCs in patients has allowed for the serial sampling and subsequent analysis of cancer cells from single patients before and after resistance develops. One study utilized this method to establish CTC-derived xenograft (CDX) models from both chemo-sensitive and chemo-resistant SCLC as well as from single patients at multiple time points throughout disease progression.96 By performing single-cell transcriptomic analyses, the authors were able to demonstrate increased heterogeneity in chemoresistant disease, as well as an increase in heterogeneity in CDXs from the same patients before and after relapse.96 Notably, this increase in heterogeneity arises at the transcriptional level, as no significant changes to the genome or an increase in tumor mutation burden were observed.96 This finding agrees with a comprehensive exome sequencing analysis of surgically resected SCLC samples which found that increased intra-tumoral heterogeneity when quantified from tumor mutational burden per cluster correlates with worse overall survival for SCLC patients.97
In addition to understanding how heterogeneity and cell states change as a response to treatment, studies have investigated how these qualities might change to drive disease progression and metastasis. Interestingly, TF subtypes can shift between primary tumors and their paired lymph node (LN) metastases, indicating a potentially advantageous role of cellular plasticity for promoting tumor dissemination.98 Notably, this shift often was from NE-high primary tumor subtypes to lower-NE subtypes in LN metastases, though this work was not able to distinguish whether this was a driver of the metastatic process or a response to the change in tumor environment. An additional study characterizing the NE phenotype of metastatic SCLC metastases almost always displayed a combination of NE and non-NE features, and that combined phenotype cases of SCLC had higher rates of drug resistance and especially poor outcomes,99 further reinforcing the potential benefit of phenotypic heterogeneity to SCLC progression. To date, no molecular subtype-specific chemo- and immunotherapy resistance mechanisms have been identified in SCLC. Rather, the prevailing hypothesis in the field is that a heterogenous tumor composition is itself a mechanism from which to develop resistance, as the tumor would be more likely to have cells primed to survive against a variety of potential stressors.100 However, further investigating what specific changes drive SCLC chemotherapy resistance and how prevalent these may be within tumors of different subtypes is an important future direction of research so that this concept can be applied to prevent resistance in SCLC patients.
SCLC Plasticity and T-Cell Responses
Recent clinical trials have shown that a subset of patients with extensive-stage SCLC have extended survival times when treated with the PD-L1 inhibitor atezolizumab in addition to standard-of-care chemotherapy.101 This has opened the question as to what sets apart patients who respond to immunotherapy, and sparked interest in whether SCLC plasticity can be utilized to shift tumors into a state more responsive to anti-tumor T cells. In accordance with this idea, accumulating evidence indicates that SCLC cells with non-NE features express higher levels of MHC I molecules on their cell surface.102 This observation has led to a search for mechanisms controlling MHC I expression in SCLC.
One mechanism that NE cancer types use to silence MHC I expression is through overexpression of the histone methyl transferase enhancer of zeste homolog 2 (EZH2). EZH2 is the catalytically active subunit of the polycomb repressive complex (PRC2) that silences gene expression by tri-methylating histone 3 lysine 27 (H3K27me3). NE cancers can co-opt an evolutionary mechanism used to prevent maternal immunity against the developing nervous system where EZH2 silences MHC I expression in select cell types.103 It had previously been established that SCLC has particularly high expression of EZH2, and that EZH2 inhibition can slow the proliferation of SCLC models.24 EZH2-mediated gene silencing is also a central mechanism by which SCLC can acquire chemotherapy resistance.104 Given its role in immune evasion in NE cancers as well as its established importance to the pathogenicity of SCLC, recent research has tested if EZH2 inhibition could slow SCLC growth and also stimulate response to immunotherapy. This idea is further supported by several studies demonstrating that EZH2 inhibition leads to an increase in MHC I and MHC II expression in various cancer types.105–107 Excitingly, inhibition of EZH2 in SCLC results in an increase in MHC I expression by potentiating the cyclic GMP–AMP synthase (cGAS)–stimulator of interferon genes (STING) pathway,102 a mechanism that has been documented in other cancer types as well.108 Drug-independent mechanisms for triggering EZH2 inhibition in combination with STING activation in SCLC have also been described, arising from endogenous retrovirus activity.109 Conversely, newer studies have identified factors such as CRACD (capping protein inhibiting regulator of actin dynamics), whose loss in SCLC promotes the NE cell state and EZH2 function, thus enhancing suppression of MHC I and immune checkpoint blockade (ICB) evasion.110 Notably, inactivation of a core PRC2 subunit, EED, is sufficient to drive a lineage switch from NE SCLC to lung adenocarcinoma and is required for the transformation from lung adenocarcinoma to SCLC in pre-clinical models,111 underscoring the central role of PRC2 in the control of cancer cell states in the lungs.
In addition to EZH2 modulation, other mechanisms with previously established importance to SCLC progression and plasticity have been discovered to modulate the immunogenicity of the SCLC microenvironment. It has now been demonstrated that inhibition of LSD1 not only activates Notch signaling to promote a non-NE transition of SCLC cells but also directly increases MHC I expression and subsequently promotes better response to ICB therapy.112,113 A comprehensive spatial multi-omic analysis of SCLC patient samples strengthened this connection by finding a correlation between lower overall NE character of a tumor with increased immune cell infiltration and better overall survival.114 Given the strong connection between Notch signaling, non-NE phenotypes, and response to ICB therapy, it has even been proposed that Notch signaling levels in patients can be a prognostic indicator for success of ICB treatment.115 Newer work studying the histone deacetylase (HDAC) class 1 inhibitor entinostat found that it was able to increase expression of MHC I and PD-L1 in SCLC cell lines. As a result, the addition of entinostat to anti-PD-1 immunotherapy led to prolonged survival and stronger responses in SCLC GEMMs.116 Concurrently, entinostat treatment shifted tumors from a predominantly NE to predominantly non-NE or NE-low phenotype,116 once again connecting a reduction of the NE phenotype to enhancement of immunotherapy response.
