")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 14
Triplet CFTR modulators: future prospects for treatment of cystic fibrosis
Authors Chaudary N
Received 27 June 2018
Accepted for publication 12 October 2018
Published 6 December 2018 Volume 2018:14 Pages 2375—2383
DOI https://doi.org/10.2147/TCRM.S147164
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Nauman Chaudary
Division of Pulmonary Disease and Critical Care Medicine, Department of Medicine, Virginia Commonwealth University, Richmond, VA, USA
Abstract: Cystic fibrosis (CF) is an autosomal recessive genetic disease characterized by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR). CFTR is a chloride channel responsible for ion flow across epithelial surfaces of lung, sinuses, pancreas, intestine, and liver. Researchers have grouped CFTR genetic mutations into various protein defects: reduced protein synthesis (class 1 mutations), abnormal protein folding and maturation (class 2 mutation), and abnormal gating (class 3 mutation). These mutations usually present as severe forms of CF due to complete absence of CFTR at cell surfaces. Milder forms (eg, protein maturation and conductance defects, classes 4–6) present as less severe forms of CF related to the presence of CFTR at the cell surface. Differences in severity are directly due to CFTR function which is based on the severity of CFTR mutation. This knowledge has proven useful for designing therapy for individual mutations and mutation classes. The discovery and US Food and Drug Administration approval of Kalydeco® (ivacaftor) in early 2011 marked the beginning of a new era of therapies that are focused on improving defective CFTR protein function. However, due to its specificity for the G551D mutation, ivacaftor only benefits ~5% of CF patients. Approximately 50% of CF patients have two copies of the F508Del mutation, while other CF patients carry only one copy of this gene. More recently, Orkambi®, a two compound medication composed of lumacaftor and ivacaftor, has provided the foundation necessary to further build on molecular concepts of: correction of trafficking, potentiation, and amplification of defective CFTR. These new concepts will form the basis of future CF therapies and extend CFTR treatment to almost 50% of CF patients. Evolving knowledge of the molecular mechanisms responsible for defective CFTR has prompted new research focused on “repair” of each phase of CFTR expression and function, thus creating a new class of combination “CFTR correctors” referred to as “triplet CFTR compounds.” This article will review how patients can be selected and treated with these newer agents that are based on specific mutations. In the future, many CF practitioners have expectations that initiation of treatment for CF patients will occur simply by use of biomarkers of CFTR expression (eg, sweat chloride, nasal potential difference, rectal organoids) rather than testing for specific mutations. As continued research identifies biomarkers with greater specificity and which predict clinical response, therapies can potentially be tailored to individual responses.
Keywords: adults, cystic fibrosis, treatment, CFTR modulators
Introduction
Discovery of CFTR modulators
