Back to Journals » International Journal of Nephrology and Renovascular Disease » Volume 7
Tubulointerstitial disease in diabetic nephropathy
Received 18 November 2013
Accepted for publication 30 January 2014
Published 21 March 2014 Volume 2014:7 Pages 107—115
DOI https://doi.org/10.2147/IJNRD.S37883
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Giancarlo Tonolo, Sara Cherchi
SC Diabetologia Aziendale ASL 2 Olbia, Hospital San Giovanni di Dio, Olbia, Italy
Abstract: Diabetes mellitus is the major cause of end stage renal disease (ESRD). We cannot predict which patient will be affected. ESRD patients suffer an extremely high mortality rate, due to a very high incidence of cardiovascular disease. Several randomized, prospective studies have been conducted to quantify the impact of strict glycemic control on morbidity and mortality, and have consistently demonstrated an association between strict glycemic control and a reduction in ESRD. Within the past 20 years, despite the implementation of treatments that were presumed to be renoprotective, diabetes mellitus has continued to rank as the leading cause of ESRD, which clearly indicates that we are still far from understanding the mechanisms involved in the initiation of ESRD. Progressive albuminuria has been considered as the sine qua non of diabetic nephropathy, but we know now that progression to diabetic nephropathy may well happen in the absence of initial microalbuminuria. The search for new biomarkers of early kidney damage has received increasing interest, since early identification of the pathways leading to kidney damage may allow us to adopt measures to prevent the development of ESRD. Most of these biomarkers are deeply influenced by environment, genetics, sex differences, and so on, making it extremely difficult to identify the ideal biomarker to target. At present, there are no new drugs that come close to providing the solutions we desire for our patients (ie, reducing complications). Even when used in combination with standard care, renal complications are, at best, only modestly reduced, at the considerable expense of additional pill burden and exposure to serious off-target effects. In this review, some of the hypothesized mechanisms of this heterogeneous disease will be considered, with particular attention to the tubule–interstitial compartment.
Keywords: TGF-ß1, ESRD, Ox-LDL, diabetes, ESRD
Introduction
Diabetes mellitus is the major cause of end stage renal disease (ESRD).1 About 20% of patients with either type 1 diabetes (T1DM) or type 2 diabetes (T2DM) develop nephropathy after many years of diabetes, but we cannot predict which patient will be affected, and the pathophysiological mechanisms that are critically involved in triggering the development of ESRD are still unclear. Within the past 20 years, despite implementation of treatments that were presumed to be renoprotective, diabetes mellitus has continued to rank as the number one cause of ESRD.1 Recently, it has become clear that diabetic nephropathy is a heterogeneous entity, conditioned by several factors, including diabetes type, genetics, social status, blood glucose and pressure levels, environment, sex, etc.
Much has been learned about the role of the vasculature and the glomerulus, including mesangial cells and podocytes, in the pathophysiology of the diabetic kidney.2,3 It remains unclear whether the tubule has a pivotal role early in the initiation of diabetic nephropathy. Because of obvious space limitations, and the wide variety of the thematic involved, it will not be possible to provide a detailed, comprehensive review of all published work on this topic. However, in this review, we will try to outline the possibly pivotal role of the kidney tubule–interstitial milieu in diabetic nephropathy, and the difficulty of targeting adequate drug therapy.
Differences between T1DM and T2DM
Renal disease in T2DM appears to be more heterogeneous than in T1DM. Usually, when proteinuria is found in T1DM, a well-defined constellation of renal structural abnormalities are expected to occur in parallel (ie, mesangial expansion, arteriolar hyalinosis, and tubulointerstitial changes).4–8 In a classical view, proteinuria is almost always associated with diabetic retinopathy. This has been discussed recently by the EURODIAB IDDM Complications study,9 which showed that diabetic retinopathy is not observed as frequently as previously believed in microalbuminuric T1DM patients, since it is strictly associated with increased circulating levels of von Willebrand factor, a marker of endothelial dysfunction. Indeed, if microalbuminuria antedates overt nephropathy in 80% of T1DM patients, this is the case in only 20%–30% of T2DM patients.10–12 Fewer than 50% of T2DM patients with overt nephropathy have diabetic retinopathy (oculo–renal syndrome), despite having micro- and even macroalbuminuria. Heterogeneity of histological lesions has been reported in T2DM.13,14 About 29% of patients have normal or near-normal renal structure; 29% have changes typical of diabetic nephropathy (as commonly seen in T1DM); and 42% have important tubulointerstitial fibrosis and/or arteriolar hyalinosis, with glomerular sclerosis and relatively trivial abnormal changes. This suggests that the tubule is important in the initiation of kidney disease, at least in some cases. Indeed, in this last group of T2DM patients, who show mainly tubulointerstitial fibrosis, an increase in kidney arteriolar resistance has been suggested.15,16 High glucose exposition and hypertension play important roles in the development of kidney damage in both T1DM and in T2DM patients. However, a genetic component is also present (since family history of kidney disease is a strong predictor of renal functional decline).17,18 Previously, we described a familial clustering of increased urinary albumin excretion (AER+), in T2DM families, with no association with hypertension.19 Our data were in keeping with what has previously been described in Pima Indian families; but, in Pima subjects, AER+ clustering was aggregately associated with hypertension – and hypertension preceding diabetes onset was a strong predictor of future renal damage.20–22 These data illustrate the importance of genetic/familial factors in the development of kidney disease in diabetes, at least in T2DM subjects.
Ethnic and social factors are also important in the development of diabetic renal disease. Native Americans, Hispanics (especially Mexican Americans), and African Americans have much higher risks of developing ESRD than non-Hispanic whites with T2DM.23 This might be the result of varying degrees of access to the health system.
