Back to Journals » Drug Design, Development and Therapy » Volume 20
Tubulin Post-Translational Modifications in Cardiovascular Diseases: Emerging Mechanisms and Therapeutic Modulation by Traditional Chinese Medicine
Authors Qi Y, Zhou Y, Ning X, Ye J, Hu Z ![]() , Li L
, Li L ![]()
Received 14 April 2026
Accepted for publication 15 June 2026
Published 22 June 2026 Volume 2026:20 616766
DOI https://doi.org/10.2147/DDDT.S616766
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Muzammal Hussain
Yangfan Qi,1 Yiyi Zhou,2 Xinyue Ning,1 Jiahao Ye,1,3 Zhixi Hu,1,3 Lin Li1,3
1School of Traditional Chinese Medicine, Hunan University of Chinese Medicine, Changsha, Hunan, People’s Republic of China; 2Medical School, Hunan University of Chinese Medicine, Changsha, Hunan, People’s Republic of China; 3Provincial Key Laboratory of TCM Diagnostics, Hunan University of Chinese Medicine, Changsha, Hunan, People’s Republic of China
Correspondence: Lin Li, Email [email protected]
Abstract: Cardiovascular diseases (CVDs) severely threaten human health, and microtubule dysfunction is one of the major contributing factors. Tubulin post-translational modifications (PTMs), such as detyrosination, acetylation, and polyglutamylation, regulate microtubule homeostasis, among which the upregulation of detyrosination is a key feature of cardiovascular diseases. Traditional Chinese Medicine (TCM) can alleviate cardiac pathologies by targeting these modifications. This review discusses the driving mechanisms of PTMs in relation to the pathological changes that occur in CVDs and summarizes the therapeutic effects of traditional Chinese Medicine targeting these modifications that have been identified in recent years. By systematically searching databases such as PubMed, it was found that PTMs primarily control the stability and contractility of the microtubule cytoskeleton in cardiomyocytes. Their dysregulation promotes the progression of diseases such as myocardial infarction (MI), atherosclerosis (AS), arrhythmias, and cardiac hypertrophy by disrupting key processes including mitochondrial energy metabolism, inflammasome activation, calcium homeostasis, and oxidative stress (OS). Certain active compounds found in TCM, such as curdione, evodiamine, and tanshinone IIA (Tan-IIA), can modulate PTMs through the synergistic regulation of multiple pathways. This review provides a theoretical basis for establishing TCM-based therapeutic strategies for CVDs that target PTMs. The diagram illustrates the modulation of microtubule post-translational modifications (PTMs) to improve heart failure. The central section shows microtubule components with processes: 1) Glutamylation is catalyzed by TTLL4/7 and removed by CCP5, whereas poly-glutamylation is catalyzed by TTLL6 and removed by CCP1/4/6. Detyrosination by VASH-SVBP and Tyrosination by TTL. 4) Acetylation (k40) by aTAT1 and deacetylation by HDAC6, SIRT2. The lower section shows improvement in heart failure, depicted by a dilated ventricle and hypertrophy, through monomeric compounds and compound formulations of traditional Chinese medicine. The text reads: ′By modulating microtubule PTMs′.Diagram of microtubule PTMs modulation improving heart failure with compounds.
Keywords: cardiovascular diseases, tubulin post-translational modifications, traditional Chinese medicine intervention
Introduction
Cardiovascular diseases (CVDs) are disorders that impair the normal function of the heart and lead to increased disease burden and death rates in many people globally. These disorders include coronary artery disease, heart failure, myocardial infarction, and arrhythmias. CVDs are more prevalent globally because of the aging population, rapid urbanization, and lifestyle changes. Cardiomyocytes are groups of cardiac cells that generate contractile force in the human heart, and their structure and normal function (normal contractile dynamics, intracellular cargo transport, and mechano-transduction) are maintained by the microtubule network. This network is composed of α-tubulin and β-tubulin heterodimers1 Various cardiovascular disorders occur when there are disruptions in the integrity of microtubules.2–5
Tubulin post-translational modifications (PTMs) maintain the balance of the microtubule network. In cardiac pathologic conditions, aberrant activation of these PTMs,6 especially detyrosination, acetylation, and polyglutamylation, leads to functional remodeling of the microtubule7 This process disrupts microtubule mechanics, dynamics, and interactions with microtubule-associated proteins, thereby affecting contractile function and intracellular signaling. Acetylation is closely associated with the stability and flexibility of microtubules, which means it can affect long-distance protein and organelle transportation8 Detyrosination makes microtubules more rigid, which improves the load-bearing capacity of cardiomyocytes9 Research conducted into these processes shows that excessively activated detyrosination leads to increased rigidity of the microtubules, which can impact the contractility of the heart, thereby causing heart failure. Consequently, proper regulation of the detyrosination modification is essential in preserving the normal function of the heart and preventing the onset of diseases2 These findings show that tubulin PTMs regulate heart biomechanics, thus offering new, potentially effective therapeutic targets for the management of CVDs.
Researchers have identified active TCM compounds that could affect the activation and expression of tubulin PTMs. These findings indicate potentially effective therapeutic approaches to treating cardiovascular disorders. Tanshinone IIA is an example of a Traditional Chinese Medicine (TCM) that regulates microtubule acetylation and inhibits the NLRP3 inflammasome. This results in reduced microtubule-dependent inflammasome assembly and vascular inflammation.10 The bioactive component of Curcuma zedoaria, curdione, another example of TCM, demonstrates substantial anticoagulant and antithrombotic properties.11
In this review, the mechanisms underlying PTM-induced cardiovascular diseases were analyzed and discussed. These findings offer valuable insights into the underlying mechanisms of PTMs, how they contribute to the initiation and progression of CVDs, and how TCM-based treatment strategies can regulate them for more effective treatment approaches.
Methods
Search Strategy
A systematic and comprehensive search of PubMed, Web of Science, Embase, and Scopus between June 2010 and July 2025 was conducted to obtain relevant English-written publications. Endnote was used to eliminate any duplications. Medical Subject Headings (MeSH) and free-text keywords were incorporated in the data search associated with cardiovascular disease, tubulin post-translational modifications, microtubule dynamics, and traditional Chinese medicine. Boolean operators (“AND”, “OR”) were used to construct the search queries across three core domains:
- Cardiovascular diseases: “Cardiovascular Diseases” OR “Heart Failure” OR “Myocardial Infarction” OR “Atherosclerosis” OR “Arrhythmias” OR “Cardiac Hypertrophy” OR “Ischemia-Reperfusion Injury” OR “Cardiac Fibrosis” OR “Ventricular Dysfunction”.
- Traditional Chinese Medicine: “Traditional Chinese Medicine” OR “Chinese Herbal Medicine” OR “Chinese Herbal Drugs” OR “Herbal Extracts” OR “Natural Compounds”.
- Microtubules and PTMs: “Microtubules” OR “Tubulin” OR “Cytoskeleton” OR “Post-translational Modification” OR “Tubulin Post-translational Modification” OR “Acetylation” OR “α-Tubulin Acetylation” OR “HDAC6” OR “αTAT1” OR “Detyrosination” OR “Tyrosination Cycle” OR “Glutamylation” OR “Glycylation” OR “MARK4” OR “VASH/SVBP Complex” OR “Microtubule Dynamics”.
Inclusion and Exclusion Criteria
The publications included in the study encompassed the following: Study type: (1) Clinical studies, in vivo animal studies, or in vitro mechanistic studies; (2) Disease relevance: Investigations conducted on cardiovascular diseases or cardiac/vascular cell models; (3) Mechanistic relevance: Investigation conducted o tubulin post-translational modifications (such as acetylation, detyrosination, glutamylation) or their regulatory enzymes; (4) Intervention relevance: Analysis of traditional Chinese medicine, including monomers, herbal extracts, or compound formulations, that influence microtubules or PTMs; (5) Availability: Full-text, peer-reviewed, original scientific publications.
The exclusion criteria for the publications encompassed the following: (1) Studies examining active ingredients derived from non-TCM sources; (2) Research investigating non-cardiovascular cellular or tissue models; (3) Investigations involving non-TCM drug combinations; (4) Studies lacking a control group; (5) Unrelated articles, reviews, case reports, book chapters, letters, conference abstracts, editorials, or non-original research.
Study Selection and Data Extraction
Two independent researchers evaluated each eligible study and collected the data listed below: (1) First author and year of publication; (2) Study models (clinical, in vivo, and/or in vitro); (3) Type of cardiovascular disease (such as myocardial infarction, atherosclerosis, arrhythmia); (4) Type of tubulin post-translational modifications (such as acetylation, detyrosination, glutamylation); (5) Types of traditional Chinese medicine interventions (monomeric compounds, compound formulations, and extracts); (6) Dosage, experimental protocol, and administration route of TCM; (7) Effects of TCM on tubulin PTMs their implications for cardiovascular functional outcomes; (8) Main conclusions of the study.A PRISMA flowchart summarizing study selection is provided in Figure 1.
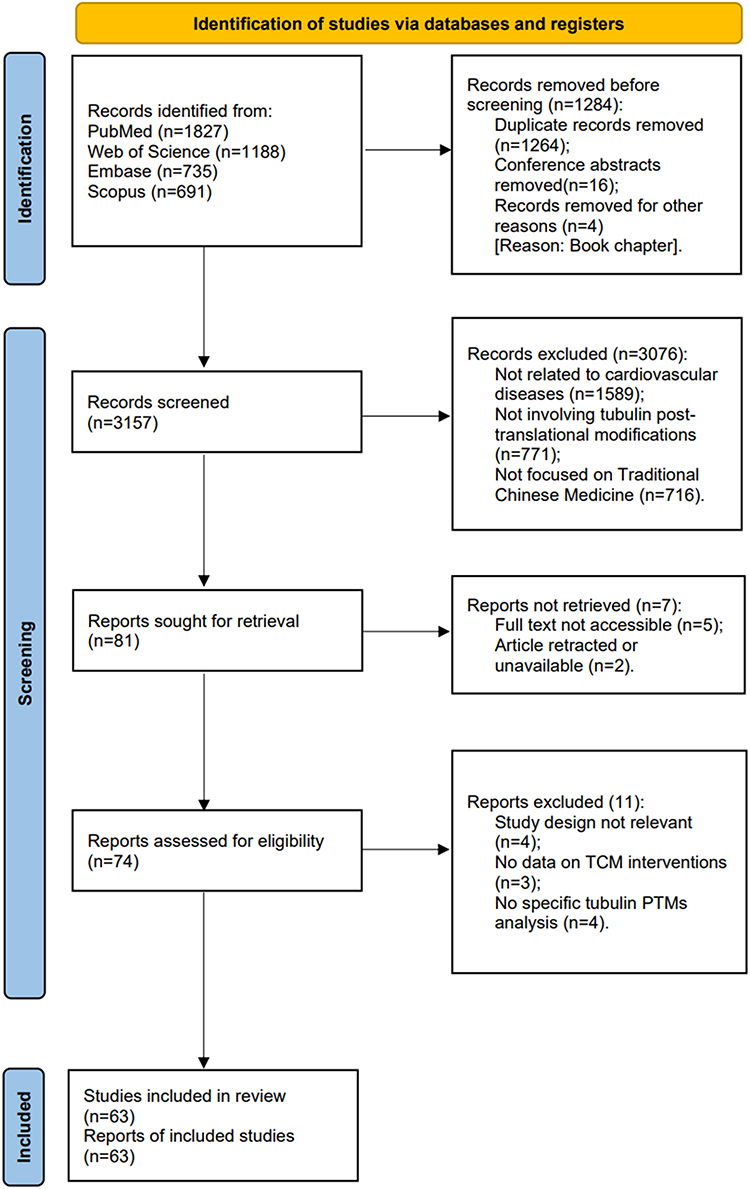 |
Figure 1 Schematic diagram of the PRISMA guideline for screening of research articles. |
The Microtubule Network in the Cardiac System
Microtubules are dynamic filamentous protein structures that make up the cellular cytoskeleton. They contribute to the structural integrity that supports cell shape, enable intracellular transport, and ensure that chromosomes accurately segregate during cell division1 Cardiomyocytes are comprised of three distinct populations that overlap.12,13 These populations are interfibrillar, cortical, and perinuclear microtubules. Interfibrillar microtubules run lengthwise along the myofibrils, forming an additional group around the nucleus14 The cortical microtubules surround the cardiomyocyte and are lined up at right angles to the myofibrils. These microtubules regulate transmembrane proteins and ion channels. They also help transmit external mechanical signals15 Lastly, perinuclear microtubules form a network around the nucleus and nearby organelles. They provide structural support and assist in the organization of these organelles6 The structural integrity and functionality of cardiomyocytes are highly dependent on the microtubule network’s dynamic stability16 Disruptions in this network lead to mechanical rigidity and instability, which are linked to cardiac conditions such as arrhythmias, cardiomyopathy, and heart failure.16,17
Tubulin Post-Translational Modifications
PTMs are dynamic and reversible processes that occur on α- and β-tubulin subunits. They cause changes that regulate the structure and function of the microtubule network.18 These changes can impact the crosstalk between the microtubules and other proteins, proper cell division, and intracellular transportation1 Furthermore, they enable coordinated communication between the network and its surrounding environment for effective response to stimuli. Consequently, preserving the proper function of these modifications is essential for supporting a healthy heart.7,15 Acetylation, detyrosination, glutamylation, and glycylation have been identified as the key types of tubulin PTMs,6 with acetylation and the detyrosination/tyrosination cycle having more significant regulatory functions. These PTMs work together and form a highly sophisticated, well-coordinated system called a “tubulin code,” which facilitates the proper function of the microtubule network and maintains microtubule integrity.
Internal and environmental stimuli modulate PTMs through enzymes and signaling pathways. When the heart sustains injury or assault, the pathological changes can alter the normal function of the enzymes and the activation of the pathways. This can further lead to the dysregulated upregulation or downregulation of some key PTMs.2,19 Glutamylation and glycylation, as well as acetylation and detyrosination, are central to microtubule regulation under cardiovascular pathology.20,21 These PTMs regulate key processes such as myocardial cytoskeletal remodeling, angiogenesis, ciliary signaling, and endothelial proliferation. It can be determined that more PTMs are involved in generating the “tubulin code” that controls cardiac and vascular function.4,22 A visual overview of these key modifications and their enzymatic regulators is presented in Figure 2.