Basic knowledge of how cystic fibrosis transmembrane conductance regulator (CFTR) is expressed and displayed on cell surfaces arose from extensive in vitro research and has enabled discovery of compounds called CFTR modulators (Figure 1).1 Detailed studies of CFTR involved development of detailed knowledge of the molecular structure of CFTR and new thinking on ways to restore function of the protein.6–9 There are two class of CFTR compounds which have received US Food and Drug Administration (FDA) approval: the so-called CFTR potentiators which increase activity of CFTR on epithelial surfaces and CFTR corrector which improves defective protein processing and trafficking. These agents (potentiators and correctors) are usually combined in treating patients with responsive mutations.
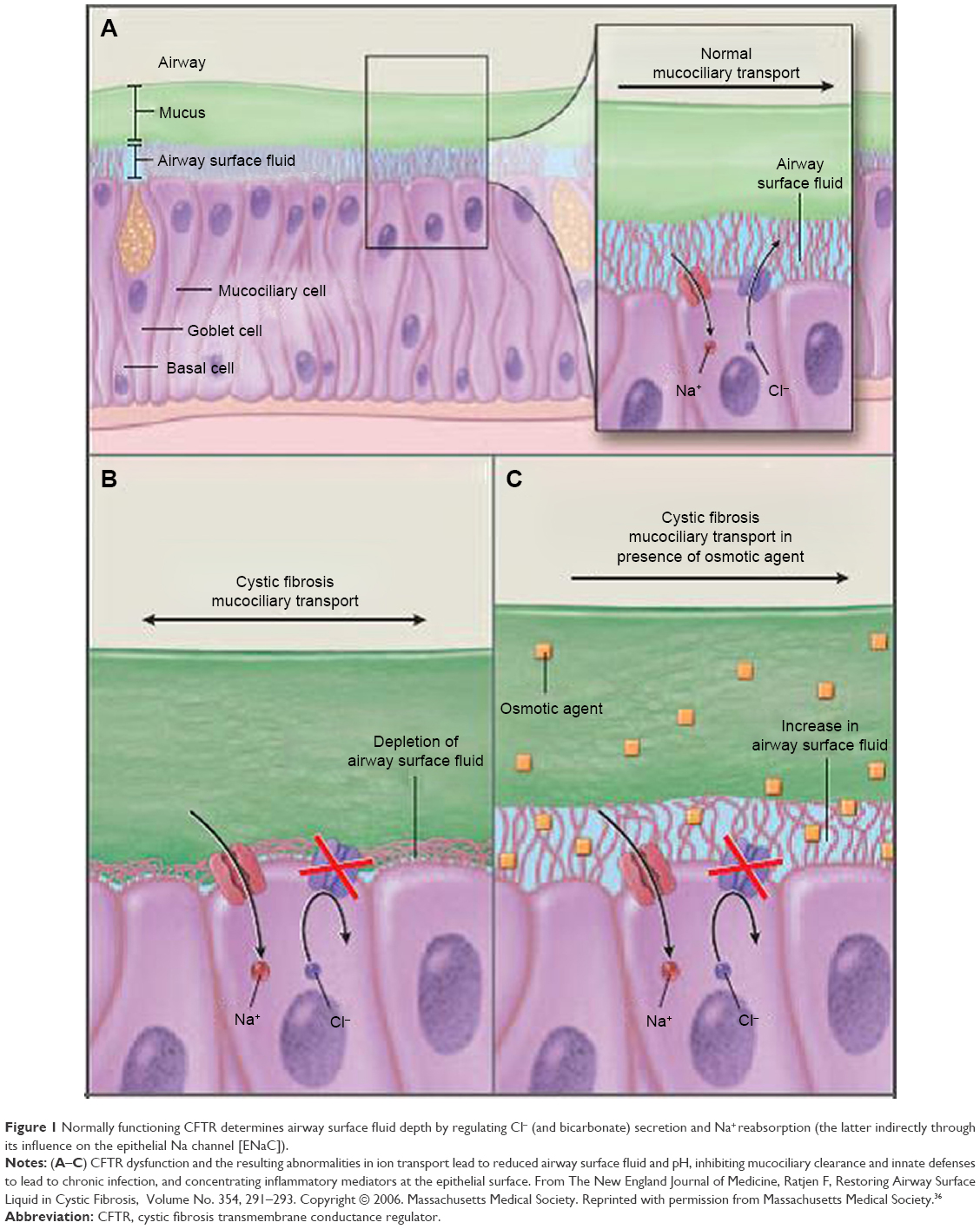 | Figure 1 Normally functioning CFTR determines airway surface fluid depth by regulating Cl− (and bicarbonate) secretion and Na+ reabsorption (the latter indirectly through its influence on the epithelial Na channel [ENaC]). |
Cystic fibrosis (CF) is the most common lethal autosomal recessive disorder in Caucasians, resulting in clinical symptoms in multiple body systems (ie, lung, sinus, gastrointestinal and reproductive tracts). Conventional therapy for CF involves monitoring pulmonary function, nutrition, airway clearance, and infection control. New oral CF treatments are providing physicians with the ability to target underlying CFTR defects which are present in specific CFTR mutations. Kalydeco® (ivacaftor, IVA; Vertex Pharmaceuticals, Boston, MA, USA) was the first agent “potentiator” that activates defective CFTR at the cell surface. IVA’s primary target is mutated CFTR protein in which glycine is replaced by aspartic acid at position 551 (G551D). This mutation disrupts chloride channel gating. IVA enabled a first step at the level of epithelial cells where there is improved ion channel flow. However, another mutation, Delta-F508 (ie, CFTRΔF508, F508del), results in a deletion of three nucleotides that span positions 507 and 508 of the CFTR gene on chromosome 7. The mutation results in loss of a single codon for amino acid phenylalanine and leads to abnormal folding and gating of the chloride channel on epithelial cell surfaces (Figure 2). Complete correction of this defect necessitates CFTR protein modification employing further molecular alterations resulting in “two-step therapy.” Orkambi® (Vertex Pharmaceuticals, Boston, MA, USA) an agent that contains two compounds (lumacaftor, IVA) has come into widespread use. Combining these two agents increases CFTR protein deposition on cell surfaces with the agents acting as molecular chaperones during protein folding, subsequently resulting in increased numbers of CFTR proteins that are trafficked to epithelial cell surfaces.1 Additional molecular correcting agents as well as molecules that will improve functionality and stability of CFTR at cell surface are currently under development. Most recently, in patients with homozygous F508Del, tezacaftor (TEZ), an agent that purportedly moves CFTR protein to its correct position on cell surfaces when combined with IVA (Symdeko®; Vertex Pharmaceuticals, Boston, MA, USA), was found to improve CF pulmonary function and reduce pulmonary exacerbation frequency. Patients heterozygous for F508del historically are difficult to correct with significant CFTR defects, occurring in folding, opening, and stability on cell membranes. Clinical trials are ongoing to study next-generation correctors where IVA is molecularly bound with TEZ/IVA. When this combined drug is used, functionality of CFTR arising from the F508del mutation is significantly improved. Clinical trials report promising results for both F508del homozygous and F508del heterozygous individuals with minimal residual function mutations.
