Back to Journals » OncoTargets and Therapy » Volume 13
Tumor-Derived Exosome-Educated Hepatic Stellate Cells Regulate Lactate Metabolism of Hypoxic Colorectal Tumor Cells via the IL-6/STAT3 Pathway to Confer Drug Resistance
Authors Li F ![]() , Zhan L, Dong Q, Wang Q
, Zhan L, Dong Q, Wang Q ![]() , Wang Y
, Wang Y ![]() , Li X, Zhang Y, Zhang J
, Li X, Zhang Y, Zhang J ![]()
Received 11 March 2020
Accepted for publication 15 July 2020
Published 7 August 2020 Volume 2020:13 Pages 7851—7864
DOI https://doi.org/10.2147/OTT.S253485
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Yao Dai
Feifei Li1 ,* Lei Zhan1 ,* Qian Dong,1 Qiwei Wang,1 Yuanhe Wang,1 Xiaoyan Li,2 Yong Zhang,2 Jingdong Zhang1
1Medical Oncology Department of Gastrointestinal Cancer, Liaoning Cancer Hospital & Institute, Cancer Hospital of China Medical University, Shenyang 110042, People’s Republic of China; 2Department of Pathology, Liaoning Cancer Hospital & Institute, Cancer Hospital of China Medical University, Shenyang 110042, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jingdong Zhang Email [email protected]
Purpose: Colorectal cancer cells spread to the liver and crosstalk with the microenvironment, and hepatic stellate cells (HSCs) are the major stromal components in the liver. However, the role of the interaction between colorectal tumor cells and HSCs in chemotherapeutic resistance remains unclear. The present study aimed to determine the mechanism of colorectal tumor cells educating the HSCs to reprogram the metabolism of adjacent tumor cells and fuel themselves in the metastatic microenvironment of the liver.
Patients and Methods: Immunohistochemistry (IHC) examined the expression of the monocarboxylate transporters 1 (MCT1) and lactate dehydrogenase B (LDHB) in colorectal liver metastases (CRLM). The Mann–Whitney U-tests analyzed the association between IL-6 levels and clinical parameters. The mechanisms of normoxic tumor-derived exosomes in the education of HSCs were investigated using IHC and ELISA. The conditioned medium of activated HSCs in the regulation of hypoxic tumor cells was analyzed by CCK-8 and cell apoptosis assays.
Results: The expression of MCT1 and LDHB was high in the liver metastases of irinotecan-resistant patients, and the high level of IL-6 in the plasma of patients with CRLM was associated with poor response to irinotecan-based chemotherapy. The colorectal tumor-derived exosomes activated HSCs to secrete excessive IL-6. Furthermore, the conditioned medium of activated HSCs enhanced the lactate metabolism of hypoxic tumor cells by activating the IL-6/STAT3 pathway and upregulating the downstream MCT1 and LDHB, in order to confer the resistance of SN38, which is the active metabolite of irinotecan.
Conclusion: Taken together, the cultured supernatant of normoxic exosome-educated HSCs enhances the lactate metabolism of hypoxic tumor cells via the IL-6/STAT3 pathway, in order to confer the SN38 resistance in a mimic liver metastatic microenvironment.
Keywords: exosomes, hepatic stellate cells, lactate metabolism, colorectal liver metastasis
Introduction
Colorectal cancer is one of the most severe gastrointestinal malignancies that threaten human health worldwide.1 Recent statistics have shown that the morbidity and mortality of colorectal cancer increase dramatically with the change in people’s lifestyles.2 Some studies have suggested that the liver is the most common organ for the metastatic spread of colorectal cancer.3 At present, approximately 20–25% of the colorectal cancer patients are known to suffer from liver metastasis at the time of diagnosis and receive intensive chemotherapy/radiotherapy.4 Liver metastasis is associated with tumor-derived inflammatory mediated fibrosis that accelerates the formation of the “pre-metastasis niche.” Furthermore, the myofibroblasts (MFs) that originate from hepatic stellate cells (HSCs) produce fibrous tumor stroma, thereby promoting the invasion, proliferation and drug resistance of tumor cells.5,6 Intercellular communication is mainly implicated in the direct cell-cell crosstalk, and the release of cytokines and exosomes that carry information.7 Exosomes are nanometer-sized membrane vesicles (30–150 nm), secreted by normal/cancer cells and that participate in the intercellular communication and tumor metastasis.8 Furthermore, these fuses with the target cell membrane and govern the behavior of receptor cells, including stromal cell activation, tumor cell invasion, metastasis and angiogenesis.9 Recent studies have highlighted that exosomes participate in regulating the tumor microenvironment and progression of colorectal cancer. Accumulating evidence revealed that tumor-derived exosomes convert fibroblasts into cancer-associated fibroblasts (CAFs), and ultimately form a drug-resistant microenvironment.10 However, the process of colorectal tumor-derived exosomes in activating HSCs has not yet been elucidated.
Another theory is that accumulated energy production and biosynthesis are conducive to maintain the viability and proliferation of tumor cells.11 Previous studies demonstrated that exosomes effectively mediate the metabolic reprogramming in the tumor microenvironment. Breast cancer cells accumulate more glucose for the invasion and metastasis through exosomal miR-122.12 CAFs inhibit the mitochondrial oxidative phosphorylation of tumor cells by secreting exosomal SNHG3 to reduce the level of miR-330-5p and elevate the expression of pyruvate kinase isozymes, thereby increasing glycolysis and enhancing the proliferation of breast tumor cells.13 At present, lactate has been under intensive research, because it disturbs the interaction between tumor and stromal cells and creates a conducive environment for rapid proliferation.14 Interestingly, some studies have shown that tumor cells can intake lactate to fuel the oxidative mitochondrial metabolism through monocarboxylate transporter 1 (MCT1) and that the expression of MCT1 and lactate dehydrogenase B (LDHB) is closely associated with poor prognosis.15,16 Blood vessels cannot meet the needs of the rapid proliferation of tumor cells, leading to oxygen deficiency occurs at the center, that is, hypoxia. Hypoxic tumor cells are a major cause of tumor recurrence and metastasis caused by resistance to conventional chemoradiotherapy. In oral squamous cell and prostate cancer, hypoxic tumor cells secrete proteins and nucleic acids to promote the invasion and metastasis of normoxic tumor cells.17,18 However, it remains to be elucidated whether colorectal tumor-derived exosomes regulate the metabolic reprogramming of hypoxic tumor cells via HSCs to confer drug resistance.
In the present study, colorectal tumor-derived exosomes activated the HSCs, and the conditioned medium of activated HSCs increased the lactate metabolism of hypoxic tumor cells by activating the IL-6/STAT3 pathway and upregulating the expression of MCT1 and LDHB. These results revealed that the mutual information exchange between colorectal cancer cells and HSCs in liver metastases provides a novel potential mechanism underlying irinotecan resistance.
Patients and Methods
Clinical Samples
Plasma samples were obtained from 78 patients with colorectal liver metastases (CRLM) in the Cancer Hospital of China Medical University (Liaoning, China) before using irinotecan between June 2017 and August 2019, and stored at −80 °C for future use. These patients served as progressive disease (PD), stable disease (SD), or partial response (PR) based on the response to irinotecan-based chemotherapy, according to the Response Evaluation Criteria in Solid Tumors. In parallel, the tissues of liver metastases were collected from the surgical specimens of patients with CRLM. These patients did not receive any chemotherapy before sampling. Furthermore, the clinicopathological features and follow-up data were also reviewed retrospectively. The present study was approved by the Ethics Committee of the Cancer Hospital of China Medical University (20170223), and an informed consent was obtained from all patients.
Cell Culture
The human colorectal cancer cell lines (HCT116 and LoVo) were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA), and cultured in RPMI-1640 medium (Gibco, Rockville, MD, USA) supplemented with 10% fetal bovine serum (FBS; Gibco, Rockville, MD, USA) and 1% penicillin/streptomycin (Gibco, Rockville, MD, USA). The human HSCs (ScienCell Research Laboratories, San Diego, CA, USA) were cultured in stellate cell medium (SteCM; ScienCell Research Laboratories, San Diego, CA, USA). All cell lines were maintained at 37 °C in a humidified incubator containing 5% CO2. In order to mimic the hypoxic environment, a chemical inducer with 100 μM of CoCl2 was used to treat the tumor cells for 48 h.