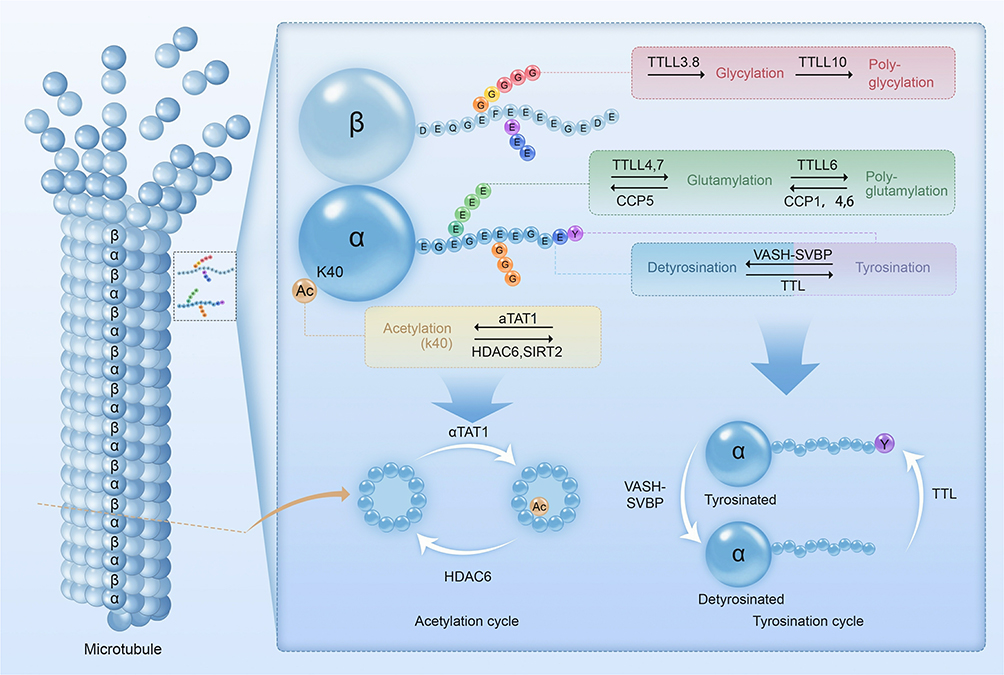 |
Figure 2 Types of Tubulin Post-Translational Modifications. Types of Tubulin Post-Translational Modifications: Microtubules dynamically assemble from head-to-tail arrays of α β-tubulin dimers. Tubulin post-translational modifications occur either at specified sites of the tubulin body (acetylation), or within the C-terminal tails (detyrosination and tyrosination, glutamylation and glycylation). These modifications are often dynamic, reversible, and catalyzed by a range of enzymes from multiple families. During the process, the functional properties of microtubules are changed by single residue alternations (acetylation, detyrosination) or modulating the non-binary signals by elongation of the side chains (polyglutamylation, polyglycylation). Abbreviations: αTAT1, α-tubulin acetyltransferase 1; CCP, cytoplasmic carboxypeptidases; HDAC 6, histone deacetylase 6; K40, lysine 40; TTL, tubulin tyrosine ligase; TTLL, tubulin tyrosine ligase-like; SIRT 2, sirtuin 2; SVBP, serpin vascular biology partner; VASH, vasohibin. |
Acetylation
This process adds an acetyl group to the α-tubulin K40 lysine residue on the luminal surface of microtubules23 The site of this acetylation is indicated in Figure 2. Its role is to stabilize microtubules and improve mechanical resilience by increasing lattice flexibility.24 Through this process, the structural integrity of the microtubules is protected from the cyclic rhythmic contraction of the heart. This results in enhanced long-distance intracellular transport, preserved network organization in cardiomyocytes, and improved self-repair.8
Acetylation of microtubules is controlled in a dynamic balance by two enzymes, namely, α-tubulin acetyltransferase 1 (αTAT1)25 and histone deacetylase 6 (HDAC6).19 αTAT1 adds acetyl groups, whereas HDAC6 removes them. This opposing enzymatic relationship is summarized in Figure 2. Moreover, HDAC6 participates in cellular plasticity and microtubule dynamics as the central regulator19 Additionally, it interacts with silent information regulator 4 (SIRT4) and regulates mitochondrial metabolism. This process creates a link between cytoskeletal remodeling and cellular energy regulation26 Studies show that when HDAC6 is silenced, the acetylation of microtubules is enhanced, which leads to increased microtubule stability and enhanced mitochondrial trafficking along microtubule tracks. This promotes intracellular energy distribution.
Microtubule acetylation, along with the actin cytoskeleton, is involved in maintaining cell morphology.27,28 This process usually involves HDAC6 interacting with cortactin (Arp2/3-stabilizer) to alter tubulin.29 Disrupting the crosstalk between the actin cytoskeleton and microtubules decreases tubulin acetylation and weakens the stability of the cellular cytoskeleton. This results in the impaired contraction of the cardiomyocytes, a feature indicative of the development of CVDs such as cardiomyopathy, coronary artery disease, and heart failure28 Therefore, analyzing the communication between HDAC6-dependent microtubule deacetylation and actin network remodeling may offer new therapeutic approaches for managing cardiovascular diseases.
Detyrosination/Tyrosination
The detyrosination and tyrosination of tubulin are among the earliest PTMs that have been extensively studied and play a crucial role in cellular physiological processes.30 Although this modification’s regulatory role has been observed in many cellular processes, tubulin modulation is one of its most important applications. It modulates tubulin by either removing or reintroducing the tyrosine residues, which control the interaction between microtubules and different regulatory proteins and the dynamics and stability of microtubule.31 The balance between detyrosination and tyrosination forms a “tubulin code,” which determines the function of microtubules and their relationship to other proteins.32
The VASH/SVBP enzyme complex initiates detyrosination in microtubules33 by acting as a carboxypeptidase and removing the C-terminal tyrosine from α-tubulin, resulting in detyrosinated α-tubulin. This enzymatic action is depicted in Figure 2. This detyrosinated form can undergo further processing to generate Δ2-tubulin. SVBP has to bind to and interact with VASH for the complex’s carboxypeptidase activity to be stabilized and enhanced. SVBP binds to VASH to stabilize and enhance the carboxypeptidase activity of the complex.34 The balance between detyrosination and tyrosination (detyrosination/tyrosination cycle) has to be maintained to preserve the normal structure of the intracellular network and the function of the microtubules. This mechanism is catalyzed by tubulin tyrosine ligase (TTL), which adds the tyrosine residue back to the C-terminal of the α-tubulin31 Circumstances that lead to the failure of the proper regulation of this modification, similar to others, result in the development of severe CVD35 Therefore, cellular mechanisms that facilitate the optimal function of cardiomyocytes depend on the balance between detyrosination and tyrosination. It has been observed that when detyrosination of microtubules is increased, the microtubule becomes more rigid, which prevents it from contracting and relaxing normally. This means that the function of the heart becomes compromised as a result36 Conversely, retyrosination of these microtubules decreases cell stiffness and increases heart muscle contractility. Therefore, maintaining the balance of the detyrosination/tyrosination cycle is a promising area of research for developing innovative heart disease treatments.9,36
Microtubule-associated proteins (MAPs), such as MAP4, control microtubule stability. MAP4 is phosphorylated by microtubule Affinity-Regulating Kinase 4 (MARK4), allowing the VASH/SVBP enzyme complex to induce detyrosination2 The function of MARK4 leads to rigid and stabilized microtubules; however, its downregulation in a mouse model showed a decrease in detyrosination, and an increase in myocyte contractility2 Furthermore, the detyrosination of microtubules can be controlled by production ofreactive oxygen species (ROS) dependent on NADPH oxidase 2 (NOX2) and calcium (Ca2⁺) channels. When a cell undergoes mechanical stretching, microtubules serve as mechanotransducers to activate NOX2, subsequently leading to the generation of ROS through the mechanically activated reactive oxygen species (X‑ROS) signaling pathway. During the detyrosination of microtubules, this process is substantially upregulated. Under normal physiological circumstances, this signaling system supports the maintenance of the heart’s normal contraction; however, disruption of the pathway can lead to the aberrant Ca2⁺ release, which can result in, which can cause arrhythmias.5 Therefore, the detyrosinated modification connects the generation of ROS in cardiomyocytes to the mechanical stress of the microtubule network.
Glutamylation
This process reversibly adds glutamate residues to the C-terminal ends of α- and β-tubulin.37 It is induced by tubulin tyrosine ligase-like (TTLL) family and cytoplasmic carboxypeptidases (CCPs) enzymes. The glutamate residues are added by the former and removed by the latter.38 The enzymes responsible for glutamylation are included in the schematic shown in Figure 2. This modification occurs in two stages, namely initiation and elongation, which are catalyzed by TTLL1,39 TTLL4,40 TTLL6,41 and TTLL7.40 The length of the glutamate chains is controlled by severing enzymes such as katanin and spastin. The variation of the chain lengths affects the characteristics of microtubules and their accessibility to microtubule-associated proteins.20,42
Glutamylation has been thoroughly investigated in various tissues, such as neurons and ciliated cells, and the findings show that it contributes to the regulation of the cardiovascular system. Microtubules are involved in modulating the directional migration and morphology of endothelial cells during angiogenic sprouting.43 CEP41 regulates ciliary tubulin glutamylation and enhances cell migration and blood vessel growth through the HIF1A-AURKA-VEGF signaling pathway44 These findings reveal the participation of glutamylation in maintaining endothelial homeostasis and facilitating vascular remodeling. According to mRNA–SNP–miRNA network analysis, CEP41-dependent glutamylation may have a role in the development of cardiovascular disorders in individuals with type II diabetes mellitus21 This suggests that this modification may be involved in cytoskeletal organization and the mechanical stability of the heart21 However, the relationship between microtubule glutamylation and the pathophysiology of cardiovascular diseases has not been well established yet. From these findings, we can determine the potential involvement of tubulin glutamylation as a regulatory mechanism for endothelial migration, angiogenesis, and cardiac structural integrity. This correlates cardiovascular physiology and disease onset with cytoskeletal post-translational modification.
Glycylation
Glycylation is a post-translational modification closely related to glutamylation, in which glycine residues are covalently added to the C-terminal tails of α- and β-tubulin. It is also catalyzed by the enzymes from the tubulin tyrosine ligase-like (TTLL) family.45 Similarly, this modification comprises the initiation and elongation stages. TTLL3 and TTLL8 catalyze initiation by attaching the first glycine through an isopeptide bond, and TTLL10 elongates the glycine chain through sequential additions.46 Figure 2 illustrates the location of glycylation alongside other tail modifications. Evidence on the deglycylation process remains limited, and the enzymes involved are yet to be determined.
The participation of glycylation in cellular movement and signaling by controlling the stability and motility of ciliary and flagellar microtubules has been investigated and established;47 however, its potential role in the cardiovascular system remains largely unexplored.48,49 The involvement of cilia biology in endothelial mechanosensation, cardiac morphogenesis, and vascular development, processes fundamental to cardiovascular homeostasis, has been extensively studied.22,50,51 Glycylation was determined to participate in the function of ciliary stability and signaling capacity. The dysregulation of this modification may impair the endothelial primary cilia which can disrupt flow sensing and angiogenic signaling, two key regulators of vascular remodeling and cardiac tissue repair.47,50 The presence of endothelial and fibroblast cilia in embryonic and adult cardiac tissues may implicate the possible involvement of tubulin glycylation in maintaining the structural integrity.52 Current research has yet to establish an association between tubulin glycylation and the development of cardiovascular disorders. However, ciliary dysfunction has been linked to cardiovascular diseases, including congenital heart defects and vascular malformations. With this evidence, future research should be aimed at establishing the potential involvement of tubulin glycylation in regulating endothelial or cardiac cilia.
Tubulin Modifications and Cardiovascular Diseases
Tubulin post-translational modifications (PTMs) are closely associated with cardiovascular physiology and pathology. The dysregulation of these modifications, which control microtubule stability, mechanics, and cellular activities, is linked to several cardiovascular disorders. The key pathological mechanisms through which specific PTMs contribute to major cardiovascular diseases are synthesized in Figure 3. The following sections describe and explore the functions of tubulin PTMs in the development of illnesses. This article methodically highlights the significance of their involvement in prevalent CVDs, with a central focus on the link between pathogenic processes and cytoskeletal remodeling.
