")
Back to Journals » International Journal of Nanomedicine » Volume 13
Triphenylphosphonium and D-α-tocopheryl polyethylene glycol 1000 succinate-modified, tanshinone IIA-loaded lipid-polymeric nanocarriers for the targeted therapy of myocardial infarction
Authors Zhang S, Li J, Hu S, Wu F, Zhang X
Received 14 February 2018
Accepted for publication 7 May 2018
Published 10 July 2018 Volume 2018:13 Pages 4045—4057
DOI https://doi.org/10.2147/IJN.S165590
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Shouwen Zhang, Jingfang Li, Shunpeng Hu, Fangfang Wu, Xianzhao Zhang
Department of Cardiology, Linyi People’s Hospital, Linyi, People’s Republic of China
Background: Cardiovascular diseases (CVDs) are the leading causes of mortality worldwide. Currently, the best treatment options for myocardial infarction focus on the restoration of blood flow as soon as possible, which include reperfusion therapy, percutaneous coronary intervention, and therapeutic thrombolytic drugs.
Materials and methods: In the present study, we report the development of lipid-polymeric nanocarriers (LPNs) for mitochondria-targeted delivery of tanshinone IIA (TN). D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS) was linked to the triphenylphosphonium (TPP) cation. The LPNs were fabricated by nanoprecipitation method. LPNs were evaluated in vitro and in vivo in comparison with free drugs and other similar nanocarriers.
Results: The mean diameter of TN/nanoparticles (NPs) was 89.6 nm, while that of TN/LPNs was 121.3 nm. The zeta potential of TN/NPs and TN/LPNs was –33.6 and –22.3 mV, respectively. Compared with free TN and TN/NPs, TN/LPNs exhibited significantly improved compatibility and therapeutic efficiency. In addition, the in vivo pharmacokinetics, biodistribution, and infarct therapy studies in Sprague Dawley rats showed that TPP-TPGS/TN/LPNs had better efficiency than their nonmodified TN/LPNs counterparts in all respects.
Conclusion: These results indicated that the TPP-TPGS/TN/LPNs were promising nanocarriers for efficient delivery of cardiovascular drugs and other therapeutic agents for the treatment of CVDs.
Keywords: myocardial infarction, mitochondria targeting, lipid-polymeric nanocarriers, D-α-tocopheryl polyethylene glycol 1000 succinate, triphenylphosphonium, tanshinone IIA
Introduction
Cardiovascular diseases (CVDs), including ischemic heart disease and myocardial infarction (MI), remain a major global health problem. CVDs are the leading causes of mortality worldwide, with an estimated 17 million deaths every year, according to the 2013 Global Burden of Disease Study.1 MI caused about one million deaths every year in the USA, resulting in more deaths than cancer.2 Currently, the best treatment options for MI focus on the restoration of blood flow as soon as possible, which include reperfusion therapy, percutaneous coronary intervention, and therapeutic thrombolytic drugs.3–5
Myocardial reperfusion injury following acute MI (AMI) is critically determined by mitochondrial dysfunction. Therefore, mitochondria-targeted therapy could be an effective strategy for limiting the MI size and decreasing harmful side effects.6 Compared to hydrophilic compounds, alkyltriphenylphosphonium cations, including methyltriphenylphosphonium (TPMP) cation and triphenylphosphonium (TPP) cation, pass easily through lipid bilayers via noncarrier-mediated transport and accumulate several hundredfold within mitochondria, because of the high negative mitochondrial membrane potential (−150 to −170 mV).7,8 Recently, several researches have focused on synthesizing new compounds based on TPP or TPMP and developing nanoparticles (NPs) which were surface-modified by TPP cations for mitochondria-targeted drug delivery systems.8–10 In order to reduce the generation of reactive oxygen species brought by myocardial ischemia reperfusion, TPMP was conjugated with antioxidants vitamin E and coenzyme Q.7
D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS, a water-soluble succinate ester of vitamin E) possesses an amphiphilic structure and could exhibit an extraordinary permeation effect to hydrophobic and hydrophilic agents.11,12 Moreover, TPGS could prolong blood circulation time, enhance the solubility of hydrophobic drugs, and improve cellular uptake.13–15 As an US FDA-approved biocompatible excipient, TPGS is widely used in nanocarriers for cancer therapy.16 TPP and TPGS conjugate could combine both advantages and was synthesized for the first time in this study.
NPs-based drug delivery systems have attracted much attention in the treatment of MI and ischemic heart disease.17,18 NPs comprising inorganic particles, lipid NPs, or polymeric micelles can be targeted toward mitochondria using mitochondriotropic moieties such as TPP and TPMP.8 Among them, lipid-polymeric nanocarriers (LPNs) have emerged as a multifunctional drug delivery platform, which combines mechanical advantages of polymeric core and biomimetic advantages of phospholipid shell into a single platform.19 Specifically, LPNs exhibit high structural integrity, stability during storage, controlled release, and high biocompatibility and bioavailability owed to the lipid layers. In our study, we designed TPP-TPGS surface-modified, tanshinone (TN)-loaded LPNs (TPP-TPGS/TN/LPNs) for the targeted therapy of MI.
TN is a major active ingredient derived from the Chinese medical herb Salvia miltiorrhiza and widely applied as a therapeutic agent for the treatment of CVDs and cerebrovascular diseases.20,21 However, its poor water solubility and low oral bioavailability have limited its clinical application. To overcome these drawbacks, NPs-based drug delivery systems have been researched including liposomes, NPs, microemulsions, cyclodextrin inclusions, solid dispersions, and so on.22,23
In the present study, we report the development of LPNs for mitochondria-targeted delivery of TN. TPGS was linked to TPP cation via lysine to form TPP-Lys-TPGS. The LPNs were fabricated by nanoprecipitation method. LPNs were evaluated in vitro by drug release behavior. In vitro drug release and stability in the fresh plasma were evaluated. In vivo pharmacokinetics, biodistribution, and pharmacodynamics (infarct size) were investigated in rats with ischemic myocardium after intravenous (iv) injection.
Materials and methods
Materials
TPGS was purchased from Eastman Chemical Company (Kingsport, TN, USA). Fmoc-Lys[Mtt]-OH was obtained from NJPeptide Biotechnology Co. Ltd. (Nanjing, People’s Republic of China). TN (≥97%, HPLC), polysorbate 80, piperidine, dimethylformamide (DMF), hydroxybenzotriazole (HOBt), N,N-diisopropylethylamine (DIPEA), dichloromethane (DCM), triisopropylsilane (Tis), trifluoroacetic acid (TFA), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide, dimethyl sulfoxide (DMSO), fetal bovine serum (FBS), Dulbecco’s Modified Eagle’s Medium (DMEM), and 3-[4,5-dimethylthiazol-2yl]-2,5-diphenyltetrazolium (MTT) were purchased from Sigma-Aldrich (St Louis, MO, USA). Poly (D,L-lactide-co-glycolide) (PLGA, 50:50, Mw 1.5–2.4 kDa) was purchased from Jinan Daigang Biomaterial Co., Ltd (Jinan, People’s Republic of China). Fluorescein isothiocyanate (FITC)-PLGA was obtained from Xi’an Ruixi Biological Technology Co., Ltd. (Xi’an, People’s Republic of China). Injectable soybean lecithin (ISL) was provided by Shanghai Taiwei Pharmaceutical Co., Ltd. (Shanghai, People’s Republic of China). All other chemicals and reagents were of analytical grade or high-performance liquid chromatography grade and used without further purification.
Synthesis of TPP-Lys-TPGS
TPP-Lys-TPGS was synthesized as shown in Figure 1. A lysine linker with two protecting groups – Fmoc and Mtt (Fmoc-Lys[Mtt]-OH) – was used, which enabled the conjugation of TPMP and TPGS.24 Firstly, the Fmoc was deprotected with 20% piperidine in DMF for 15 min at room temperature. Then, 3 equivalents of Lys[Mtt]-OH was added to 5 equivalents of HOBt and 5 equivalents of DIPEA in DMF. The mixture was agitated for 5 h at room temperature. Subsequently, 5 equivalents of TPP-conjugated carboxylic acid (TPP+C10COOH) was coupled to the lysine via HOBt (5 equivalents) and DIPEA (5 equivalents) in DMF for 6 h at room temperature as described above. The Mtt group on the lysine was then deprotected by incubation with 94% DCM, 5% Tis, and 1% TFA for 20 min. Next, 3 equivalents of TPGS was coupled to the lysine via HOBt (5 equivalents) and DIPEA (5 equivalents) in DMF for 6 h at room temperature.