Role of tubule in diabetic nephropathy
While diabetic glomerulopathy is present in the early stages of diabetic nephropathy,24,25 tubulointerstitial fibrosis is typical only of advanced disease. This often heralds the onset of progression towards ESRD,26,27 after which the diabetic milieu, and the prolonged interaction of albuminuria (and other factors in the glomerular filtrate) with the tubular system, triggers renal oxidative stress and cortical interstitial inflammation, with resulting hypoxia and tubulointerstitial fibrosis, which determines, to a great extent, the progression of renal disease.4,28–30 Tubular damage might well be the trigger of the disease. A recent study showed that regression of microalbuminuria in patients with T1DM is associated with lower levels of urinary tubular injury biomarkers (kidney injury molecule-1 and N-acetyl-β-d-glucosaminidase), which is consistent with the notion that tubular dysfunction is a critical component of the early course of diabetic nephropathy.31
Poor glucose control, and the renin–angiotensin aldosterone system (RAS), are well established risk factors for the development of nephropathy, both in T1DM and T2DM patients.
When renal epithelial cells in culture are exposed to high glucose concentrations, they produce increased amounts of type I and type IV collagens,32 leading to fibrosis. The profibrogenic cytokine transforming growth factor beta 1 (TGF-β1) has been implicated in the development of renal fibrosis in a number of kidney diseases; it is closely linked to the development and progression of diabetic nephropathy.33 In murine mesangial cells in culture, increased levels of glucose stimulate the production of TGF-β1, which then acts, through autocrine mechanisms, to enhance collagen production.34 The same mechanisms apply to human proximal tubule cells exposed in vitro to high glucose concentrations; the expression of high levels of TGF-β1, in the presence of platelet-derived growth factor, leads to subsequent synthesis of type I and type IV collagens.35
The local RAS in kidney tissues clearly play a role in the pathophysiology of diabetic nephropathy.36 The widespread use of RAS inhibitors, in order to reduce AER+ in diabetic patients, is indicative of the role of intrarenal RAS in the progression of diabetic nephropathy. It has been reported that levels of urinary angiotensinogen increase before glomerular injury in diabetic patients,37 as well as in rodents,38 which suggests that urinary angiotensinogen might be a biomarker of RAS activation in the tubule, in diabetes. RAS inhibitors ameliorate intrarenal RAS activation,39,40 reduce tubular angiotensinogen and decrease tubular injury, with no increase in urinary α1-microglobulin (an established early marker of proximal tubule dysfunction in diabetic nephropathy).41 Despite the widespread use of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers, kidney function decrease in diabetic patients continues to have a significant prevalence. Angiotensin II is able to induce TGF-β1; all intracellular signaling activated by glucose entering the cell is able to increase transcription of this profibrogenic cytokine. TGF-β1 can influence the concentration of small, noncoding, single-stranded RNA molecules called microRNAs (miRNAs) in the tubular compartment, as well as at the glomerular level.42,43 These miRNAs, which are synthesized from longer precursors, through a multistep maturation process, act as translational repressors of specific target mRNAs.44 Animal models, using TGF-β1 antibodies, were able to slow the progression of diabetic nephropathy. Together with recent studies on miRNA in experimental diabetic nephropathy,45–47 this opens the future possibility that TGF-β1 might become a candidate for developing new drugs to prevent or reduce the impact of kidney disease – in particular, tubular damage in diabetic patients. Although animal models would be extremely useful in this regard,48 so far, unfortunately, real animal models that resemble tubular damage in induced diabetes are lacking.49
Unlike T1DM, wherein insulin deficiency is due to massive destruction of pancreatic beta cells, relative insulin deficiency in T2DM is secondary to insulin resistance, which is often associated with obesity. In the early stages of obesity, the action of insulin is preserved, or even enhanced, as far as action on lipogenesis and adipogenesis is concerned, particularly in liver and skeletal muscle. Ectopic fat accumulation damages metabolic responses to insulin in muscle and liver, and may lead to impaired insulin synthesis and secretion in pancreatic beta cells. At this stage, overt diabetes may occur, lipogenesis and adipogenesis become stationary, and lipolysis is activated.50 Obesity is related to progression of the rate of ESRD, independent of metabolic control of glucose, blood pressure, and baseline AER+, in T2DM subjects.12 But, little information is available with regard to fat accumulation in the kidney before the overt development of diabetes in obese patients.
It has been proposed that glucose overload, at the kidney level, may contribute to an observed glycogen deposit not associated with evident accumulation of intrarenal fat.51 In diabetic subjects, renal glucose uptake is inversely correlated with free fatty acid (FFA) uptake,51,52 which suggests the existence of a renal glucose–FFA cycle. Even if the absolute uptake of FFA by the kidney is not increased in T2DM patients, studies using diabetic animals have shown that FFA, entrapped in the mitochondrial matrix, leads to production of mitochondrial reactive oxygen species (ROS), lipid peroxidation, and mitochondrial damage and dysfunction.51,53 In renal tissue, as well as in the overall systemic vascular bed, the generation of oxidized lipids can modulate the formation of macrophage foam cells. Oxidized low-density lipid (LDL) (ox-LDL) encompasses various oxidation-specific neoepitopes, such as malondialdehyde-modified LDL, and the group of phosphorylcholine-headed, oxidized phospholipids. These neoepitopes are recognized both by adaptive, lymphocyte T cell-dependent, and by innate, T cell-independent mechanisms.53 Scavenger receptors of macrophages play a central role in lipotoxicity in tubulointerstitial tissue. CXCL16 is one of the main receptors for the uptake of ox-LDL in podocytes; CD36 plays this role in tubular renal cells, as well as in macrophages.54 Heme oxygenase is induced in epithelial cells following exposure to ox-LDL, and is accompanied by worsening of several indices of cytotoxicity, in vitro. The uncanny resemblance of the behavior of renal tubular epithelial cells to endothelial cells, after exposure to ox-LDL, both in terms of oxidant-mediated toxicity and expression of heme oxygenase, leads to speculation that the paradigmatic role of ox-LDL in atherogenesis may apply to tubular injury as well.55 Cellular iron is assigned a central importance in the toxicity of ox-LDL in tubular cells.56 In proteinuric renal diseases, abnormal amounts of iron are present in the kidney.57 Pretreatment with an iron chelator blocks the cytotoxicity of ox-LDL and attenuates upregulation of heme oxygenase. The mechanisms for oxidation of LDL in proteinuric renal disease are not completely understood. LDL is vulnerable to oxidative attack in the urinary space, where it may be endocytosed by tubular cells. Alternatively, LDL may be oxidized within tubular cells, and in the microvascular bed that surrounds tubulointerstitial tissue. This hypothesis is supported by the observation that tubular damage can be found at a stage at which loss of glomerular permselectivity does not yet permit LDL to enter the urinary space.56,57 This argument has previously been discussed extensively.58 Indeed, we have shown previously that treatment with statins is able to reverse urinary albumin excretion in T2DM patients;59 a meta-analysis indicated that lipid reduction may preserve kidney function in diabetic patients.60