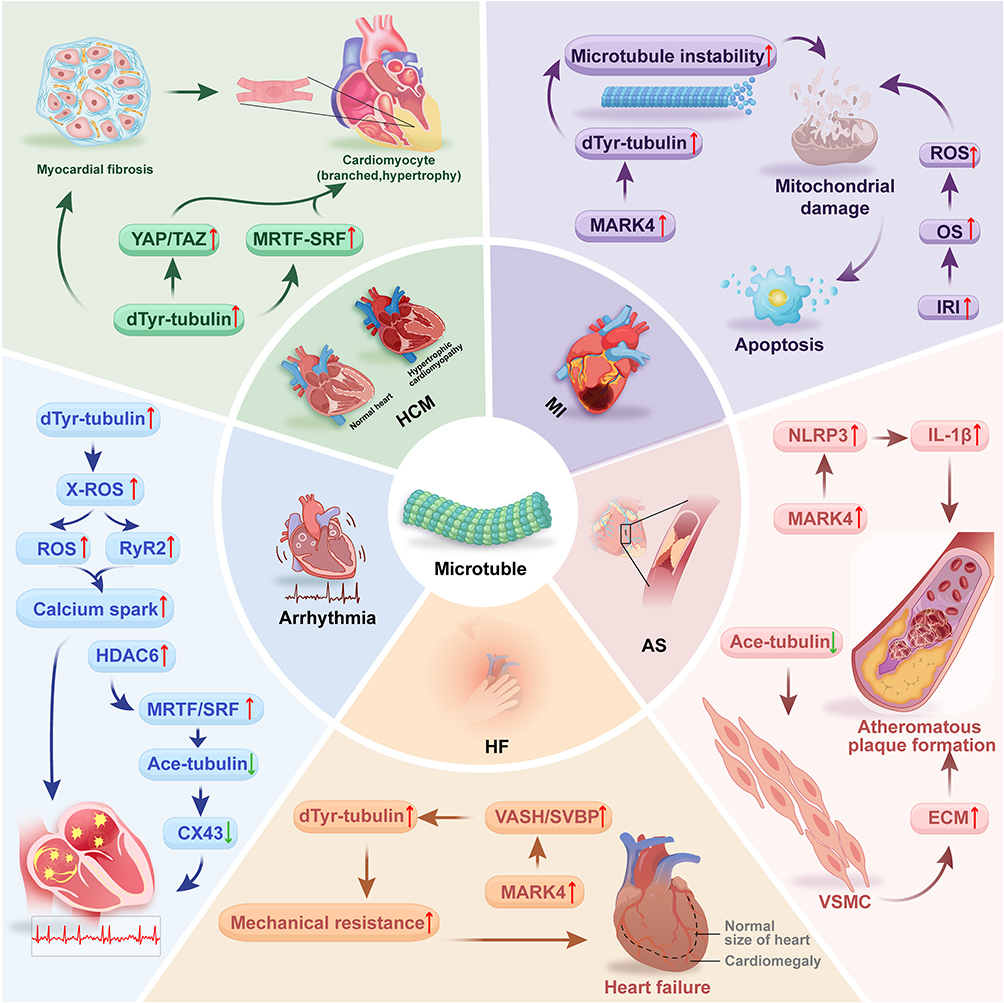 |
Figure 3 Tubulin Modifications and Cardiovascular Diseases. Arrow interpretation: In this figure, upward arrows (↑) indicate upregulation, increased activity, or aggravated pathological processes. Downward arrows (↓) indicate downregulation, decreased activity, or impairment of protective mechanisms.The figure systematically summarizes the roles of PTMs in the core pathological processes of cardiovascular diseases: (1) MI: Under ischemic stress, upregulated MARK4 kinase activity causes a significant increase in microtubule detyrosination. This hyper-modification enhances microtubule network stability and mechanical resistance, subsequently inducing mitochondrial damage and ultimately leading to cardiomyocyte apoptosis and fibrosis. (2) AS: MARK4-mediated detyrosinated microtubules provide a platform for NLRP3 inflammasome assembly and activation, promoting the release of inflammatory factors such as IL-1β. In addition, reduced microtubule acetylation diminishes microtubule stability, driving pathological migration and phenotypic switching of VSMCs, thereby accelerating arterial plaque formation. (3) Arrhythmias: Detyrosinated microtubules amplify the X-ROS signaling pathway through efficient mechanotransduction, causing abnormal RyR2 calcium channel opening and calcium Sparks that trigger afterdepolarizations. Moreover, Downregulated acetylation resulting from HDAC6 overexpression or LMNA mutations leads to compromised microtubule stability, which interferes with normal Cx43 trafficking and localization and impairs electrical conduction. (4) HCM: Under high pressure overload, progressively accumulating detyrosinated microtubules reduce diastolic compliance of cardiomyocytes and directly activate mechanosensitive transcription factors, which include YAP/TAZ and MRTF-SRF. This results in activating pro-hypertrophic genetic mechanisms. (5) HF: The VASH/SVBP complex interacts with MARK4 to induce pathological elevation of microtubule detyrosination in cardiomyocytes. The hyper-stabilized microtubule network forms tight associations with Z-discs of sarcomeres, generating substantial intrinsic mechanical resistance that severely compromises cardiac diastolic function, thus creating a key pathological environment for heart failure. Abbreviations: Ace-tubulin, Acetylated microtubules; Cx43, Gap junction protein 43; DTyr-tubulin, Detyrosinated microtubules; ECM, Extracellular matrix; HCM, Hypertrophic cardiomyopathy; HDAC6, Histone deacetylase 6; IL-1β, Interleukin-1β; IRI, Ischemia-Reperfusion Injury; MARK4, Microtubule affinity-regulating kinase 4; MRTF/SRF, Myocardin-related transcription factor A/serum response factor; NLRP3, Nucleotide-binding oligomerization domain-like receptor protein 3; OS, Oxidative Stress; ROS, Reactive oxygen species; RyR2, Ryanodine receptor 2; SVBP, Serpin vascular biology partner; VASH, Vasohibin; VSMC, Vascular Smooth Muscle Cell; X-ROS, Mechanically activated reactive oxygen species. |
Tubulin Modifications and Myocardial Infarction
Myocardial infarction (MI) is a condition that develops due to the occurrence of acute ischemia, which causes metabolic stress and substantial cell death. This condition initiates a cascade of complex changes inside the heart, usually known as cardiac remodeling.53 The advancement of MI may result in the impaired function of the microtubule network, as it leads to myocardial stiffness, contractile dysfunction, and impaired mitochondrial transport.54–56 PTMs are regulated by specific enzymes and pathways to maintain the functions of the microtubule network and control its interaction with microtubule-associated protein.17 However, when MI develops, this process becomes disrupted, changing the microenvironment of the myocardium to an oxidative and mechanically stressful one and compromising the integrity and function of the microtubule network.57
Detyrosination significantly alters lattice mechanics, and its significant increase in microtubules is a characteristic feature of post-infarction cytoskeletal remodeling.2,9 Acute ischemia triggers the activation of MARK4, which phosphorylates MAP4. Following the phosphorylation of MAP4, the VASH2/SVBP complex can access the microtubules, where it catalyzes the detyrosination of α-tubulin2 As outlined in Figure 3 (MI panel), this MARK4-driven detyrosination axis causes dysfunction in mitochondrial transport, decreases respiratory chain activity, impairs metabolic flexibility, and increases the production of reactive oxygen species54 The combined impact of microtubule stiffness and mitochondrial dysfunction promotes further maladaptive remodeling, including fibrosis, matrix deposition, and cardiomyocyte apoptosis.58
Yu et al studied these effects using an MI mouse model. The study discovered that mice that were MARK4-deficient showed negligible reductions in left ventricular ejection fraction after the mice developed MI, whereas wild-type mice with similar infarct sizes showed a significant decrease in left ventricular ejection fraction.2 The difference suggests that a key determinant of systolic failure is pathological detyrosination rather than just infarct size9 Furthermore, inhibition of detyrosination restores mitochondrial positioning, improves mechanochemical coupling within myofibrils, attenuates maladaptive inflammatory signaling, and reduces fibrotic remodeling of the infarct border zone.35
These results show that in ischemic injury, detyrosinated microtubes trigger mechanical dysfunction, bioenergetic impairment, and progressive structural deterioration2 As a result, to treat or manage MI, therapeutic approaches that target the MARK4-VASH2-detyrosination pathway may be very effective as they can alter post-MI remodeling, maintain the contractile capacity of the myocytes, and enhance long-term recovery.
Tubulin Modifications and Atherosclerosis
Atherosclerosis (AS) is a chronic inflammatory disease characterized by the accumulation of lipids, the infiltration of immune cells, and the abnormal activation and migration of vascular smooth muscle cells (VSMCs).59 Current investigations have shown the pathological effect the dynamic remodeling of the cytoskeleton exerts, particularly alterations in the stability and function of the microtubule network3 Post-translational modifications of tubulin are the major regulators of microtubule function, and their dysregulation is tightly associated with the advancement of AS.3
Atherosclerotic lesions induce a significant remodeling of the microtubule network. These changes disrupt the homeostatic balance between microtubule detyrosination and acetylation. Results from clinical and pathological studies indicate that detyrosinated α-tubulin (dTyr-tubulin) levels are substantially elevated in human atherosclerotic plaque tissues.3 Additionally, changes in microtubule acetylation exhibit cell-specific patterns; in vascular smooth muscle cells (VSMCs), acetylation levels are inversely correlated with the ability of the cells to migrate.4
The abnormal changes that occur in PTMs are tightly controlled by key regulatory enzymes. MARK4 is one of these enzymes. Its function is to promote detyrosination,2 and it is found to be significantly upregulated in human AS plaques.3 It directly drives the detyrosination process through the phosphorylation of microtubule-associated proteins. This then allows for the VASH/SVBP complex to remove tyrosine from the C-terminus of microtubules.34 Other pathways have been linked to the regulation of PTMs.4 The knockdown of ULK1 in VSMCs inhibits autophagic flux, resulting in the increased levels of histone acetyltransferase KAT2A/GCN5, which in turn raises acetylation at the Lys40 position of α-tubulin.4
The dysregulated PTMs aggravate the pathology of AS by activating different downstream molecular mechanisms. MARK4-driven microtubule detyrosination facilitates the spatial organization of the NLRP3 inflammasome by generating a stable platform.3 The detyrosinated microtubule network facilitates the clustering of NLRP3, ASC, and caspase-1 at the microtubule-organizing center, resulting in the formation of ASC specks. When this occurs, caspase-1 becomes activated, leading to the secretion of mature pro-inflammatory cytokines such as interleukin-1β (IL-1β) and IL-18. This results in a significantly amplified local inflammatory response in the vascular wall.60,61 As summarized in the AS section of Figure 3, in an in vivo study, the silencing of the MARK4 gene led to the suppression of NLRP3 inflammasome activation and a reduction of atherosclerotic lesions in mice.3 On the other hand, KAT2A-mediated microtubule acetylation makes the microtubule cytoskeleton more stable, restricting the directional migration ability of VSMCs. As a result, this slows their migration into the intima and subsequent neointimal hyperplasia. This mechanism has been investigated and validated in vascular injury models.4
Targeting the regulatory network of microtubule post-translational modifications (PTMs) reveals significant therapeutic promise based on the previously mentioned mechanisms. Inhibiting the activity of MARK42 or interfering with the function of the VASH/SVBP34 complex anti-inflammatory and anti-AS effects.34 However, the pharmacological enhancement of microtubule acetylation34 or suppression of aberrant detyrosination36 could be a possible approach to stabilizing the VSMC phenotype and preventing their pathological migration. In summary, tubulin PTMs play a crucial role in the onset and progression of AS by tightly regulating vascular inflammation and VSMC activity. Therapeutic intervention that target key regulatory factors in this network open new avenues for developing effective targeted therapy for AS.
Tubulin Modifications and Arrhythmias
Cardiac arrhythmias are heart disorders that disrupt the heart’s electrical system, leading to the irregular generation, frequency, or distribution of the electrical impulses. In serious conditions, sudden death can occur. Tubule PTMs and the microtubule network are linked to the onset of this illness16 Pathological conditions such as pressure overload, ischemia, or gene alterations such as LMNA increased microtubule detyrosination and led to unbalanced tubule PTMs.2,5 Detyrosination increases microtubule stability and mechanical stability, which elevates their load-bearing capabilities and resistance to force. However, in disease states, this modification can become detrimental.35,36
Furthermore, the specific enzymes that drive tubule PTMs can also be activated in pathological states, thus leading to the progression of the disease. For example, the activation of MARK4 phosphorylates MAP4 from microtubules,2 which allows the VASH/SVBP complex to remove the tyrosine residue at the C-terminus of tubulin. Under pathological states, increased detyrosination is harmful34 HDAC6 is yet another enzyme that, when present in abnormally high levels, reduces α-tubulin acetylation, resulting in the destabilization of the microtubule network62 LMNA mutations trigger dilated cardiomyopathy, and the unique signaling pathway from the nucleus to the cytoplasm plays a significant role. Myocardin-related transcription factor A (MRTF-A)–serum response factor (SRF) complex transcriptional activity is decreased by the ERK/Cofilin pathway because of the structural disruption of the nuclear lamina. Then, the microtubule acetylation levels decrease as a result of the downregulation of αTAT1 levels.63
As summarized in the Arrhythmias section of Figure 3, through different but complementary downstream pathways, the changes in these PTMs together contribute to the arrhythmogenic electrophysiological substrate. The heavily detyrosinated microtubule network serves as a powerful amplifier of the X-ROS signaling pathway.5 During diastole, microtubules act as load-bearing structures that efficiently transmit mechanical stretch to activate the NOX2 enzyme, leading to an increased production of Ca2+ and ROS. The elevated increase in Ca2+ from the sarcoplasmic reticulum ryanodine receptor 2 (RyR2) causes spontaneous calcium sparks and calcium waves. These conditions are the source of afterdepolarizations and aberrant activity, which eventually result in fatal arrhythmias.5,64,65 In addition, the downregulated activity of microtubule acetylation substantially disrupts the normal function of the intracellular transport system.8 In LMNA mutation models, the decrease in acetylated microtubules directly interferes with the transport and precise localization of the gap junction protein Cx43 between cardiomyocytes. Its accumulation at the intercalated discs is then prevented by this interference, which leads to its abnormal distribution along the lateral membrane. The abnormal distribution of Cx43 significantly impacts its function. This leads to delayed longitudinal conduction velocity of electrical impulses and increased Cx43 heterogeneity, thus creating a conducive environment for the development of recurrent arrhythmias.63,66
In conclusion, microtubule PTMs play a major role in the development of abnormal contractions of the heart by dysregulating calcium cycling and intercellular electrical connection between heart cells. Therefore, strategically targeting key enzymes that regulate microtubule PTMs to restore cytoskeletal dynamic equilibrium and normal function is a more promising route of treatment compared to suppressing or stabilizing microtubules.
Tubulin Modifications and Cardiac Hypertrophy
Myocardial hypertrophy is an adaptive mechanism to chronic conditions, which results in increased cardiomyocyte size and thicker ventricular walls.67 Although this adaptation can sustain cardiac output during the initial phases, ongoing progression results in compromised diastolic function, myocardial fibrosis, and electrical irregularities, eventually advancing to heart failure and severe arrhythmias.9 The modification of the cardiomyocyte cytoskeleton, including structural and functional changes in the microtubule network, has received growing consideration in the investigation of pathophysiology and the course of pathological cardiac hypertrophy.16 According to current clinical and experimental research, tubulin post-translational modifications have been determined to significantly participate in the progression of myocardial hypertrophy, with the abnormal increase in detyrosination being particularly prominent.2,36
Myocardial tissues from patients with cardiac hypertrophy and pressure overload-induced animal models show a clear rise in microtubule network density, along with significantly elevated levels of detyrosinated α-tubulin.68,69 According to an HFpEF (heart failure with preserved ejection) study conducted by Schulz and et al on a ZSF1 obese rat model, both total α-tubulin and its detyrosinated form were consistently elevated in hypertrophied left ventricular tissue at 20 weeks and 32 weeks stages of the disease. The results from this study suggest a strong association between this modification and the development of cardiac hypertrophy.68 They also reveal the potential impact of detyrosination in the structural and functional deterioration of cardiac hypertrophy as the central factor, rather than an accompanying event.36
TTL and the VASH/SVBP complex regulate the detyrosination/tyrosination cycle. The VASH/SVBP complex is responsible for regulating detyrosination, whereas TTL is in charge of regulating tyrosination.30,34 In cardiac hypertrophy models, higher VASH1/SVBP activity or lower TTL activity raises levels of detyrosinated microtubules. This creates higher microtubule stability and mechanical stiffness.2 In a study conducted by Pietsch et al, myocardial dTyr-tubulin levels were markedly decreased, and cardiac function improved when AAV9-mediated TTL was overexpressed in mice with myocardial hypertrophy. The study demonstrated the enzyme system’s principal regulatory function in regulating cardiac hypertrophy in the field of reverse genetics.70,71
Abnormally high levels of detyrosinated microtubules aggravate cardiac dysfunction via a number of downstream pathways. During heart contractions, these rigid, altered microtubules “buckle” instead of bending. Then the diastolic function becomes damaged by mechanical resistance to sarcomere shortening9 Additionally, they can function as intracellular signaling platforms that activate mechanosensitive transcription factors, including MRTF-SRF and YAP/TAZ, which in turn initiate a program of pro-hypertrophic gene expression.70,71 Moreover, detyrosinated microtubules interfere with the location of calcium-handling proteins and mitochondrial transport, which exacerbates the imbalance in calcium homeostasis and energy metabolism, creating a pathological loop.5,54 The cascade from microtubule detyrosination to diastolic dysfunction and transcriptional activation in hypertrophy is captured in the HCM section of Figure 3.