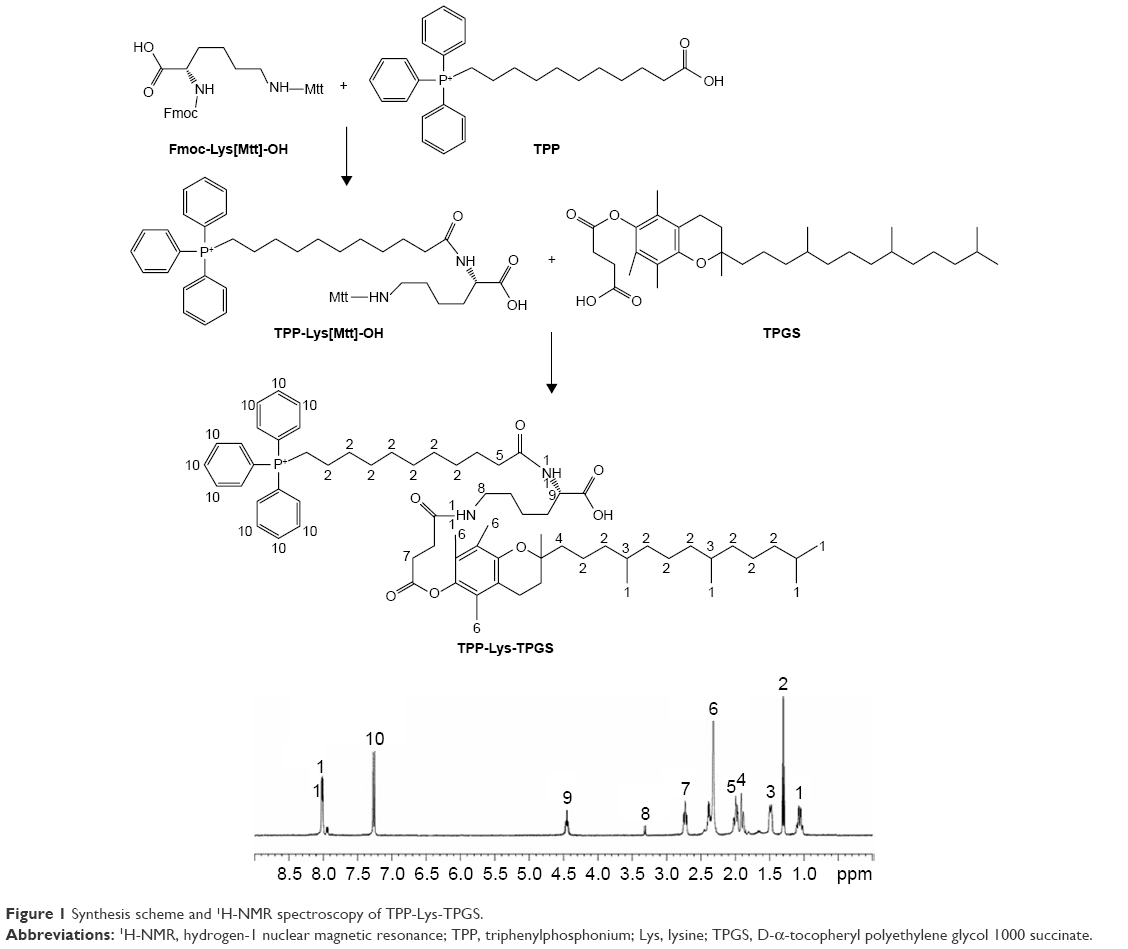 | Figure 1 Synthesis scheme and 1H-NMR spectroscopy of TPP-Lys-TPGS. |
Preparation of TPP-TPGS/TN/LPNs
TPP-TPGS/TN/LPNs was fabricated by nanoprecipitation method.25 TPP-Lys-TPGS and ISL were dispersed in distilled water to form the aqueous phase. TN, PLGA, and polysorbate 80 (0.5%) were dissolved in acetone to form the oil phase. The oil phase was added drop by drop into the aqueous phase under gentle stirring (300 rpm) at room temperature for 6 h until complete evaporation of the organic solvent to get the TPP-TPGS/TN/LPNs (Figure 2).
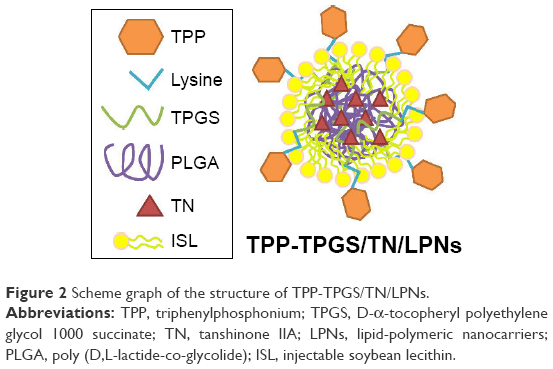 | Figure 2 Scheme graph of the structure of TPP-TPGS/TN/LPNs. |
TPP-TPGS-modified, blank LPNs (TPP-TPGS/LPNs) were prepared in the same way, by dissolving PLGA (without TN) in acetone to form the oil phase.
Non-TPP-TPGS-modified, TN-loaded LPNs (TN/LPNs) were prepared in the same way, by dispersing ISL (without TPP-Lys-TPGS) in distilled water to form the aqueous phase.
Non-TPP-TPGS-modified, TN-loaded polymeric NPs (TN/NPs) were prepared in the same way, by using distilled water (without ISL and TPP-Lys-TPGS) as the aqueous phase.
For preparation of fluorescently tagged FITC-containing LPNs and NPs, regular PLGA was replaced with FITC-PLGA. LPNs and NPs were separated by ultrafiltration from the suspensions to remove the free drug. They were stored at 2°C–8°C until use.
Mean diameter and zeta potential determination
Mean diameter and zeta potential were determined with a Zetasizer Nano ZS (Malvern Instruments Ltd, Malvern, UK). Measurements were carried out in triplicate at 25°C on three independent preparations.26
Drug loading and entrapment efficiency evaluation
The entrapment efficiency (EE) of TN-loaded LPNs and NPs was measured using a Sephadex-G25 column (15.0×1.5 cm2).23 UV5 ultraviolet spectrophotometer (Mettler-Toledo GmbH, Greifensee, Switzerland) was used to analyze TN; the assay of TN was in the linear range with concentrations of 1.0–50.0 μg/mL. Using physiological saline as the eluent, entrapped TN and free TN were collected at continuous volume intervals of 4.0 mL, followed by their analysis. The drug loading (DL) and EE of the nanocarriers were calculated by dividing the entrapped TN in the nanocarriers by the total amounts of TN following the below equations:
|

|
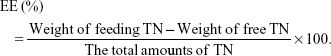
Stability of TPP-TPGS/TN/LPNs
Stability of TN-loaded LPNs and NPs was evaluated by measuring mean diameter, zeta potential, DL, and EE for 120 days.27 The suspensions were stored at 2°C–8°C. The results were calculated, and the analyses were performed at 0, 15, 30, 60, 90, and 120 days.
In vitro drug release of TPP-TPGS/TN/LPNs
The amounts of drug released from LPNs and NPs were measured using the dialysis method.28 TN-loaded LPNs and NPs were placed in dialysis bags (MWCO, 3.5 kDa), separately. After sealing of both ends, the bag was immersed in 50 mL of phosphate buffer solution (PBS) in the presence of 10% FBS (pH 7.4). For sampling, 1 mL of medium was collected at various time points and replaced with the same volume of fresh medium. The concentrations of released TN were quantified as described in the section “Drug loading and entrapment efficiency evaluation”.