Given the understanding that the proximal tubule plays a vital role in the pathophysiology of the diabetic kidney, we are beginning to better understand the molecular basis of complex interactions between the diabetic milieu, proximal tubule, and tubulointerstitium. Tubular uptake of glucose is important in the detrimental renal effects of diabetes, as well as in glucose homeostasis. Inhibition of glucose reabsorption in the proximal tubule is a promising approach to lowering blood glucose levels. Glucose is taken into the proximal tubule primarily through two sodium-glucose transporter proteins. In the early part of the proximal tubule, glucose is taken up by a high-capacity, low-affinity sodium-glucose cotransporter SGLT2 (SLC5A2). In the later portions of the proximal tubule, glucose uptake occurs primarily through SGLT1 (SLC5A1), a high-affinity, low-capacity sodium-glucose cotransporter. mRNA levels of both SGLT1 and SGLT2 are increased in the renal cortex of obese Zucker rats,61 being upregulated by high glucose levels. Proximal62 and interstitial63 cells respond to high levels of glucose, with production of profibrotic mediators before an increase in filtered albumin, or any measurable change in glomerular levels. In a streptozotocin rat model of diabetic nephropathy, insulin administration was able to reverse increased mRNA levels of proximal tubular procollagen alpha 1 before any measurable change at the glomerular level.64 This unique early growth phenotype of the proximal tubule might be identified as a potential target for the prevention of early glomerular hyperfiltration and increased tubular reabsorption, as well as an early link to tubulointerstitial inflammation, fibrosis, oxidative stress, hypoxia, and renal failure in diabetes. Differences in the capacity of the tubule to respond to hyperglycemia might explain why some diabetic patients, in response to glucose overload, develop diabetic nephropathy, while others do not. The tubule’s SGTL2 protein is upregulated in T2DM patients, increasing glucose reabsorption, resulting in a net renal glucose uptake. Thus, drugs that target this mechanism are promising in diabetes therapy. Although the long-term efficacy and safety of SGLT2 inhibitors remain under study, this class represents a novel therapeutic approach, with potential for treatment of both type 2 and type 1 diabetes, as recently reviewed.65 TGF-β1 regulates SGLT2 expression in response to high glucose in proximal tubular cells in culture. Empagliflozin, a SGLT2 inhibitor, reduces high glucose-induced inflammatory and fibrotic markers, by blocking glucose transport, without a compensatory increase in SGLT1 and glucose transporter 2 (GLUT2) expression.66
Other new drugs for diabetes therapy appear to have possible tubular-protective effects, besides glucose-lowering effects. The use of incretin-based therapies, such as dipeptidyl peptidase-4 (DPP-4) inhibitors and glucagon-like peptide-1 (GLP-1) analogs, is becoming widespread in T2DM patients. Besides optimization of glycemic control in a wider range of patients, these new classes of drugs have shown pleiotropic effects at the kidney level. GLP-1 stimulates glucose-dependent insulin secretion from pancreatic beta cells, and suppresses glucagon release from alpha cells, thereby improving glucose control. Besides its action on the pancreas, GLP-1 has direct effects on the heart, vessels, and kidney, mainly via the GLP-1 receptor. A GLP-1 receptor agonist, in streptozotocin-induced diabetic rats, is able to inhibit the advanced glycation end products and their generation of receptor-mediated asymmetric dimethylarginine (ADMA) (an endogenous inhibitor of nitric oxide synthase) – a mechanism that is involved in tubular dysfunction and diabetic nephropathy.67–69 This effect is mediated by suppressing protein arginine methyltransferase-1, an enzyme that generates mainly ADMA in human proximal tubular cells, via inhibition of ROS generation, thereby protecting against the development and progression of diabetic nephropathy.70 DDP-4 inhibitors act to reduce the degradation of incretins, such as GLP-1 and gastric inhibitory polypeptide. Other molecules that have proven renal and cardiovascular effects are DPP-4 substrates, such as brain natriuretic peptide/atrial natriuretic peptide, neuropeptide Y, peptide YY, and stromal cell-derived factor 1alpha. It is not surprising that these drugs might have different pleiotropic effects at the tubular level, as well as the vascular level. Through a mechanism yet not fully understood, the DPP-4 inhibitor vildagliptin was able to functionally protect the kidney in a rat model of unilateral renal ischemia. Reduced tubular necrosis observed was associated with antiapoptotic, immunological and antioxidative changes.71 Studies have suggested possible direct renoprotective effects of DPP-4 inhibition that may go beyond its glucose-lowering potential.72,73
Another important factor that contributes to tubulointerstitial injury is chronic hypoxia. In animal models, renal hypoxia can be due to enhanced tubular oxygen consumption, as shown ex vivo in cortical and medullar tubular cells of streptozotocin-diabetic rats.74 Modification of vasoactive factors, such as nitric oxide and angiotensin II, may induce interstitial vascular rarefaction via postglomerular blood flow modifications, impairing peritubular blood flow and oxygen delivery to the tubules. This mechanism may be maintained and enhanced through the concurrent action of anemia, appearing in impaired renal function,75 and through oxygen consumption in the remnant nephrons.76 In vitro studies in human proximal tubular epithelial cells showed that hypoxia increased total collagen production,77 with decreased collagen IV mRNA levels and increased collagen I mRNA levels, which suggested the induction of interstitial collagen. Although hypoxia did stimulate TGF-β1 production, it did not appear to mediate the profibrogenic stimulus of hypoxia.77,78 These findings link both hypoxia and oxidative stress to the tubulointerstitial accumulation of extracellular matrix, and fibrosis in the diabetic kidney.