The mechanisms described above for activating MHC I expression involve proteins for which inhibitors have been developed and are available for use in clinical trials, exemplifying how understanding the mechanisms that drive diversity of cell states can directly influence how we develop therapeutic approaches and combination treatment plans for patients with SCLC. Notably, it has been found that how immune “hot” or “cold” the SCLC tumor microenvironment is may correlate with molecular subtype, with SCLC-A tumors having lower infiltration of CD8+ T-lymphocytes than SCLC-P tumors.117 Other studies comparing SCLC-A to SCLC-N tumors found that they contain different compositions of T-cell populations and that N-subtype tumors have fewer cytotoxic T-cells and more regulatory T-cells, which confers less anti-tumor T-cell activity overall.118 Such findings reinforce the importance of understanding the molecular features of a tumor in order to identify patients who may experience increased therapeutic benefit from ICB. There is great interest in finding combination therapeutic strategies utilizing known mechanisms of modulating the immune microenvironment of SCLC (outlined in Figure 3) such as EZH2, LSD1, or HDAC inhibition to enhance the response of tumors to immunotherapy. By directly inhibiting factors which suppress antigen presentation and immune receptors, small molecule inhibitors in combination with immunotherapy might prove to be an effective new avenue to increase the only modest response to ICB seen among SCLC patients at large.
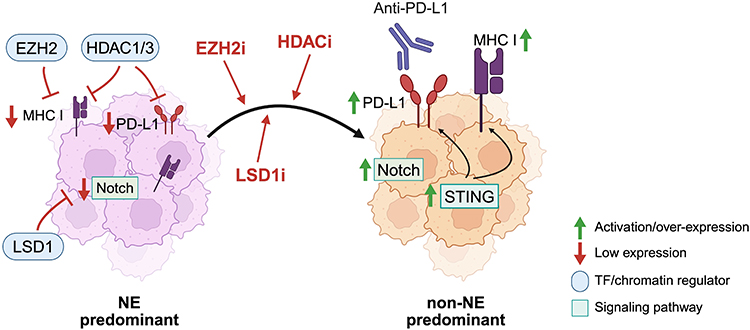 |
Figure 3 Alterations to the SCLC tumor landscape that influence cell signaling pathway activity, cancer cell neuroendocrine character, and the immune microenvironment. In NE-cell predominant SCLC cells, EZH2 inhibits MHC I expression, and baseline levels of Notch signaling are low. Upon inhibition of EZH2 or LSD1, Notch signaling is activated, tumors shift more towards non-NE cell predominance with increased expression of MHC I and PD-L1, including via activation of the STING pathway. Created in BioRender. Duronio, G. (2025) https://BioRender.com/t4ukx3t. |
Conclusions and Future Directions
SCLC continues to have abysmally low survival statistics that have not kept pace with the drastic improvements seen for many other cancers over the past few decades. Rapid growth and metastasis and the development of therapy resistance continue to make SCLC a particularly challenging clinical target. Understanding the diversity of cell states within SCLC tumors and how these states change during metastasis and in the face of therapy is crucial for developing new, effective treatments. There has been great effort within the field to define molecular subtypes of SCLC that are distinguished at the level of TF expression, rather than by distinct genetic signatures or mutational patterns. There is a need to further define the known transcriptional states and identify new states, but also to gain a better understanding of what factors regulate how SCLC cell states interconvert between one another. It is also currently largely unknown whether the spatial organization of different cancer cell states in SCLC, for which there is emerging evidence,119 influences plasticity and response to treatment.
As discussed, subtype-specific therapies are beginning to emerge for SCLC treatment, such as the newly FDA-approved BiTE Tarlatamab targeting the SCLC-A-specific surface antigen DLL3. As these things come into clinical practice, it is likely we will in turn see subtype-specific mechanisms of therapeutic resistance emerge. For Tarlatamab, such mechanisms could include silencing of DLL3 expression or transition from SCLC-A to other SCLC subtypes that do not express DLL3. Another example may be the future utilization of SWI/SNF inhibitors following the discovery that SCLC-P relies on the function of this complex to maintain POU2F3 expression and proliferate. Loss of POU2F3 expression in the setting of such treatment may promote subtype switching to NE-SCLC not reliant on POU2F3 expression. Given the great propensity of SCLC for phenotypic plasticity, the employment of subtype-specific therapeutics will likely require careful monitoring of both the clinical and molecular response of tumors in order to adapt treatment in the face of phenotypic change and emergence of resistance.