Isolation of primary cardiac cells
Primary cardiac cells (PC cells) were isolated as follows:29 Wistar rats were sacrificed by decapitation, and their ventricles were excised and cut into small pieces. The tissue pieces were enzymatically digested by incubating at 37°C with gentle mixing for 2 h in a solution containing 100 mM NaCl, 10 mM KCl, 1.2 mM KH2PO4, 4.0 mM MgSO4, 50 mM taurine, 20 mM glucose, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 2 mg/mL collagenase type 2, 2 mg/mL pancreatin, and 1% penicillin–streptomycin. Cells in suspension were collected by centrifugation at 2,000 rpm for 5 min. The supernatant and the top layer containing damaged cells were discarded, and the remaining cells were suspended in DMEM supplemented with 10% FBS. The cells were preplated for 1 h onto cell culture plates to enrich for cardiomyocytes, after which the unattached cells (enriched cardiomyocytes) were plated at a desired density. On the next day, the medium was replaced with a complete serum-free medium.
Cellular uptake of TPP-TPGS/TN/LPNs
PC cells were grown in DMEM supplemented with 10% FBS.30 The cells were maintained in a humidified incubator at 37°C and 5% CO2 at an initial density of 1×104 cells/well in black clear-bottom 96-well plates. The cells were incubated in fresh medium after 24 h of incubation and treated with fluorescently tagged FITC-containing LPNs and NPs. After 4 and 48 h of incubation, the cells were washed with PBS, and fresh cell culture medium was added. The fluorescence intensity was measured at 489/535 nm using an inversion fluorescence microscope (ZX71; Olympus, Tokyo, Japan), and the image was captured. After fluorescence measurement, flow cytometry analysis was performed on PC cells.31 The cells were washed once with 1 mL of PBS and were detached with trypsin/ethylenediamine tetraacetic acid. Then, the cells were centrifuged at 1,500 rpm for 5 min 4°C, the supernatant was discarded, and the cells were washed once with 1 mL of PBS. The cells were centrifuged again (1,500 rpm for 5 min at 4°C), the supernatant was discarded, and the cells were resuspended in 300 μL of PBS and directly introduced to a BD FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA).
In vitro cytotoxicity of TPP-TPGS/TN/LPNs
The cytotoxicity of LPNs and NPs on PC cells was measured using the MTT bioassay.32 Cells were seeded in 96-well plates at a density of 1×104 cells/well. The spent medium was discarded and substituted with 100 μL of fresh DMEM with LPNs or NPs, or free TN of different concentrations, while the culture medium without FBS was used as the control group. After incubation for 24 h, the medium was replaced with 20 μL of MTT solution in PBS (5 mg/mL). The media was removed, and then 100 μL of DMSO was added to each well and oscillated for 10 min to dissolve the MTT formazan generated by live cells after incubation for 4 h. The relative cell viability (%) was measured by comparing the absorbance of each well at 490 nm with the control well by using a microplate reader (Bio-Rad Laboratories, Hercules, CA, USA). The cell viability was calculated following the equation:
|
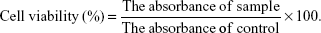
Induction of AMI animal model
Sprague Dawley rats (SD rats, 220–250 g) were purchased from Nanjing Junke Biological Engineering Co., Ltd (Nanjing, People’s Republic of China) and housed under controlled conditions (temperature of 20°C±2°C and a 12-h light/12-h dark cycle). AMI was induced as follows: Rats were anesthetized with a combination of ketamine (40 mg/kg) and xylazine (10 mg/kg), incubated, and mechanically ventilated. The chest was opened by left thoracotomy, the pericardium was removed, and the proximal left coronary artery was permanently occluded with an intramural stitch.33 All the animal experiments were approved by the Medical Ethics Committee of Linyi People’s Hospital (No 201706121077) and were performed according to the National Institutes of Health guide for the care and use of laboratory animals (NIH Publications No 8023, revised 1978).
Group design and drug administration
Rats were randomly divided into seven groups with eight rats per group as follows:34 1) sham-operated group, in which the rats underwent identical surgery except for the coronary artery ligation and were injected with physiological saline; 2) AMI group, in which the AMI rats received physiological saline; 3) free TN group, in which the rats were treated with 50 mg/kg of free TN (TN dissolved in ethanol); 4) TN/NPs group, in which the rats were treated with TN/NPs containing 50 mg/kg of TN; 5) TN/LPNs group, in which the rats were treated with TN/LPNs containing 50 mg/kg of TN; 6) TPP-TPGS/LPNs group, in which the rats were treated with TPP-TPGS/LPNs containing 50 mg/kg of TN; and 7) TPP-TPGS/TN/LPNs group, in which the rats were treated with TPP-TPGS/TN/LPNs containing 50 mg/kg of TN. The samples were physiologically injected every other day for 14 days.
In vivo pharmacokinetics evaluation
Blood samples (1.5 mL) of free TN group, TN/NPs group, TN/LPNs group, and TPP-TPGS/TN/LPNs group (eight rats per group) were collected into heparinized centrifuge tubes just before iv administration (0 h) and after 15 and 30 min, and 1, 2, 4, 6, 8, 12, 16, 24, 36, and 48 h after iv administration.35 The blood samples were separated immediately by centrifugation at 4,000 rpm for 5 min and stored at −20°C for further analysis. Concentrations of TN in rat plasma were determined by the method described in the section “Drug loading and entrapment efficiency evaluation”.
In vivo biodistribution study
At 15 min after iv administration, rats of free TN group, TN/NPs group, TN/LPNs group, and TPP-TPGS/TN/LPNs group (eight rats per group) were sacrificed. Blood samples were collected, and the heart, liver, spleen, lung, and kidney of rats were removed, washed, weighed, and homogenized.5 All of the samples were stored at −20°C for further analysis. Concentrations of TN in rat plasma were determined by the method described in the section “Drug loading and entrapment efficiency evaluation”.
In vivo infarct therapy effect
All rats (eight rats per group) were anesthetized with intraperitoneal injection of pentobarbital sodium (40 mg/kg).36 Then, heart samples were immediately measured through the aorta, and physiological saline was used for washing. The coronary artery was ligated after 6 h, and the left ventricle was placed at −80°C for 20 min. Heart samples were sliced into 2 mm thick sections. Infarct size of heart sample was measured with 1% 2,3,5-triphenyltetrazolium chloride for 30 min in the dark. The area of MI was calculated using ImageJ (National Institutes of Health, Bethesda, MD, USA) software for analyzing the stained or unstained areas of the heart tissue. The size of the infarcted area was evaluated as a percentage of whole size of the left ventricle.
Statistical analysis
Statistical comparison between two groups was performed using an unpaired t-test with SPSS software version 20.0 (IBM Corp, Armonk, NY, USA). Results were expressed as mean ± SD. *P < 0.05 and **P < 0.01 were considered statistically significant.
Results
Characterization of TPMP-Lys-TPGS
1H nuclear magnetic resonance spectroscopy of TPMP-Lys-TPGS in DMSO-d6 (600 mHz) was carried out, and each proton peak was identified according to the structural formula: δ 1.06 (1, −CH3); 1.31 (2, −CH2−); 1.49 (3, −CH−); 1.82 (4, −CH2−C−); 2.06 (5, −CH2−C=O−N); 2.31 (6, Ph−CH3); 2.75 (7, −CH2−C=O−O); 3.32 (8, −CH2−N−C=O); 4.46 (9, −CH−N−C=O); 7.23 (10, −Ph3−P); 8.02 (11, −NH−C=O) (Figure 1).
LNPs and NPs characterization
The diameter of the LPNs and NPs was measured and is shown in Figure 3A. The mean diameter of TN/NPs was 89.6±2.6 nm, while that of TN/LPNs was 121.3±3.1 nm. Diameters of TPP-TPGS/LPNs and TPP-TPGS/TN/LPNs were about 140 nm. The polydispersity index of TN/NPs, TN/LPNs, TPP-TPGS/LPNs, and TPP-TPGS/TN/LPNs were 0.13±0.02, 0.12±0.02, 0.15±0.03, and 0.16±0.03, respectively.