Identification of the mechanisms leading to tubule–interstitial damage is complicated by the general lack of clinical attention to male–female differences, although the impact of sex on diabetic nephropathy has been reviewed previously.79–81 In general, women show greater protection than men against the development and progression of renal disease. However, the advantage is less evident in diabetic nephropathy than in non-diabetic kidney diseases.82,83 When type 1 diabetes is first diagnosed during adolescence, a sex-related difference is present between men and women for diabetic kidney disease, which suggests a role for sex hormones in mediating this difference.84 A simple concept arising from this observation is, regarding susceptibility to kidney disease, that 17β-estradiol (E2) is good, and testosterone is bad. However, the underlying pathways by which E2 and testosterone confer their effects are complex. Under good metabolic control, diabetic women are more likely to develop diabetic nephropathy than diabetic men, whereas the situation is reversed under poor metabolic control. In females; protection from diabetic kidney disease via E2 occurs, in part, by regulation of TGF-β1. TGF-β1 serum levels are higher in men than in women;85 rodent animal models with low E2 levels (both diabetic and non-diabetic) show increased renal expression of TGF-β1.86 In streptozotocin-induced diabetic rats, E2 supplementation provides renoprotectivity by reducing extracellular matrix synthesis, and by increasing degradation and attenuating tubulointerstitial fibrosis.87 E2 treatment has the same effects on TGF-β1 at the glomerular level in other rat models,88,89 suggesting that the mechanism might be non-peculiar, or that it is not the first initiating event in tubular damage. There are several mechanisms proposed for E2’s effects on TGF-β1 expression that have been exhaustively described.90
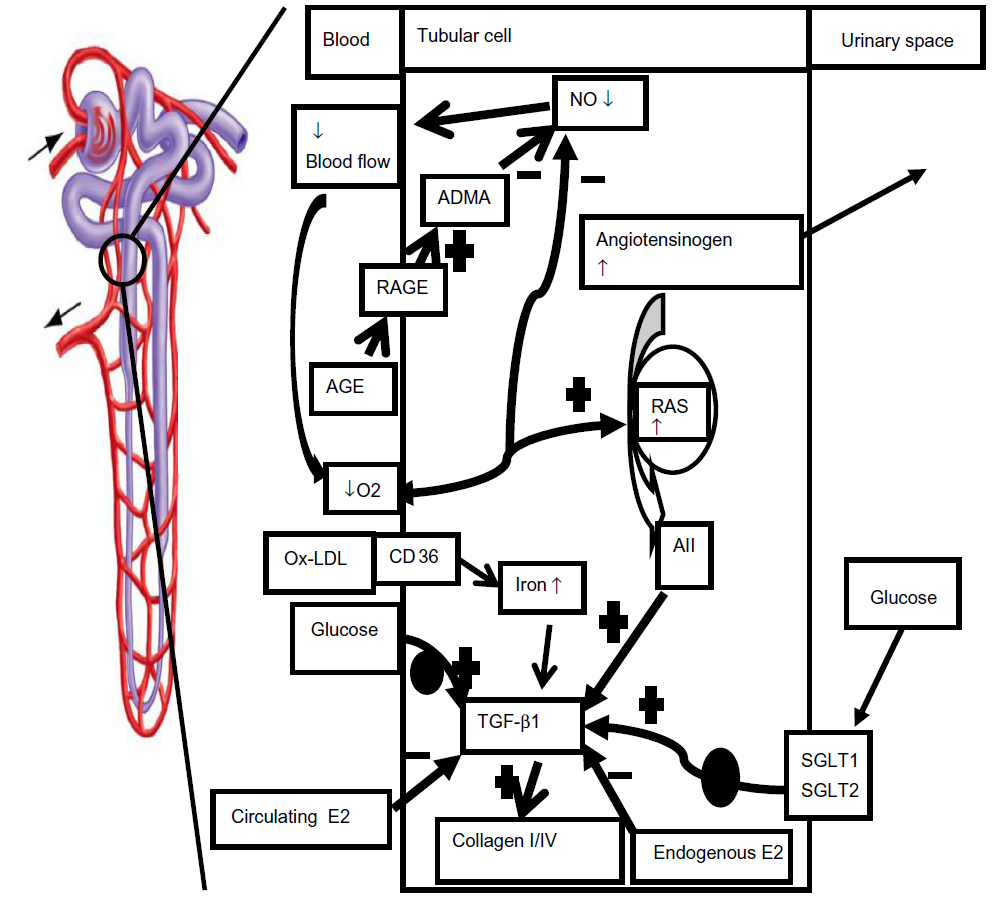 | Figure 1 Proposed mechanisms of tubular dysfunction in diabetes mellitus |
If E2 protects the kidney from diabetic damage by interfering with TGF-β1 signaling, an obvious extension of these studies would be to administer E2 to diabetic patients, to prevent or ameliorate diabetic kidney damage. Since exogenous estrogen’s protection against the development and progression of cardiovascular disease has been questioned,91–94 a clear answer to the possible protective role of E2 on kidney function should come from large-scale, randomized trials (which are not planned, so far). There are several theoretical limitations within the concept that E2 might be directly responsible for the observed sex difference in diabetic nephropathy. Firstly, diabetic men already have higher levels of E2 than non-diabetics.95 In male diabetic rats, overexpression of kidney aromatase induces a higher rate of conversion of testosterone to E2, leading to higher intrarenal E2 levels.96 Secondly, oral contraceptives that contain high doses of estrogens promote the risk of diabetic nephropathy, whereas lower-estrogen doses have no influence on renal function; thus, the effects of estrogens might be variable, depending on the dose.83 Thirdly, the kidney is able to synthesize steroid hormones; local variation in the concentrations E2 and testosterone in the kidney could affect the abovementioned expression, and signaling of TGF-β1, independently of serum hormone concentrations.