The discovery that many epigenetic regulatory proteins are central to the pathogenesis and plasticity of SCLC has opened exciting avenues for novel therapeutic strategies, as many targeted inhibitors have been developed against these epigenetic factors. Examples of this include the loss of NE cell identity and subsequent expression of antigen presentation machinery by inhibiting LSD2, EZH2, or class 1 HDACs. However, it can be anticipated that the inherent intra-tumoral heterogeneity of SCLC will again allow for resistant clones to predominate in the face of novel epigenetic inhibitor treatment. By further elucidating the molecular pathways driving SCLC heterogeneity and evolution, these discoveries will hopefully be leveraged to inhibit SCLC plasticity and drive tumors to a cell state more amendable to therapeutic eradication. The future of SCLC treatment likely lies in a combination therapeutic approach that simultaneously engages multiple facets of SCLC vulnerability to mitigate the impact of heterogeneity on the development of resistance and relapse. Importantly, knowledge gained in SCLC may be relevant to a larger group of tumors with neuroendocrine features, as well as other cancers known to be composed of highly heterogenous and plastic cell populations.
Abbreviations
SCLC, small-cell lung cancer; NE, neuroendocrine, ICB, immune checkpoint blockade; BiTE, bispecific T-cell engager; GEMM, genetically engineered mouse model; TF, transcription factor.
Acknowledgments
The authors thank members of the lab for useful discussions.
Funding
This work was funded by the NIH (R35 CA231997 and R01 CA285740 to J.S., and T32 GM145402 to G.N.D.), a Mark Foundation Endeavor Award (J.S.), and the Ludwig Cancer Institute (J.S.).
Disclosure
J.S. has equity in, and is an advisor for, DISCO Pharmaceuticals. G.N.D declares no competing interests in this work.
References
1. Mehta A, Stanger BZ. Lineage plasticity: the new cancer hallmark on the block. Cancer Res. 2024;84(2):184–191. doi:10.1158/0008-5472.CAN-23-1067
2. Gargiulo G, Serresi M, Marine JC. Cell states in cancer: drivers, passengers, and trailers. Cancer Discov. 2024;14(4):610–614. doi:10.1158/2159-8290.CD-23-1510
3. Tabassum DP, Polyak K. Tumorigenesis: it takes a village. Nat Rev Cancer. 2015;15(8):473–483. doi:10.1038/nrc3971
4. Rudin CM, Brambilla E, Faivre-Finn C, Sage J. Small-cell lung cancer. Nat Rev Dis Primers. 2021;7(1):3. doi:10.1038/s41572-020-00235-0
5. Zugazagoitia J, Osma H, Baena J, Ucero AC, Paz-Ares L. Facts and hopes on cancer immunotherapy for small cell lung cancer. Clin Cancer Res. 2024;30(14):2872–2883. doi:10.1158/1078-0432.CCR-23-1159
6. Megyesfalvi Z, Gay CM, Popper H, et al. Clinical insights into small cell lung cancer: tumor heterogeneity, diagnosis, therapy, and future directions. CA Cancer J Clin. 2023;73(6):620–652. doi:10.3322/caac.21785
7. Howlader N, Forjaz G, Mooradian MJ, et al. The effect of advances in lung-cancer treatment on population mortality. N Engl J Med. 2020;383(7):640–649. doi:10.1056/NEJMoa1916623
8. George J, Lim JS, Jang SJ, et al. Comprehensive genomic profiles of small cell lung cancer. Nature. 2015;524(7563):47–53. doi:10.1038/nature14664
9. Rudin CM, Durinck S, Stawiski EW, et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nature Genet. 2012;44(10):1111–1116. doi:10.1038/ng.2405
10. Peifer M, Fernández-Cuesta L, Sos ML, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nature Genet. 2012;44(10):1104–1110. doi:10.1038/ng.2396
11. Febres-Aldana CA, Chang JC, Ptashkin R, et al. Rb tumor suppressor in small cell lung cancer: combined genomic and IHC analysis with a description of a distinct Rb-proficient subset. Clin Cancer Res. 2022;28(21):4702–4713. doi:10.1158/1078-0432.CCR-22-1115
12. Sivakumar S, Moore JA, Montesion M, et al. Integrative analysis of a large real-world cohort of small cell lung cancer identifies distinct genetic subtypes and insights into histologic transformation. Cancer Discovery. 2023;13(7):1572–1591. doi:10.1158/2159-8290.CD-22-0620
13. Wildey G, Shay AM, McColl KS, et al. Retinoblastoma expression and targeting by CDK4/6 inhibitors in small cell lung cancer. Mol Cancer Ther. 2023;22(2):264–273. doi:10.1158/1535-7163.MCT-22-0365
14. Pal Choudhuri S, Girard L, Lim JYS, et al. Acquired cross-resistance in small cell lung cancer due to extrachromosomal DNA amplification of MYC paralogs. Cancer Discov. 2024;14(5):804–827. doi:10.1158/2159-8290.CD-23-0656
15. George J, Maas L, Abedpour N, et al. Evolutionary trajectories of small cell lung cancer under therapy. Nature. 2024;627(8005):880–889. doi:10.1038/s41586-024-07177-7
16. Chen H, Biel MA, Borges MW, et al. Tissue-specific expression of human achaete-scute homologue-1 in neuroendocrine tumors: transcriptional regulation by dual inhibitory regions. Cell Growth Differ. 1997;8(6):677–686.