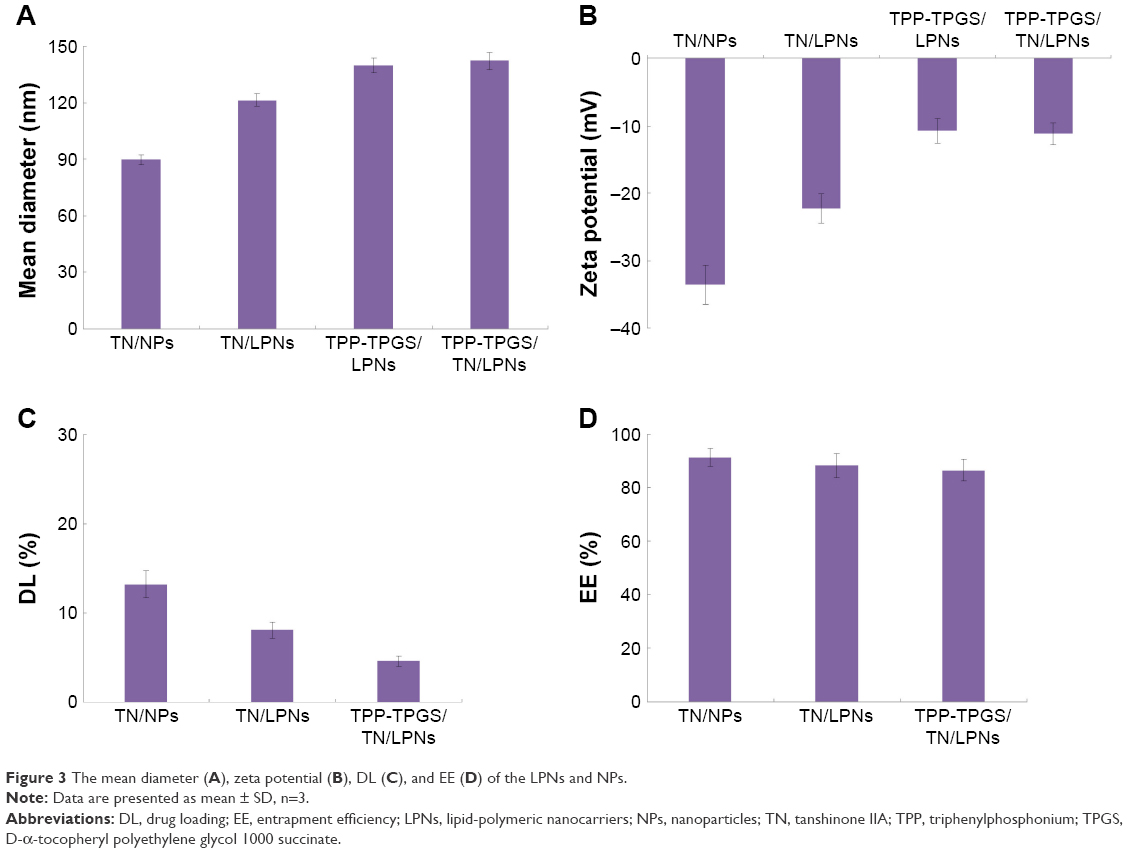 | Figure 3 The mean diameter (A), zeta potential (B), DL (C), and EE (D) of the LPNs and NPs. |
The zeta potential of polymeric TN/NPs was −33.6±2.9 mV, owing to the presence of negatively charged PLGA. TN/LPNs had a potential of −22.3±2.2 mV. After TPP-TPGS modification, the surface charge of TPP-TPGS/LPNs and TPP-TPGS/TN/LPNs decreased to around −10 mV (Figure 3B).
The DL (Figure 3C) varied among the different systems due to the amount of ingredients used, and the EE of the TN-loaded nanocarriers was about 90% (Figure 3D).
Stability of LNPs and NPs
The stability of TN-loaded LPNs and NPs was evaluated over a period of 120 days of storage at 2°C–8°C. For LPNs and NPs, the mean diameter remained almost constant throughout the period (Figure 4A). There were no significant changes with time in the zeta potentials of all the formulations tested (Figure 4B). For all the samples tested, DL and EE remained stable at all the time points during the 120 days of storage (Figure 4C and D).
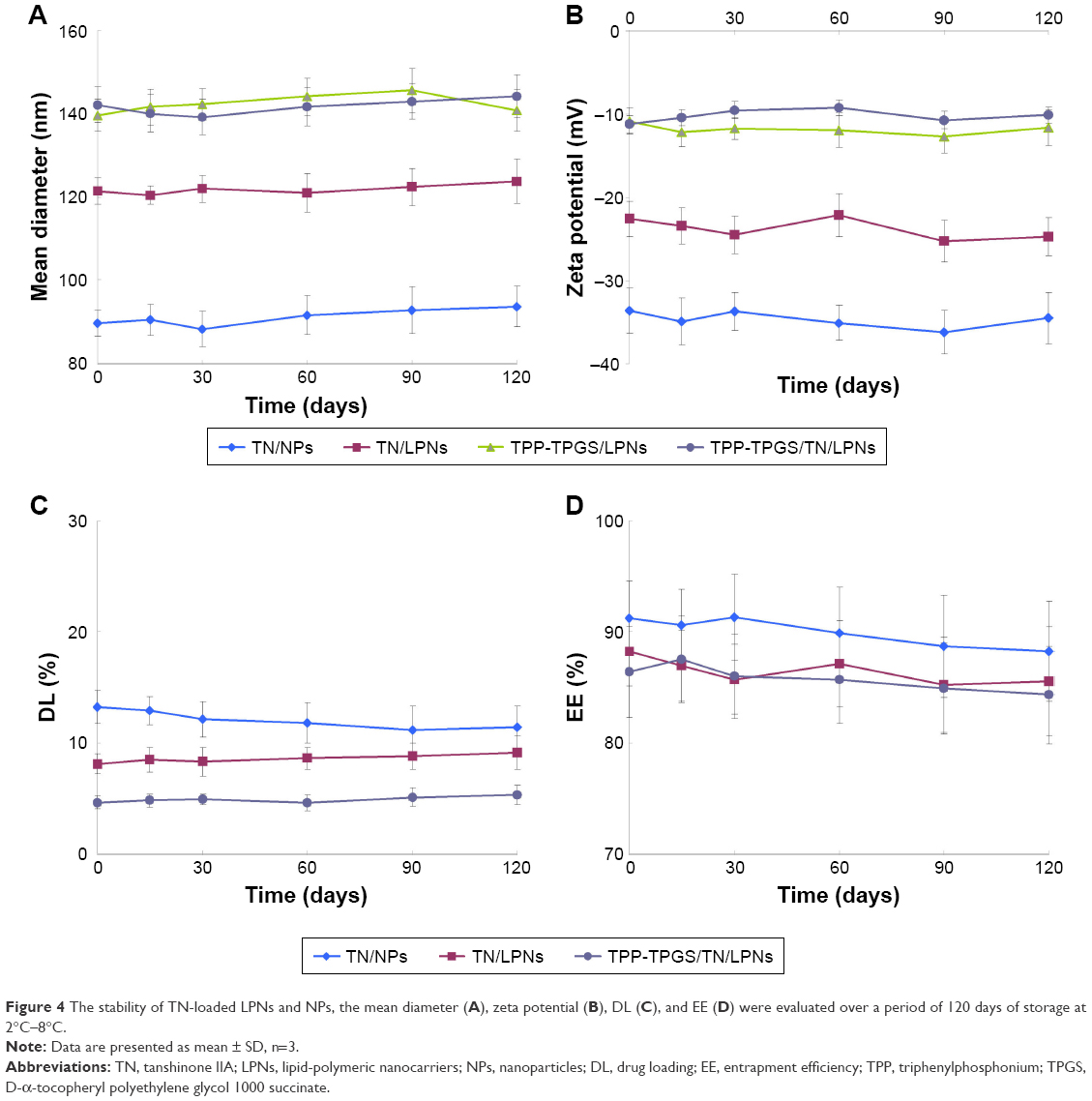 | Figure 4 The stability of TN-loaded LPNs and NPs, the mean diameter (A), zeta potential (B), DL (C), and EE (D) were evaluated over a period of 120 days of storage at 2°C–8°C. |
In vitro drug release
The in vitro TN release profiles of LPNs and NPs are depicted in Figure 5. The results showed that the release of TN from polymeric NPs was faster than from LPNs. TPP-TPGS/TN/LPNs exhibited more sustained release than the nonmodified TN/LPNs. The time taken by TN/NPs, TN/LPNs, and TPP-TPGS/TN/LPNs to completely release TN (over 80%) was 24, 48, and 60 h, respectively.