Sexual hormones also significantly affect the RAS system; this might well be another way by which sexual hormones can condition the development and progression of diabetic nephropathy differently in the two sexes.97–99 The problem is far more complicated, since sex differences interact with the environment and genetic background. For example, the M235T polymorphism in the angiotensinogen gene increases the incidence of diabetic nephropathy in males but not in females, both in T2DM100 and T1DM101 patients.
Conclusion
Rather than the orthodox view (that glomerular injury is primary in diabetic nephropathy, with the tubular injury secondary), it is evident that the kidney tubule plays a critical role in the genesis of diabetic nephropathy, as it is also that progression of diabetic nephropathy is best correlated with the degree of tubulointerstitial disease. There are many biomarkers of human tubular injury available at the moment. The main problem is that most of the biomarkers are still at an intermediate phenotype level, which is too distant from the gene level; therefore there is a high risk of influence by other gene products or the environment. Integrative analysis of the proteomic and transcriptomic features at different levels, among different categories of patients affected by diabetic nephropathy, could finally lead to the identification of new, early markers, closer to the gene level. Better identification of the first molecules involved in tubular damage might enable therapies to target the mitigation of initial injury to the proximal tubule, and the subsequent inflammation and fibrosis that lead to diabetic nephropathy. Therefore, additional effort is required to understand the wide network of biochemical pathways linking diabetes to eventual tubular damage. Despite current therapy that aims to improve glucose control and lower intraglomerular pressure by controlling systemic blood pressure, and even with inhibitors of the renin–angiotensin system, the prevalence of diabetic nephropathy continues to increase. Several pharmaceutical approaches in humans: modulating lipid metabolism and insulin action (ie, statins, metformin, peroxisome proliferator-activated receptor gamma, and nicotinamide adenine dinucleotide phosphate oxidase inhibitors), as well as chemokine expression and function, the new drug classes of incretins, and DPP-4 and SGLT2 inhibitors, might be helpful to understanding these issues, and may be more successful in slowing the progression of diabetic nephropathy, which is the single most important contributor to the epidemic of end stage kidney disease worldwide.
Disclosure
The authors report no conflicts of interest in this work.
References
de Boer IH, Rue TC, Hall YN, et al. Temporal trends in the prevalence of diabetic kidney disease in the United States. JAMA. 2011;305(24):2532–2539. | |
Kanwar YS, Wada J, Sun L, et al. Diabetic nephropathy: mechanisms of renal disease progression. Exp Biol Med (Maywood). 2008;233(1):4–11. | |
Vallon V, Komers R. Pathophysiology of the diabetic kidney. Compr Physiol. 2011;1(3):1175–1232. | |
Mauer M. Structural-functional correlations of diabetic nephropathy. Kidney Int. 1994;45(2):612–622. | |
Parving HH, Hommel E, Mathiesen E, et al. Prevalence of microalbuminuria, arterial hypertension, retinopathy and neuropathy in patients with insulin dependent diabetes. Br Med J (Clin Res Ed). 1988;296(6616):156–160. | |
Viberti GC, Hill RD, Jarret RJ, et al. Microalbuminuria as a predictor of clinical nephropathy in insulin-dependent diabetes mellitus. Lancet. 1982;1(8287):1430–1432. | |
Parving HH, Oxenboll B, Svendsen PA, Christiansen JS, Andersen AR. Early detection of patients at risk of developing nephropathy. A longitudinal study of urinary albumin excretion. Acta Endocrinol (Copenh). 1982;100(4):550–552. | |
Mogensen CE, Christensen CK. Predicting diabetic nephropathy in insulin-dependent patients. N Engl J Med. 1986;331(2):89–93. | |
Karamanos B, Fuller JH, Stevens LK. Short duration of IDDM with microangiopathy vs long duration without risk: determinants and protective factors. Diabetologia. 1996;39(Suppl 1):A26. | |
Retnakaran R, Cull CA, Thome KI, et al. Risk factors for renal dysfunction in type 2 diabetes: UK Prospective Diabetes Study 74. Diabetes. 2006;55(6):1832–1839. | |
MacIsaac RJ, Panagiotopoulos S, McNeil KJ, et al. Is non albuminuric renal insufficiency in type 2 diabetes related to an increase in intrarenal vascular disease? Diabetes Care. 2006;29(7):1560–1566. | |
Rossi MC, Nicolucci A, Pellegrini F, et al. Obesity and changes in urine albumin/creatinine ratio in patients with type 2 diabetes: the DEMAND Study. Nutr Metab Cardiovasc Dis. 2010;20(2):110–116. | |
Fioretto P, Mauer M, Brocco E, et al. Patterns of renal injury in type 2 NIDDM patients with microalbuminuria. Diabetologia.1996;39(12):1569–1576. | |
Fioretto P, Stehouwer CD, Mauer M, et al. Heterogeneous nature of microalbuminuria in NIDDM: studies of endothelial function and renal structure. Diabetologia. 1998;41(2):233–236. | |
Nosadini R, Velussi M, Brocco E, et al. Altered transcapillary escape of albumin and microalbuminuria reflects two different pathogenetic mechanisms. Diabetes. 2005;54(1):228–233. | |