17. Westerman BA, Breuer RH, Poutsma A, et al. Basic helix-loop-helix transcription factor profiling of lung tumors shows aberrant expression of the proneural gene atonal homolog 1 (ATOH1, HATH1, MATH1) in neuroendocrine tumors. Int J Biol Markers. 2007;22(2):114–123. doi:10.1177/172460080702200205
18. Calbo J, van Montfort E, Proost N, et al. A functional role for tumor cell heterogeneity in a mouse model of small cell lung cancer. Cancer Cell. 2011;19(2):244–256. doi:10.1016/j.ccr.2010.12.021
19. Kashiwagi K, Ishii J, Sakaeda M, et al. Differences of molecular expression mechanisms among neural cell adhesion molecule 1, synaptophysin, and chromogranin A in lung cancer cells. Pathol Int. 2012;62(4):232–245. doi:10.1111/j.1440-1827.2011.02781.x
20. Ishii J, Sato H, Sakaeda M, et al. POU domain transcription factor BRN2 is crucial for expression of ASCL1, ND1 and neuroendocrine marker molecules and cell growth in small cell lung cancer. Pathol Int. 2013;63(3):158–168. doi:10.1111/pin.12042
21. Krohn A, Ahrens T, Yalcin A, et al. Tumor cell heterogeneity in small cell lung cancer (SCLC): phenotypical and functional differences associated with epithelial-mesenchymal transition (EMT) and DNA methylation changes. PLoS One. 2014;9(6):e100249. doi:10.1371/journal.pone.0100249
22. Osborne JK, Guerra ML, Gonzales JX, McMillan EA, Minna JD, Cobb MH. NeuroD1 mediates nicotine-induced migration and invasion via regulation of the nicotinic acetylcholine receptor subunits in a subset of neural and neuroendocrine carcinomas. Mol Biol Cell. 2014;25(11):1782–1792. doi:10.1091/mbc.E13-06-0316
23. Borromeo MD, Savage TK, Kollipara RK, et al. ASCL1 and NEUROD1 reveal heterogeneity in pulmonary neuroendocrine tumors and regulate distinct genetic programs. Cell Rep. 2016;16(5):1259–1272. doi:10.1016/j.celrep.2016.06.081
24. Poirier JT, Gardner EE, Connis N, et al. DNA methylation in small cell lung cancer defines distinct disease subtypes and correlates with high expression of EZH2. Oncogene. 2015;34(48):5869–5878. doi:10.1038/onc.2015.38
25. Horie M, Saito A, Ohshima M, Suzuki HI, Nagase T. YAP and TAZ modulate cell phenotype in a subset of small cell lung cancer. Cancer Sci. 2016;107(12):1755–1766. doi:10.1111/cas.13078
26. Ito T, Matsubara D, Tanaka I, et al. Loss of YAP1 defines neuroendocrine differentiation of lung tumors. Cancer Sci. 2016;107(10):1527–1538. doi:10.1111/cas.13013
27. McColl K, Wildey G, Sakre N, et al. Reciprocal expression of INSM1 and YAP1 defines subgroups in small cell lung cancer. Oncotarget. 2017;8(43):73745. doi:10.18632/oncotarget.20572
28. Huang Y-H, Klingbeil O, He X-Y, et al. POU2F3 is a master regulator of a tuft cell-like variant of small cell lung cancer. Genes Dev. 2018;32(13–14):915–928. doi:10.1101/gad.314815.118
29. Rudin CM, Poirier JT, Byers LA, et al. Molecular subtypes of small cell lung cancer: a synthesis of human and mouse model data. Nat Rev Cancer. 2019;19(5):289–297. doi:10.1038/s41568-019-0133-9
30. Baine MK, Hsieh M-S, Lai WV, et al. SCLC subtypes defined by ASCL1, NEUROD1, POU2F3, and YAP1: a comprehensive immunohistochemical and histopathologic characterization. J Thorac Oncol. 2020;15(12):1823–1835. doi:10.1016/j.jtho.2020.09.009
31. Kumar S, Singh N, Gupta P, Kumar R, Mitra S, Bal A. Prognostic significance of immunohistochemical surrogate molecular sub‐typing of small cell lung carcinoma. APMIS. 2025;133(6):e70035. doi:10.1111/apm.70035
32. Ng J, Cai L, Girard L, et al. Molecular and pathologic characterization of YAP1-expressing small cell lung cancer cell lines leads to reclassification as SMARCA4-deficient malignancies. Clin Cancer Res. 2024;30(9):1846–1858. doi:10.1158/1078-0432.CCR-23-2360
33. Go SI, Yang JW, Jeong EJ, et al. Redefining YAP1 in small cell lung cancer: shifting from a dominant subtype marker to a favorable prognostic indicator. Transl Lung Cancer Res. 2024;13(8):1768–1779. doi:10.21037/tlcr-24-317
34. Gay CM, Stewart CA, Park EM, et al. Patterns of transcription factor programs and immune pathway activation define four major subtypes of SCLC with distinct therapeutic vulnerabilities. Cancer Cell. 2021;39(3):346–360.e7. doi:10.1016/j.ccell.2020.12.014
35. Nabet BY, Hamidi H, Lee MC, et al. Immune heterogeneity in small-cell lung cancer and vulnerability to immune checkpoint blockade. Cancer Cell. 2024;42(3):429–443e4. doi:10.1016/j.ccell.2024.01.010
36. Redin E, Quintanal-Villalonga A, Rudin CM. Small cell lung cancer profiling: an updated synthesis of subtypes, vulnerabilities, and plasticity. Trends Cancer. 2024;10(10):935–946. doi:10.1016/j.trecan.2024.07.008
37. Catozzi A, Peiris Pages M, Humphrey S, et al. Functional characterization of the ATOH1 molecular subtype indicates a pro-metastatic role in small cell lung cancer. Cell Rep. 2025;44(5):115603. doi:10.1016/j.celrep.2025.115603
38. Wang Z, Liu C, Zheng S, et al. Molecular subtypes of neuroendocrine carcinomas: a cross-tissue classification framework based on five transcriptional regulators. Cancer Cell. 2024;42(6):1106–1125e8. doi:10.1016/j.ccell.2024.05.002