Exosome Isolation and Analyses
The exosomes were extracted from tumor cells cultured in exosome-depleted complete medium for 48 h by ultracentrifugation. Briefly, the supernatant was obtained by centrifuging at 2000 ×g for 30 min and 10,000 ×g for 30 min at 4 °C. Then, this was filtered using a 0.22-μm filter, followed by ultracentrifugation at 120,000 ×g for 70 min at 4 °C. Finally, the exosome-enriched pellets were resuspended in a volume of 50 μL of the suitable buffer.19 These exosomes were observed under a transmission electron microscope (FEI Tecnai G2 Spirit; Thermo Scientific, Waltham, MA, USA), and the size determined by Nano Tracking System Analysis (NTA) 300 (UK).
Cellular Internalization of Exosomes
In order to determine the interaction between tumor cell exosomes and HSCs, the tumor cells were pretreated using DiO (Beyotime, Shanghai, China). Then, the exosomes (50 μg/mL) were isolated from the cultured supernatant, according to the method described above, and co-cultured with DiI (Beyotime, Shanghai, China)-labeled HSCs for 48 h. Subsequently, the exosomes and HSCs were analyzed using a fluorescence microscope (DMi8; Leica, Wetzlar, Germany).
Western Blot
The whole-cell protein was extracted using lysis buffer (Beyotime, Shanghai, China) and estimated using a Bicinchoninic acid (BCA) Protein Assay Kit (Beyotime, Shanghai, China). The equivalent of protein was separated using 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a polyvinylidene fluoride (PVDF) membrane (Millipore, Burlington, MA, USA). Then, these membranes were blocked with 5% non-fat milk for 1 h at room temperature and probed with primary antibodies (Table S1) overnight at 4 °C, followed by incubation with the fluorescein-conjugated secondary antibody for 1 h at room temperature and detection using an Enhanced Chemiluminescence (ECL) Detection kit (Millipore, Burlington, MA, USA).
Quantitative Real-Time PCR (qPCR)
TRIzol (Takara, Tokyo, Japan) was used to extract the total RNA from HSCs, LoVo and HCT116, and the mRNA level of each group was examined. A RT-PCR kit (Takara, Tokyo, Japan) was used to reverse transcribe the mRNA into complementary DNA (cDNA) according to the manufacturer’s instructions. qPCR was conducted using SYBR Green PCR Master Mix (Takara, Tokyo, Japan) on a CFX96 Real-Time PCR Detection System (Bio-Rad, Hercules, CA, US). The PCR program was as follows: 95 °C for 30 s, then 40 cycles of denaturation at 95 °C for 5 s, annealing at 60 °C for 30 s, and final extension at 95 °C for 10 s. The GAPDH mRNA was used as an internal control and the relative expression level of the target genes was calculated using 2−ΔΔCt method. The primer sequences used for real-time PCR are listed in Table S2.
Immunofluorescence Staining
Cells were grown in 6-well plates (5×104 cells/well), fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, blocked with 2% BSA, and incubated with primary antibodies (Table S1) overnight at 4 °C. Then, these samples were incubated with Alexa Fluor 488-conjugated secondary antibody (1:200; Invitrogen, Carlsbad, CA, USA) at room temperature for 1 h in the dark, and the nuclei were stained with DAPI (1:300; Invitrogen, Carlsbad, CA, USA). Finally, the images of these cells were captured using a fluorescence microscope DMi8.
Cell Viability Assay
LoVo and HCT116 cells were seeded into 96-well culture plates (5×103 cells/well) in 100 µL of RPMI-1640 medium containing 10% FBS for 48 h. Then, SN38, CoCl2 and conditioned medium of HSCs or aHSCs were added to the cells and incubated for an additional 48 h. The cell viability was assessed using Cell Counting Kit-8 (CCK-8) assay (Dojindo Laboratories, Kumamoto, Japan), according to the manufacturer’s instructions. The optical density was recorded at 450 nm.
Cell Apoptosis Analysis
LoVo and HCT116 cells were seeded into 6-well culture plates (2×105 cells/well). Subsequently, the cells were harvested after pretreatment and stained with 5µL FITC-Annexin V and 5 µL PI for 15 min in the dark according to the manufacturer’s instructions of the Apoptosis Detection Kit (BD PharmingenTM, USA), and quantified using the FlowJo software (Version 10.2).
ATP Assay
LoVo and HCT116 cells were seeded in 6-well culture plates (2×105 cells/well) and collected after pretreatment. The concentration of ATP was measured using an ATP assay kit, according to manufacturer’s instructions (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
Enzyme-Linked Immunosorbent Assay (ELISA)
The cellular debris was removed from the conditioned media of activated hepatic stellate cells (aHSCs) and human plasma by centrifugation. The IL-6 concentration was quantified using an ELISA kit (BioLegend, San Diego, CA, USA), according to the manufacturer’s protocol. Briefly, the conditioned media of aHSCs and human plasma were collected, and the standard and sample were placed in a microplate pre-coated with IL-6 antibody. Then, the horseradish peroxidase (HRP) substrate was added to each well, and the level of IL-6 was measured at 450 nm.
Small-Interference RNAs (siRNAs)
The siRNAs that targeted the human MCT1 mRNA, LDHB mRNA and negative control siRNA (siControl) were designed and synthesized by Ruibobio (Guangzhou, China). The most effective siRNAs used for further experiments were as follows: MCT1-1: 5ʹ-GCAGTATCCTGGTGAATAA-3ʹ; MCT1-2: 5ʹ-CTAGCACCTTTATCCACTA-3ʹ;
LDHB-1: 5ʹ-CAACTGGGCTATTGGATTA-3ʹ; LDHB-2: 5ʹ-CAGTCCTGATTGCATCATA-3ʹ. Human STAT3 pool siRNA (L-003544-00-0005) and scrambled siRNA (D-001810-01-05) were obtained from Dharmacon. Briefly, 5×104 cells/well of LoVo and HCT116 cells were transfected with siRNA (50 nM) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA), according to the manufacturer’s protocol. After 48 h, the transfected cells were treated with SN38, CoCl2 and conditioned medium of HSCs or aHSCs.
Immunohistochemistry (IHC)
The IHC staining was conducted on 4-μm paraffin-embedded tissue sections to detect the expression levels of HIF-1α, α-SMA, IL-6, MCT1 and LDHB proteins. Briefly, the slides were probed with HIF-1α, α-SMA, IL-6, MCT1 and LDHB antibodies at 4 °C overnight, and incubated with HRP-conjugated secondary antibodies at 37 °C for 30 min. Finally, these sections were stained with DAB and counterstained with hematoxylin (Sigma–Aldrich, St. Louis, MO, USA). Afterwards, the images were captured using a microscope DMi8.
Statistical Analysis
All statistical data were analyzed using the SPSS 19.0 software. The statistical comparisons between the two groups were made by two-tailed Student’s t-test, and one-way analysis of variance (ANOVA) with Dunnett’s test was applied to analyze multiple comparisons among more than three groups. Mann–Whitney U-test was used to estimate the association between IL-6 levels and clinical parameters. P-value <0.05 was considered statistically significant.
Results
Plasma IL-6 Level and the Expression of MCT1 and LDHB in CRLM are High in Irinotecan-Resistant Patients
For advanced colorectal cancer, irinotecan-based chemotherapy is the primary treatment. However, the effective rate of this first-line treatment is merely 50%, while that of the second-line therapy is only 4%.20,21 Hypoxia plays a critical role in tumor-resistant microenvironment.22 A high expression of HIF-1α was detected in CRLM (Figure S1A). MCT1 and LDHB are highly expressed in the liver metastases of irinotecan-resistant patients as compared to sensitive patients (Figure 1A, P<0.05). In order to determine whether IL-6 is associated with the progression of patients with CRLM and resistance to irinotecan, the clinicopathological features of 78 patients were analyzed. The plasma IL-6 levels of patients with PD after ≤4 cycles of irinotecan treatment were significantly higher than those in patients who were able to maintain SD or PR (Figure 1B, P<0.0001). Further analysis revealed that the upregulated IL-6 in plasma was correlated with high CA199, extrahepatic metastases, and multiple liver metastases in advanced colorectal cancer (Table 1 and Figure 1C–E, P<0.05). These results indicated that plasma IL-6 level and the expression of MCT1 and LDHB in CRLM were high in irinotecan-resistant patients, and high plasma IL-6 levels were significantly related to poor prognosis.