Nosadini R, Velussi M, Brocco E, et al. Increased renal arterial resistance predicts the course of renal function in type 2 diabetes with microalbuminuria. Diabetes. 2006;55(1):234–239. | |
UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet. 1998;352(9131):837–853. | |
de Boer IH, Rue TC, Cleary PA, et al. Long-term renal outcomes of patients with type 1 diabetes mellitus and microalbuminuria: an analysis of the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications cohort. Arch Intern Med. 2011;171(5):412–420. | |
Faronato PP, Maioli M, Tonolo G, et al. Clustering of albumin excretion rate abnormalities in Caucasian patients with NIDDM. The Italian NIDDM Nephropathy Study Group. Diabetologia. 1997;40(7):816–823. | |
Kunzelman CL, Knowler WC, Pettitt DJ, Bennett PH. Incidence of nephropathy in type 2 diabetes mellitus in the Pima Indians. Kidney Int. 1989;35:681–687. | |
Nelson RG, Pettitt DJ, Baird HR. Pre-diabetic blood pressure predicts urinary albumin excretion after the onset of type 2 (non-insulin-dependent) diabetes mellitus in Pima Indians. Diabetologia. 1993;33(10):998–1001. | |
Nelson RG, Pettitt DJ, de Courten MP, et al. Parental hypertension and proteinuria in Pima Indians with NIDDM. Diabetologia. 1996;39(4):433–438. | |
Sibley SD, Thomas W, de Boer I, Brunzell JD, Steffes MW. Gender and elevated albumin excretion in the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) cohort: role of central obesity. Am J Kidney Dis. 2006;47(2):223–232. | |
Fioretto P, Mauer M. Histopathology of diabetic nephropathy. Semin Nephrol. 2007;27(2):195–207. | |
Gellman DD, Pirani CL, Soothill JF, Muehrcke RC, Kark RM. Diabetic nephropathy: a clinical and pathologic study based on renal biopsies. Medicine (Baltimore). 1959;38:321–367. | |
Fioretto P, Caramori ML, Mauer M. The kidney in diabetes: dynamic pathways of injury and repair. The Camillo Golgi Lecture 2007. Diabetologia. 2008;51(8):1347–1355. | |
Bohle A, Wehrmann M, Bogenschutz O, et al. The pathogenesis of chronic renal failure in diabetic nephropathy. Investigation of 488 cases of diabetic glomerulosclerosis. Pathol Res Pract. 1991;187(2–3):251–259. | |
Marcussen N. A tubular glomeruli and the structural basis for chronic renal failure. Lab Invest. 1992;66:265–284. | |
Singh DK, Winocour P, Farrington K. Mechanisms of disease: the hypoxic tubular hypothesis of diabetic nephropathy. Nat Clin Pract Nephrol. 2008;4(4):216–226. | |
D’Amico G, Ferrario F, Rastaldi MP. Tubulointerstitial damage in glomerular diseases: its role in the progression of renal damage. Am J Kidney Dis. 1995;26(1):124–132. | |
Vaidya VS, Niewczas MA, Ficociello LH, et al. Regression of microalbuminuria in type 1 diabetes is associated with lower levels of urinary tubular injury biomarkers, kidney injury molecule-1, and N-acetyl-beta-d-glucosaminidase. Kidney Int. 2011;79(4):464–470. | |
Ziyadeh FN, Snipes ER, Watanabe M, et al. High glucose induces cell hypertrophy and stimulates collagen gene transcription in proximal tubule. Am J Physiol. 1990;259(4 Pt 2):F704–F714. | |
Zhu Y, Usui HK, Sharma K. Regulation of transforming growth factor beta in diabetic nephropathy: implications for treatment. Semin Nephrol. 2007;27(2):153–160. | |
Ziyadeh FN, Sharma K, Ericksen M, Wolf G. Stimulation of collagen gene expression and protein synthesis in murine mesangial cells by high glucose is mediated by autocrine activation of transforming growth factor-beta. J Clin Invest. 1994;93(2):536–542. | |
Fraser D, Brunskill N, Ito T, Phillips A. Long-term exposure of proximal tubular epithelial cells to glucose induces transforming growth factor-beta 1 synthesis via an autocrine PDGF loop. Am J Pathol. 2003;163(6):2565–2574. | |
Roksnoer LC, Verdonk K, van den Meiracker AH, et al. Urinary markers of intrarenal renin-angiotensin system activity in vivo. Curr Hypertens Rep. 2013;15(2):81–88. | |
Saito T, Urushihara M, Kotani Y, Kagami S, Kobori H. Increased urinary angiotensinogen is precedent to increased urinary albumin in patients with type 1 diabetes. Am J Med Sci. 2009;338(6):478–480. | |
Kamiyama M, Zsombok A, Kobori H. Urinary angiotensinogen as a novel early biomarker of intrarenal renin-angiotensin system activation in experimental type 1 diabetes. J Pharmacol Sci. 2012;119(4):314–323. | |
Hong CY, Hughes K, Chia KS, Ng V, Ling SL. Urinary alpha1-microglobulin as a marker of nephropathy in type 2 diabetic Asian subjects in Singapore. Diabetes Care. 2003;26(2):338–342. | |
Korpinen E, Teppo AM, Hukkanen L, et al. Urinary transforming growth factor-beta1 and alpha1-microglobulin in children and adolescents with type 1 diabetes. Diabetes Care. 2000;23(5):664–668. | |
Terami T, Wada J, Inoue K, et al. Urinary angiotensinogen is a marker for tubular injuries in patients with type 2 diabetes. Int J Nephrol Renovasc Dis. 2013;6:233–240. | |
Kato M, Putta S, Wang M, et al. TGF-beta activates Akt kinase through a microRNA-dependent amplifying circuit targeting PTEN. Nat Cell Biol. 2009;11(7):881–889. | |
Hata A, Davis BN. Control of microRNA biogenesis by TGFbeta signaling pathway: a novel role of Smads in the nucleus. Cytokine Growth Factor Rev. 2009;20(5–6):517–521. | |