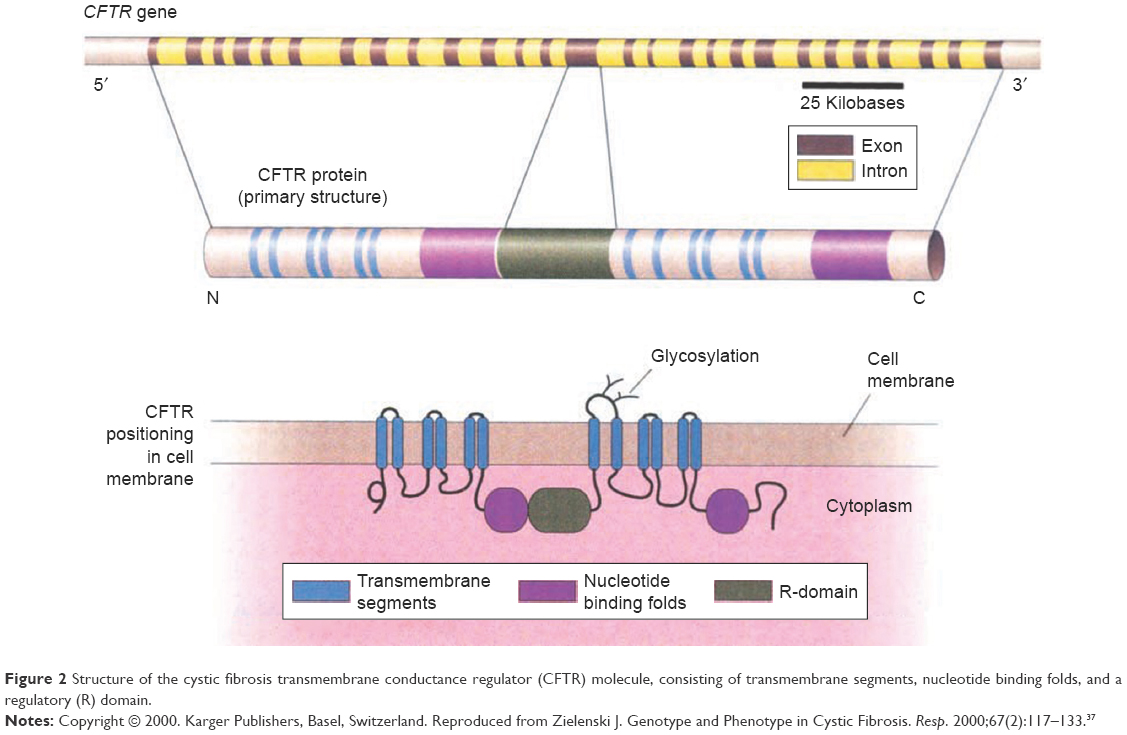 | Figure 2 Structure of the cystic fibrosis transmembrane conductance regulator (CFTR) molecule, consisting of transmembrane segments, nucleotide binding folds, and a regulatory (R) domain. |
Initial research as well as molecular modeling prompted belief by many CF physicians and researchers that mutations fitting into specific classes respond to all pharmacological CFTR compounds in similar fashions. However, when these agents were employed in clinical trials as well as post-FDA approval, results fell short of expectations.3 CFTR compounds can be tailored to individuals based on their cell culture response arrived at by in vitro laboratory testing using a process called “theratyping.” In the coming years, hopes are that theratyping will facilitate patient-specific treatments that are based on novel biomarkers. This review will provide a focused review of current federal FDA-approved CFTR modulator therapies and research which extend CFTR therapy to the larger CF population.
Patient selection and future directions in CFTR modification
There are numerous therapies currently being advanced for CFTR modification. Clinicians treating CF will increasingly encounter opportunities for evaluation of patients for “CFTR modification therapy” and be able to monitor short-term and long-term response. In use of these agents, physicians must carefully monitor consequences such as drug interactions, liver function alteration, and cataract formation in younger patients. Furthermore, chest tightness is a recognized common adverse reaction, especially in patients with severe lung dysfunction.
Clinical development of new CFTR modulators for treatment of CF will continue and will be extended to newer patient populations, including those with less firmly established disease. Expectations are that these therapies will be extended to dysfunctional CFTR arising from other gene mutations. Several next-generation F508Del corrector agents are in early phase development where the focus is suppression of premature stop codons as well molecular amplifiers designed to further increase quantities of available CFTR protein substrate thus enabling subsequent modification by other drugs.11,12,17–20
Advances in molecular knowledge are essential to the understanding of “theratyping.” In theratyping, an important goal is the acquisition of important new molecular knowledge that will enable understanding of other CFTR gene mutations not specifically approved currently for treatment. As knowledge of new mutations becomes available, theratyping will advance treatment of CF patient populations previously untreatable.