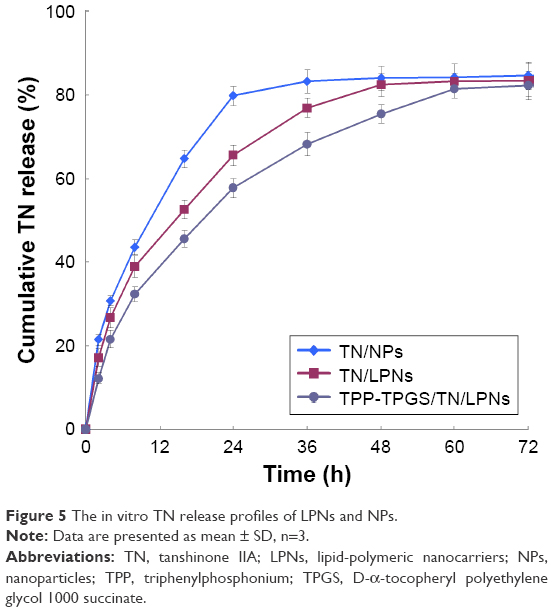 | Figure 5 The in vitro TN release profiles of LPNs and NPs. |
Cellular uptake
The PC cells treated with TPP-TPGS-modified LPNs exhibited significantly higher fluorescence intensity as compared to the cells that underwent other treatments at both 4 and 48 h posttreatment (Figure 6A). Flow cytometry analysis showed that TPP-TPGS-modified LPNs had higher cell uptake than nonmodified TN/LPNs (P < 0.05). Higher cellular uptake efficiency was observed in LPNs groups as compared to polymeric NPs group (P < 0.05). An increase in fluorescence intensity at 48 h was observed compared with 4 h post-administration for all the groups tested (Figure 6B).
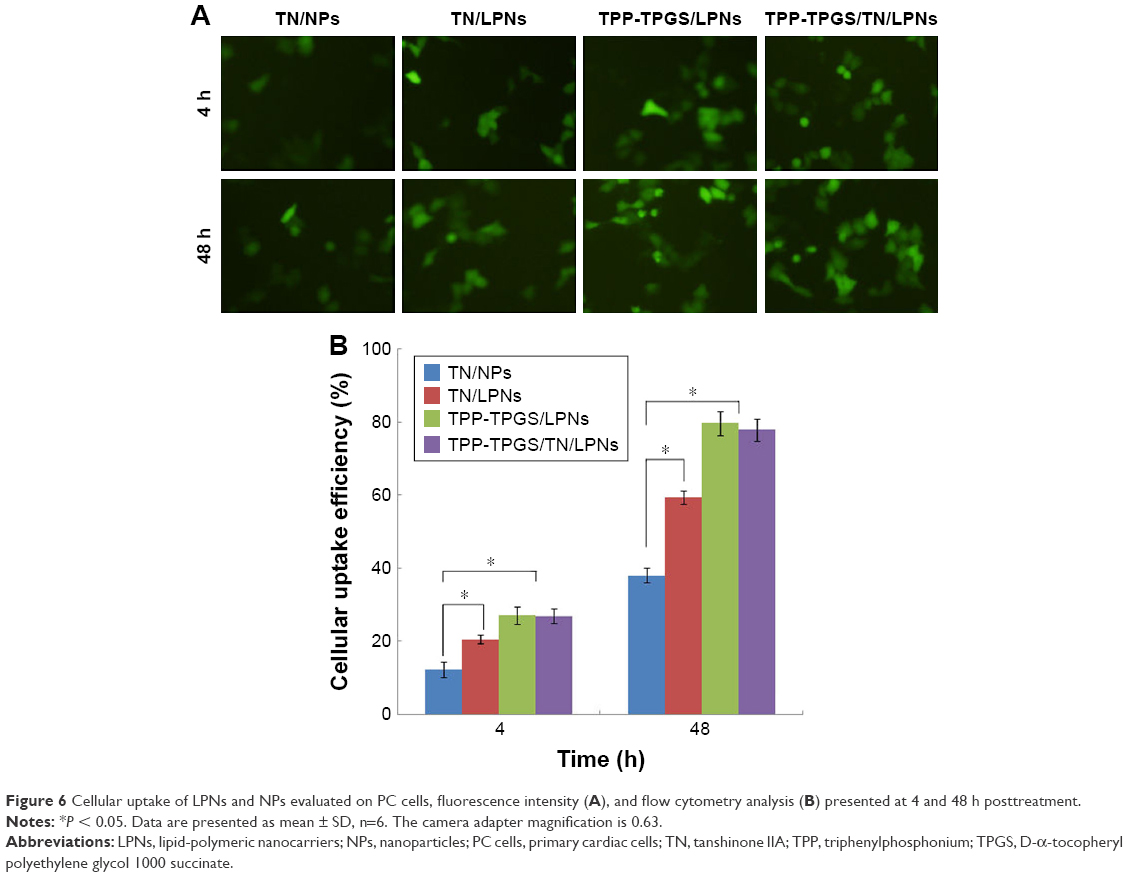 | Figure 6 Cellular uptake of LPNs and NPs evaluated on PC cells, fluorescence intensity (A), and flow cytometry analysis (B) presented at 4 and 48 h posttreatment. |
In vitro cytotoxicity
The MTT assay was performed to investigate the in vitro cytotoxic activity of LPNs and NPs against cardiac cells. The cell viability of the LPNs and NPs groups over the concentration range from 1 to 100 μM was over 80% compared with the controls (Figure 7). The cell viability of free TN group reduced with the concentration, and the group showed obvious higher cytotoxicity than the nanocarriers in high concentrations (100 and 200 μM) (P < 0.05). The drug-free TPP-TPGS/LPNs exhibited the lowest toxicity at all concentrations studied (1–200 μM).
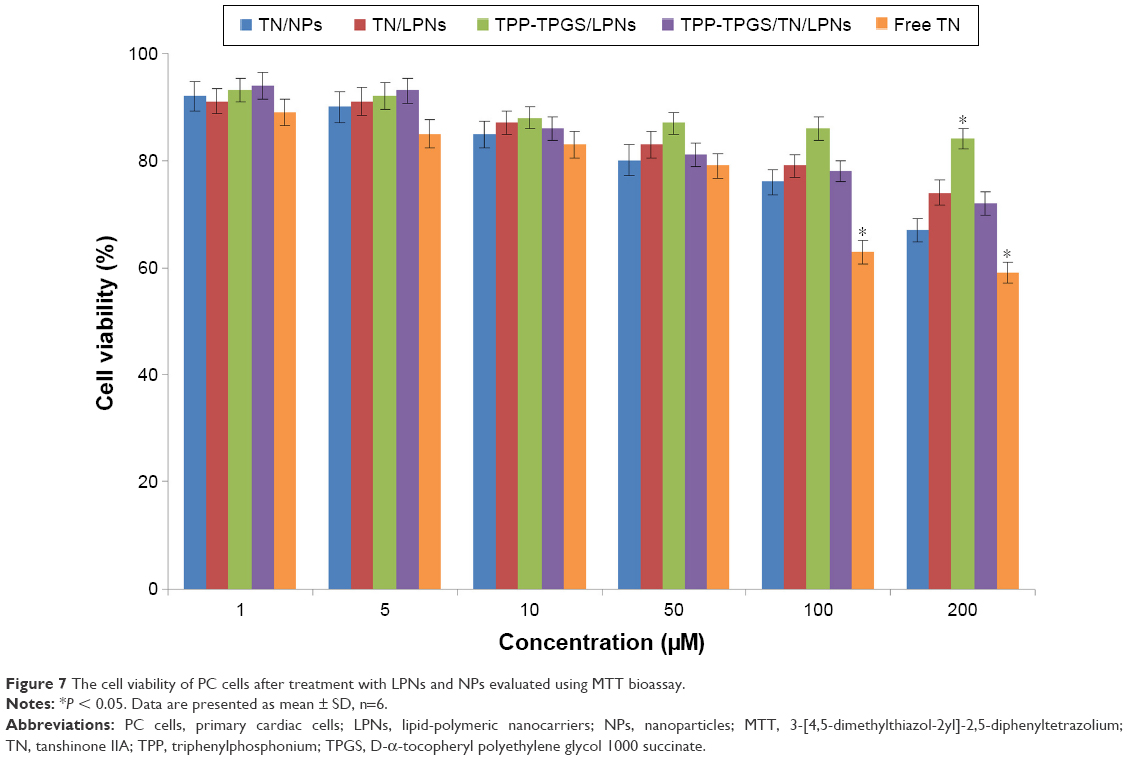 | Figure 7 The cell viability of PC cells after treatment with LPNs and NPs evaluated using MTT bioassay. |
In vivo pharmacokinetics
The mean plasma drug concentration–time profile and the main pharmacokinetics parameters are shown in Figure 8 and Table 1, respectively. It can be seen from the plasma drug concentration–time profile that the free TN was rapidly cleared from the circulation within 6 h, whereas TN-loaded LPNs and NPs exhibited a prolonged plasma circulation time. The half-lives of TN-loaded LPNs and NPs were shorter than that of the free TN (P < 0.05). TPP-TPGS/TN/LPNs exhibited the maximum area under the plasma drug concentration–time curve (AUC) (129.27±3.37 mg/L·h) followed by TN/LPNs (42.27±2.65 mg/L·h) and TN/NPs (20.46±1.95 mg/L·h). The plasma clearance and volume of distribution of TPP-TPGS/TN/LPNs were significantly higher than those of nonmodified TN/LPNs, TN/NPs, and free TN (P < 0.05).