Kim VN, Han J, Siomi MC. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol. 2009;10(2):126–139. | |
Wang Q, Wang Y, Minto AW, et al. MicroRNA-377 is up-regulated and can lead to increased fibronectin production in diabetic nephropathy. FASEB J. 2008;22(12):4126–4135. | |
Zhang Z, Peng H, Chen J, et al. MicroRNA-21 protects from mesangial cell proliferation induced by diabetic nephropathy in db/db mice. FEBS Lett. 2009;583(12):2009–2014. | |
Fu Y, Zhang Y, Wang Z, et al. Regulation of NADPH oxidase activity is associated with miRNA-25-mediated NOX4 expression in experimental diabetic nephropathy. Am J Nephrol. 2010;32(6):581–589. | |
Brosius FC, Alpers CE, Bottinger EP, et al. Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2009;20(12):2503–2512. | |
Soler MJ, Riera M, Batlle D. New experimental models of diabetic nephropathy in mice models of type 2 diabetes: efforts to replicate human nephropathy. Exp Diabetes Res. 2012;2012:616313. | |
Hopps E, Noto D, Caimi G, Averna MR. A novel component of the metabolic syndrome: the oxidative stress. Nutr Metab Cardiovasc Dis. 2010;20(1):72–77. | |
Meyer C, Gerich JE. Role of the kidney in hyperglycemia in type 2 diabetes. Curr Diab Rep. 2002;2(3):237–241. | |
Woerle HJ, Popa E, Dostou J, et al. Exogenous insulin replacement in type 2 diabetes reverses excessive hepatic glucose release, but not excessive renal glucose release and impaired free fatty acid clearance. Metabolism. 2002;51(11):1494–1500. | |
Wang Z, Jiang T, Li J, Proctor G, McManam JL, Lucia S et al. Regulation of renal lipid metabolism, lipid accumulation, and glomerulosclerosis in FVBdb/db mice with type 2 diabetes. Diabetes. 2005;54(8):2328–2335. | |
Park YM, Febbraio M, Silverstein RL. CD36 modulates migration of mouse and human macrophages in response to oxidized LDL and may contribute to macrophage trapping in the arterial intima. J Clin Invest. 2009;119(1):136–145. | |
Agarwal A, Balla J, Balla G, et al. Renal tubular epithelial cells mimic endothelial cells upon exposure to oxidized LDL. Am J Physiol. 1996;271(4 Pt 2):F814–F823. | |
Abrass CK. Cellular lipid metabolism and the role of lipids in progressive renal disease. Am J Nephrol. 2004;24(1):46–53. | |
Santini E, Lupi R, Baldi S, et al. Effects of different LDL particles on inflammatory molecules in human mesangial cells. Diabetologia. 2008;51(11):2117–2225. | |
Nosadini R, Tonolo G. Role of oxidized low density lipoproteins and free fatty acids in the pathogenesis of glomerulopathy and tubulointerstitial lesions in type 2 diabetes. Nutr Metab Cardiovasc Dis. 2011;21(2):79–85. | |
Tonolo G, Ciccarese M, Brizzi P, et al. Reduction of albumin excretion rate in normotensive microalbuminuric type II diabetic patients during long-term simvastatin treatment. Diabetes Care. 1997;20(12):1891–1895. | |
Fried LF, Orchard TJ, Kasiske BL. Effect of lipid reduction on the progression of renal disease: a meta-analysis. Kidney Int. 2001;59(1):260–269. | |
Tabatabai NM, Sharma M, Blumenthal SS, Petering DH. Enhanced expressions of sodium-glucose cotransporters in the kidneys of diabetic Zucker rats. Diabetes Res Clin Pract. 2009;83(1):e27–e30. | |
Ziyadeh FN, Snipes ER, Watanabe M, et al. High glucose induces cell hypertrophy and stimulates collagen gene transcription in proximal tubule. Am J Physiol. 1990;259(4 Pt 2):F704–F714. | |
Polhill TS, Saad S, Poronnik P, Fulcher GR, Pollock CA. Short-term peaks in glucose promote renal fibrogenesis independently of total glucose exposure. Am J Physiol Renal Physiol. 2004;287(2):F268–F273. | |
Ihm CG, Lee GS, Nast CC, et al. Early increased renal procollagen alpha 1(IV) mRNA levels in streptozotocin induced diabetes. Kidney Int. 1992;41(4):768–777. | |
DeFronzo RA, Davidson JA, Del Prato S. The role of the kidneys in glucose homeostasis: a new path towards normalizing glycaemia. Diabetes Obes Metab. 2012;14(1):5–14. | |
Panchapakesan U, Pegg K, Gross S, et al. Effects of SGLT2 inhibition in human kidney proximal tubular cells – renoprotection in diabetic nephropathy? PLoS One. 2013;8(2):e54442. | |
Chiarelli F, Gaspari S, Marcovecchio ML. Role of growth factors in diabetic kidney disease. Horm Metab Res. 2009;41(8):585–593. | |
Bucala R, Tracey KJ, Cerami A. Advanced glycosylation products quench nitric oxide and mediate defective endothelium-dependent vasodilatation in experimental diabetes. J Clin Invest. 1991;87(2):432–438. | |
Ziyadeh FN, Wolf G. Pathogenesis of the podocytopathy and proteinuria in diabetic glomerulopathy. Curr Diabetes Rev. 2008;4(1):39–45. | |
Ojima A, Ishibashi Y, Matsui T, et al. Glucagon-like peptide-1 receptor agonist inhibits asymmetric dimethylarginine generation in the kidney of streptozotocin-induced diabetic rats by blocking advanced glycation end product-induced protein arginine methyltranferase-1 expression. Am J Pathol. 2013;182(1):132–141. | |
Glorie LL, Verhulst A, Matheeussen V, et al. DPP4 inhibition improves functional outcome after renal ischemia-reperfusion injury. Am J Physiol Renal Physiol. 2012;303(5):F681–F688. | |
Hocher B, Reichetzeder C, Alter ML. Renal and cardiac effects of DPP4 inhibitors – from preclinical development to clinical research. Kidney Blood Press Res. 2012;36(1):65–84. | |