Availability of certain CF disease cell lines and research investigating newly discovered disease-specific biomarkers that arise from this work will lead to federal FDA approval of new CFTR drugs. Biomarkers derived from certain cellular sources (ie, bronchial epithelium, rectal organelles, nasal brushings, sweat chloride, and nasal potential difference studies) are driving this research. Based upon prior knowledge derived from years of intensive molecular research, IVA/lumacaftor therapy was extended to 23 additional “responsive mutations” without the FDA requiring additional clinical trials.25–29 The FDA arrived at this conclusion from further important research. When bronchial epithelial cells which possessed “non-G551D mutations” were exposed to IVA/lumacaftor in vitro, CFTR responses were found to be similar to those observed in cells which possessed the G551D mutation that arose from the original research.4,5
F508Del homozygous patients
Orkambi is FDA approved for use in F508Del homozygous patients. The corrector molecule component of Orkambi (ie, lumacaftor) improves and stabilizes interactions between membrane portions of CFTR and a nucleotide blocking domain (NBD1). This domain suppresses CFTR protein folding defects which occur in this particular CFTR mutation. This knowledge has provided the insight that additional CFTR correction can be accomplished by addition of a second corrector. The rationale for using the lumacaftor/IVA combination is based on their complimentary mechanisms of action (ie, corrector and potentiator). In this two-step process, correction of CFTR protein folding and channel opening is required for treatment of patients with F508DEL. With the use of lumacaftor/IVA, clinicians caring for patients with the mutation may see up to a 3% absolute increase in FEV1% and a remarkable reduction in sweat chloride values. From a patient’s perspective, a more pronounced effect of lumacaftor/ivacaftor combination is on disease stability (ie, reduced pulmonary exacerbations).
The safety and efficacy of lumacaftor/IVA were evaluated in a Phase III clinical trial. The TRAFFIC clinical trial (NCT01807923) was a randomized, double-blind, placebo-controlled study (RCT) in 559 CF patients who were homozygous for F508Del mutation. The study duration was 24 weeks. The primary end point was a change in lung function. The TRANSPORT clinical trial (NCT01807949) was another RCT in 563 patients with CF, homozygous for the F508Del CFTR mutation. The study duration was 24 weeks and the primary end point was change in lung function. Based on an analysis of pooled study data, patients who received combination therapy showed statistically significant improvements in lung function, as well as weight gain and reductions in pulmonary exacerbations (all end points compared to placebo). Use of intravenous antibiotics and hospitalizations were reduced. Patients gained weight over time with improved body mass index (BMI). Adverse events in the lumacaftor/IVA combination were generally respiratory in nature with dyspnea and chest tightness. Elevations in levels of hepatic transaminases to greater than three times the upper limit of normal were observed in 5.2% of placebo patients and 5.2% of lumacaftor/IVA treated patients. As a result of these findings, monitoring of liver function is advised for the first year. Further, careful dosing of azole-type drugs and rifampin dosing is advised if these agents are used.
TEZ/IVA (Symdeko) was most recently approved for patients having two copies of the F508Del mutation. In this Phase III study, TEZ/IVA improved lung function by ~3%–4% and reduced pulmonary exacerbation. Symdeko is believed to have less drug interactions and fewer side effects such as chest tightness when compared to Orkambi.2 Symdeko is also approved with IVA responsive CFTR gating mutations. TEZ/IVA assists in F508Del “availability.” Combining Symdeko with IVA facilitates CFTR channel opening on epithelial surfaces.
F508Del heterozygous patients
As of this writing, no FDA-approved CFTR modulator therapy for one copy of F508del or other CFTR gene mutations with minimal function is available. Rowe et al reported measurable reductions in sweat chloride along with improved pulmonary symptoms in patients with one F508del copy and a second minimal function mutation.6 In a Phase III study of TEZ/IVA vs IVA alone vs placebo in patients having one copy of F508del and second mutation with residual function, a 6.8% improvement in FEV1% was observed for TEZ/IVA vs a 4.7% for IVA alone (compared to placebo).2
Patients possessing G551D and other gating mutation
Kalydeco is FDA approved for use in patients with the G551D mutation and for CFTR with 35 additional mutations. Having been approved in 2012, clinicians have the most experience with this agent. Kalydeco dosing is recommended every 12 hours. Patients are advised to take their dosages with fatty finger foods such as cheese or yogurt. Kalydeco improves FEV1% by ~10%–15% in days to weeks. Body mass indices improve in patients on Kalydeco over weeks to months. Symptoms such as cough and sputum production were significantly reduced. Kalydeco reduced declines in lung function and stabilized CF disease. Sweat chloride values measured in CF patients treated with this agent were dramatically reduced by 50 points.4 Some evidence exists suggesting that Kalydeco improves pancreatic function in younger patients as well as improves airway smooth muscle tone. IVA was the first CFTR modulator/potentiator approved by the FDA for use in patients with CF and certain genetic mutations. IVA is believed to increase the time that activated CFTR channels at the cell surface remain open. This action would correct the functioning of CFTR in patients with CF.