In conclusion, tubulin PTMs participate in the onset and progression of ventricular hypertrophy, and detyrosination plays a more significant role. These enzyme-regulated modifications impair the mechanical properties of cardiomyocytes, intracellular signal transmission, and microtubule stability, thereby leading to diastolic dysfunction and pathological remodeling.2,9 Therefore, research involving how PTMs restore microtubule dynamic balance can be a new, effective therapeutic approach to treating myocardial hypertrophy.
Tubulin Modifications and Heart Failure
The final stage of many CVDs is heart failure. At this stage, the heart fails to pump blood adequately to meet the body’s basic metabolic needs72 Furthermore, the cardiomyocyte cytoskeleton undergoes significant and frequently irreversible alterations16 In particular, microtubule stability becomes impaired.
Increased detyrosination stiffens the microtubules, causing diastolic dysfunction and reduced contractile efficiency.2,9 This phenomenon was observed in a study involving heart pathologies and heart failure animal models. The results showed a notable and consistent elevation of tubulin detyrosination levels.9,73 The increased activation of this PTM is not an isolated event; rather, it represents the combination of several of the previously discussed pathological events (such as ischemia and pressure overload).2,9 For instance, activated MARK4 kinase phosphorylates MAP4 during post-myocardial infarction remodeling, which subsequently establishes catalytic conditions for the VASH2/SVBP complex.2 Whereas mechanical stress directly causes the detyrosinated microtubule network to expand in pressure-overload hypertrophy.36 Therefore, in heart failure, the microtubule network acts like a stiff scaffold that carries marks from many past diseases. Its upregulated detyrosination serves as downstream manifestation of converging upstream signaling pathways.
This stiffened structure results in cardiomyocyte impairment. The close association of highly detyrosinated microtubules with the Z-discs of sarcomeres creates significant internal mechanical resistance within the cell, greatly limiting the complete expansion of myofibrils during diastole and thus increasing diastolic stiffness.9,73 This is a key feature in heart failure pathophysiology and is characteristic of most in HFpEF cases.68 Clinical data reveal that cellular diastolic stiffness and the amount of detyrosinated microtubules in pathological hearts are positively correlated.9,73 Additionally, the stiffened network causes direct mechanical blockage as well as impairs the normal intracellular transport, resulting in mislocalization of critical proteins, like as connexin 43 (Cx43), and increasing electrical conduction heterogeneity. This paves the way for heart failure with arrhythmias.5,63
As synthesized in Figure 3 (HF panel), detyrosination is central to the pathogenesis of heart failure; hence, controlling its activation may be a significant approach to treatment.2,73 Current research reveals that therapeutic approaches targeting the detyrosination/tyrosination cycle significantly enhance the mechanical performance and diastolic function of cardiomyocytes. This offers a new treatment option for heart failure that treats more than just standard nerve and hormone control9 As a result, expanding therapeutic targets to the cytoskeleton mechanisms of the microtubules can mitigate cardiac “stiffness” at the basic level of cellular mechanics.16 Consequently, future studies focusing on the regulatory interventions that target tubule PTMs may both supplement current treatments and provide new targeted interventions for heart failure phenotypes characterized primarily by diastolic dysfunction. This means that assessing the tightly regulated restoration mechanism of microtubule dynamic stability in cardiomyocytes may lead to new ways to treat heart failure. This approach can shift the focus from symptom relief to reversing the structural and functional damage.
Traditional Chinese Medicine and Tubulin Modifications
The latest investigations show that several cardiovascular pathogenic processes, such as cardiac hypertrophy, fibrosis, and endothelial dysfunction, are closely correlated with dysregulated microtubule and aberrant PTMs.2,9,68,71 Conventional microtubule-targeting agents (MTAs) like colchicine and paclitaxel can alleviate these pathogenic processes; however, their long-term clinical application is restricted due to considerable toxicity.74,75
Compared to conventional forms of treatment, TCM restores microtubule homeostasis and cytoskeletal integrity through its multi-component, multi-target mechanisms of action.76 Monomeric TCM compounds, such as Cinnamaldehyde, Icariin, and Tanshinone IIA, have primarily been the main subject of research.10 They have been observed to affect the regulation of microtubule polymerization, stability, and specific post-translational modifications (such as acetylation, detyrosination). This allows them to protect cardiovascular tissues against oxidative stress and inflammatory injury.77
However, limited research has been done to identify these components and their formulations and reveal their mechanism of action, although it is gradually becoming more prevalent.78 Some TCM compounds, such as Guizhi Gancao Decoction, Tongyang Huoxue Decoction, and Zishen Huoxue Decoction, were determined to change the structure of β-tubulin and the expression of tubule PTM in cardiovascular models.79,80 According to these preliminary results, TCM compounds and their formulations may protect the heart by controlling microtubule dynamics and PTMs.81 However, additional studies are needed to determine their systematic mechanisms and key targets.
The current TCM analyses and the therapeutic influence on microtubule PTMs remain primary at the monomeric level remains predominantly at the monomeric level. In the following section¸ the effects and underlying mechanisms of representative TCM monomers and their regulation of tubulin PTMs within the cardiovascular system are discussed.
Natural Compounds
Natural compounds serve as the main active ingredients in compound formulas. They have well-defined chemical structures and relatively focused target specificity, making them crucial entry points for investigating PTM regulation of tubulin by traditional Chinese medicine.10,76 In recent years, numerous bioactive monomers derived from natural medicines have demonstrated directly or indirectly regulation of microtubule polymerization status, stability, and PTM levels, thereby participating in both pathological regulation and protective processes within the cardiovascular system (Table 1).
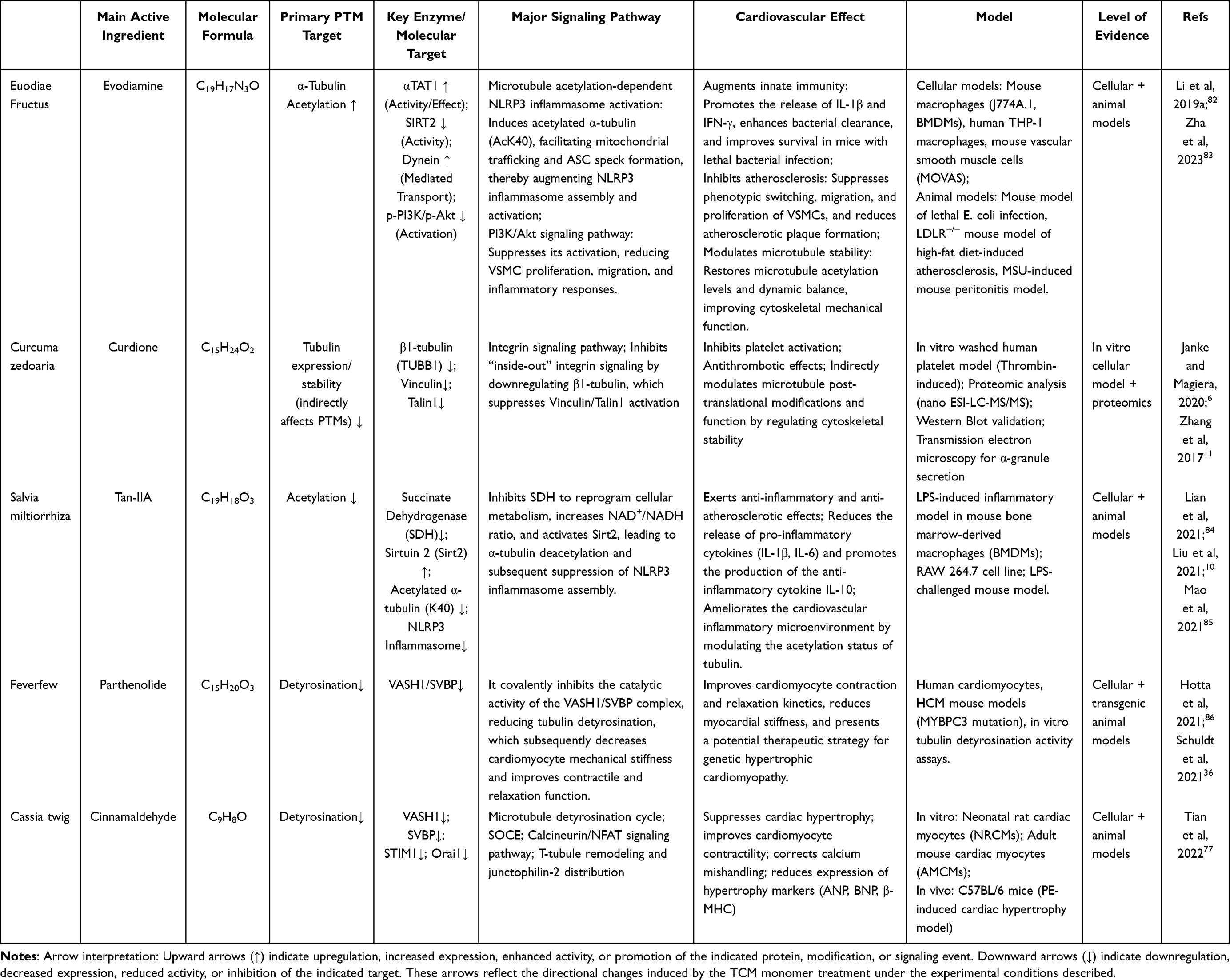 |
Table 1 The Effects of Microtubule-Targeting TCM Monomers on Cardiovascular Diseases |
Evodiamine
Evodia rutaecarpa is a Chinese herb derived from the dried, nearly ripe fruit of several Evodia species, such as Evodia rutaecarpa, Evodia officinalis, or Evodia melanophylla (Rutaceae).87 For centuries, it has been used to treat conditions such as digestive system disorders and cancer.88,89 Evodiamine is the main active compound in Evodia rutaecarpa. In recent years, it has drawn more interest for its therapeutic ability to treat heart and blood vessel disease. Specifically, its therapeutic effects have been observed in tubulin PTMs82 Recent studies demonstrate that evodiamine may regulate the balance between tubulin acetylation and detyrosination. This slows the progression of CVDs (such as atherosclerosis) by improving the cytoskeletal stability and function of vascular smooth muscle cells.82,83
Li and et al studied the underlying mechanism of this herb using mouse macrophage and vascular smooth muscle cell models.82 They also constructed acute infection and peritonitis mouse models by intraperitoneally injecting monosodium urate crystals or a fatal dosage of Escherichia coli, thus replicating immunological stress and inflammation in vivo.82 In the experimental design, evodiamine was administered orally at 10–20 mg/kg, while cells were pretreated with 1–10 μM of evodiamine. The experimental models were then exposed to LPS + ATP or oxidized low-density lipoprotein to examine. Following this, the therapeutic effects of this compound on microtubule structure and function were examined.82 Acetylation level at the Lys40 site of α-tubulin, ASC speck formation, caspase-1 activity, and interleukin-1β release were measured, and the results showed that evodiamine significantly upregulated microtubule acetylation in macrophages. This treatment improved the body’s capacity to eradicate bacterial infections, enhanced the perinuclear clustering of mitochondria and the assembly of the NLRP3 inflammasome, and increased survival rates82 The mechanism of action was dependent on the activation of αTAT1, which can be reversed by acetylation inhibitors. The mechanism is as follows “evodiamine → αTAT1 → microtubule acetylation → NLRP3 inflammasome activation → antibacterial immunity”.82,90
An investigation conducted by Zha et al developed an oxidized low-density lipoprotein (ox-LDL)-stimulated vascular smooth muscle cell (VSMC) mouse model and an atherosclerosis LDLR−⁄− mouse model caused by a high-fat diet. In this study, the mechanisms underlying the anti-atherosclerotic effects of evodiamine were investigated.83 Following treatment with 10 mg/kg/day of evodiamine, the development of aortic plaques in the mouse models was substantially reduced.83 An extensive study of the mechanism of evodiamine showed that its therapeutic action involves regulating microtubule stability in vascular smooth muscle cells. When exposed to ox-LDL, VSMCs underwent a pathological phenotypic shift in response, which is marked by decreased expression of contractile markers (Myh11, Acta2) and increased proliferative (upregulated PCNA expression) and migratory (upregulated MMP2 expression) capacity. However, administering evodiamine restored the imbalanced microtubule acetylation levels induced by ox-LDL in vascular smooth muscle cells.83 Microtubule acetylation maintains microtubule mechanical stability and cellular transport functions.23,24 Evodiamine improved microtubule stability by increasing acetylation levels. This suppressed the pathology induced in VSMCs by microtubule dynamic instability and eventually impeded the development of atherosclerosis.83
Although ongoing research is still in the preclinical phase, the mechanism by which evodiamine modulates microtubule post-translational modifications to address cardiovascular diseases has established a basic therapeutic framework.82,83 To provide more substantial experimental proof for clinical translation, future research should reinforce its validity in complex animal models and investigate its potential for combined therapy with current cardiovascular medications.