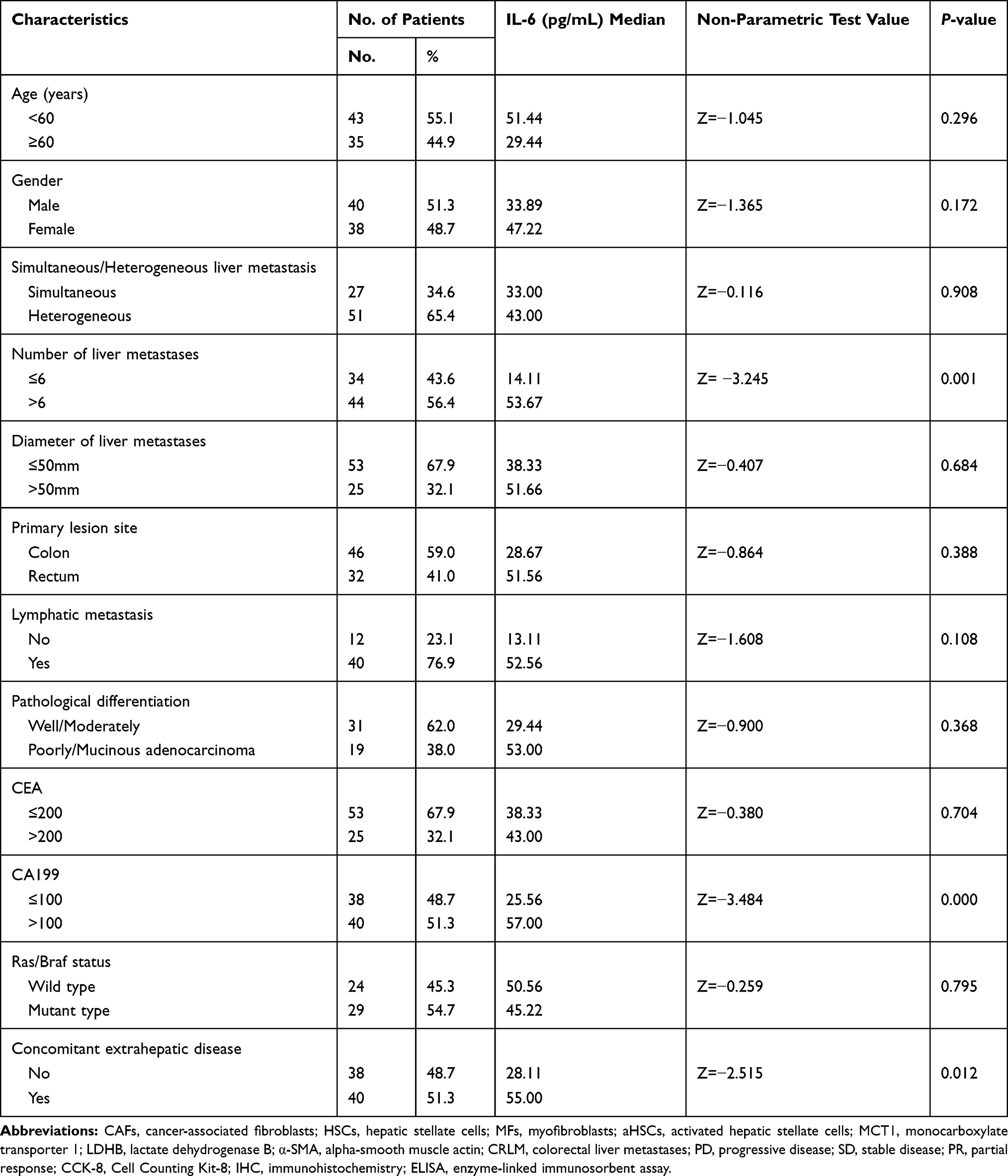 |
Table 1 The Relationship Between Plasma IL-6 Levels and Clinicopathologic Features (N=78) |
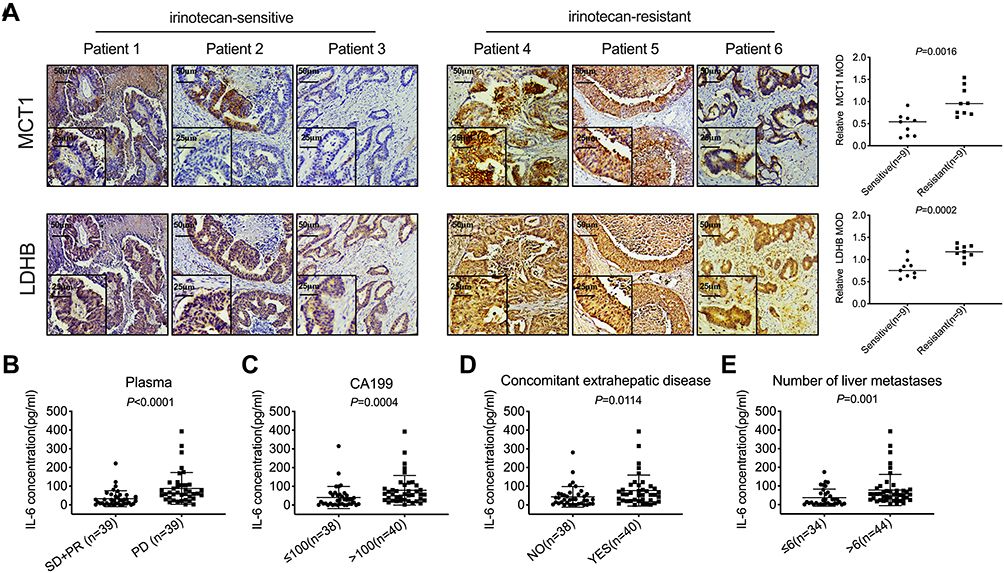 |
Figure 1 The expression of MCT1 and LDHB, and the plasma IL-6 level in CRLM was elevated in irinotecan-resistant patients. (A) IHC was used to analyze MCT1 and LDHB in the liver metastases of irinotecan-sensitive and irinotecan-resistant patients. (B) ELISA was used to detect the concentration of IL-6 in the plasma of patients with PD (n=39) and SD or PR (n=39). (C–E) The correlation between the plasma IL-6 concentration and the level of CA199, the extrahepatic metastases state, and the number of liver metastases. |
Tumor-Derived Exosomes Induce the Activation of HSCs
Exosomes are extracellular vesicles released by various cells and mediate intercellular communication in the metastatic niche.23 First, a typical cup-shaped morphology of colorectal tumor exosomes was identified by electron microscopy (Figure 2A). Concurrently, the size (30–150 nm) and number of exosomes and the expression of exosomal marker proteins (CD9 and TSG101) in cells and exosomes were analyzed (Figure 2B and C). Then, tumor-derived exosomes labeled with DiO (green) were taken up by HSCs labeled with Dil (red) (Figure 2D), indicating that the exosomes secreted by these tumor cells were transmitted to HSCs. Next, tumor cell exosome-treated HSCs expressed α-SMA, which is a typical marker of aHSCs at both the mRNA and protein levels (Figure 2E and F, P<0.05). In addition, the liver metastases of colorectal cancer patients showed a high expression of α-SMA (Figure 2G). Therefore, it was concluded that colorectal tumor-derived exosomes activate HSCs.