The FDA approved IVA for use in patients with the following genetic mutations: G551D, R117H, G178R, S549N, S549R, G551S, G1244E, S1251N, S1255P, and G1349D. The efficacy and safety of IVA were evaluated in Phase III clinical studies. The STRIVE clinical trial was an RCT in 161 adolescent/adult patients with the G551D mutation. The study duration was 48 weeks and use of IVA was associated with statistically significant improvements in lung function (FEV1), fewer pulmonary exacerbations, and reduced respiratory symptoms.7 ENVISION14 was another RCT in 52 pediatric patients (6–11 years old) with the G551D mutation. The study duration was 48 weeks and patients who received IVA demonstrated a statistically significant improvement in lung function. Of note, the ENVISION population was younger and healthier (FEV1 values were near normal) than the patients from STRIVE.8,9 Patients who completed the RCTs were able to continue in an open-label, long-term extension (PERSIST) for up to 96 weeks. The investigators observed sustained clinical benefits in study participants.11 A longitudinal observational cohort study GOAL15 followed 151 patients taking IVA. Of these patients, 29% (26/89) who were culture positive for Pseudomonas aeruginosa the year prior to IVA use were culture negative the year following treatment. In addition, 88% (52/59) of patients who were culture negative remained uninfected. The odds of a positive P. aeruginosa culture in the year following IVA compared to the year prior were reduced by 35% (P<0.05).10 Individual case reports showed increased IVA efficacy and CFTR functionality in homozygous G551D patients when compared to heterozygous G551D patients. For patients on Kalydeco, liver function studies should be checked quarterly for the first year and yearly thereafter. Younger patients should have annual eye exams to monitor for cataract formation.
R117H patients
Kalydeco is FDA approved for R117H, a mixed gating and conduction mutation along with several other rare mutations that show in vitro response as measured in the laboratory.7 The splicing of R117H is affected by noncoding portions of the CFTR gene called the poly-T-tact. CF is more likely when that tract is shorter (called 5T) rather than 7T or 9T.
Can patients stop inhaled aerosols and airway clearance therapies while on a CFTR modulator?
There is no evidence that CF patients can stop or alter their current standard of care while on CFTR modulators. Once more sensitive biomarkers are available, alterations in care may become clearer as to which potential therapies can be discontinued while on fundamental specific modulator therapy.
Triplet CFTR modulators
The success of the therapies mentioned earlier along with a more fundamental understanding of the underlying molecular pathology in CFTR synthesis, processing, trafficking, and deposition on cell surfaces has identified the fact that CFTR may be present on cell surfaces but may not function normally. The hypothesis that triple-combination therapy will provide greater efficacy in the F508Del patient population is currently being explored. Phase II studies to evaluate the “next-generation correctors” with research agents VX-440, VX-152, and VX-659 (Vertex Pharmaceuticals) in triple-combination regimens with TEZ and IVA in CF patients are underway. The Phase II study of VX-440 is designed to evaluate the safety and efficacy of a 4-week dosing regimen of VX-440 in combination with TEZ/IVA in CF patients having one F508del mutation and one minimal function mutation as well as patients with two copies of the F508del mutation. TEZ/IVA, VX-440, VX-152, and VX-659 thus far report efficacy in the range of 9.6%–12% improvement in FEV1 at the highest dosage in trials ranging from 2 to 4 weeks for both F508Del homozygous and heterozygous patient populations.35 These data highlight the potential for small-molecule “triple-combination therapy” in the difficult-to-treat F508Del population. The newer triple-combination agents that are in early Phase II trials are believed to have additive biological effects to the already established effectiveness of TEZ/IVA boosting CFTR effectiveness and availability on epithelial surfaces.16 Preliminary results with next-generation CFTR modulators show promise in both F508Del homozygous and heterozygous patients possessing a second nonfunctional mutation. If these triplet compounds show success in future Phase III clinical trials, physicians caring for CF patients will have new options for therapy in virtually 90% of patients with CF.
Current strategies in patients with the F508Del mutation are focused on development of amplifier compounds capable of increasing the quantity of cell surface CFTR molecules available as therapeutic targets.18 Next-generation correctors focus on stabilizing portions of the F508Del gene to overcome multiple folding defects that are present in F508Del patients.19–21 Success of these therapies depends upon stabilization of defects found in F508Del mutations.22 Several next-generation correctors are currently tested in triple-combination therapies together with TEZ/IVA in preclinical models and early phase clinical trials (Table 1).23 According to recent studies, significant availability of F508Del CFTR on cell surfaces are possible with triple-combination correctors not achieved with TEZ/IVA alone.17 TEZ/IVA therapy served as the foundation of triple-combination therapy. In in vitro studies, the next-generation correctors, including VX-440, VX-152, and VX-659, were also characterized with TEZ/IVA. The in vitro efficacy of the triple combination in F508Del/F508Del human bronchial epithelial (HBE) cells is similar to that obtained with IVA in F508Del/G551D-HBE cells, while that on F508Del/MF (minimal function) HBE cells is higher than that obtained with IVA in F508Del/F508Del HBE cells. Triple-combination treatment improves chloride transport, fluid transport, and increases ciliary beat frequency to a greater extent than the dual combination of TEZ/IVA. If these findings continue to be present in Phase III trials which are ongoing, such triple-combination therapies may enhance clinical efficacy substantially in F508Del homozygous patients to levels similar to improvements observed for IVA in patients with G551D and gating mutations. These triple-combination CFTR modulator therapies may rescue F508Del function promoting clinical benefits in F508Del heterozygous patients who have associated minimal function mutation on the second allele. Triplet therapy generation correctors may therefore expand the fraction of CF patients that can be treated with CFTR modulator therapies to up to 90% of the total patient population.