39. Dammert MA, Bragelmann J, Olsen RR, et al. MYC paralog-dependent apoptotic priming orchestrates a spectrum of vulnerabilities in small cell lung cancer. Nat Commun. 2019;10(1):3485. doi:10.1038/s41467-019-11371-x
40. Kim DW, Wu N, Kim YC, et al. Genetic requirement for Mycl and efficacy of RNA Pol I inhibition in mouse models of small cell lung cancer. Genes Dev. 2016;30(11):1289–1299. doi:10.1101/gad.279307.116
41. Mollaoglu G, Guthrie MR, Bohm S, et al. MYC drives progression of small cell lung cancer to a variant neuroendocrine subtype with vulnerability to aurora kinase inhibition. Cancer Cell. 2017;31(2):270–285. doi:10.1016/j.ccell.2016.12.005
42. Romero OA, Torres-Diz M, Pros E, et al. MAX inactivation in small cell lung cancer disrupts MYC-SWI/SNF programs and is synthetic lethal with BRG1. Cancer Discov. 2014;4(3):292–303. doi:10.1158/2159-8290.CD-13-0799
43. Semenova EA, Kwon MC, Monkhorst K, et al. Transcription factor NFIB is a driver of small cell lung cancer progression in mice and marks metastatic disease in patients. Cell Rep. 2016;16(3):631–643. doi:10.1016/j.celrep.2016.06.020
44. Denny SK, Yang D, Chuang CH, et al. Nfib promotes metastasis through a widespread increase in chromatin accessibility. Cell. 2016;166(2):328–342. doi:10.1016/j.cell.2016.05.052
45. Böttger F, Semenova EA, Song J-Y, et al. Tumor heterogeneity underlies differential cisplatin sensitivity in mouse models of small-cell lung cancer. Cell Rep. 2019;27(11):3345–3358.e4. doi:10.1016/j.celrep.2019.05.057
46. Wu N, Jia D, Ibrahim AH, Bachurski CJ, Gronostajski RM, MacPherson D. NFIB overexpression cooperates with Rb/p53 deletion to promote small cell lung cancer. Oncotarget. 2016;7(36):57514–57524. doi:10.18632/oncotarget.11583
47. Simpson KL, Rothwell DG, Blackhall F, Dive C. Challenges of small cell lung cancer heterogeneity and phenotypic plasticity. Nat Rev Cancer. 2025;25(6):447–462. doi:10.1038/s41568-025-00803-0
48. Zhang J, Zeng X, Guo Q, et al. Small cell lung cancer: emerging subtypes, signaling pathways, and therapeutic vulnerabilities. Exp Hematol Oncol. 2024;13(1):78. doi:10.1186/s40164-024-00548-w
49. Lo YC, Rivera-Concepcion J, Vasmatzis G, Aubry MC, Leventakos K. Subtype of SCLC is an intrinsic and persistent feature through systemic treatment. JTO Clin Res Rep. 2023;4(9):100561. doi:10.1016/j.jtocrr.2023.100561
50. Yu Z, Zou J, Xu F. The molecular subtypes of small cell lung cancer defined by key transcription factors and their clinical significance. Lung Cancer. 2024;198:108033. doi:10.1016/j.lungcan.2024.108033
51. Grenda A, Krawczyk P, Obara A, Gajek L, Lomza-Laba A, Milanowski J. Transitioning to a personalized approach in molecularly subtyped small-cell lung cancer (SCLC). Int J Mol Sci. 2024;25(8):4208. doi:10.3390/ijms25084208
52. Goel S, Bergholz JS, Zhao JJ. Targeting CDK4 and CDK6 in cancer. Nat Rev Cancer. 2022;22(6):356–372. doi:10.1038/s41568-022-00456-3
53. Palmer CL, Boras B, Pascual B, et al. CDK4 selective inhibition improves preclinical anti-tumor efficacy and safety. Cancer Cell. 2025;43(3):464–481e14. doi:10.1016/j.ccell.2025.02.006
54. Freeman-Cook K, Hoffman RL, Miller N, et al. Expanding control of the tumor cell cycle with a CDK2/4/6 inhibitor. Cancer Cell. 2021;39(10):1404–1421e11. doi:10.1016/j.ccell.2021.08.009
55. Daniel D, Kuchava V, Bondarenko I, et al. Trilaciclib prior to chemotherapy and atezolizumab in patients with newly diagnosed extensive-stage small cell lung cancer: a multicentre, randomised, double-blind, placebo-controlled Phase II trial. Int J Cancer. 2021;148(10):2557–2570. doi:10.1002/ijc.33453
56. Ding J, Yeong C. Advances in DLL3-targeted therapies for small cell lung cancer: challenges, opportunities, and future directions. Front Oncol. 2024;14:1504139. doi:10.3389/fonc.2024.1504139
57. Ahn MJ, Cho BC, Felip E, et al. Tarlatamab for patients with previously treated small-cell lung cancer. N Engl J Med. 2023;389(22):2063–2075. doi:10.1056/NEJMoa2307980
58. Mountzios G, Sun L, Cho BC, et al. Tarlatamab in small-cell lung cancer after platinum-based chemotherapy. N Engl J Med. 2025;393(4):349–361. doi:10.1056/NEJMoa2502099
59. Casarosa S, Fode C, Guillemot F;. Research Support, Non-US Gov’t. Mash1 regulates neurogenesis in the ventral telencephalon. Development. 1999;126(3):525–534. doi:10.1242/dev.126.3.525
60. Henke RM, Meredith DM, Borromeo MD, Savage TK, Johnson JE. Ascl1 and Neurog2 form novel complexes and regulate Delta-like3 (Dll3) expression in the neural tube. Dev Biol. 2009;328(2):529–540. Research Support, N.I.H. Extramural. doi:10.1016/j.ydbio.2009.01.007
61. Duplaquet L, So K, Ying AW, et al. Mammalian SWI/SNF complex activity regulates POU2F3 and constitutes a targetable dependency in small cell lung cancer. Cancer Cell. 2024;42(8):1352–1369e13. doi:10.1016/j.ccell.2024.06.012
62. Reddy HG, Qin A, Kalemkerian GP. Emerging drugs for small cell lung cancer: a focused review on immune checkpoint inhibitors. Expert Opin Emerg Drugs. 2020;25(3):353–366. doi:10.1080/14728214.2020.1798929
63. Scott SC, Hann CL. Immunotherapy for small cell lung cancer: established applications and novel approaches. Clin Adv Hematol Oncol. 2021;19(10):654–663.