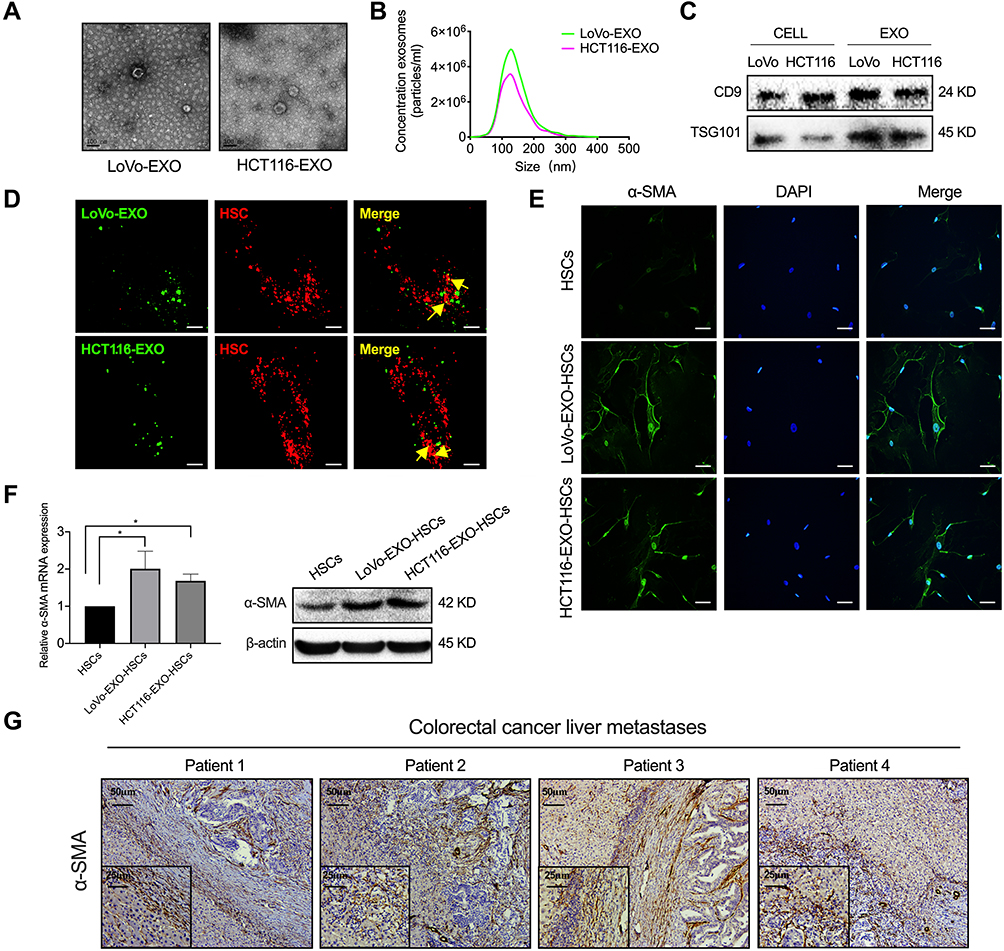 |
Figure 2 Tumor-derived exosomes induce the activation of HSCs. (A) Typical electron microscopy images of exosomes secreted by colorectal cancer (LoVo and HCT116; Scale bar, 100 nm). (B) NanoSight particle-tracking analysis of exosomes from LoVo and HCT116. (C) The total cell lysates and exosomes of LoVo and HCT116 were analyzed by Western blot using the antibody against exosomal proteins (CD9, TSG101). (D) The imaging reveals the delivery of Dio-labeled exosomes (green) to Dil-labeled HSCs (red). The yellow arrows indicate the delivered exosomes, and the representative images are presented (Scale bar, 10 μm). (E) Immunofluorescence staining for the α-SMA expression of HSCs and tumor-derived exosomes pretreated HSCs. Scale bar, 20 μm. (F) PCR and Western blot analyses of the expression of α-SMA in aHSC. (G) IHC was performed to analyze α-SMA in the liver metastases of colorectal cancer patients. N=3; *P<0.05. |
p-ERK and p-AKT in Tumor-Derived Exosomes Regulate IL-6 Secretion
CAFs frequently regulate the inflammatory microenvironment by altering the pro-inflammatory signaling pathway and secreting cytokines during tumorigenesis. Herein, the IL-6 concentration in the conditioned medium of aHSCs was found to be significantly higher than that of HSCs measured by ELISA, while exosomes increased the expression of IL-6 in HSCs at both mRNA and protein levels (Figure 3A and B, P<0.05). In addition, the liver metastases of colorectal cancer patients had a high expression of IL-6, which was co-localization with α-SMA in aHSCs (Figure 3C). Consistently, the level of NF-κB in the nucleus of aHSCs was higher than that of HSCs (Figure 3D). Furthermore, HSCs were simultaneously treated with NF-κB inhibitor (BAY11-7082) and exosomes, and it was found that the expression of IL-6 in aHSCs and that secreted by aHSCs were significantly reduced, which did not differ markedly as compared to HSCs (Figures 3E and S2A, B, P>0.05). Collectively, it can be concluded that tumor-derived exosomes promoted IL-6 secretion by activating NF-κB in aHSCs.
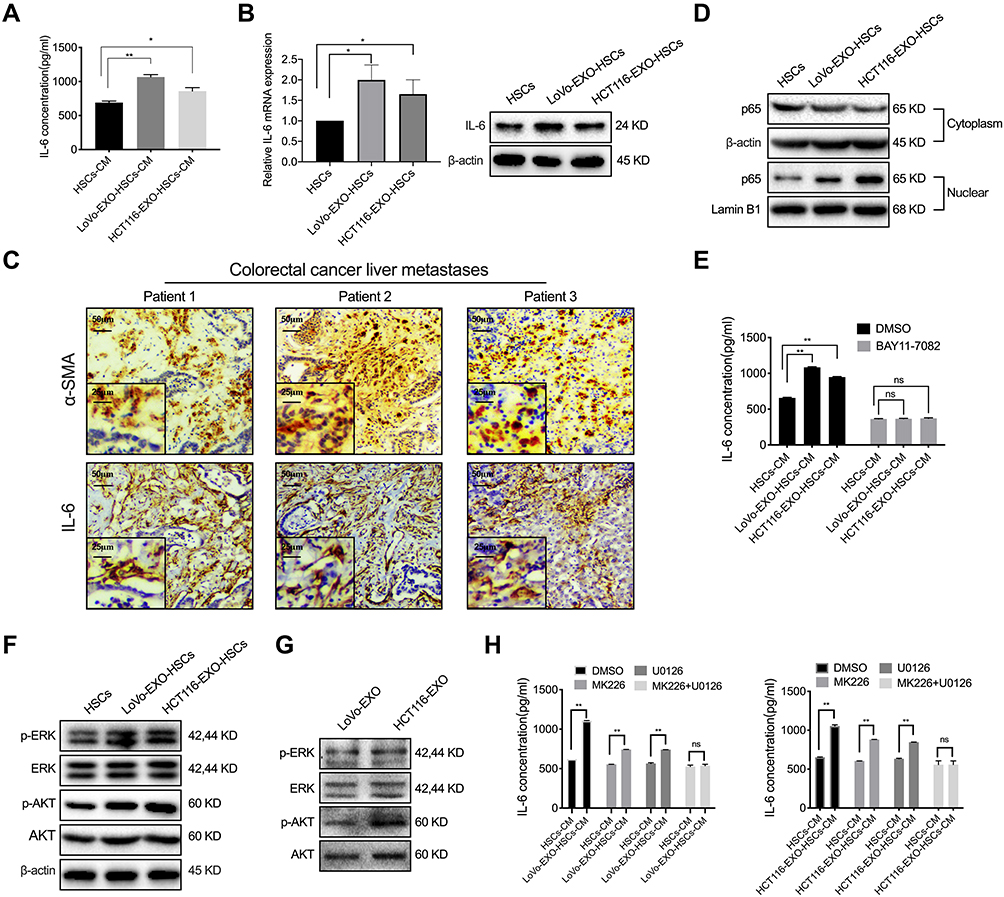 |
Figure 3 P-ERK and p-AKT in tumor-derived exosomes regulate IL-6 secretion. (A) ELISA was used to detect the concentration of IL-6 in the cultured medium of HSCs and aHSCs (LoVo and HCT116 exosome-treated HSCs). (B) PCR and Western blotting for the IL-6 expression of HSCs and tumor-derived exosomes pre-treated HSCs. (C) IHC was performed to analyze α-SMA and IL-6 in the liver metastases of colorectal cancer patients. (D) Western blotting analysis of the expression level of p65 in the cytoplasm and nucleus of HSCs and aHSCs. (E) ELISA was used to detect the concentration of IL-6 in the medium of HSCs and aHSCs, with or without NF-κB inhibitors (BAY11-7082). (F) Western blotting analysis of the expression of p-ERK, ERK, p-AKT and AKT in HSCs and aHSCs. (G) Western blotting analysis of the expression of p-ERK, ERK, p-AKT and AKT in the exosomes of LoVo and HCT116. (H) ELISA was used to detect the concentration of IL-6 in the cultured medium of HSCs and aHSCs treated with MK226 and U0126. N=3; ns, no significant difference; *P<0.05; **P<0.001. |
In the present study, the expression of p-ERK and p-AKT increased significantly in aHSCs as compared to HSCs (Figure 3F). Next, the p-ERK and p-AKT proteins were detected in colorectal tumor-derived exosomes (Figure 3G). In order to further validate the regulatory effects of p-AKT and p-ERK on aHSCs, MK226 and U0126 were employed to inhibit the p-AKT and p-ERK in aHSCs. Consequently, we observed that both MK226 and U0126 inhibits the expression of NF-κB in the nucleus of aHSCs, and also inhibits the expression of IL-6 in aHSCs at both the mRNA and protein levels (Figure S2C–E, P>0.05). Furthermore, MK226 or U0126 decreased the IL-6 secretion through aHSCs, but the combined inhibitory effect was better than that of the single-use, resulting in no significant difference as compared to HSCs (Figure 3H, P>0.05). Therefore, it was concluded that colorectal tumor-derived exosomal p-AKT and p-ERK regulated the NF-κB-dependent IL-6 production in aHSCs.