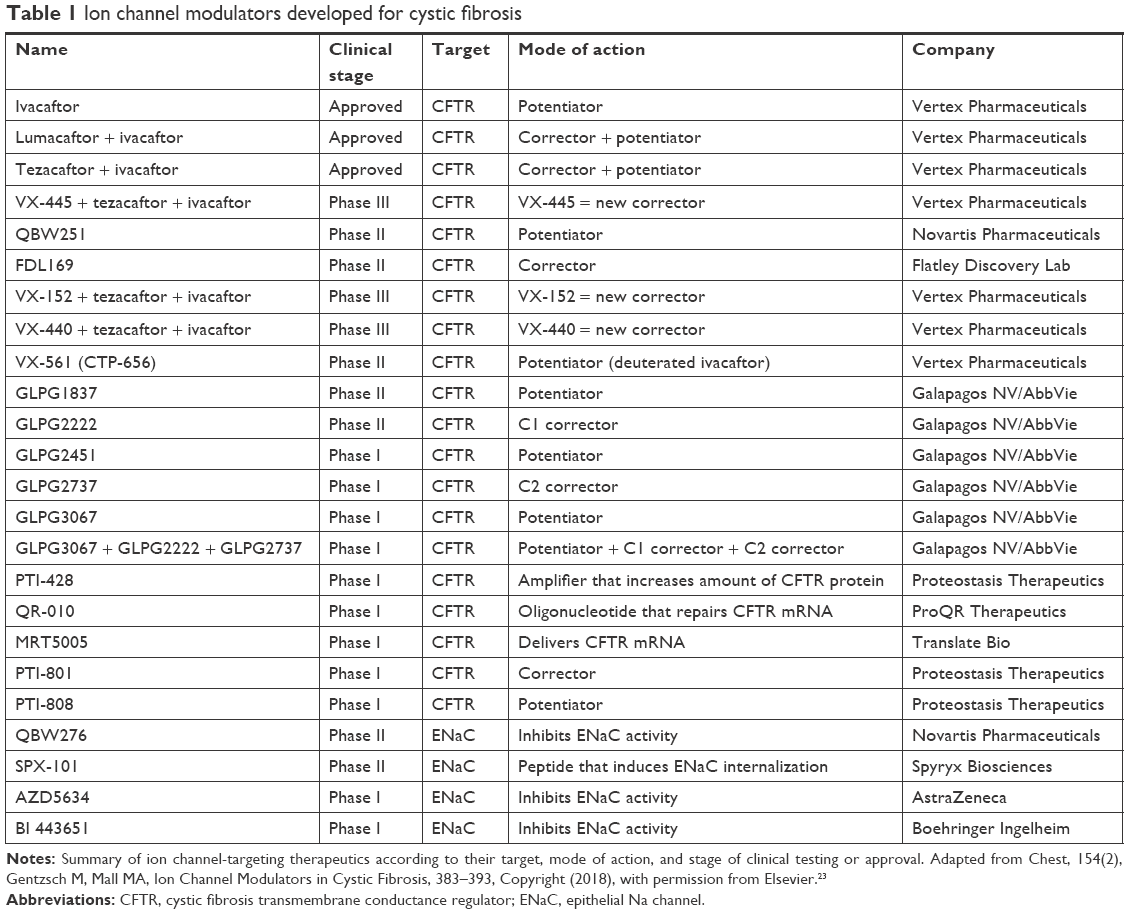 | Table 1 Ion channel modulators developed for cystic fibrosis |
Patient-specific biomarkers for precision medicine in triplet therapy
Patients having one or two copies of F508Del or two untreatable CFTR mutations are still unable to be treated with CFTR modulator therapy.24 Designing therapy for these patients will require addressing multiple defects employing combined therapies. Treating these patients may require combinations to be patient specific. Newer CFTR triplet modulator compounds will require reliable screening tools and personalized medicine approaches that are capable of predicting drug efficacy. Nasal and bronchial epithelial cultures and rectal tissues from individual CF patients have allowed drug discovery and testing using in vitro assays such as electrophysiological measurements of CFTR activity and the evaluation of ion and fluid movement. These studies have employed “organoid cultures” which enable prediction of patient-specific responses.25–29 Measuring Cl ion transport in bronchial epithelial cultures and nasal epithelial cells is widely used for evaluating CFTR-targeting therapeutics. Studying CFTR function in rectal biopsies using intestinal current measurements has detected in vivo responses to IVA-treated G551D and F508Del homozygous patients treated with lumacaftor/IVA combination therapy.30,31 A functional fluid-movement assay using spherical organoid cultures derived from CF intestinal tissue has been used increasingly for in vitro testing of CFTR modulator effects on specific CFTR mutations.32 Conditional “reprogramming” of spheroid cultures from nasal and bronchial specimens has allowed expanding the pool of patient-derived cells available for assessment of therapeutic responses.33 Airway cultures created from skin and blood cells may be employed in the near future to evaluate individual pharmacological responses.34 These patient-derived experimental systems provide prediction for drug responses in individual patients with variable CFTR mutations. However, long-term usefulness of these models in predicting clinical benefits remains unknown.