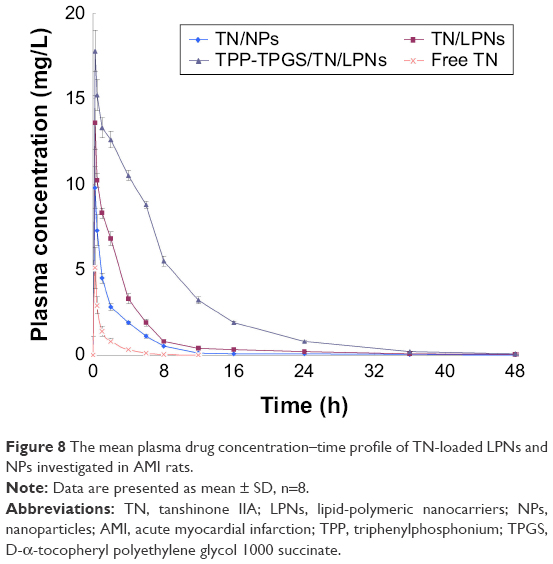 | Figure 8 The mean plasma drug concentration–time profile of TN-loaded LPNs and NPs investigated in AMI rats. |
 | Table 1 Pharmacokinetic parameters after iv administration in rats (n=8) |
In vivo tissue distribution
In vivo tissue distribution of TN-loaded LPNs, NPs, and free TN was investigated in AMI rats (Figure 9). TN distribution of TN-loaded LPNs and NPs was higher in the heart than free TN (P < 0.05). TPP-TPGS/TN/LPNs exhibited the highest TN distribution in the heart compared with TN/LPNs and TN/NPs (P < 0.05). TPP-TPGS/TN/LPNs and TN/LPNs distributed significantly more than free TN (P < 0.01).
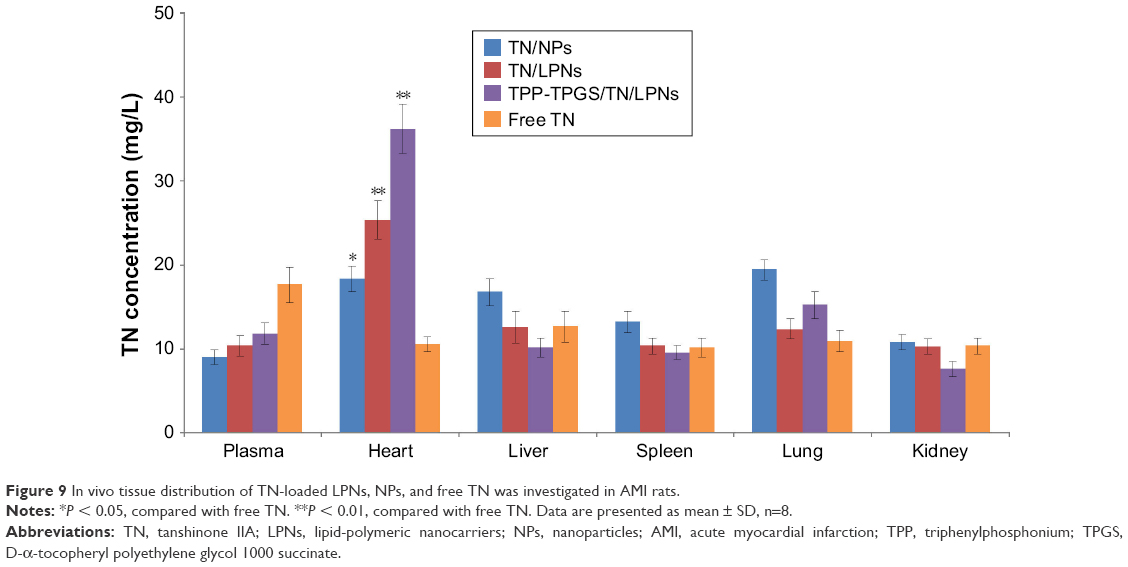 | Figure 9 In vivo tissue distribution of TN-loaded LPNs, NPs, and free TN was investigated in AMI rats. |
In vivo infarct therapy effect
The infarct size was significantly decreased in free TN group (51%±1.8%) compared with the AMI group (62%±2.3%; P < 0.05) (Figure 10). Furthermore, the infarct size was significantly decreased in drugs-loaded LPNs and NPs groups than the free TN group (P < 0.05). The most obvious efficiency was exhibited by TPP-TPGS/TN/LPNs group (28%±1.9%) than the TN/LPNs group (37%±1.7%; P < 0.05), TN/NPs groups (46%±2.1%; P < 0.05), and free TN group (P < 0.01).
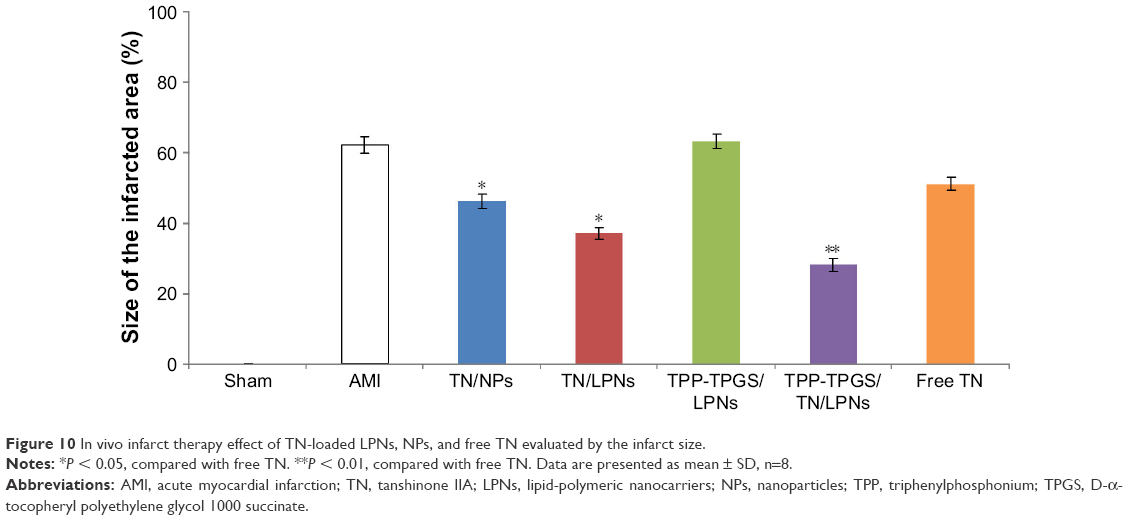 | Figure 10 In vivo infarct therapy effect of TN-loaded LPNs, NPs, and free TN evaluated by the infarct size. |
Discussion
In the present study, we report the development of LPNs for mitochondria-targeted delivery of TN. TPGS was linked to the TPP cation as a targeted ligand. The LPNs were fabricated by nanoprecipitation method. The mean diameter of TN/LPNs (121 nm) was larger than that of TN/NPs (90 nm). This could be explained by the lipid shell on the polymeric core that increased the size of the nanocarriers. Compared with non-TPP-TPGS-modified TN/LPNs, the diameters of TPP-TPGS/LPNs and TPP-TPGS/TN/LPNs increased by about 20 nm, which may be due to the TPP-TPGS ligands coated over TN/LPNs. This could also be taken as an evidence that TPP-TPGS was successfully modified on the outer layer of the system.37 Zeta potential is a significant factor to determine the stability of NPs in suspension through the electrostatic repulsion between particles.38 PLGA is a negatively charged polymer; the negative zeta potential of polymeric TN/NPs (−34 mV) could be due to PLGA. TN/LPNs had a higher zeta potential (−22 mV), which could be explained by that the lipid shell at the surface of nanocarriers was able to shield the surface negative charge of PLGA.39 After TPP-TPGS modification, the surface charge of TPP-TPGS/LPNs and TPP-TPGS/TN/LPNs kept on increasing (−10 mV), indicating that the surface charge of the NPs became positive due to the existence of the positively charged TPP-TPGS. The EE of all systems was high and showed no significant difference. These results indicated that LPNs and NPs had good drug entrapment capacity.