Activated HSC-Secreted IL-6 Confers SN38 Resistance to Hypoxic Tumor Cells
In order to determine whether aHSCs regulate the resistance of colorectal cancer cells to irinotecan, the conditioned medium of aHSCs or HSCs was used to culture LoVo and HCT116 cells. The CCK-8 assay revealed that the conditioned medium of aHSCs increased the viability of LoVo and HCT116 in response to SN38, while hypoxic LoVo and HCT116 cells exhibited an obvious increase as compared to the normoxic cells (Figure 4A). Additionally, we found that the conditioned medium of aHSCs educed the apoptosis of hypoxic LoVo and HCT116 as compared to the conditioned medium of HSCs (Figures 4B and S3A, P<0.001). Furthermore, the expression of the pro-apoptotic protein (Bax) decreased while the expression of the anti-apoptotic protein (Bcl-2) increased in the conditioned medium of aHSCs as compared to HSCs (Figure 4C). These findings demonstrated that HSCs activated by tumor-derived exosomes conferred SN38 resistance to hypoxic tumor cells by inhibiting apoptosis.
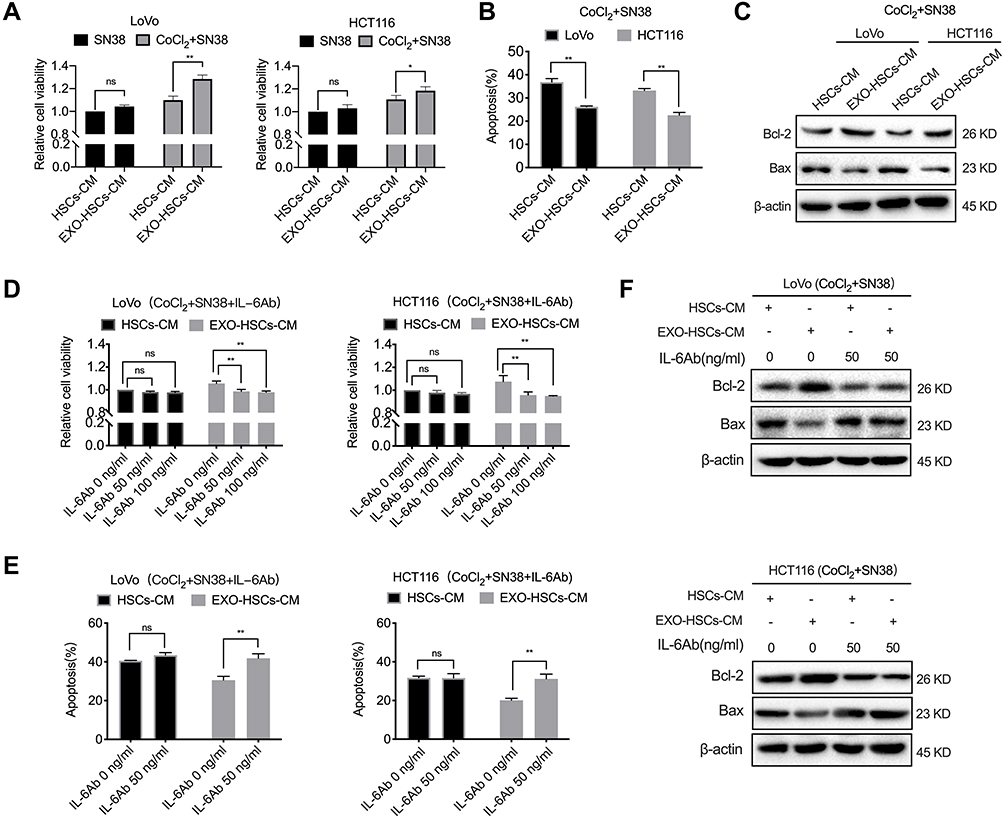 |
Figure 4 Activated HSC-secreted IL-6 confers the SN38 resistance to hypoxic tumor cells. (A) Cell viability assays with LoVo and HCT116 at 48 h after co-culture with the HSC medium or aHSC medium, and 80 nM of SN38 with or without 100 μM of CoCl2. (B) Flow cytometric analysis of the apoptosis in LoVo and HCT116 cells after 48 h of the indicated treatment. (C) Western blot analysis of the expression level of Bcl-2 and Bax in LoVo and HCT116 cells after 48 h post-treatment. (D) Cell viability assays with LoVo and HCT116 after the co-culture with the HSC medium or aHSC medium and CoCl2, SN38 and IL-6 neutralizing antibodies. (E) Flow cytometry analysis of the apoptosis in LoVo and HCT116 cells after 48 h of the indicated treatment. (F) Western blot analysis of the expression level of Bcl-2 and Bax in LoVo and HCT116 cells after 48 h of the indicated treatment. N=3; ns, no significant difference; *P<0.05; **P<0.001. |
Next, it was determined whether IL-6 is involved in promoting drug resistance in hypoxic tumor cells. The CCK-8 and flow cytometric analysis revealed that the IL-6 neutralizing antibody eliminated the altered viability and apoptosis in SN38-stimulated hypoxic tumor cells (Figures 4D–F and S3B, P<0.001). Furthermore, it was observed that exosome-activated HSCs promoted the drug resistance of hypoxic tumor cells by secreting IL-6.
IL-6 Regulates the Hypoxic Tumor Cell Lactate Metabolism Involved in SN38 Resistance
Tumor cells have high metabolic plasticity, and lactate plays a major role in the metabolic symbiosis among tumor cells due to the uneven supply of oxygen and glucose inside the tumor tissues.24 MCT1 is a critical transporter of lactate in tumor cells, and LDHB is a key enzyme that converts lactate into pyruvate, which provides energy to these cells.25,26 In the present study, the expression of MCT1 and LDHB was increased at both the mRNA and protein levels in hypoxic LoVo and HCT116 cells treated with the conditioned medium of aHSCs (Figures 5A and S4A). The ATP assay revealed that these cells released more ATP as compared to the control group (Figure 5B, P<0.001). However, it was observed that the treatment with MCT1 siRNA or LDHB siRNA restored the production of ATP in LoVo cells treated with the conditioned medium of aHSCs, and the combined effect of the two siRNAs was rather distinct (Figures 5C and S4B, P<0.001). In addition, the treatment with MCT1 and LDHB siRNAs eliminated the alteration of the viability and apoptosis in SN38-stimulated hypoxic tumor cells, as assessed by CCK-8 and flow cytometry (Figures 5D, E and S4C, D, P<0.001). These results revealed that aHSCs help hypoxic tumor cells in consuming lactate by upregulating MCT1 and LDHB, in order to generate energy against SN38. Furthermore, the IL-6 neutralizing antibody treatment significantly decreased the expression of MCT1 and LDHB of SN38-stimulated hypoxic tumor cells in the conditioned medium of aHSCs at both mRNA and protein levels; also, the production of ATP significantly decreased (Figures 5F, G and S4E, P<0.05). These findings demonstrated that the IL-6 secretion of aHSCs regulates the lactate metabolism of hypoxic tumor cells to confer drug resistance.