64. Goldman JW, Dvorkin M, Chen Y, et al. Durvalumab, with or without tremelimumab, plus platinum-etoposide versus platinum-etoposide alone in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): updated results from a randomised, controlled, open-label, Phase 3 trial. Lancet Oncol. 2021;22(1):51–65. doi:10.1016/S1470-2045(20)30539-8
65. Rudin C, Kim H, Navarro A, et al. OA12. 06 first-line pembrolizumab or placebo combined with etoposide and platinum for ES-SCLC: KEYNOTE-604 long-term follow-up results. J Thorac Oncol. 2022;17(9):S33–S34. doi:10.1016/j.jtho.2022.07.063
66. Cheng Y, Han L, Wu L, et al. Serplulimab, a novel anti-PD-1 antibody, plus chemotherapy versus chemotherapy alone as first-line treatment for extensive-stage small-cell lung cancer: an international randomized phase 3 study. Am Soc Clin Oncol. 2022;40(16_suppl):8505. doi:10.1200/JCO.2022.40.16_suppl.8505
67. Spigel DR, Cheng Y, Cho BC, et al. ADRIATIC: durvalumab (D) as consolidation treatment (tx) for patients (pts) with limited-stage small-cell lung cancer (LS-SCLC). Am Soc Clin Oncol. 2024;42(17_suppl):LBA5–LBA5. doi:10.1200/JCO.2024.42.17_suppl.LBA5
68. Yazawa T, Kamma H, Fujiwara M, et al. Lack of class II transactivator causes severe deficiency of HLA-DR expression in small cell lung cancer. J Pathol. 1999;187(2):191–199. doi:10.1002/(SICI)1096-9896(199901)187:2<191::AID-PATH206>3.0.CO;2-3
69. Doyle A, Martin WJ, Funa K, et al. Markedly decreased expression of class I histocompatibility antigens, protein, and mRNA in human small-cell lung cancer. J Exp Med. 1985;161(5):1135–1151. doi:10.1084/jem.161.5.1135
70. Traversari C, Meazza R, Coppolecchia M, et al. IFN-γ gene transfer restores HLA-class I expression and MAGE-3 antigen presentation to CTL in HLA-deficient small cell lung cancer. Genet Ther. 1997;4(10):1029–1035. doi:10.1038/sj.gt.3300489
71. Ahn HM, Park SY, Choi Y, Kim J, Lee Y. Molecular subtype changes after acquiring resistance to tarlatamab in small cell lung cancer. Drug Resist Updat. 2024;79:101198. doi:10.1016/j.drup.2024.101198
72. Ferone G, Lee MC, Sage J, Berns A. Cells of origin of lung cancers: lessons from mouse studies. Genes Dev. 2020;34(15–16):1017–1032. doi:10.1101/gad.338228.120
73. Oser MG, MacPherson D, Oliver TG, Sage J, Park KS. Genetically-engineered mouse models of small cell lung cancer: the next generation. Oncogene. 2024;43(7):457–469. doi:10.1038/s41388-023-02929-7
74. Ireland AS, Hawgood SB, Xie DA, et al. Basal cell of origin resolves neuroendocrine-tuft lineage plasticity in cancer. bioRxiv;2024.
75. Yang D, Denny SK, Greenside PG, et al. Intertumoral heterogeneity in SCLC is influenced by the cell type of origin. Cancer Discovery. 2018;8(10):1316–1331. doi:10.1158/2159-8290.CD-17-0987
76. Carney DN, Gazdar AF, Bepler G, et al. Establishment and identification of small cell lung cancer cell lines having classic and variant features. Cancer Res. 1985;45(6):2913–2923.
77. Gazdar AF, Carney DN, Nau MM, Minna JD. Characterization of variant subclasses of cell lines derived from small cell lung cancer having distinctive biochemical, morphological, and growth properties. Cancer Res. 1985;45(6):2924–2930.