The storage stability of LPNs and NPs was evaluated over a period of 120 days in terms of mean diameter, zeta potential, DL, and EE. The average particle diameter of both LPNs and NPs remained almost constant throughout the period. This could be evidence that no aggregate was formed. The value of the zeta potential also indicated the stability of nanocarriers in suspension.40 For both formulations, the zeta potential was negative and showed no major changes during the 120 days of storage. The DL and EE of drug-loaded LPNs and NPs remained stable at all the time points tested in the research, indicating that the systems were stable within 4 months.
The in vitro release of TN from LPNs was slower than polymeric NPs. This difference may be due to the lipid shell on the surface of LPNs which hindered the drug release.41 More sustained release behavior exhibited by the profile of TPP-TPGS/TN/LPNs than that of the nonmodified TN/LPNs may be explained by the TPP-TPGS modification, which prolonged the release of drugs from the systems. The reason behind this prolongation of release time can be attributed to slow degradation of the nanomaterials; drugs were diffused from the matrix in a sustained manner. Moreover, the lipid shell on the outside of the polymer core also protected the drugs and allowed them to release in a more sustained manner. In addition, in the presence of serum, the release of drugs was not affected, indicating the stability of LNPs in circulation.
Cellular uptake research could provide some circumstantial evidence to display the advantages of the nanocarriers in entering the cells. The cellular uptake of LNPs and NPs by PC cells was studied by using FITC-containing nanocarriers. The cells were treated with different formulations for 4 and 48 h. The cellular uptake efficiency of TPP-TPGS-modified LNPs was significantly higher than nonmodified LNPs. This could be attributed to the ability of TPP to pass easily through lipid bilayers via the cells and accumulate in the cells, and to prolong blood circulation time, enhance the solubility of hydrophobic drugs, and improve cellular uptake.42
In vitro cytotoxicity of LPNs to the cardiac cells could be an evidence of the safety of the system. The cytotoxicity of all samples conformed to a concentration-dependent pattern.43 TPP-TPGS/LPNs exhibited the lowest toxicity at all concentrations studied. This could be explained by the excellent compatibility of the lipid shell to the cell membranes, which could reduce the toxicity effect to the cardiac cells. This is good for the delivery system and could be safe for administration. Free TN group showed obvious higher cytotoxicity than the nanocarriers in high concentrations, which means the encapsulation of TN in the carriers could somehow reduce the toxicity of the drug. The viability of the LPNs over the concentration range studied (below 100 μM) was over 80%; therefore, a concentration below 100 μM may be safe for the use of this nanosystem.
In vivo pharmacokinetics and biodistribution in rats were assessed. The plasma drug concentration–time curves of three formulations were all fitted with the two-compartment open model, which was chosen to analyze the pharmacokinetics data. Pharmacokinetics and biodistribution results showed similar longer circulating characteristics of LNPs and NPs than that of free drug, which proved that nanocarriers elongate the circulation time of drugs in serum and have a great potential to accumulate in the MI area.44 Particularly, LPNs formulations showed better AUC and heart distribution than polymeric NPs, which could be a proof of their better efficiency. Furthermore, TPP-TPGS/TN/LPNs showed obvious heart accumulation than nonmodified TN/LPNs, which is in accordance with the aim of the modification – to deliver more nanocarriers to the infarct zone. This phenomenon could be due to the ligands-mediated targeting of TPP-TPGS-modified LPNs.45
In vivo infarct therapy effect was evaluated by measuring the infarct size. Infarct size is conceived as one of the critical indices for evaluating the cardiac damage in the generation of ischemic heart disease.46 In this study, compared with free TN and TN/NPs, TN/LPNs exhibited obviously better efficiency in infarct therapy. Moreover, TPP-TPGS/TN/LPNs group showed the smallest infarct size compared with TN/LPNs group and all other groups studied. Thus, TPP-TPGS/TN/LPNs is more suitable to treat acute myocardial ischemia than other similar carriers and free drugs.
Conclusion
In summary, we synthesized TPP-Lys-TPGS as a ligand and prepared TPP-TPGS/TN/LPNs as a promising drug delivery carrier due to the targeting ability and core–shell structure of the LPNs. Compared with free TN and TN/NPs, TN/LPNs exhibited significantly improved compatibility and therapeutic efficiency. In addition, the in vivo pharmacokinetics, biodistribution, and infarct therapy studies in SD rats showed that TPP-TPGS/TN/LPNs had better efficiency than their nonmodified TN/LPNs counterparts in all respects. These results indicated that the TPP-TPGS/TN/LPNs were promising nanocarriers for efficient delivery of cardiovascular drugs and other therapeutic agents for the treatment of CVDs.
Disclosure
The authors report no conflicts of interest in this work.