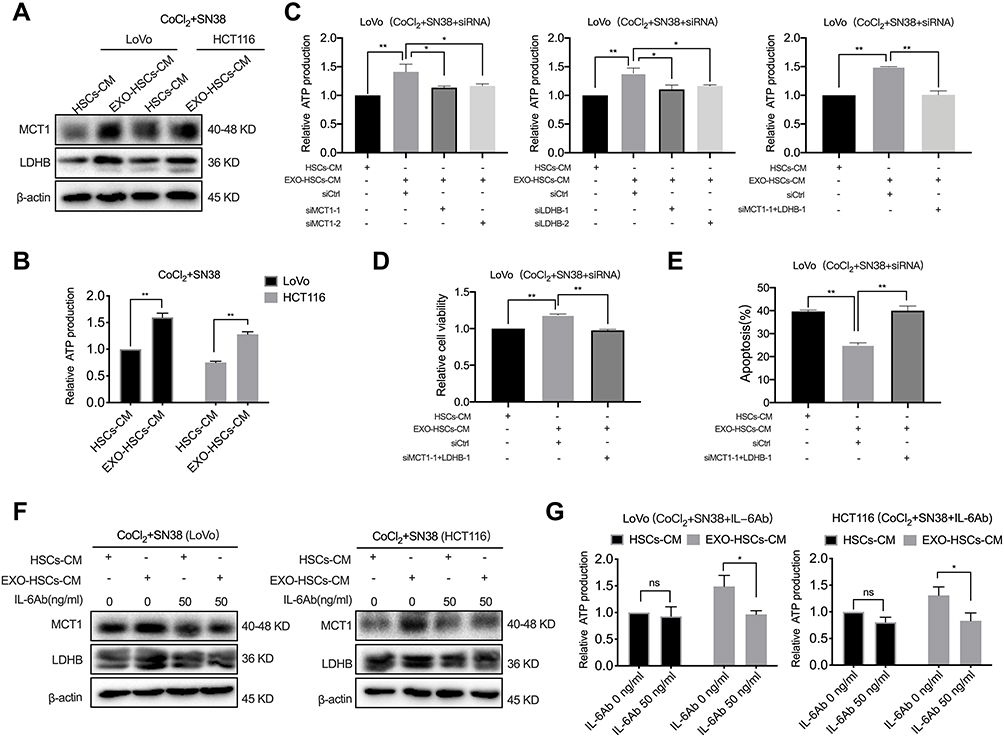 |
Figure 5 IL-6 is involved in SN38 resistance by regulating the lactate metabolism of tumor cells. (A) Western blot analysis of the expression level of MCT1 and LDHB in LoVo and HCT116 cells at 48 h after co-culture with HSC medium or aHSC medium with CoCl2 and SN38. (B) The relative production of ATP by LoVo and HCT116 cells after 48 h of the indicated treatment. (C) The relative production of ATP by LoVo treated with MCT1 siRNA and LDHB siRNA after co-culture with HSC medium or aHSCs medium with CoCl2 and SN38. (D) Cell viability assay with LoVo after 48 h of the indicated treatment. (E) Flow cytometric analysis of the apoptosis in LoVo after 48 h of the indicated treatment. (F) Western blot analysis of the expression level of MCT1 and LDHB in LoVo and HCT116 cells after co-culture with the HSC medium or aHSC medium and CoCl2, SN38, and IL-6 neutralizing antibodies. (G) The relative production of ATP by LoVo and HCT116 cells after 48 h of the indicated treatment. N=3; ns, no significant difference; *P<0.05; **P<0.001. |
Activated HSCs Enhance Lactate Metabolism via the IL-6/STAT3 Pathway in Tumor Cells
Our previous study revealed that p-STAT3 is a crucial transcription factor in the resistance to vemurafenib in colorectal cancer cells with BRAF mutations.27 However, the correlation between STAT3 and resistance to irinotecan in patients with CRLM remains unclear. In the present study, the level of p-STAT3 in hypoxic LoVo and HCT116 cells was upregulated in the conditioned medium of aHSCs and that the IL-6 neutralizing antibody decreased the expression of p-STAT3 (Figure 6A and B). Next, 2 μM and 5 μM of p-STAT3 inhibitor (Stattic) was used to treat the SN38-stimulated hypoxic LoVo and HCT116 cells, and 5 μM of Stattic was sufficient to inhibit the expression of p-STAT3 and restrain the viability of these cells (Figure 6C and D, P<0.001). Similarly, when SN38-stimulated LoVo and HCT116 cells were treated with Stattic, the apoptosis of hypoxic cells decreased in the conditioned medium of aHSCs (Figures 6E, F and S5A, P<0.001). Therefore, it can be considered that the activation of the IL-6/STAT3 pathway confers the drug resistance of hypoxic tumor cells.
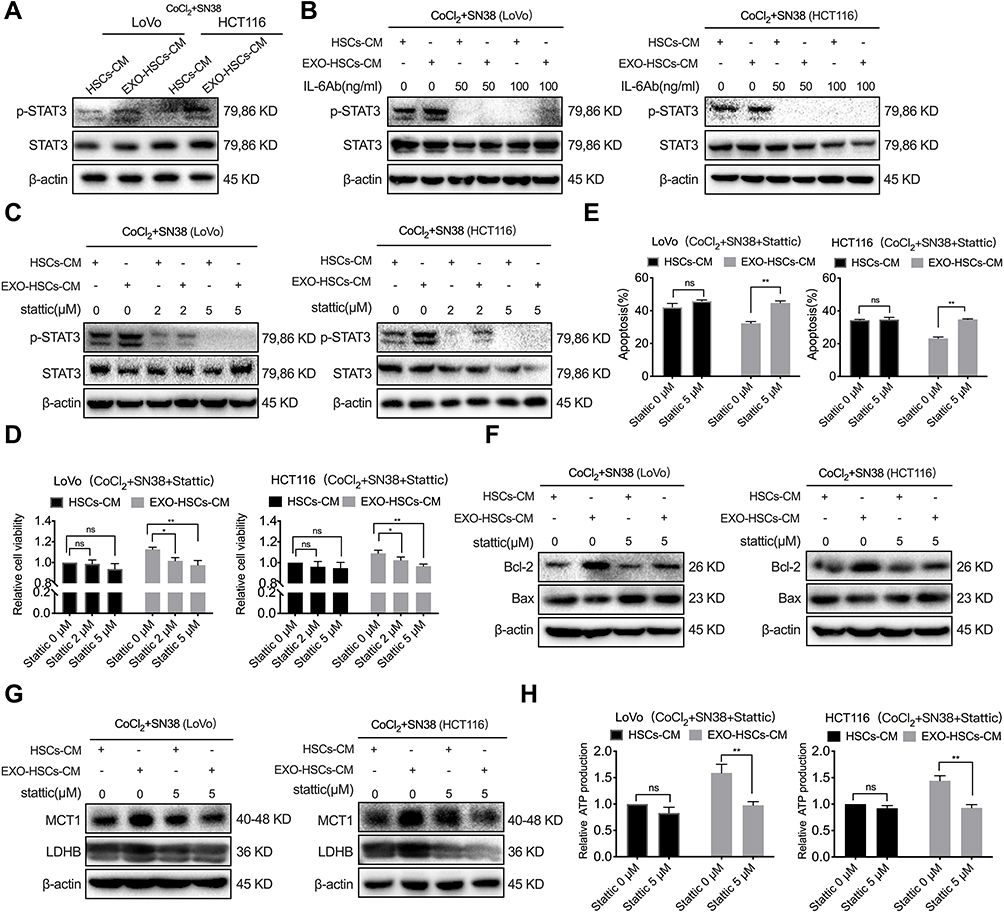 |
Figure 6 Exosome-activated HSCs enhance the lactate metabolism by activating the IL-6/STAT3 pathway in tumor cells. (A) Western blot analysis of the expression level of p-STAT3 and STAT3 in LoVo and HCT116 cells at 48 h after co-culture with HSCs medium or aHSCs medium with CoCl2 and SN38. (B) Western blot analysis of the expression level of p-STAT3 and STAT3 in LoVo and HCT116 cells at 48 h after co-culture with the HSCs medium or aHSCs medium and CoCl2, SN38 and IL-6 neutralizing antibodies. (C) Western blot analysis of the expression level of p-STAT3 and STAT3 in LoVo and HCT116 cells at 48 h after co-culture with the HSCs medium or aHSCs medium and CoCl2, SN38 and Stattic. (D) Cell viability assays with LoVo and HCT116 cells after 48 h of the indicated treatment. (E) Flow cytometric analysis of the apoptosis of LoVo and HCT116 cells after 48 h of the indicated treatment. (F) Western blot analysis of the expression level of Bcl-2 and Bax in LoVo and HCT116 cells after 48 h of the indicated treatment. (G) Western blot analysis of the expression level of MCT1 and LDHB in LoVo and HCT116 cells after 48 h of the indicated treatment. (H) The relative production of ATP by LoVo and HCT116 after 48 h of the indicated treatment. N=3; ns, no significant difference; *P<0.05; **P<0.001. |
STAT3 has been shown to regulate LDHB in various tumor cells and MCT1 in cervical cancer.28,29 The Western blot and PCR analyses revealed that Stattic and STAT3 siRNAs can decrease the expression of LDHB and MCT1 in SN38-stimulated hypoxic tumor cells in the conditioned medium of aHSCs at both mRNA and protein levels (Figures 6G and S5B–D). In addition, Stattic and STAT3 siRNAs eliminated the increase in ATP production (Figures 6H and S5E, P<0.001). Thus, it can be considered that activated HSCs regulate the lactate metabolism via IL-6/STAT3 pathway and upregulate the level of MCT1 and LDHB to promote the resistance of hypoxic tumor cells against SN38.