78. Ravi RK, Thiagalingam A, Weber E, McMahon M, Nelkin BD, Mabry M. Raf-1 causes growth suppression and alteration of neuroendocrine markers in DMS53 human small-cell lung cancer cells. Am J Respir Cell Mol Biol. 1999;20(4):543–549. doi:10.1165/ajrcmb.20.4.3406
79. Caeser R, Hulton C, Costa E, et al. MAPK pathway activation selectively inhibits ASCL1-driven small cell lung cancer. iScience. 2021;24(11):103224. doi:10.1016/j.isci.2021.103224
80. Sriuranpong V, Borges MW, Ravi RK, et al. Notch signaling induces cell cycle arrest in small cell lung cancer cells. Cancer Res. 2001;61(7):3200–3205.
81. Kunnimalaiyaan M, Vaccaro AM, Ndiaye MA, Chen H, Research Support, N.I.H. Extramural Research Support, Non-U.S. Overexpression of the NOTCH1 intracellular domain inhibits cell proliferation and alters the neuroendocrine phenotype of medullary thyroid cancer cells. Journal of Biological Chemistry. 2006;281(52):39819–39830. doi:10.1074/jbc.M603578200
82. Lim JS, Ibaseta A, Fischer MM, et al. Intratumoural heterogeneity generated by Notch signalling promotes small-cell lung cancer. Nature. 2017;545(7654):360–364. doi:10.1038/nature22323
83. Charbat MA, Abdulhalim YH, Alrabeei MA, Hassan WA. Role of Notch1 signaling pathway in small cell lung carcinoma. Iran J Pathol. 2024;19(4):365. doi:10.30699/ijp.2024.2013339.3184
84. Ku S-Y, Wang Y, Garcia MM, et al. Notch signaling suppresses neuroendocrine differentiation and alters the immune microenvironment in advanced prostate cancer. J Clin Invest. 2024;134(17). doi:10.1172/JCI175217
85. Shue YT, Drainas AP, Li NY, et al. A conserved YAP/Notch/REST network controls the neuroendocrine cell fate in the lungs. Nat Commun. 2022;13(1):2690. doi:10.1038/s41467-022-30416-2
86. Hong D, Knelson EH, Li Y, et al. Plasticity in the absence of NOTCH uncovers a RUNX2-dependent pathway in small cell lung cancer. Cancer Res. 2022;82(2):248–263. doi:10.1158/0008-5472.CAN-21-1991
87. Pearsall SM, Williamson SC, Humphrey S, et al. Lineage plasticity in SCLC generates non-neuroendocrine cells primed for vasculogenic mimicry. J Thorac Oncol. 2023;18(10):1362–1385. doi:10.1016/j.jtho.2023.07.012
88. Shue YT, Lim JS, Sage J. Tumor heterogeneity in small cell lung cancer defined and investigated in pre-clinical mouse models. Transl Lung Cancer Res. 2018;7(1):21–31. doi:10.21037/tlcr.2018.01.15
89. Ireland AS, Micinski AM, Kastner DW, et al. MYC drives temporal evolution of small cell lung cancer subtypes by reprogramming neuroendocrine fate. Cancer Cell. 2020;38(1):60–78.e12. doi:10.1016/j.ccell.2020.05.001
90. Groves SM, Ireland A, Liu Q, et al. Cancer hallmarks define a continuum of plastic cell states between small cell lung cancer archetypes. BioRxiv. 2021;
91. Gopal P, Petty A, Rogacki K, et al. Multivalent state transitions shape the intratumoral composition of small cell lung carcinoma. Sci Adv. 2022;8(50):eabp8674. doi:10.1126/sciadv.abp8674
92. Takumida H, Saito A, Okabe Y, et al. Integrative epigenome and transcriptome analyses reveal transcriptional programs differentially regulated by ASCL1 and NEUROD1 in small cell lung cancer. Oncogene. 2025;44(1):1–13. doi:10.1038/s41388-024-03174-2
93. Seo J, Kumar M, Mason J, et al. Plasticity of circulating tumor cells in small cell lung cancer. Sci Rep. 2023;13(1):11775. doi:10.1038/s41598-023-38881-5
94. Mohammad HP, Smitheman KN, Kamat CD, et al. A DNA hypomethylation signature predicts antitumor activity of LSD1 inhibitors in SCLC. Cancer Cell. 2015;28(1):57–69. doi:10.1016/j.ccell.2015.06.002
95. Duplaquet L, Li Y, Booker MA, et al. KDM6A epigenetically regulates subtype plasticity in small cell lung cancer. Nat Cell Biol. 2023;25(9):1346–1358. doi:10.1038/s41556-023-01210-z
96. Stewart CA, Gay CM, Xi Y, et al. Single-cell analyses reveal increased intratumoral heterogeneity after the onset of therapy resistance in small-cell lung cancer. Nat Cancer. 2020;1(4):423–436. doi:10.1038/s43018-019-0020-z
97. Zhou H, Hu Y, Luo R, et al. Multi-region exome sequencing reveals the intratumoral heterogeneity of surgically resected small cell lung cancer. Nat Commun. 2021;12(1):5431. doi:10.1038/s41467-021-25787-x
98. Csende K, Ferencz B, Boettiger K, et al. Comparative profiling of surgically resected primary tumors and their lymph node metastases in small-cell lung cancer. ESMO open. 2025;10(4):104514. doi:10.1016/j.esmoop.2025.104514
99. Lissa D, Takahashi N, Desai P, et al. Heterogeneity of neuroendocrine transcriptional states in metastatic small cell lung cancers and patient-derived models. Nat Commun. 2022;13(1):2023. doi:10.1038/s41467-022-29517-9