Given the recently reported clinical results of the Vertex’s triple-combination therapy in F508Del patients and the promise for additional triple-combination therapies to emerge from other companies, it is likely that multiple therapeutic options with transformative potential will be forthcoming for the treatment of the largest population of CF patients in the near future.
RNA antisense therapy
CFTR modulators are pills taken by mouth that help improve CFTR protein availability and functionality at epithelium. Another approach is using inhaled RNA antisense therapy that helps to make RNA work more effectively and produce a functional CFTR protein. ProQR has studied this therapy in a small group of patients who inhaled three times per week and showed improvements in lung functions as well as symptoms.
Future developments
CFTR correctors, potentiators, and activators are being studied in CF patients to extend therapies to new patients as well improving current FDA-approved CFTR modulators. Novel correctors of F508Del added on to potentiators and possibly drugs that are capable of improving function of premature stop codon mutations and possibly molecular amplifiers that create greater epithelial CFTR available for modulators to act on are promising therapeutic options in CF clinical care in future. Future CF care will require novel biomarkers to become available in order to prescribe correct modulator therapy. Clear guidelines for using this future therapy will be required for selection of these agents. Establishing patient partnerships will continue to be an integral component in modulator selection in patients together with continuing maintenance therapies. Also, in employing these future agents, a lung clearance index will be a useful tool for assessment of clinical responses.
Theratyping
Biomarkers will assist in theratyping where known paired responses in one mutation class are compared with responses in a new mutation class. The response to TEZA/IVA combination in patients with F508Del and P67L is possible with knowledge of molecular underpinnings of modulators and genetic defects of CFTR.
Conclusion
The increased number of CFTR modulators for treatment of CF and now with ~50% of CF patients being treated with CFTR modulators makes it imperative for physicians caring for CF patients to be familiar with current and future trends in management. Drugs such as Kalydeco, Orkambi, and Symdeko target the most common CFTR mutation and address the underlying molecular basis of CFTR dysfunction. Combination therapy with next-generation correctors in addition to background TEZA/IVA holds promise for treating CFTR rare functional mutations. Novel biomarkers and lung clearance index will provide detailed understanding of the impact of these therapies.
Acknowledgments
The author is grateful to Dr Michael Schechter and Dr Alpha Fowler for their support and guidance in writing of this manuscript.
Disclosure
Dr Chaudary is Associate Professor and Director of the Adult Cystic Fibrosis Center at the Virginia Commonwealth University Medical Center. The author reports no other conflicts of interest in this work.
References
Wainwright CE, Elborn JS, Ramsey BW, et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med. 2015;373(3):220–231. | ||
Taylor-Cousar JL, Munck A, Mckone EF, et al. Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N Engl J Med. 2017;377(21):2013–2023. | ||
Veit G, Avramescu RG, Chiang AN, et al. From CFTR biology toward combinatorial pharmacotherapy: expanded classification of cystic fibrosis mutations. Mol Biol Cell. 2016;27(3):424–433. | ||
de Boeck K, Munck A, Walker S, et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J Cyst Fibros. 2014;13(6):674–680. | ||
Sawicki GS, Mckone EF, Pasta DJ, et al. Sustained Benefit from ivacaftor demonstrated by combining clinical trial and cystic fibrosis patient registry data. Am J Respir Crit Care Med. 2015;192(7):836–842. | ||
Wainwright CE, Elborn JS, Ramsey BW, et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med. 2015;373(3):220–231. | ||
Tabcharani JA, Chang XB, Riordan JR, Hanrahan JW. Phosphorylation-regulated Cl-channel in CHO cells stably expressing the cystic fibrosis gene. Nature. 1991;352(6336):628–631. | ||
Rich DP, Anderson MP, Gregory RJ, et al. Expression of cystic fibrosis transmembrane conductance regulator corrects defective chloride channel regulation in cystic fibrosis airway epithelial cells. Nature. 1990;347(6291):358–363. | ||
Protasevich I, Yang Z, Wang C, et al. Thermal unfolding studies show the disease causing F508del mutation in CFTR thermodynamically destabilizes nucleotide-binding domain 1. Protein Sci. 2010;19(10):1917–1931. | ||