References
GBD 2013 Mortality and Causes of Death Collaborators. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;385(9963):117–171. | ||
Lloyd-Jones D, Adams RJ, Brown TM, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Executive summary: heart disease and stroke statistics – 2010 update: a report from the American Heart Association. Circulation. 2010;121(7):948–954. | ||
Writing Committee Members, Yancy CW, Jessup M, Bozkurt B, et al; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation. 2013;128(16):e240–e327. | ||
Lundy DJ, Chen KH, Toh EK, Hsieh PC. Distribution of systemically administered nanoparticles reveals a size-dependent effect immediately following cardiac ischaemia-reperfusion injury. Sci Rep. 2016;6:25613. | ||
Zhang S, Wang J, Pan J. Baicalin-loaded PEGylated lipid nanoparticles: characterization, pharmacokinetics, and protective effects on acute myocardial ischemia in rats. Drug Deliv. 2016;23(9):3696–3703. | ||
Murphy MP, Smith RA. Drug delivery to mitochondria: the key to mitochondrial medicine. Adv Drug Deliv Rev. 2000;41(2):235–250. | ||
Smith RA, Porteous CM, Gane AM, Murphy MP. Delivery of bioactive molecules to mitochondria in vivo. Proc Natl Acad Sci U S A. 2003;100(9):5407–5412. | ||
Ong SB, Lu S, Katwadi K, Ismail NI, Kwek XY, Hausenloy DJ. Nanoparticle delivery of mitoprotective agents to target ischemic heart disease. Future Cardiol. 2017;13(3):195–198. | ||
Kelso GF, Porteous CM, Coulter CV, et al. Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. J Biol Chem. 2001;276(7):4588–4596. | ||
Neuzil J, Widén C, Gellert N, et al. Mitochondria transmit apoptosis signalling in cardiomyocyte-like cells and isolated hearts exposed to experimental ischemia-reperfusion injury. Redox Rep. 2007;12(3):148–162. | ||
Singh RP, Sharma G, Sonali, et al. Vitamin E TPGS conjugated carbon nanotubes improved efficacy of docetaxel with safety for lung cancer treatment. Colloids Surf B Biointerfaces. 2016;141:429–442. | ||
Cheng W, Liang C, Xu L, et al. TPGS-functionalized polydopamine-modified mesoporous silica as drug nanocarriers for enhanced lung cancer chemotherapy against multidrug resistance. Small. 2017;13(29). | ||
Jin X, Yang Q, Zhang Y. Synergistic apoptotic effects of apigenin TPGS liposomes and tyroservatide: implications for effective treatment of lung cancer. Int J Nanomedicine. 2017;12:5109–5118. | ||
Dong K, Yan Y, Wang P, et al. Biodegradable mixed MPEG-SS-2SA/TPGS micelles for triggered intracellular release of paclitaxel and reversing multidrug resistance. Int J Nanomedicine. 2016;11:5109–5123. | ||
Xu G, Yu X, Zhang J, et al. Robust aptamer-polydopamine-functionalized M-PLGA-TPGS nanoparticles for targeted delivery of docetaxel and enhanced cervical cancer therapy. Int J Nanomedicine. 2016;11:2953–2965. | ||
Tan S, Zou C, Zhang W, Yin M, Gao X, Tang Q. Recent developments in d-α-tocopheryl polyethylene glycol-succinate-based nanomedicine for cancer therapy. Drug Deliv. 2017;24(1):1831–1842. | ||
Ho YT, Poinard B, Kah JC. Nanoparticle drug delivery systems and their use in cardiac tissue therapy. Nanomedicine (Lond). 2016;11(6):693–714. | ||
Maranhão RC, Tavares ER. Advances in non-invasive drug delivery for atherosclerotic heart disease. Expert Opin Drug Deliv. 2015;12(7):1135–1147. | ||
Zhu B, Yu L, Yue Q. Co-delivery of vincristine and quercetin by nanocarriers for lymphoma combination chemotherapy. Biomed Pharmacother. 2017;91:287–294. | ||
Zhou L, Zuo Z, Chow MS. Danshen: an overview of its chemistry, pharmacology, pharmacokinetics, and clinical use. J Clin Pharmacol. 2005;45(12):1345–1359. | ||
Yuan X, Jing S, Wu L, Chen L, Fang J. Pharmacological postconditioning with tanshinone IIA attenuates myocardial ischemia-reperfusion injury in rats by activating the phosphatidylinositol 3-kinase pathway. Exp Ther Med. 2014;8(3):973–977. | ||
Cai Y, Zhang W, Chen Z, Shi Z, He C, Chen M. Recent insights into the biological activities and drug delivery systems of tanshinones. Int J Nanomedicine. 2016;11:121–130. | ||
Zhu H, Hao J, Chen H, et al. Nanovesicles system for rapid-onset sublingual delivery containing sodium tanshinone IIA sulfonate: in vitro and in vivo evaluation. J Pharm Sci. 2013;102(7):2332–2340. | ||
Mossalam M, Soto J, Lim CS, Abel ED. Solid phase synthesis of mitochondrial triphenylphosphonium-vitamin E metabolite using a lysine linker for reversal of oxidative stress. PLoS One. 2013;8(1):e53272. | ||
Zhang X, Li X, Hua H, et al. Cyclic hexapeptide-conjugated nanoparticles enhance curcumin delivery to glioma tumor cells and tissue. Int J Nanomedicine. 2017;12:5717–5732. | ||
Dufaÿ Wojcicki A, Hillaireau H, Nascimento TL, et al. Hyaluronic acid-bearing lipoplexes: physico-chemical characterization and in vitro targeting of the CD44 receptor. J Control Release. 2012;162(3):545–552. | ||
Chen C, You P. A novel local anesthetic system: transcriptional transactivator peptide-decorated nanocarriers for skin delivery of ropivacaine. Drug Des Devel Ther. 2017;11:1941–1949. | ||
Yu D, Li W, Zhang Y, Zhang B. Anti-tumor efficiency of paclitaxel and DNA when co-delivered by pH responsive ligand modified nanocarriers for breast cancer treatment. Biomed Pharmacother. 2016;83:1428–1435. | ||
Ferreira MPA, Ranjan S, Kinnunen S, et al. Drug-loaded multifunctional nanoparticles targeted to the endocardial layer of the injured heart modulate hypertrophic signaling. Small. 2017;13(33). | ||
Lomis N, Gaudreault F, Malhotra M, Westfall S, Shum-Tim D, Prakash S. A novel milrinone nanoformulation for use in cardiovascular diseases: preparation and in vitro characterization. Mol Pharm. Epub 2017 Aug 24. | ||
Yu W, Liu C, Ye J, Zou W, Zhang N, Xu W. Novel cationic SLN containing a synthesized single-tailed lipid as a modifier for gene delivery. Nanotechnology. 2009;20(21):215102. | ||
Yang L, Zhang Z, Hou J, et al. Targeted delivery of ginsenoside compound K using TPGS/PEG-PCL mixed micelles for effective treatment of lung cancer. Int J Nanomedicine. 2017;12:7653–7667. | ||
Qiu J, Cai G, Liu X, Ma D. α(v)β(3) integrin receptor specific peptide modified, salvianolic acid B and panax notoginsenoside loaded nanomedicine for the combination therapy of acute myocardial ischemia. Biomed Pharmacother. 2017;96:1418–1426. | ||
Liu X, Gu J, Fan Y, Shi H, Jiang M. Baicalin attenuates acute myocardial infarction of rats via mediating the mitogen-activated protein kinase pathway. Biol Pharm Bull. 2013;36(6):988–994. | ||
Wei Y, Pi C, Yang G, et al. LC-UV determination of baicalin in rabbit plasma and tissues for application in pharmacokinetics and tissue distribution studies of baicalin after intravenous administration of liposomal and injectable formulations. Molecules. 2016;21(4):444. | ||
Sun SJ, Wu XP, Song HL, Li GQ. Baicalin ameliorates isoproterenol-induced acute myocardial infarction through iNOS, inflammation, oxidative stress and P38MAPK pathway in rat. Int J Clin Exp Med. 2015;8(12):22063–22072. | ||
Tao W, Zeng X, Wu J, et al. Polydopamine-based surface modification of novel nanoparticle-aptamer bioconjugates for in vivo breast cancer targeting and enhanced therapeutic effects. Theranostics. 2016;6(4):470–484. | ||
Zeng X, Tao W, Mei L, Huang L, Tan C, Feng SS. Cholic acid-functionalized nanoparticles of star-shaped PLGA-vitamin E TPGS copolymer for docetaxel delivery to cervical cancer. Biomaterials. 2013;34(25):6058–6067. | ||
Zou W, Liu C, Chen Z, Zhang N. Studies on bioadhesive PLGA nanoparticles: a promising gene delivery system for efficient gene therapy to lung cancer. Int J Pharm. 2009;370(1–2):187–195. | ||
Yang CC, Hung CF, Chen BH. Preparation of coffee oil-algae oil-based nanoemulsions and the study of their inhibition effect on UVA-induced skin damage in mice and melanoma cell growth. Int J Nanomedicine. 2017;12:6559–6580. | ||
Wang C, Su L, Wu C, Wu J, Zhu C, Yuan G. RGD peptide targeted lipid-coated nanoparticles for combinatorial delivery of sorafenib and quercetin against hepatocellular carcinoma. Drug Dev Ind Pharm. 2016;42(12):1938–1944. | ||
Yu A, Lv J, Yuan F, et al. mPEG-PLA/TPGS mixed micelles via intranasal administration improved the bioavailability of lamotrigine in the hippocampus. Int J Nanomedicine. 2017;12:8353–8362. | ||
Han Y, Zhang Y, Li D, Chen Y, Sun J, Kong F. Transferrin-modified nanostructured lipid carriers as multifunctional nanomedicine for codelivery of DNA and doxorubicin. Int J Nanomedicine. 2014;9:4107–4116. | ||
Dong Z, Guo J, Xing X, Zhang X, Du Y, Lu Q. RGD modified and PEGylated lipid nanoparticles loaded with puerarin: formulation, characterization and protective effects on acute myocardial ischemia model. Biomed Pharmacother. 2017;89:297–304. | ||
Hao J, Tong T, Jin K, et al. Folic acid-functionalized drug delivery platform of resveratrol based on Pluronic 127/D-α-tocopheryl polyethylene glycol 1000 succinate mixed micelles. Int J Nanomedicine. 2017;12:2279–2292. | ||
Qi Q, Lu L, Li H, et al. Spatiotemporal delivery of nanoformulated liraglutide for cardiac regeneration after myocardial infarction. Int J Nanomedicine. 2017;12:4835–4848. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.