100. Ying Q, Fan R, Shen Y, et al. Small cell lung cancer—an update on chemotherapy resistance. Curr Treatment Options Oncol. 2024;25(8):1112–1123. doi:10.1007/s11864-024-01245-w
101. Reck M, Dziadziuszko R, Sugawara S, et al. Five-year survival in patients with extensive-stage small cell lung cancer treated with atezolizumab in the Phase III IMpower133 study and the Phase III IMbrella A extension study. Lung Cancer. 2024;196:107924. doi:10.1016/j.lungcan.2024.107924
102. Mahadevan NR, Knelson EH, Wolff JO, et al. Intrinsic immunogenicity of small cell lung carcinoma revealed by its cellular plasticity. Cancer Discov. 2021;11(8):1952–1969. doi:10.1158/2159-8290.CD-20-0913
103. Burr ML, Sparbier CE, Chan KL, et al. An evolutionarily conserved function of polycomb silences the MHC class I antigen presentation pathway and enables immune evasion in cancer. Cancer Cell. 2019;36(4):385–401.e8. doi:10.1016/j.ccell.2019.08.008
104. Gardner EE, Lok BH, Schneeberger VE, et al. Chemosensitive relapse in small cell lung cancer proceeds through an EZH2-SLFN11 axis. Cancer Cell. 2017;31(2):286–299. doi:10.1016/j.ccell.2017.01.006
105. DuCote TJ, Song X, Naughton KJ, et al. EZH2 inhibition promotes tumor immunogenicity in lung squamous cell carcinomas. Canc Res Commun. 2024;4(2):388–403. doi:10.1158/2767-9764.CRC-23-0399
106. Piunti A, Meghani K, Yu Y, et al. Immune activation is essential for the antitumor activity of EZH2 inhibition in urothelial carcinoma. Sci Adv. 2022;8(40):eabo8043. doi:10.1126/sciadv.abo8043
107. James JL, Taylor BC, Axelrod ML, et al. Polycomb repressor complex 2 suppresses interferon-responsive MHC-II expression in melanoma cells and is associated with anti-PD-1 resistance. J ImmunoTher Cancer. 2023;11(11):e007736. doi:10.1136/jitc-2023-007736
108. Xu T, Dai J, Tang L, et al. EZH2 inhibitor enhances the STING agonist‒induced antitumor immunity in melanoma. J Invest Dermatol. 2022;142(4):1158–1170.e8. doi:10.1016/j.jid.2021.08.437
109. Canadas I, Thummalapalli R, Kim JW, et al. Tumor innate immunity primed by specific interferon-stimulated endogenous retroviruses. Nat Med. 2018;24(8):1143–1150. doi:10.1038/s41591-018-0116-5
110. Seo Y, Zhang S, Jang J, et al. Actin dysregulation induces neuroendocrine plasticity and immune evasion: a vulnerability of small cell lung cancer. bioRxiv;2024.
111. Li Y, Laimon YN, Cho H, et al. EED maintains the small cell lung cancer neuroendocrine phenotype and drives lung cancer histological transformation. bioRxiv. 2025. doi:10.1101/2025.07.07.663486
112. Hiatt JB, Sandborg H, Garrison SM, et al. Inhibition of LSD1 with bomedemstat sensitizes small cell lung cancer to immune checkpoint blockade and T-cell killing. Clin Cancer Res. 2022;28(20):4551–4564. doi:10.1158/1078-0432.CCR-22-1128
113. Nguyen EM, Taniguchi H, Chan JM, et al. Targeting lysine-specific demethylase 1 rescues major histocompatibility complex class I antigen presentation and overcomes programmed death-ligand 1 blockade resistance in SCLC. J Thorac Oncol. 2022;17(8):1014–1031. doi:10.1016/j.jtho.2022.05.014
114. Jin Y, Wu Y, Reuben A, et al. Single-cell and spatial proteo-transcriptomic profiling reveals immune infiltration heterogeneity associated with neuroendocrine features in small cell lung cancer. Cell Discovery. 2024;10(1):93. doi:10.1038/s41421-024-00703-x
115. Roper N, Velez MJ, Chiappori A, et al. Notch signaling and efficacy of PD-1/PD-L1 blockade in relapsed small cell lung cancer. Nat Commun. 2021;12(1):3880. doi:10.1038/s41467-021-24164-y
116. Ghafoor A, Zhu L, Weaver Z, et al. HDAC inhibition unlocks tumor plasticity and enhances immunotherapy response in Myc-driven small cell lung cancer. bioRxiv. 2025;
117. Vigdorovits A, Olteanu G-E, Pascalau A-V, Pirlog R, Berindan-Neagoe I, Pop O-L. Novel immunohistochemical profiling of small-cell lung cancer: correlations between tumor subtypes and immune microenvironment. Diagnostics. 2024;14(23):2660. doi:10.3390/diagnostics14232660
118. Chan JM, Quintanal-Villalonga Á, Gao VR, et al. Signatures of plasticity, metastasis, and immunosuppression in an atlas of human small cell lung cancer. Cancer Cell. 2021;39(11):1479–1496.e18. doi:10.1016/j.ccell.2021.09.008
119. Rovira-Clave X, Drainas AP, Jiang S, et al. Spatial epitope barcoding reveals clonal tumor patch behaviors. Cancer Cell. 2022;40(11):1423–1439e11. doi:10.1016/j.ccell.2022.09.014
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.