Curdione
Curdione is an established active ingredient of the traditional Chinese medicinal herb Curcuma phaeocaulis. According to research, curdione has strong anti-platelet activation properties. Its mechanism is strongly linked to the direct targeting of tubulin and the regulation of cytoskeletal remodeling.11
Significant experimental proof was shown in the Zhang et al study to clarify its mode of action. This study employed a washed human platelet model. A thrombin-induced platelet activation system was implemented using 0.3 U of thrombin. The system was pre-incubated with 100 μM curdione to evaluate its therapeutic effects. The results from the transmission electron microscopy revealed that curdione pretreatment significantly prevented thrombin-induced α-granule release, thus demonstrating its efficient inhibition of platelet activation at the morphological level11 Researchers examined platelet proteins utilizing a quantitative proteomic analysis and used nano ESI-LC-MS/MS to identify molecular targets. They discovered that 22 proteins were differentially upregulated in response to thrombin induction, while only Talin1 and β1-tubulin were effectively downregulated following curdione therapy. These results suggest that β1-tubulin could be a key target for curdione’s actions.11
Additionally, Western blot analysis was used to verify the results from the proteomic analysis and identify downstream signaling pathways involved. It was observed that β1-tubulin was downregulated by curdione, which further inhibited the activation of its downstream cytoskeletal linker proteins, vinculin and talin1.11 A bioinformatic study illustrated that the integrin signaling pathway involves both β1-tubulin and Talin1. This implies that curdione may influence integrin-mediated “inside-out” signal transduction by modulating the “β1-tubulin–Vinculin–Talin1” axis, which would ultimately prevent platelet activation and aggregation.11 To validate these findings, the researchers used targeted inhibitors. ABT-751 was chosen for β1-tubulin, and melittin for vinculin. Both inhibitors were observed to replicate similar effects to curdione. They inhibited platelet activation and reduced the expression of associated proteins, thus functionally confirming the key involvement of this pathway.11
β1-tubulin is an essential component of microtubules. Its expression levels in specific PTMs, such as detyrosination, greatly impact the dynamics and stability of microtubules.6 The downregulation of β1-tubulin expression by curdione suggests that there is a potential route through which modified tubulin isotype composition may indirectly impact global microtubule PTM balance and function. This provides a fresh perspective on its antithrombotic mechanism. However, very little direct enzymological evidence exists regarding the regulation of specific PTMs, such as acetylation and detyrosination.1
It should be noted that cellular models and in vitro platelet evaluations continue to be the main forms of investigating the cardiovascular protective properties of curdione.11 Nonetheless, clinical evidence on its safety and effectiveness is notably absent, as well as the direct proof of its regulation of microtubule PTMs in whole-animal models.
Tanshinone IIA
Salvia miltiorrhiza is highly valued for its therapeutic properties in TCM. It is widely used in treating a wide range of cardiovascular diseases and promotes the circulation of blood.91,92 Tanshinone IIA (Tan-IIA) is the primary therapeutic compound in Salvia miltiorrhiza. It possesses strong antioxidant and anti-inflammatory properties, which lead to reduced myocardial and short-term cardiac damage risk.84,85 Its anti-inflammatory properties in the cardiovascular system are associated with the regulation of tubulin acetylation.10
These findings were validated by an experimental study conducted by Liu et al10 In this study, LPS-treated bone marrow-derived macrophages from mice were used as an inflammation model to replicate the activated state of immune cells in cardiovascular inflammation in vivo. Following treatment with LPS, macrophages experienced metabolic reprogramming, which was marked by succinate accumulation and increased succinate dehydrogenase (SDH) activity.93,94 This subsequently leads to a significant increase in the production of mitochondrial ROS, which stabilizes the expression of HIF-1α protein as well as promotes glycolytic metabolic reprogramming. This establishes an environment conducive to the assembly of the NLRP3 inflammasome.95
Treatment with Tan-IIA suppresses the transmission of this inflammatory signaling at its metabolic source. According to the study, Tan-IIA functions by inhibiting SDH activity in a concentration-dependent manner (IC50 ≈ 4.47 μM), lowers succinate levels, decreases the burden of mitochondrial ROS, and subsequently the stability and nuclear translocation of HIF-1α.10 Moreover, Tan-IIA restored the activity of the downstream deacetylase Sirtuin2 (Sirt2) by raising the NAD⁺/NADH ratio. It dramatically reduced the acetylation level at the Lys40 location of α-tubulin by enhancing the interaction between Sirt2 and α-tubulin, as demonstrated by co-immunoprecipitation findings.10 The acetylation status of α-tubulin serves as a critical regulatory factor for the spatial assembly of the NLRP3 inflammasome.96 Microtubules function as scaffolding to encourage the activation of caspase-1 and the aggregation of NLRP3 and ASC when they are hyperacetylated. Tan-IIA, on the other hand, disrupts this assembly platform by encouraging Sirt2-mediated deacetylation, which prevents IL-1β from maturing and being released.10 The knockout of the Sirt2 gene, along with treatment using the AGK-2 inhibitor, reversed the previously mentioned effects of Tan-IIA, demonstrating how crucial the Sirt2-microtubule acetylation axis is to this regulating mechanism.
It is worth noting that in some cases, Tan-IIA protects by suppressing the activation of tubulin acetylation, a modification that has been established to play a protective role in cardiomyocytes.10,24 This contradiction reveals that the activity of tubulin PTM may be dependent on the type of cell, stimulation, and environment. Nevertheless, acetylation serves a protective role in cardiomyocytes by increasing microtubule flexibility to withstand mechanical stress.24 However, in M1-polarized macrophages, acetylation drives detrimental inflammatory responses by providing a “molecular scaffold” of highly acetylated microtubules for NLRP3 inflammasome assembly.96 This mechanism does not generally disrupt the cytoskeleton, but Tan-IIA-induced deacetylation specifically reverses the pathologically increased acetylation levels in immune cells that arise from overactivation, removing the crucial physical substrate needed for inflammasome.10 This precisely exemplifies the “tubulin code”. PTMs can regulate a tubulin molecule to trigger various cellular processes. This is highly dependent on factors such as the specific cell type, the environmental conditions, and the presence of other interacting proteins.
The currently available evidence that supports the therapeutic role of Tan-IIA in regulating tubulin PTMs is mainly derived from models of macrophage inflammation.10 However, its role in electrophysiological stability, pathological hypertrophy suppression, or inhibiting migration in cardiovascular cells (cardiomyocytes and vascular smooth muscle cells) through similar tubulin deacetylation mechanisms has not been determined. As a result, this research gap is important and warrants further study.
Parthenolide
Parthenolide is a compound obtained from the feverfew plant (Tanacetum parthenium).97 It is a type of TCM that has been used to treat rheumatoid arthritis, migraines, and fever. Current research has shown that parthenolide has a variety of therapeutic properties, including anti-inflammatory effects, apoptosis induction, and prevention of microtubule detyrosination.86,97,98 Studies have focused on determining parthenolide’s tubulin PTM-related mechanisms in treating CVDs. This interest comes from increasing evidence of its mechanisms involved in cytoskeletal remodeling associated with these conditions.99
In cardiovascular disease conditions, high levels of microtubule detyrosination cause rigid cardiomyocytes, which lead to weakened contractile function.9,36 This process is predominantly mediated by the VASH/SVBP complex.34 Using structural biology research and functional studies, parthenolide was determined to significantly inhibit the VASH/SVBP complex99 A study conducted by Li et al, using crystallographic analyses to determine the chemical process, determined that parthenolide inhibits VASH1’s carboxypeptidase activity by forming a covalent adduct between its α-methylene group and the cysteine residue (C169) in the catalytic core via Michael addition.99 The results revealed that parthenolide inhibited microtubule detyrosination in a concentration-dependent manner in a co-incubation system containing recombinant human VASH1-SVBP protein with either microtubules or C-terminal tubulin peptides. In a cellular model using HeLa cells, a study by Li et al (2019) showed the therapeutic effects of parthenolide in reversing the effects of α-tubulin detyrosination induced by the exogenous overexpression of VASH1-SVBP99 In yet another study, these findings were verified using a genetically engineered mouse model of hypertrophic cardiomyopathy.36 Their study used a novel gene-knockin HCM mouse model carrying the MYBPC32373insG mutation. These mice exhibited thickening of the left ventricle wall, decreased diastolic function, a significant rise in detyrosinated microtubule levels in cardiomyocytes, and altered systolic/diastolic dynamics. Their findings showed that following treatment with parthenolide, detyrosinated microtubule levels substantially decreased, and the abnormal systolic and diastolic durations were restored to wild-type levels observed in intact cardiomyocytes isolated from this model.36
From these findings, it can be suggested that parthenolide protects the heart by targeting the VASH/SVBP-microtubule detyrosination axis.99 However, there are still several limitations in this area. First, the majority of research is still in the preclinical phase, with a deficiency of organized clinical trials to assess its safety and effectiveness in human subjects.98 Second, parthenolide may affect pathways other than VASH/SVBP inhibition due to its ability to target multiple factors. As a result, more research is needed to determine the specificity of its protective effects.100,101 Future research should be aimed at developing more selective small-molecule inhibitors of VASH/SVBP to mitigate potential off-target effects associated with parthenolide. Moreover, a new generation of lead drugs that target microtubule PTMs to treat cardiovascular disorders may be identified through the combination of organoid models or patient-derived cardiomyocytes for high-throughput screening.
Cinnamaldehyde
Cinnamomum cassia (Lauraceae) is the dried young twig of the cinnamon tree, known as Gui Zhi in traditional Chinese medicine, with common medicinal uses.102,103 Cinnamaldehyde, the core active component of Cinnamomum cassia, ameliorates pathological cardiac hypertrophy through direct modulation of tubulin detyrosination.77 According to a study by Tian et al (2022), cinnamaldehyde remedies pathological microtubule hyperstabilization and restores dynamic equilibrium in cardiac hypertrophy models by significantly suppressing abnormally elevated α-tubulin detyrosination levels. This process occurs through the downregulation of the key detyrosinase VASH1/SVBP complex.34,77
Tian et al conducted both in vitro and in vivo experimental models. The study used phenylephrine (PE) to establish a cardiomyocyte hypertrophy model in neonatal rat cardiomyocytes (NRCMs) and adult mouse cardiomyocytes (AMCMs). Cinnamonaldehyde was then used as an intervention.77 It was observed that exposure to PE significantly raised the level of α-tubulin detyrosination and the protein expression of two key detyrosinating enzymes, VASH1 and SVBP.34 Treatment with cinnamaldehyde effectively inhibited the overexpression of VASH1 and SVBP induced by PE, consequently decreasing the accumulation of detyrosinated microtubules and restoring the dynamic equilibrium of the microtubule network.77 Further studies on its mechanism of action showed that cinnamaldehyde lowers VASH1/SVBP expression, thus preventing the abnormal translocation of the STIM1/Orai1 complex to the plasma membrane, which was induced by excessive detyrosination. This inhibited the aberrant activation of store-operated calcium entry (SOCE), as well as the nuclear translocation of NFAT transcription factors and the production of hypertrophy-associated genes (ANP, BNP, β-MHC) by directly decreasing the activity of the downstream calcineurin pathway.77 Osmotic minipumps were subcutaneously implanted for continuous PE infusion, and cinnamaldehyde intervention was administered contemporaneously. This was done to induce heart hypertrophy in mice in in vivo studies. The results showed that cinnamaldehyde significantly reduced the mRNA expression levels of ANP, BNP, and β-MHC in cardiac tissue. Furthermore, it considerably reduced PE-induced morphological changes, such as ventricular wall thickening and increased cardiomyocyte cross-sectional area.77 Additionally, myocardial tissue immunofluorescence and Western blot analyses verified that cinnamaldehyde successfully reversed the PE-induced rise in microtubule detyrosination levels and downregulated the expression of VASH1 and SVBP proteins, confirming its regulatory role in microtubule stability in vivo.77
In summary, the available data clearly indicates a causal pathway through which cinnamaldehyde regulates microtubule stability by targeting the VASH1/SVBP–dTyr–tubulin axis. This enhances calcium homeostasis and cellular architecture, which in turn reduces cardiac hypertrophy.77 While this mechanism has predominantly been validated in models of cardiac hypertrophy, its possible therapeutic function as a natural microtubule-stabilizing agent presents innovative opportunities for cardiovascular therapies targeting PTMs. Future research should be aimed at establishing fundamental mechanisms in a wider range of cardiovascular disease models and findings that are easier to apply in clinical settings.
Other Monomeric Compounds
In addition to the TCM monomers discussed above, other monomeric substances have also been shown to affect PTM-related enzyme activity or microtubule integrity. For instance, resveratrol indirectly regulates α-tubulin acetylation levels by activating the deacetylase SIRT1.104–106 Additionally, Berberine has been demonstrated to directly induce α-tubulin acetylation in p53-deficient HL-60 leukemia cells.107 Lastly, In a primary rat cortical neuron model of homocysteine-induced injury, icariin significantly restored the suppressed expression levels of both acetylated α-tubulin and tyrosinated α-tubulin.76 These findings further support the potential role of TCM monomers in regulating tubulin post-translational modifications.
Traditional Chinese Medicine Formula and Tubulin Post-Translational Modifications
Compound formulations of traditional Chinese medicine (TCM) exert complex regulatory effects on cellular structure and function in cardiovascular diseases through their multi-component, multi-target mechanisms of action. Recent research has gradually shown that their effects exceed those observed in macroscopic processes like signal transduction, metabolic regulation, and anti-inflammatory responses. They may also affect the cytoskeletal system to help maintain cellular mechanical homeostasis, especially by modifying tubulin dynamic stability and spatial organization.79–81 However, overall, the research field on TCM compound formulations targeting the microtubule system for the treatment of cardiovascular diseases remains relatively underexplored, and the existing evidence is limited both in scope and mechanistic depth. Nevertheless, research on Guizhi Gancao Decoction (GGD)80 provides fairly direct and systematic mechanistic evidence for how TCM compound formulas can directly regulate the cardiomyocyte microtubule PTMs network, making it worthy of focused discussion.