Rowe SM, Verkman AS. Cystic fibrosis transmembrane regulator correctors and potentiators. Cold Spring Harb Perspect Med. 2013;3(7):1–15. | ||
Rowe SM, Daines C, Ringshausen FC, et al. Â Tezacaftor-Ivacaftor in Residual-Function Heterozygotes with Cystic Fibrosis. N Engl J Med. 2017;377(21):2024–2035. | ||
Moss RB, Flume PA, Elborn JS, et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis who have an Arg117His-CFTR mutation: a double-blind, randomised controlled trial. Lancet Resp Med. 2015;3(7):524–533. | ||
Ramsey BW, Davies J, Mcelvaney NG, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365(18):1663–1672. | ||
Davies JC, Wainwright CE, Canny GJ, et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am J Respir Crit Care Med. 2013;187(11):1219–1225. | ||
Heltshe SL, Mayer-Hamblett N, Burns JL, et al. Pseudomonas aeruginosa in cystic fibrosis patients with G551D-CFTR treated with ivacaftor. Clin Infect Dis. 2015;60(5):703–712. | ||
Mckone EF, Borowitz D, Drevinek P, et al. Long-term safety and efficacy of ivacaftor in patients with cystic fibrosis who have the Gly551Asp-CFTR mutation: a phase 3, open-label extension study (PERSIST). Lancet Respir Med. 2014;2(11):902–910. | ||
Vertex Press Release; 2018. Available from: http://investors.vrtx.com/releasedetail.cfm?ReleaseID=1055958. Accessed October 26, 2018. | ||
Molinski SV, Ahmadi S, Ip W, et al. Orkambi® and amplifier co-therapy improves function from a rare CFTR mutation in gene-edited cells and patient tissue. EMBO Mol Med. 2017;9(9):1224–1243. | ||
Rabeh WM, Bossard F, Xu H, et al. Correction of both NBD1 energetics and domain interface is required to restore ΔF508 CFTR folding and function. Cell. 2012;148(1–2):150–163. | ||
Okiyoneda T, Veit G, Dekkers JF, et al. Mechanism-based corrector combination restores ΔF508-CFTR folding and function. Nat Chem Biol. 2013;9(7):444–454. | ||
Mendoza JL, Schmidt A, Li Q, et al. Requirements for efficient correction of ΔF508 CFTR revealed by analyses of evolved sequences. Cell. 2012;148(1–2):164–174. | ||
Meng X, Clews J, Kargas V, Wang X, Ford RC. The cystic fibrosis transmembrane conductance regulator (CFTR) and its stability. Cell Mol Life Sci. 2017;74(1):23–38. | ||
Gentzsch M, Mall MA. Ion Channel Modulators in Cystic Fibrosis. Chest. 2018;154(2):383–393. | ||
Grasemann H. CFTR Modulator Therapy for Cystic Fibrosis. N Engl J Med. 2017;377(21):2085–2088. | ||
Cholon DM, Gentzsch M. Recent progress in translational cystic fibrosis research using precision medicine strategies. J Cyst Fibros. 2018;17(2S):S52–S60. | ||
Dekkers JF, Wiegerinck CL, de Jonge HR, et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat Med. 2013;19(7):939–945. | ||
Dekkers JF, Berkers G, Kruisselbrink E, et al. Characterizing responses to CFTR-modulating drugs using rectal organoids derived from subjects with cystic fibrosis. Sci Transl Med. 2016;8(344):344ra84. | ||
Brewington JJ, Filbrandt ET, Larosa FJ, et al. Detection of CFTR function and modulation in primary human nasal cell spheroids. J Cyst Fibros. 2018;17(1):26–33. | ||
Guimbellot JS, Leach JM, Chaudhry IG, et al. Nasospheroids permit measurements of CFTR-dependent fluid transport. JCI Insight. 2017;2(22). pii: 95734. | ||
Graeber SY, Hug MJ, Sommerburg O, et al. Intestinal Current Measurements Detect Activation of Mutant CFTR in Patients with Cystic Fibrosis with the G551D Mutation Treated with Ivacaftor. Am J Respir Crit Care Med. 2015;192(10):1252–1255. | ||
Graeber SY, Dopfer C, Naehrlich L, et al. Effects of Lumacaftor/Ivacaftor Therapy on CFTR Function in Phe508del Homozygous Patients with Cystic Fibrosis. Am J Respir Crit Care Med. 2018;197(11):1433–1442. | ||
Dekkers JF, Wiegerinck CL, de Jonge HR, et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat Med. 2013;19(7):939–945. | ||
Gentzsch M, Boyles SE, Cheluvaraju C, et al. Pharmacological Rescue of Conditionally Reprogrammed Cystic Fibrosis Bronchial Epithelial Cells. Am J Respir Cell Mol Biol. 2017;56(5):568–574. | ||
Crane AM, Kramer P, Bui JH, et al. Targeted correction and restored function of the CFTR gene in cystic fibrosis induced pluripotent stem cells. Stem Cell Reports. 2015;4(4):569–577. | ||
Tullis E, Colombo C, Davies JC, et al. Preliminary safety and efficacy of triple combination CFTR modulator regiments in CF. Poster 777. In: The 31st Annual North American Cystic Fibrosis Conference; November 2–4, 2017; Indianapolis, IN. | ||
Ratjen F. Restoring Airway Surface Liquid in Cystic Fibrosis. N Engl J Med. 2006;354:291–293. | ||
Zielenski J. Genotype and Phenotype in Cystic Fibrosis. Respiration. 2000;67(2):117–133. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.