Scirica BM, Bhatt DL, Braunwald E, et al. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. SAVOR-TIMI 53 Steering Committee and Investigators. N Engl J Med. 2013;369(14):1317–1326. | |
Palm F, Cederberg J, Hansell P, Liss P, Carlsson PO. Reactive oxygen species cause diabetes-induced decrease in renal oxygen tension. Diabetologia. 2003;46(6):1153–1160. | |
Bahlmann FH, Fliser D. Erythropoietin and renoprotection. Curr Opin Nephrol Hypertens. 2009;18(1):15–20. | |
Nath KA, Croatt AJ, Hostetter TH. Oxygen consumption and oxidant stress in surviving nephrons. Am J Physiol. 1990;258(5 Pt 2):F1354–F1362. | |
Orphanides C, Fine LG, Norman JT. Hypoxia stimulates proximal tubular cell matrix production via a TGF-beta1-independent mechanism. Kidney Int. 1997;52(3):637–647. | |
Lin CL, Wang FS, Kuo YR, et al. Ras modulation of superoxide activates ERK-dependent fibronectin expression in diabetes-induced renal injuries. Kidney Int. 2006;69(9):1593–1600. | |
Seliger SL, Davis C, Stehman-Breen C. Gender and the progression of renal disease. Cur Opin Nephrol Hypertens. 2001;10(2):219–225. | |
Diamond-Stanic MK, You YH, Sharma K. Sugar, sex, and TGF-β in diabetic nephropathy. Semin Nephrol. 2012;32(3):261–268. | |
Franconi F, Campesi I, Occhioni S, Tonolo G. Sex-gender differences in diabetes vascular complications and treatment. Endocr Metab Immune Disord Drug Targets. 2012;12(2):179–196. | |
Schulb CJ, Konopelska-Bahu T, Dalton RN, et al. Microalbuminuria prevalence varies with age, sex, and puberty in children with type 1 diabetes followed from diagnosis in a longitudinal study. Oxford Regional Prospective Study Group. Diabetes Care. 1999;22(3):495–502. | |
Maric C. Sex, diabetes and the kidney. Am J Physiol Renal Physiol. 2009;296(4):F680–F688. | |
Harjutsalo V, Maric C, Forsblom C, et al. Sex-related differences in the long-term risk of microvascular complication by age at onset of type 1 diabetes. Diabetologia. 2011;54(8):1992–1999. | |
Lin Y, Nakachi K, Ito Y, et al. Variations in serum transforming growth factor-beta1 levels with gender, age and lifestyle factors of healthy Japanese adults. Dis Markers. 2009;27(1):23–28. | |
Elliot SJ, Berho M, Korach K, et al. Gender-specific effects of endogenous testosterone: female alpha-estrogen receptor-deficient C57BI/6J mice develop glomerulosclerosis. Kidney Int. 2007;72(4):464–472. | |
Mankhey RW, Bhatti F, Maric C. 17beta-Estradiol replacement improves renal function and pathology associated with diabetic nephropathy. Am J Physiol Renal Physiol. 2005;288(2):F399–F405. | |
Catanuto P, Doublier S, Lupia E, et al. 17 beta-estradiol and tamoxifen upregulate estrogen receptor beta expression and control podocyte signaling pathways in a model of type 2 diabetes. Kidney Int. 2009;75(11):1194–1201. | |
Dixon A, Maric C. 17beta-Estradiol attenuates diabetic kidney disease by regulating extracellular matrix and transforming growth factor-beta protein expression and signaling. Am J Physiol Renal Physiol. 2007;293(5):F1678–F1690. | |
Yanes LL, Sartori-Valinotti JC, Reckelhoff JF. Sex steroids and renal disease: lessons from animal studies. Hypertension. 2008;51(4):976–981. | |
Kannel WB, Hjortland MC, McNamara PM, Gordon T. Menopause and risk of cardiovascular disease: the Framingham study. Ann Intern Med. 1976;85(4):447–452. | |
Hulley S, Grady D, Bush T, et al. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. Heart and Estrogen/progestin Replacement Study (HERS) Research Group. JAMA. 1998;280(7):605–613. | |
Rossouw JE, Prentice RL, Manson JE, et al. Postmenopausal hormone therapy and risk of cardiovascular disease by age and years since menopause. JAMA. 2007;297(13):1465–1477. | |
Manson JE, Allison MA, Rossouw JE, et al. Estrogen therapy and coronary-artery calcification. N Engl J Med. 2007;356(25):2591–2602. | |
Maric C, Forsblom C, Thorn L, Waden J, Groop PH. Association between testosterone, estradiol and sex hormone binding globulin levels in men with type 1 diabetes with nephropathy. Steroids. 2010;75(11):772–778. | |
Prabhu A, Xu Q, Manigrasso MB, et al. Expression of aromatase, androgen and estrogen receptors in peripheral target tissues in diabetes. Steroids. 2010;75(11):779–787. | |
Cherney DZ, Sochett EB, Miller JA. Gender differences in renal responses to hyperglycemia and angiotensin-converting enzyme inhibition in diabetes. Kidney Int. 2005;68(4):1722–1728. | |
Hajjar I, Kotchen TA. Trends in prevalence, awareness, treatment, and control of hypertension in the United States. 1988–2000. JAMA. 2003;290(2):199–206. | |
Thoenes M, Neuberger HR, Volpe M, et al. Antihypertensive drug therapy and blood pressure control in men and women: an international perspective. J Hum. Hypertens. 2010;24(5):336–344. | |
Freire MB, Ji L, Onuma T, et al. Gender-specific association of M235T polymorphism in angiotensinogen gene and diabetic nephropathy in NIDDM. Hypertension. 1998;31(4):896–899. | |
Pettersson-Fernholm K, Frojdo S, Fagerudd J, et al. The AT2 gene may have a gender-specific effect on kidney function and pulse pressure in type I diabetic patients. Kidney Int. 2006;69(10):1880–1884. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.