Guizhi Gancao Decoction (GGD), composed of Cinnamomum cassia and Glycyrrhiza uralensis, has been traditionally used to warm and unblock heart-yang and restore pulse regularity. In a mouse model of pressure overload-induced heart failure, Chen et al demonstrated that GGD significantly ameliorated cardiac hypertrophy, myocardial fibrosis, and contractile dysfunction.80 Importantly, its cardioprotective effect is closely linked to the precise regulation of tubulin post-translational modifications, extending beyond mere hemodynamic modulation.80 At the mechanistic level, pressure overload triggers aberrant cytoskeletal remodeling in cardiomyocytes, characterized by excessive detyrosination and reduced acetylation of α-tubulin. These modification abnormalities impair microtubule dynamics, increase cytoskeletal stiffness, and compromise myocardial compliance.8,24,36 GGD counteracts these pathological changes through a dual regulatory pathway: First, it significantly suppresses tubulin detyrosination, a process potentially achieved by modulating yet-to-be-fully-identified carboxypeptidase activity or its upstream signaling.80 Second, it effectively inhibits HDAC6, thereby enhancing the acetylation level at the Lys40 site of α-tubulin.19,80 Acetylation stabilizes microtubules and maintains their functional flexibility,24 while reduced detyrosination alleviates excessive microtubule polymerization and cytoskeletal rigidity.36 It is important to note that the downregulation of TTL expression observed does not signify an initial target of GGD. However, it indicates a recovery of intracellular microtubule homeostasis and a compensatory alleviation of stress that occurs after the effective upstream suppression of the detyrosination cascade.80 This coordinated restoration of tubulin PTM homeostasis helps preserve sarcomere structural integrity, reduces pathological collagen deposition, and improves both systolic and diastolic cardiac function.23,35
Furthermore, other compound formulations, such as Zishen Huoxue Decoction (ZSHX), reduces ischemic cardiac damage through the Sirt5-β-tubulin-mediated “mitophagy–UPR” synergistic pathway. This suggests that this drug formulation can alter β-tubulin and contribute to cytoskeletal stability and cellular stress response.79 Tongyang Huoxue Decoction (TYHX) regulates β-tubulin expression in sinoatrial node cells, ameliorating calcium homeostasis imbalance induced by hypoxia/reoxygenation and thereby maintaining rhythm stability.81
In summary, existing research indicates that TCM compound formulations, represented by Guizhi Gancao Decoction,80 can precisely intervene in key enzymes within the microtubule PTM network through multi-component synergy. This results in the comprehensive regulation of modification states like acetylation and detyrosination, thereby restoring cardiomyocyte cytoskeletal homeostasis and improving cardiac function. However, current research on the regulation of tubulin post-translational modifications by traditional Chinese medicine compound formulations remains preliminary, with clarity and mechanistic depth far behind that of monomeric compounds. This represents a major bottleneck in the field. First, evidence for the effects of most compound formulas on various PTM types is still lacking, and studies have been highly focused on only a few formulations, with conclusions lacking systematic validation across models and modification types. Second, and more critically, even for formulations with preliminary clues (such as Guizhi Gancao Decoction), the active component profile, the dominant constituents, and their interactions remain unclear. This makes it difficult to construct a precise causal chain linking the compound formula to specific PTM enzyme targets and functional phenotypes, and to clarify potential synergistic or antagonistic effects among components. Third, the research paradigm itself has limitations, as current evidence relies almost entirely on preclinical models and has not yet been connected to human pathophysiology. These gaps collectively lead to a fundamental problem: we are still unable to map out a clear pathway from the multi-component input of a compound formula, through precise regulation of the tubulin PTM network, to the eventual improvement of specific cardiovascular functions (such as enhancing myocardial mechano-transduction or calcium handling). Therefore, future research should aim to penetrate this complex system. The focus should not only be on expanding the screening of compound formulas and PTM types but also on adopting strategies such as spectrum-effect relationship analysis, simplified artificial formulation design, and high-throughput target screening to decipher the compositional logic of compound formulas. Furthermore, efforts should be made to advance research toward clinically relevant models and biomarker validation. This will help transform TCM compound formulations from empirical “modulators” into mechanism-defined “targeted regulators.”
Clinical Translation and Safety Considerations
In experimental settings, TCM has demonstrated the ability to modify microtubule PTMs, consequently enhancing cardiac remodeling and function. Extensive research in cardiomyocytes and hypertrophic hearts from mouse models has determined that trans-cinnamaldehyde reduces pathogenic microtubule detyrosination, thereby relieving structural remodeling. This work highlights the axis defined by detyrosination, microtubule function, and mechanical remodeling as a potential therapeutic target for therapies derived from traditional Chinese medicine.77 Furthermore, Guizhi Gancao Decoction directly demonstrates that multi-component TCM formulations may coordinately modify PTMs in pressure-overload heart failure by restoring aberrant α-tubulin detyrosination and acetylation and improving both systolic and diastolic performance.80
Despite these advances, the majority of existing evidence is still derived from cell and animal studies, and there is a notable lack of clinical studies that directly examine the regulation of tubulin post-translational modifications by traditional Chinese medicine in cardiovascular diseases. Several factors contribute to this gap. First, from a technical perspective, the definitive assessment of PTMs currently requires myocardial biopsy. Non-invasive surrogate biomarkers, such as circulating microtubule-associated proteins or PTM enzymes within exosomes, have not yet been established or clinically validated. Second, the field itself is relatively nascent. The pathological significance of PTMs in CVDs has only been widely recognized in the last 5–8 years, and mechanistic studies of TCM targeting these modifications have emerged even more recently. Third, the inherent complexity of TCM, particularly the undefined active components and diverse pharmacological targets within multi-herb compound formulations, poses substantial challenges for designing early-stage clinical trials with well-defined pharmacodynamic endpoints. Nevertheless, direct evidence from failing human myocardium has already validated the pathophysiological relevance of this axis. Specifically, Chen et al demonstrated that failing human cardiomyocytes exhibit a dense, highly detyrosinated microtubule network, and that pharmacological inhibition of detyrosination restored 40–50% of the lost contractile function.73 Caporizzo et al further showed that detyrosinated microtubules directly contribute to diastolic viscoelastic resistance in human failing myocardium, and that either microtubule depolymerization or inhibition of detyrosination reduces diastolic stiffness in isolated human myocardial trabeculae.9 These human tissue studies confirm that detyrosinated microtubules represent a functionally relevant and therapeutically targetable node in human CVDs, thereby providing a strong mechanistic rationale for TCM interventions that modulate this PTM axis. Strikingly, this mechanistic framework is not without clinical precedent. A recent randomized controlled trial in Parkinson’s disease demonstrated that Modified Zhigancao Decoction significantly reduced serum levels of α-tubulin and microtubule-associated protein-2 in patients, providing direct human evidence that a TCM compound formulation can act on the microtubule system.108 Although this study is not in the cardiovascular field, its mechanistic core shares fundamental commonalities with the PTM-based strategies articulated in this review. Thus, this cross-disease proof-of-concept does far more than confirm target accessibility; it strongly suggests that the translational gap in cardiovascular medicine is not a matter of principle, but rather one of priority. Based on the mechanistic insights articulated thus far and ex vivo validation in human tissues, well-designed clinical studies to evaluate TCM interventions targeting tubulin PTMs in patients with cardiovascular diseases are not only justified but also eminently feasible.
Given the current evidence gaps, we propose a phased translational roadmap to bridge preclinical findings of TCM with future clinical applications. In the short term, priority should be given to utilizing existing human cardiac tissues, such as discarded left ventricular specimens from heart transplant recipients or surgical biopsies, for ex vivo validation. This would involve testing whether TCM monomers, herbal extracts, or medicated serum can reduce dTyr-tubulin levels,9 modulate HDAC619/αTAT125 activity, or alter the VASH/SVBP33 complex in human cardiomyocytes. In the medium term, efforts should focus on developing non-invasive circulating biomarkers, such as plasma detyrosinated microtubule fragments, exosomal HDAC6,19 or VASH/SVBP33 enzymatic activity, as pharmacodynamic readouts of TCM interventions. Such biomarkers would facilitate dose-finding and proof-of-mechanism studies without the need for repeated myocardial biopsies. In the long term, leveraging the extensive clinical safety record of TCM compound formulations, such as Guizhi Gancao Decoction,80 small-scale proof-of-concept clinical trials should be designed incorporating dual endpoints: conventional echocardiographic parameters of diastolic and systolic function, alongside PTM-related biomarkers.
Importantly, the long-term clinical use of TCM not only provides a safety record but also offers a rich repository of empirically validated therapeutic paradigms awaiting mechanistic decoding. This mechanistic reinterpretation transforms traditional empirical knowledge into testable hypotheses, offering immediate translational insights for clinical practice. First, disease-specific PTM signatures can guide patient stratification. Given that HFpEF myocardium exhibits a distinct viscoelastic/PTM profile compared to HFrEF,68 measuring myocardial or circulating levels of dTyr-tubulin, HDAC6 activity, or VASH/SVBP complex abundance could help identify patient populations most likely to benefit from PTM-targeting TCM therapies. Second, these PTM parameters themselves can serve as pharmacodynamic biomarkers to monitor treatment response and optimize dosing regimens in future trials, transforming empirical formula modification into evidence-based precision medicine. Third, the differential involvement of PTMs across distinct CVD etiologies suggests that TCM compound formulations could be tailored to the dominant PTM pathology in each patient, analogous to how modern targeted therapies are guided by molecular subtyping. Thus, the established clinical experience of TCM, combined with recent mechanistic insights into the regulation of PTMs, positions TCM as a promising source for developing novel microtubule-modulating therapies for CVDs.
In addition to clinical translation considerations, the safety of TCM use must also be taken into account. The intrinsic complexity, batch variability, and chemical instability of TCM preparations present additional difficulties when compared to the high dosage sensitivity of microtubule PTMs. Moderate suppression of detyrosination leads to a decrease in viscous resistance within cardiomyocytes; however, excessive stabilization of microtubules correlates with diastolic stiffness, diminished compliance, and electrophysiological irregularities.9,73,109 Therefore, well-defined exposure-response and exposure-toxicity relationships across the continuum from effective exposure to PTM response and finally to functional outcome are essential for the clinical translation of PTM-targeting TCM beyond standard quality control.
It is also important to carefully investigate any potential interactions between TCM and widely used cardiovascular drugs. Certain phytochemicals can affect CYP450 enzymes or P-glycoprotein, altering the pharmacokinetics of β-blockers, anticoagulants, and other cardiovascular drugs, and indirectly affecting microtubule homeostasis. For example, when combined with warfarin, Salvia miltiorrhiza preparations may enhance the anticoagulant effect and increase the risk of bleeding.110 The representative calcium channel blocker nifedipine is primarily metabolized by CYP3A4. Compound formulations containing Glycyrrhiza uralensis may decrease its plasma concentration, as glycyrrhizic acid can induce CYP3A4 activity.111 The shortage of systematic pharmacokinetic, pharmacodynamic, and safety data for combination therapy represents another major limitation.
Overall, translating TCM-based treatment to clinical settings requires extensive pharmacokinetic studies, safety, and standardization of formulations, and robust clinical validation.
Knowledge Gaps and Future Directions
More evaluations are still required to increase our understanding of the underlying mechanisms, regardless of the advancements made in learning about microtubule PTMs in cardiovascular disease. First, each biological effect of PTMs can vary greatly depending on the microenvironment. For example, although acetylation generally improves microtubule mechanical resilience and flexibility,24 it may interfere with sarcomere–microtubule coupling in some pathological conditions.10 Additionally, although detyrosination is closely linked to diastolic dysfunction and cardiac stiffness,96 it physiological functions in preserving baseline mechanical homeostasis are still poorly understood. Therefore, it is essential to compare PTM functions systematically across cell types, disease stages, and mechanical conditions.
Second, the rapid and transient PTM dynamics occurring throughout every cardiac cycle cannot be well captured by existing methods. Microtubule subpopulations cannot be fully resolved by proteomics, and traditional immunostaining lacks the temporal resolution to identify structural alterations at the microsecond scale. Advances in super-resolution imaging and spatial proteomics7 may enable the construction of a comprehensive “spatiotemporal atlas” of microtubule PTMs in the myocardium, providing new insight into their dynamic regulatory roles.
Third, other PTMs, such as glutamylation, glycylation, and phosphorylation, have remained underinvestigated compared to acetylation and detyrosination in cardiovascular biology. Systematic characterization of these underinvestigated modifications has the potential to significantly expand existing models of microtubule regulation.
Fourth, mechanistic research on TCM modulation of microtubule PTMs is still in its infancy. Although cinnamaldehyde77 and Guizhi Gancao Decoction80 provide preliminary data, the multi-component, multi-target feature of TCM formulae restricts mechanistic interpretation. To determine the mechanism through which TCM controls PTMs across molecular and cellular levels, integrated methods integrating network pharmacology, single-cell omics, and in vivo functional validation are required.
Fifth, bridging the gap between the empirical practice of TCM and modern mechanism-based medicine requires the coordinated advancement of clinical research on PTMs. The long-standing clinical experience accumulated with TCM compound formulations should be systematically re-examined through the lens of PTM mechanisms, thereby generating translatable knowledge to guide patient selection, treatment monitoring, and individualized therapy in CVDs.
Future research should target these key areas: (1) Frameworks that compare PTM functions across disease stages and mechanical conditions. (2) High-resolution imaging tools and real-time monitors for PTM studies. (3) Cardiovascular significance of understudied PTMs such as glutamylation. (4) Multi-omics and network pharmacology integration to map PTM networks influenced by TCM formulas. (5) Translating PTM-guided TCM empirical knowledge into clinical practice through clinical research.
Conclusion
Cardiovascular diseases are often linked to problems inside cells, especially with the microtubule network. Post-translational modifications of tubulin, such as acetylation and detyrosination, are key ways to control how this network works. This review connects the key role of PTMs in cardiovascular disease with the potential of traditional Chinese medicine, including its active monomers and compound formulas, to regulate this system. So, it points to a new way to treat diseases by targeting the cytoskeleton’s “modification code.” Studies show that some TCM ingredients can act on key enzymes for PTMs, affecting microtubule stability and function. So, in model systems, they can improve cardiac hypertrophy, reduce vascular inflammation, or stabilize electrical signals. This gives a new molecular way to explain how traditional drugs like Tanshinone IIA and evodiamine work. Also, it shows a possible link between TCM’s holistic approach and changes at the cytoskeleton level.
As a review that brings ideas together, this paper’s main value is in suggesting this cross-disciplinary framework and giving early evidence. But, how deep we can discuss it is limited by what basic research has found so far. There are two main limits. First, most of the mechanisms cited for how TCM regulates PTMs come from scattered, early experimental studies. These results have not yet been fully tested or put together in more complex animal models or disease settings. Second, when discussing TCM compound formulas, because related studies are few and their mechanisms are not deeply explored, we could not detail how different components in a formula work together to regulate the PTM network. So, the discussion still stays at the level of “overall association.”
Future research based on this framework should focus on two things. First, it needs more strict tests to prove the cause-and-effect link between “component – PTM target – functional outcome.” Second, it needs new models and methods to study how TCM multi-component systems work. Doing this could move this promising field from the stage of just having ideas to the stage of truly finding mechanisms. Third, timely translation of PTM-guided TCM insights into clinical practice enables the reinterpretation of long-accumulated empirical knowledge through mechanistic understanding, thereby guiding patient stratification, treatment monitoring, and individualized therapy. Doing so will advance this promising field from the stage of merely having ideas to the stage of truly delivering value.