Discussion
The reprogramming of cellular metabolism is a hallmark of various tumors.30 Metabolic programs play a crucial role in sustaining the growth, progression and immune evasion of tumor cells. Several studies have delineated that metabolic rearrangements were effectively facilitated by oncogene activation and tumor suppressor loss. These results indicated that colorectal tumor-derived exosomes activate the HSCs, and that the IL-6 secreted by HSCs induced chemotherapeutic resistance by enhancing lactate metabolism. HSCs are the major components of the liver microenvironment, and the activation and migration of HSCs play a vital role in the progression of hepatocellular carcinomas.31,32 Pancreatic cancer has a significant characteristic of metastasizing to the liver, and pancreatic cancer cells secrete metalloproteinases-1 to activate HSCs and create a pre-metastatic niche.33 Furthermore, exosomes have attracted considerable attention in mediating the crosstalk between cells and the cellular microenvironment.34 Previous studies found that hepatocellular tumor-derived exosomes activate HSCs and accelerate tumor progression.35 However, it remains unclear whether colorectal tumor-derived exosomes sustain tumor cell survival by activating HSCs (Figure 7A). In the present study, it was found that HSCs take up colorectal tumor-derived exosomes and highly express α-SMA. Activated HSCs pave the way for the invasion, metastasis and colonization of tumor cells through paracrine factors, which consist of chemokines, cytokines and angiogenic factors. CAFs facilitate cell proliferation and angiogenesis in colorectal tumors through paracrine IL-6.36 However, the correlation between activated HSCs and IL-6 secretion in the metastatic liver microenvironment is yet to be elucidated.
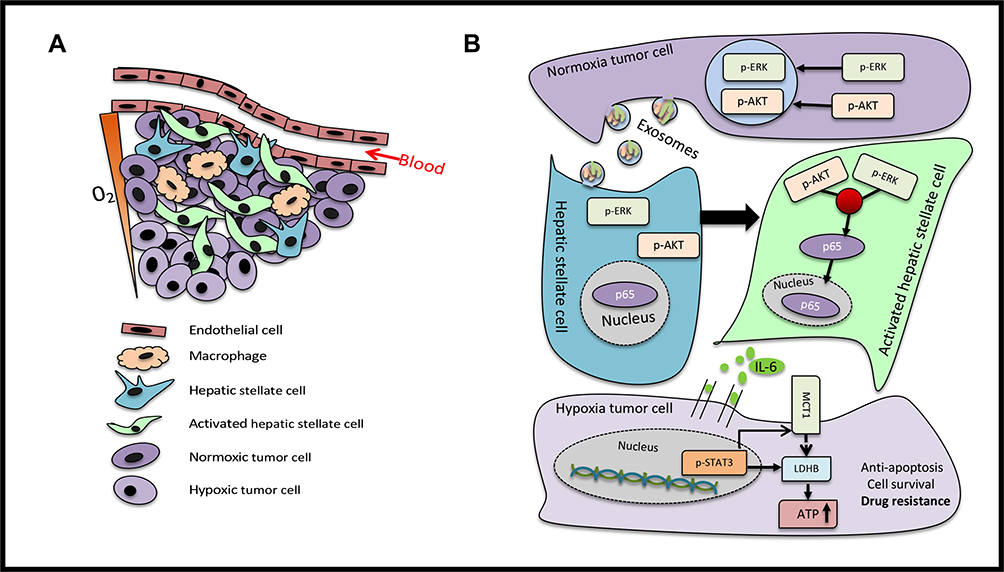 |
Figure 7 Scheme of the proposed mechanisms (A) The tumor microenvironment of the liver metastases mainly comprises of endothelial cells, macrophages, HSCs, activated HSCs and tumor cells. The tumor cells around the blood vessels were oxygenous, while those farther away from the blood vessels were in a hypoxic state. (B) Normoxic tumor cells secreted exosomes that carried p-ERK and p-AKT, in order to convert HSCs into aHSCs. The p-ERK and p-AKT signaling pathways were activated to target p65 to increase the secretion of IL-6. The cultured supernatant of aHSCs enhanced the lactate metabolism of tumor cells by activating the IL-6/STAT3 pathway and upregulating MCT1 and LDHB to produce ATP to confer drug resistance. |
Tumor-induced inflammation serves as a critical component and key driver in the tumor microenvironment. IL-6 is a pleiotropic cytokine that regulates a variety of cellular functions, including cell proliferation, cell differentiation, immune defense mechanisms and inflammatory responses.37 A previous study revealed that IL-6 affects tumor progression by interfering with cell adhesion, motility, thrombosis and tumor-specific antigen expression.38 In addition, IL-6 is correlated to the survival of advanced colorectal cancer and can be used as a potential biomarker for colorectal cancer diagnosis.39,40 However, the association of IL-6 and irinotecan resistance in colorectal tumors has not been widely studied. In the present study, normoxic tumor-derived exosomes delivered p-ERK and p-AKT to activate HSCs and increased the secretion of IL-6. Furthermore, the conditioned medium of activated HSCs activates the IL-6/STAT3 pathways to confer the drug resistance to tumor cells. The IL-6 expression was significantly increased in the plasma of irinotecan-resistant patients and correlated with poor prognosis. CAFs promoted proliferation, migration, and invasion in melanoma and non-small cell lung cancer by regulating oxidative mitochondrial metabolism.41,42 In this study, the data indicated that aHSCs activate the IL-6/STAT3 pathways and upregulate the downstream MCT1 and LDHB to take full advantage of the lactate, in order to generate ATP as a response to chemotherapy drugs. In addition, MCT1 effectively mediates the passive transport of lactate, and LDHB is a key enzyme for reprograming the metabolism in the tumor microenvironment.43,44 Also, the expression of MCT1 and LDHB in liver metastases and SN38-resistant patients with colorectal cancer was significantly upregulated. Therefore, it was speculated that MCT1 and LDHB could be combined as predictive markers for the diagnosis of colorectal cancer.
Conclusion
The results of the present study indicated that normoxic tumor-derived exosomes deliver p-ERK and p-AKT to activate HSCs and increase the secretion of IL-6. The conditioned medium of activated HSCs enhances lactate metabolism by stimulating the IL-6/STAT3 pathway to confer drug resistance in colorectal cancer cells (Figure 7B). These findings illuminate a novel mechanism underlying the reprogramming of metabolism between tumor and stromal cells, wherein IL-6 and lactate metabolism are the key contributors, deeming these as potential biomarkers and therapeutic targets for the resistance of advanced colorectal cancer.
Acknowledgments
We thank Dr. Ting Hong, Dr. Liqun Chen and all members of the Central Laboratory of Cancer Hospital of China Medical University for the technical assistance. This work was supported by Science and Technology Planning Project of Shenyang (No.191124088), Science and Technology Planning Project of Liaoning Province of China (No.201800449), National Key R&D Program of China (Grant #2018YFC1311600), Scientific Research Foundation For The introduction of Talents, Liaoning Cancer Hospital & Institute (No.Z1702), The National Natural Science Foundation of China (No.81372532) and Science and Technology Planning Project of Shenyang (No. F15-139-9-27).
Disclosure
The authors declare that there are no competing interests.
References
1. Mattiuzzi C, Sanchis-Gomar F, Lippi G. Concise update on colorectal cancer epidemiology. Ann Transl Med. 2019;7(21):609. doi:10.21037/atm.2019.07.91
2. Jiao Q, Ren Y, Ariston Gabrie AN, et al. Advances of immune checkpoints in colorectal cancer treatment. Biomed Pharmacother. 2020;123:109745. doi:10.1016/j.biopha.2019.109745
3. Sun L, Liu X, Pan B, et al. Serum exosomal miR-122 as a potential diagnostic and prognostic biomarker of colorectal cancer with liver metastasis. J Cancer. 2020;11(3):630–637. doi:10.7150/jca.33022
4. Biasco G, Derenzini E, Grazi G, et al. Treatment of hepatic metastases from colorectal cancer: many doubts, some certainties. Cancer Treat Rev. 2006;32(3):214–228. doi:10.1016/j.ctrv.2005.12.011
5. Gascard P, Tlsty TD. Carcinoma-associated fibroblasts: orchestrating the composition of malignancy. Genes Dev. 2016;30(9):1002–1019. doi:10.1101/gad.279737.116
6. Rhee H, Kim H-Y, Choi J-H, et al. Keratin 19 expression in hepatocellular carcinoma is regulated by fibroblast-derived HGF via a MET-ERK1/2-AP1 and SP1 Axis. Cancer Res. 2018;78(7):1619–1631. doi:10.1158/0008-5472.CAN-17-0988
7. Sato S, Weaver AM. Extracellular vesicles: important collaborators in cancer progression. Essays Biochem. 2018;62(2):149–163. doi:10.1042/EBC20170080
8. Becker A, Thakur BK, Weiss JM, Kim HS, Peinado H, Lyden D. Extracellular vesicles in cancer: cell-to-cell mediators of metastasis. Cancer Cell. 2016;30(6):836–848. doi:10.1016/j.ccell.2016.10.009
9. Wu K, Xing F, Wu S-Y, Watabe K. Extracellular vesicles as emerging targets in cancer: recent development from bench to bedside. Biochim Biophys Acta Rev Cancer. 2017;1868(2):538–563. doi:10.1016/j.bbcan.2017.10.001
10. Steinbichler TB, Dudás J, Skvortsov S, Ganswindt U, Riechelmann H, Skvortsova -I-I. Therapy resistance mediated by exosomes. Mol Cancer. 2019;18(1). doi:10.1186/s12943-019-0970-x
11. Korshunov DA, Kondakova IV, Shashova EE. Modern perspective on metabolic reprogramming in malignant neoplasms. Biochemistry (Moscow). 2019;84(10):1129–1142. doi:10.1134/S000629791910002X
12. Fong MY, Zhou W, Liu L, et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat Cell Biol. 2015;17(2):183–194. doi:10.1038/ncb3094
13. Li Y, Zhao Z, Liu W, Li X. SNHG3 functions as miRNA sponge to promote breast cancer cells growth through the metabolic reprogramming. Appl Biochem Biotechnol. 2020.