Declaration of Generative AI and AI-Assisted Technologies in the Writing Process
During the preparation of this manuscript, the authors partially utilized generative artificial intelligence tools, such as ChatGPT and DeepSeek, to refine certain minor sections of the text and improve its overall readability. However, the majority of the work was conducted without any involvement of generative artificial intelligence. After using this tool, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
Abbreviations
AS, Atherosclerosis; αTAT1, α-tubulin acetyltransferase 1; CCP, Cytoplasmic carboxypeptidases; CEP41, Centrosomal Protein 41; CVDs, Cardiovascular diseases; Cx43, Connexin 43; dTyr-tubulin, Detyrosinated microtubules; ECM, Extracellular matrix; GGD, Guizhi Gancao Decoction; HCM, Hypertrophic cardiomyopathy; HDAC6, Histone deacetylase 6; HF, Heart Failure; IL-1β, Interleukin-1β; IRI: Ischemia-reperfusion injury; K40, Lysine 40; LMNA, Lamin A/C; LPS, Lipopolysaccharide; MARK4, Microtubule affinity-regulating kinase 4; MI, Myocardial infarction; MTAs, Microtubule-targeting agents; NOX2, NADPH oxidase 2; NLRP3, Nucleotide-binding oligomerization domain-like receptor protein 3; OS: Oxidative stress; PTMs, Post-translational modifications; ROS, Reactive oxygen species; RyR2, Ryanodine receptor 2; SDH, Succinate dehydrogenase; SIRT2, Sirtuin 2; SOCE, Store-operated calcium entry; SVBP, Serpin vascular biology partner; TCM, Traditional Chinese Medicine; TTL, Tubulin tyrosine ligase; TTLL, Tubulin tyrosine ligase-like; TYHX, Tongyang Huoxue Decoction; ULK1, Unc-51-like autophagy activating kinase 1; VASH, Vasohibin; VSMC, Vascular Smooth Muscle Cell; X‑ROS, Mechanically activated reactive oxygen species; ZSHX, Zishen Huoxue Decoction.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This project was supported by the National Natural Science Foundation of China (grant number 82305092, 82274411), and Hunan Youth Science and Technology Innovation Talent Project (2024RC3199). Excellent Young Scholars Research Fund Project of Hunan University of Chinese Medicine (Z2023XJYQ03).
Disclosure
The authors declare no competing interests.
References
1. McKenna ED, Sarbanes SL, Cummings SW, Roll-Mecak A. The tubulin code, from molecules to health and disease. Annu Rev Cell Dev Biol. 2023;39(1):331–26. doi:10.1146/annurev-cellbio-030123-032748
2. Yu X, Chen X, Amrute-Nayak M, et al. MARK4 controls ischaemic heart failure through microtubule detyrosination. Nature. 2021;594(7864):560–565. doi:10.1038/s41586-021-03573-5
3. Qin YS, Li H, Wang SZ, Wang ZB, Tang CK. Microtubule affinity regulating kinase 4: a promising target in the pathogenesis of atherosclerosis. J Cell Physiol. 2022;237(1):86–97. doi:10.1002/jcp.30530
4. Ouyang C, Li J, Zheng X, et al. Deletion of Ulk1 inhibits neointima formation by enhancing KAT2A/GCN5-mediated acetylation of TUBA/α-tubulin in vivo. Autophagy. 2021;17(12):4305–4322. doi:10.1080/15548627.2021.1911018
5. Joca HC, Coleman AK, Ward CW, Williams GSB. Quantitative tests reveal that microtubules tune the healthy heart but underlie arrhythmias in pathology. J Physiol. 2020;598(7):1327–1338. doi:10.1113/jp277083
6. Janke C, Magiera MM. The tubulin code and its role in controlling microtubule properties and functions. Nat Rev Mol Cell Biol. 2020;21(6):307–326. doi:10.1038/s41580-020-0214-3
7. Liu C, Chen Y, Xie Y, Xiang M. Tubulin post-translational modifications: potential therapeutic approaches to heart failure. Front Cell Dev Biol. 2022;10:872058. doi:10.3389/fcell.2022.872058
8. Nekooki-Machida Y, Hagiwara H. Role of tubulin acetylation in cellular functions and diseases. Med Mol Morphol. 2020;53(4):191–197. doi:10.1007/s00795-020-00260-8
9. Caporizzo MA, Chen CY, Bedi K, Margulies KB, Prosser BL. Microtubules increase diastolic stiffness in failing human cardiomyocytes and myocardium. Circulation. 2020;141(11):902–915. doi:10.1161/circulationaha.119.043930
10. Liu Q-Y, Zhuang Y, Song X-R, et al. Tanshinone IIA prevents LPS-induced inflammatory responses in mice via inactivation of succinate dehydrogenase in macrophages. Acta Pharmacol Sin. 2021;42(6):987–997. doi:10.1038/s41401-020-00535-x
11. Zhang D, Qiao W, Zhao Y, Fang H, Xu D, Xia Q. Curdione attenuates thrombin-induced human platelet activation: β1-tubulin as a potential therapeutic target. Fitoterapia. 2017;116:106–115. doi:10.1016/j.fitote.2016.11.016
12. Scarborough EA, Uchida K, Vogel M, et al. Microtubules orchestrate local translation to enable cardiac growth. Nat Commun. 2021;12(1):1547. doi:10.1038/s41467-021-21685-4
13. Uchida K, Scarborough EA, Prosser BL. Cardiomyocyte microtubules: control of mechanics, transport, and remodeling. Annu Rev Physiol. 2022;84:257–283. doi:10.1146/annurev-physiol-062421-040656
14. Gadadhar S, Bodakuntla S, Natarajan K, Janke C. The tubulin code at a glance. J Cell Sci. 2017;130(8):1347–1353. doi:10.1242/jcs.199471
15. Roll-Mecak A. The tubulin code in microtubule dynamics and information encoding. Dev Cell. 2020;54(1):7–20. doi:10.1016/j.devcel.2020.06.008
16. Caporizzo MA, Prosser BL. The microtubule cytoskeleton in cardiac mechanics and heart failure. Nat Rev Cardiol. 2022;19(6):364–378. doi:10.1038/s41569-022-00692-y
17. Caporizzo MA, Chen CY, Prosser BL. Cardiac microtubules in health and heart disease. Exp Biol Med. 2019;244(15):1255–1272. doi:10.1177/1535370219868960
18. Wloga D, Joachimiak E, Fabczak H. Tubulin post-translational modifications and microtubule dynamics. Int J Mol Sci. 2017;18(10). doi:10.3390/ijms18102207
19. Pulya S, Amin SA, Adhikari N, Biswas S, Jha T, Ghosh B. HDAC6 as privileged target in drug discovery: a perspective. Pharmacol Res. 2021;163:105274. doi:10.1016/j.phrs.2020.105274
20. Szczesna E, Zehr EA, Cummings SW, et al. Combinatorial and antagonistic effects of tubulin glutamylation and glycylation on katanin microtubule severing. Dev Cell. 2022;57(21):2497–2513.e6. doi:10.1016/j.devcel.2022.10.003
21. Fan Z, Peng W, Wang Z, Zhang L, Liu K. Identification of biomarkers associated with metabolic cardiovascular disease using mRNA-SNP-miRNA regulatory network analysis. BMC Cardiovasc Disord. 2021;21(1):351. doi:10.1186/s12872-021-02166-4
22. Djenoune L, Berg K, Brueckner M, Yuan S. A change of heart: new roles for cilia in cardiac development and disease. Nat Rev Cardiol. 2022;19(4):211–227. doi:10.1038/s41569-021-00635-z
23. Delgado ILS, Carmona B, Nolasco S, Marques R, Gonçalves J, Soares H. Tubulin acetylation: a critical regulator of microtubule function. Results Probl Cell Differ. 2025;75:91–140. doi:10.1007/978-3-031-91459-1_4
24. Lu YM. Reinterpreting the effects of α-tubulin K40 acetylation on microtubule stability and cellular functions. J Cell Sci. 2025;138(14). doi:10.1242/jcs.263431
25. Friedmann DR, Aguilar A, Fan J, Nachury MV, Marmorstein R. Structure of the α-tubulin acetyltransferase, αTAT1, and implications for tubulin-specific acetylation. Proc Natl Acad Sci U S A. 2012;109(48):19655–19660. doi:10.1073/pnas.1209357109
26. Bergmann L, Lang A, Bross C, et al. Subcellular localization and mitotic interactome analyses identify sirt4 as a centrosomally localized and microtubule associated protein. Cells. 2020;9(9). doi:10.3390/cells9091950
27. Valderrama F, Durán JM, Babià T, Barth H, Renau-Piqueras J, Egea G. Actin microfilaments facilitate the retrograde transport from the Golgi complex to the endoplasmic reticulum in mammalian cells. Traffic. 2001;2(10):717–726. doi:10.1034/j.1600-0854.2001.21006.x
28. Yoshimura A, Miserey-Lenkei S, Coudrier E, Goud B. Branched actin maintains acetylated microtubule network in the early secretory pathway. Cells. 2021;11(1). doi:10.3390/cells11010015
29. Moise K, Arun KM, Pillai M, et al. Endothelial cell elongation and alignment in response to shear stress requires acetylation of microtubules. Front Physiol. 2024;15:1425620. doi:10.3389/fphys.2024.1425620
30. Nieuwenhuis J, Brummelkamp TR. The tubulin detyrosination cycle: function and enzymes. Trends Cell Biol. 2019;29(1):80–92. doi:10.1016/j.tcb.2018.08.003
31. Bak J, Brummelkamp TR, Perrakis A. Decoding microtubule detyrosination: enzyme families, structures, and functional implications. FEBS Lett. 2024;598(12):1453–1464. doi:10.1002/1873-3468.14940
32. Girão H, Macário-Monteiro J, Figueiredo AC, et al. α-tubulin detyrosination fine-tunes kinetochore-microtubule attachments. Nat Commun. 2024;15(1):9720. doi:10.1038/s41467-024-54155-8
33. Wang N, Bosc C, Ryul Choi S, et al. Structural basis of tubulin detyrosination by the vasohibin-SVBP enzyme complex. Nat Struct Mol Biol. 2019;26(7):571–582. doi:10.1038/s41594-019-0241-y
34. Nieuwenhuis J, Adamopoulos A, Bleijerveld OB, et al. Vasohibins encode tubulin detyrosinating activity. Science. 2017;358(6369):1453–1456. doi:10.1126/science.aao5676
35. Sanyal C, Pietsch N, Ramirez Rios S, Peris L, Carrier L, Moutin MJ. The detyrosination/re-tyrosination cycle of tubulin and its role and dysfunction in neurons and cardiomyocytes. Semin Cell Dev Biol. 2023;137:46–62. doi:10.1016/j.semcdb.2021.12.006
36. Schuldt M, Pei J, Harakalova M, et al. Proteomic and functional studies reveal detyrosinated tubulin as treatment target in sarcomere mutation-induced hypertrophic cardiomyopathy. Circ Heart Fail. 2021;14(1):e007022. doi:10.1161/circheartfailure.120.007022
37. Ruse CI, Chin HG, Pradhan S. Polyglutamylation: biology and analysis. Amino Acids. 2022;54(4):529–542. doi:10.1007/s00726-022-03146-4
38. Mahalingan KK, Grotjahn DA, Li Y, Lander GC, Zehr EA, Roll-Mecak A. Structural basis for α-tubulin-specific and modification state-dependent glutamylation. Nat Chem Biol. 2024;20(11):1493–1504. doi:10.1038/s41589-024-01599-0
39. Ping Y, Ohata K, Kikushima K, et al. Tubulin polyglutamylation by TTLL1 and TTLL7 regulate glutamate concentration in the mice brain. Biomolecules. 2023;13(5). doi:10.3390/biom13050784
40. Zhang X, Li X, Chen W, et al. The distinct initiation sites and processing activities of TTLL4 and TTLL7 in glutamylation of brain tubulin. J Biol Chem. 2023;299(7):104923. doi:10.1016/j.jbc.2023.104923
41. Janke C, Rogowski K, Wloga D, et al. Tubulin polyglutamylase enzymes are members of the TTL domain protein family. Science. 2005;308(5729):1758–1762. doi:10.1126/science.1113010
42. Vemu A, Szczesna E, Roll-Mecak A. In vitro reconstitution assays of microtubule amplification and lattice repair by the microtubule-severing enzymes katanin and spastin. Methods Mol Biol. 2020;2101:27–38. doi:10.1007/978-1-0716-0219-5_3
43. Bayless KJ, Johnson GA. Role of the cytoskeleton in formation and maintenance of angiogenic sprouts. J Vasc Res. 2011;48(5):369–385. doi:10.1159/000324751
44. Ki SM, Kim JH, Won SY, et al. CEP41-mediated ciliary tubulin glutamylation drives angiogenesis through AURKA-dependent deciliation. EMBO Rep. 2020;21(2):e48290. doi:10.15252/embr.201948290
45. Wloga D, Webster DM, Rogowski K, et al. TTLL3 Is a tubulin glycine ligase that regulates the assembly of cilia. Dev Cell. 2009;16(6):867–876. doi:10.1016/j.devcel.2009.04.008
46. Rogowski K, Juge F, van Dijk J, et al. Evolutionary divergence of enzymatic mechanisms for posttranslational polyglycylation. Cell. 2009;137(6):1076–1087. doi:10.1016/j.cell.2009.05.020
47. Gadadhar S, Dadi H, Bodakuntla S, et al. Tubulin glycylation controls primary cilia length. J Cell Biol. 2017;216(9):2701–2713. doi:10.1083/jcb.201612050
48. Kubo T, Sasaki R, Oda T. Tubulin glycylation controls ciliary motility through modulation of outer-arm dyneins. Mol Biol Cell. 2024;35(7):ar90. doi:10.1091/mbc.E24-04-0154
49. Gadadhar S, Alvarez Viar G, Hansen JN, et al. Tubulin glycylation controls axonemal dynein activity, flagellar beat, and male fertility. Science. 2021;371(6525). doi:10.1126/science.abd4914
50. Gabriel GC, Young CB, Lo CW. Role of cilia in the pathogenesis of congenital heart disease. Semin Cell Dev Biol. 2021;110:2–10. doi:10.1016/j.semcdb.2020.04.017
51. Yang Y, Ren K, Shi X, Luan Y. Primary cilia orchestrate cardiac pathogenesis: a central nexus of remodeling, signaling, and repair. Cell Prolif. 2025;58(10):e70113. doi:10.1111/cpr.70113
52. Pigino G. Intraflagellar transport. Curr Biol. 2021;31(10):R530–r536. doi:10.1016/j.cub.2021.03.081
53. Thygesen K, Alpert JS, Jaffe AS, et al. Fourth universal definition of myocardial infarction (2018). J Am Coll Cardiol. 2018;72(18):2231–2264. doi:10.1016/j.jacc.2018.08.1038
54. Kuznetsov AV, Javadov S, Grimm M, Margreiter R, Ausserlechner MJ, Hagenbuchner J. Crosstalk between mitochondria and cytoskeleton in cardiac cells. Cells. 2020;9(1). doi:10.3390/cells9010222
55. Morris RL, Hollenbeck PJ. Axonal transport of mitochondria along microtubules and F-actin in living vertebrate neurons. J Cell Biol. 1995;131(5):1315–1326. doi:10.1083/jcb.131.5.1315
56. Vale RD, Funatsu T, Pierce DW, Romberg L, Harada Y, Yanagida T. Direct observation of single kinesin molecules moving along microtubules. Nature. 1996;380(6573):451–453. doi:10.1038/380451a0
57. Borbolis F, Kteniadaki M, Palikaras K. MEC-12/alpha tubulin regulates mitochondrial distribution and mitophagy during oxidative stress in C. elegans. MicroPubl Biol. 2024. doi:10.17912/micropub.biology.001232
58. Bagur R, Tanguy S, Foriel S, et al. The impact of cardiac ischemia/reperfusion on the mitochondria-cytoskeleton interactions. Biochim Biophys Acta. 2016;1862(6):1159–1171. doi:10.1016/j.bbadis.2016.03.009
59. Fan J, Watanabe T. Atherosclerosis: known and unknown. Pathol Int. 2022;72(3):151–160. doi:10.1111/pin.13202
60. Liaqat A, Asad M, Shoukat F, Khan AU. A spotlight on the underlying activation mechanisms of the nlrp3 inflammasome and its role in atherosclerosis: a review. Inflammation. 2020;43(6):2011–2020. doi:10.1007/s10753-020-01290-1
61. Wang L, Pu W, Wang C, Lei L, Li H. Microtubule affinity regulating kinase 4 promoted activation of the NLRP3 inflammasome-mediated pyroptosis in periodontitis. J Oral Microbiol. 2022;14(1):2015130. doi:10.1080/20002297.2021.2015130
62. Li P, Ge J, Li H. Lysine acetyltransferases and lysine deacetylases as targets for cardiovascular disease. Nat Rev Cardiol. 2020;17(2):96–115. doi:10.1038/s41569-019-0235-9
63. Le Dour C, Chatzifrangkeskou M, Macquart C, et al. Actin-microtubule cytoskeletal interplay mediated by MRTF-A/SRF signaling promotes dilated cardiomyopathy caused by LMNA mutations. Nat Commun. 2022;13(1):7886. doi:10.1038/s41467-022-35639-x
64. Medvedev RY, Afolabi SO, Turner DGP, Glukhov AV. Mechanisms of stretch-induced electro-anatomical remodeling and atrial arrhythmogenesis. J Mol Cell Cardiol. 2024;193:11–24. doi:10.1016/j.yjmcc.2024.05.011
65. Limbu S, Prosser BL, Lederer WJ, Ward CW, Jafri MS. X-ROS signaling depends on length-dependent calcium buffering by troponin. Cells. 2021;10(5). doi:10.3390/cells10051189
66. Yan J, Killingsworth C, Walcott G, et al. Molecular remodeling of Cx43, but not structural remodeling, promotes arrhythmias in an arrhythmogenic canine model of nonischemic heart failure. J Mol Cell Cardiol. 2021;158:72–81. doi:10.1016/j.yjmcc.2021.05.012
67. Shimizu I, Minamino T. Physiological and pathological cardiac hypertrophy. J Mol Cell Cardiol. 2016;97:245–262. doi:10.1016/j.yjmcc.2016.06.001
68. Schulz L, Werner S, Böttner J, et al. Tubulin expression and modification in heart failure with preserved ejection fraction (HFpEF). Sci Rep. 2022;12(1):15734. doi:10.1038/s41598-022-19766-5
69. Algül S, Dorsch LM, Sorop O, et al. The microtubule signature in cardiac disease: etiology, disease stage, and age dependency. J Comp Physiol B. 2023;193(5):581–595. doi:10.1007/s00360-023-01509-1
70. Pietsch N, Chen CY, Kupsch S, et al. Chronic activation of tubulin tyrosination in HCM mice and human iPSC-engineered heart tissues improves heart function. bioRxiv. 2024. doi:10.1101/2023.05.25.542365
71. Pietsch N, Chen CY, Kupsch S, et al. Chronic activation of tubulin tyrosination improves heart function. Circ Res. 2024;135(9):910–932. doi:10.1161/circresaha.124.324387
72. Fragin J, Stephens M. An update on heart failure: new definitions and treatment. Prim Care. 2024;51(1):171–178. doi:10.1016/j.pop.2023.08.004
73. Chen CY, Caporizzo MA, Bedi K, et al. Suppression of detyrosinated microtubules improves cardiomyocyte function in human heart failure. Nat Med. 2018;24(8):1225–1233. doi:10.1038/s41591-018-0046-2
74. Sheibani M, Zamani N, Gerami AH, Akhondi H, Hassanian-Moghaddam HC. Laboratory, and electrocardiographic findings in colchicine toxicity: 10 years of experience. Front Med. 2022;9:872528. doi:10.3389/fmed.2022.872528
75. Al-Mahayri ZN, AlAhmad MM, Ali BR. Current opinion on the pharmacogenomics of paclitaxel-induced toxicity. Expert Opin Drug Metab Toxicol. 2021;17(7):785–801. doi:10.1080/17425255.2021.1943358
76. Li XA, Ho YS, Chen L, Hsiao WL. The protective effects of icariin against the homocysteine-induced neurotoxicity in the primary embryonic cultures of rat cortical neurons. Molecules. 2016;21(11). doi:10.3390/molecules21111557
77. Tian J, Shan XL, Wang SN, et al. Trans-cinnamaldehyde suppresses microtubule detyrosination and alleviates cardiac hypertrophy. Eur J Pharmacol. 2022;914:174687. doi:10.1016/j.ejphar.2021.174687
78. Wu Q, Wang Y, Liu J, et al. Microtubules and cardiovascular diseases: insights into pathology and therapeutic strategies. Int J Biochem Cell Biol. 2024;175:106650. doi:10.1016/j.biocel.2024.106650
79. Chang X, Zhou S, Huang Y, et al. Zishen Huoxue decoction (ZSHX) alleviates ischemic myocardial injury (MI) via Sirt5-β-tubulin mediated synergistic mechanism of “mitophagy-unfolded protein response” and mitophagy. Chin J Nat Med. 2025;23(3):311–321. doi:10.1016/s1875-5364(25)60838-7
80. Chen HH, Zhao P, Tian J, et al. The effects of guizhi gancao decoction on pressure overload-induced heart failure and posttranslational modifications of tubulin in mice. Evid Based Complement Alternat Med. 2017;2017:2915247. doi:10.1155/2017/2915247
81. Zhang X, Zhou Y, Chang X, Wu Q, Liu Z, Liu R. Tongyang Huoxue decoction (TYHX) ameliorating hypoxia/reoxygenation-induced disequilibrium of calcium homeostasis via regulating β-tubulin in rabbit sinoatrial node cells. J Ethnopharmacol. 2024;318(Pt B):117006. doi:10.1016/j.jep.2023.117006
82. Li CG, Zeng QZ, Chen MY, et al. Evodiamine Augments NLRP3 inflammasome activation and anti-bacterial responses through inducing α-tubulin acetylation. Front Pharmacol. 2019;10:290. doi:10.3389/fphar.2019.00290
83. Zha Y, Yang Y, Zhou Y, Ye B, Li H, Liang J. Dietary evodiamine inhibits atherosclerosis-associated changes in vascular smooth muscle cells. Int J Mol Sci. 2023;24(7). doi:10.3390/ijms24076653
84. Lian B, Zeng R, Chen Y, Liao P, Guo L, Zhang M. Sodium Tanshinone IIA sulfonate for acute myocardial infarction: a systematic review and Meta-analysis. J Tradit Chin Med. 2021;41(1):26–35. doi:10.19852/j.cnki.jtcm.2021.01.004
85. Mao S, Wang L, Zhao X, et al. Efficacy of sodium tanshinone iia sulfonate in patients with non-st elevation acute coronary syndrome undergoing percutaneous coronary intervention: results from a multicentre, controlled, randomized trial. Cardiovasc Drugs Ther. 2021;35(2):321–329. doi:10.1007/s10557-020-07077-8
86. Hotta T, Haynes SE, Blasius TL, et al. Parthenolide destabilizes microtubules by covalently modifying tubulin. Curr Biol. 2021;31(4):900–907.e6. doi:10.1016/j.cub.2020.11.055
87. Sun Q, Xie L, Song J, Li X. Evodiamine: a review of its pharmacology, toxicity, pharmacokinetics and preparation researches. J Ethnopharmacol. 2020;262:113164. doi:10.1016/j.jep.2020.113164
88. Zhou Z, Zhou Y, Zhang Z, et al. Progress on the effects and underlying mechanisms of evodiamine in digestive system diseases, and its toxicity: a systematic review and meta-analysis. Phytomedicine. 2024;132:155851. doi:10.1016/j.phymed.2024.155851
89. Panda M, Tripathi SK, Zengin G, Biswal BK. Evodiamine as an anticancer agent: a comprehensive review on its therapeutic application, pharmacokinetic, toxicity, and metabolism in various cancers. Cell Biol Toxicol. 2023;39(1):1–31. doi:10.1007/s10565-022-09772-8
90. Ding W, Ding Z, Wang Y, et al. Evodiamine attenuates experimental colitis injury via activating autophagy and inhibiting nlrp3 inflammasome assembly. Front Pharmacol. 2020;11:573870. doi:10.3389/fphar.2020.573870
91. Guo R, Li L, Su J, et al. Pharmacological activity and mechanism of tanshinone iia in related diseases. Drug Des Devel Ther. 2020;14:4735–4748. doi:10.2147/dddt.S266911
92. Ansari MA, Khan FB, Safdari HA, et al. Prospective therapeutic potential of Tanshinone IIA: an updated overview. Pharmacol Res. 2021;164:105364. doi:10.1016/j.phrs.2020.105364
93. Ishida K, Nagatake T, Saika A, et al. Induction of unique macrophage subset by simultaneous stimulation with LPS and IL-4. Front Immunol. 2023;14:1111729. doi:10.3389/fimmu.2023.1111729
94. Mills EL, Kelly B, Logan A, et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell. 2016;167(2):457–470.e13. doi:10.1016/j.cell.2016.08.064
95. Elliott EI, Sutterwala FS. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol Rev. 2015;265(1):35–52. doi:10.1111/imr.12286
96. Misawa T, Takahama M, Kozaki T, et al. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol. 2013;14(5):454–460. doi:10.1038/ni.2550
97. Freund RRA, Gobrecht P, Fischer D, Arndt HD. Advances in chemistry and bioactivity of parthenolide. Nat Prod Rep. 2020;37(4):541–565. doi:10.1039/c9np00049f
98. Wang M, Li Q. Parthenolide could become a promising and stable drug with anti-inflammatory effects. Nat Prod Res. 2015;29(12):1092–1101. doi:10.1080/14786419.2014.981541
99. Li F, Hu Y, Qi S, Luo X, Yu H. Structural basis of tubulin detyrosination by vasohibins. Nat Struct Mol Biol. 2019;26(7):583–591. doi:10.1038/s41594-019-0242-x
100. Yao Y, Song L, Zuo Z, et al. Parthenolide attenuates hypoxia-induced pulmonary hypertension through inhibiting STAT3 signaling. Phytomedicine. 2024;134:155976. doi:10.1016/j.phymed.2024.155976
101. Mathema VB, Koh Y-S, Thakuri BC, Sillanpää M. Parthenolide, a sesquiterpene lactone, expresses multiple anti-cancer and anti-inflammatory activities. Inflammation. 2012;35(2):560–565. doi:10.1007/s10753-011-9346-0
102. Zhang C, Fan L, Fan S, et al. Cinnamomum cassia Presl: a review of its traditional uses, phytochemistry, pharmacology and toxicology. Molecules. 2019;24(19). doi:10.3390/molecules24193473
103. Han R, Li X, Gao X, Lv GC. Cinnamaldehyde: pharmacokinetics, anticancer properties and therapeutic potential (Review). Mol Med Rep. 2024;30(3). doi:10.3892/mmr.2024.13287
104. Parsamanesh N, Asghari A, Sardari S, et al. Resveratrol and endothelial function: a literature review. Pharmacol Res. 2021;170:105725. doi:10.1016/j.phrs.2021.105725
105. Ding X, Zhu C, Wang W, Li M, Ma C, Gao B. SIRT1 is a regulator of autophagy: implications for the progression and treatment of myocardial ischemia-reperfusion. Pharmacol Res. 2024;199:106957. doi:10.1016/j.phrs.2023.106957
106. Hosoda R, Nakashima R, Yano M, et al. Resveratrol, a SIRT1 activator, attenuates aging-associated alterations in skeletal muscle and heart in mice. J Pharmacol Sci. 2023;152(2):112–122. doi:10.1016/j.jphs.2023.04.001
107. Khan M, Giessrigl B, Vonach C, et al. Berberine and a Berberis lycium extract inactivate Cdc25A and induce alpha-tubulin acetylation that correlate with HL-60 cell cycle inhibition and apoptosis. Mutat Res. 2010;683(1–2):123–130. doi:10.1016/j.mrfmmm.2009.11.001
108. Wei YF, Lv FR, Yao J, et al. Clinical observation of modified zhigancao tang in treating patients with liver and kidney deficiency of parkinson’s disease and its effect on neuronal signal-related proteins. Chin J Exp Traditional Med Formulae. 2025;31(4):166–173. doi:10.13422/j.cnki.syfjx.20241525
109. Salomon AK, Phyo SA, Okami N, et al. Desmin intermediate filaments and tubulin detyrosination stabilize growing microtubules in the cardiomyocyte. Basic Res Cardiol. 2022;117(1):53. doi:10.1007/s00395-022-00962-3
110. Chan TY. Interaction between warfarin and danshen (Salvia miltiorrhiza). Ann Pharmacother. 2001;35(4):501–504. doi:10.1345/aph.19029
111. Tu JH, He YJ, Chen Y, et al. Effect of glycyrrhizin on the activity of CYP3A enzyme in humans. Eur J Clin Pharmacol. 2010;66(8):805–810. doi:10.1007/s00228-010-0814-5
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.