14. de la Cruz-lopez KG, Castro-Munoz LJ, Reyes-Hernandez DO, Garcia-Carranca A, Manzo-Merino J. Lactate in the regulation of tumor microenvironment and therapeutic approaches. Front Oncol. 2019;9:1143. doi:10.3389/fonc.2019.01143
15. Hong CS, Graham NA, Gu W, et al. MCT1 modulates cancer cell pyruvate export and growth of tumors that co-express MCT1 and MCT4. Cell Rep. 2016;14(7):1590–1601. doi:10.1016/j.celrep.2016.01.057
16. Wu G, Yuan S, Chen Z, et al. The KLF14 transcription factor regulates glycolysis by downregulating LDHB in colorectal cancer. Int J Biol Sci. 2019;15(3):628–635. doi:10.7150/ijbs.30652
17. Li L, Li C, Wang S, et al. Exosomes derived from hypoxic oral squamous cell carcinoma cells deliver miR-21 to normoxic cells to elicit a prometastatic phenotype. Cancer Res. 2016;76(7):1770–1780. doi:10.1158/0008-5472.CAN-15-1625
18. Ramteke A, Ting H, Agarwal C, et al. Exosomes secreted under hypoxia enhance invasiveness and stemness of prostate cancer cells by targeting adherens junction molecules. Mol Carcinog. 2015;54(7):554–565. doi:10.1002/mc.22124
19. Lässer C, Eldh M, Lötvall J. Isolation and characterization of RNA-containing exosomes. J Vis Exp. 2012;59:e3037.
20. Shigeta K, Hasegawa H, Okabayashi K, et al. Randomized Phase II trial of TEGAFIRI (tegafur/uracil, oral leucovorin, irinotecan) compared with FOLFIRI (folinic acid, 5-fluorouracil, irinotecan) in patients with unresectable/recurrent colorectal cancer. Int J Cancer. 2016;139(4):946–954. doi:10.1002/ijc.30127
21. Tournigand C, André T, Achille E, et al. FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: a randomized GERCOR study. J Clin Oncol. 2004;22(2):229–237. doi:10.1200/JCO.2004.05.113
22. Jing X, Yang F, Shao C, et al. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol Cancer. 2019;18(1). doi:10.1186/s12943-019-1089-9.
23. Li I, Nabet BY. Exosomes in the tumor microenvironment as mediators of cancer therapy resistance. Mol Cancer. 2019;18(1):32. doi:10.1186/s12943-019-0975-5
24. Feron O. Pyruvate into lactate and back: from the Warburg effect to symbiotic energy fuel exchange in cancer cells. Radiother Oncol. 2009;92(3):329–333. doi:10.1016/j.radonc.2009.06.025
25. Vegran F, Boidot R, Michiels C, Sonveaux P, Feron O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF- B/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011;71(7):2550–2560. doi:10.1158/0008-5472.CAN-10-2828
26. Urbańska K, Orzechowski A. Unappreciated role of LDHA and LDHB to control apoptosis and autophagy in tumor cells. Int J Mol Sci. 2019;20(9):2085. doi:10.3390/ijms20092085
27. Wang K, Li Y, Song N, et al. Signal transducer and activator of transcription 3 inhibition enhances vemurafenib sensitivity in colon cancers harboring the BRAF (V600E) mutation. J Cell Biochem. 2019;120(4):5315–5325. doi:10.1002/jcb.27808
28. Zha X, Wang F, Wang Y, et al. Lactate dehydrogenase B is critical for hyperactive mTOR-mediated tumorigenesis. Cancer Res. 2011;71(1):13–18. doi:10.1158/0008-5472.CAN-10-1668
29. Silva LS, Goncalves LG, Silva F, et al. STAT3:FOXM1 and MCT1 drive uterine cervix carcinoma fitness to a lactate-rich microenvironment. Tumour Biol. 2016;37(4):5385–5395. doi:10.1007/s13277-015-4385-z
30. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi:10.1016/j.cell.2011.02.013
31. Levada K, Omelyanchik A, Rodionova V, Weiskirchen R, Bartneck M. Magnetic-assisted treatment of liver fibrosis. Cells. 2019;8(10):1279. doi:10.3390/cells8101279
32. Ding X, He M, Chan AWH, et al. Genomic and epigenomic features of primary and recurrent hepatocellular carcinomas. Gastroenterology. 2019;157(6):1630–1645.e6. doi:10.1053/j.gastro.2019.09.005
33. Grünwald B, Harant V, Schaten S, et al. Pancreatic premalignant lesions secrete tissue inhibitor of metalloproteinases-1, which activates hepatic stellate cells via CD63 signaling to create a premetastatic niche in the liver. Gastroenterology. 2016;151(5):1011–1024.e1017. doi:10.1053/j.gastro.2016.07.043
34. Han L, Lam EWF, Sun Y. Extracellular vesicles in the tumor microenvironment: old stories, but new tales. Mol Cancer. 2019;18(1). doi:10.1186/s12943-019-0980-8
35. Dai X, Chen C, Xue J, et al. Exosomal MALAT1 derived from hepatic cells is involved in the activation of hepatic stellate cells via miRNA-26b in fibrosis induced by arsenite. Toxicol Lett. 2019;316:73–84. doi:10.1016/j.toxlet.2019.09.008
36. Heichler C, Scheibe K, Schmied A, et al. STAT3 activation through IL-6/IL-11 in cancer-associated fibroblasts promotes colorectal tumour development and correlates with poor prognosis. Gut. 2019.
37. Jones SA, Jenkins BJ. Recent insights into targeting the IL-6 cytokine family in inflammatory diseases and cancer. Nat Rev Immunol. 2018;18(12):773–789. doi:10.1038/s41577-018-0066-7
38. Nguyen DP, Li J, Tewari AK. Inflammation and prostate cancer: the role of interleukin 6 (IL-6). BJU Int. 2014;113(6):986–992. doi:10.1111/bju.12452
39. Read JA, Beale PJ, Volker DH, Smith N, Childs A, Clarke SJ. Nutrition intervention using an eicosapentaenoic acid (EPA)-containing supplement in patients with advanced colorectal cancer. Effects on nutritional and inflammatory status: a phase II trial. Support Care Cancer. 2006;15(3):301–307. doi:10.1007/s00520-006-0153-3
40. Xu J, Ye Y, Zhang H, et al. Diagnostic and prognostic value of serum interleukin-6 in colorectal cancer. Medicine. 2016;95(2). doi:10.1097/MD.0000000000004864.
41. Luo M, Luo Y, Mao N, et al. Cancer-associated fibroblasts accelerate malignant progression of non-small cell lung cancer via connexin 43-formed unidirectional gap junctional intercellular communication. Cell Physiol Biochem. 2018;51(1):315–336. doi:10.1159/000495232
42. Zhang D, Wang Y, Shi Z, et al. Metabolic reprogramming of cancer-associated fibroblasts by IDH3α downregulation. Cell Rep. 2015;10(8):1335–1348. doi:10.1016/j.celrep.2015.02.006
43. Zhang G, Zhang Y, Dong D, et al. MCT1 regulates aggressive and metabolic phenotypes in bladder cancer. J Cancer. 2018;9(14):2492–2501. doi:10.7150/jca.25257
44. Cheng A, Zhang P, Wang B, et al. Aurora-A mediated phosphorylation of LDHB promotes glycolysis and tumor progression by relieving the substrate-inhibition effect. Nat Commun. 2019;10(1):5566. doi:10.1038/s41467-019-13485-8
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.