Back to Journals » Vascular Health and Risk Management » Volume 12
Triglyceride-rich lipoproteins as a causal factor for cardiovascular disease
Authors Toth P
Received 15 January 2016
Accepted for publication 15 March 2016
Published 6 May 2016 Volume 2016:12 Pages 171—183
DOI https://doi.org/10.2147/VHRM.S104369
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Daniel Duprez
Peter P Toth1,2
1Ciccarone Center for the Prevention of Cardiovascular Disease, Johns Hopkins University School of Medicine, Baltimore, MD, 2Preventive Cardiology, CGH Medical Center, Sterling, IL, USA
Abstract: Approximately 25% of US adults are estimated to have hypertriglyceridemia (triglyceride [TG] level ≥150 mg/dL [≥1.7 mmol/L]). Elevated TG levels are associated with increased cardiovascular disease (CVD) risk, and severe hypertriglyceridemia (TG levels ≥500 mg/dL [≥5.6 mmol/L]) is a well-established risk factor for acute pancreatitis. Plasma TG levels correspond to the sum of the TG content in TG-rich lipoproteins (TRLs; ie, very low-density lipoproteins plus chylomicrons) and their remnants. There remains some uncertainty regarding the direct causal role of TRLs in the progression of atherosclerosis and CVD, with cardiovascular outcome studies of TG-lowering agents, to date, having produced inconsistent results. Although low-density lipoprotein cholesterol (LDL-C) remains the primary treatment target to reduce CVD risk, a number of large-scale epidemiological studies have shown that elevated TG levels are independently associated with increased incidence of cardiovascular events, even in patients treated effectively with statins. Genetic studies have further clarified the causal association between TRLs and CVD. Variants in several key genes involved in TRL metabolism are strongly associated with CVD risk, with the strength of a variant's effect on TG levels correlating with the magnitude of the variant's effect on CVD. TRLs are thought to contribute to the progression of atherosclerosis and CVD via a number of direct and indirect mechanisms. They directly contribute to intimal cholesterol deposition and are also involved in the activation and enhancement of several proinflammatory, proapoptotic, and procoagulant pathways. Evidence suggests that non-high-density lipoprotein cholesterol, the sum of the total cholesterol carried by atherogenic lipoproteins (including LDL, TRL, and TRL remnants), provides a better indication of CVD risk than LDL-C, particularly in patients with hypertriglyceridemia. This article aims to provide an overview of the available epidemiological, clinical, and genetic evidence relating to the atherogenicity of TRLs and their role in the progression of CVD.
Keywords: triglycerides, non-highdensity, lipoprotein cholesterol, hypertriglyceridemia, lipoprotein lipase, chylomicrons, very low-density lipoproteins
Introduction
Robust clinical evidence demonstrates that statin-induced reductions in low-density lipoprotein cholesterol (LDL-C) lead to substantial reductions in cardiovascular disease (CVD) risk in both the primary and secondary prevention settings.1 Therefore, in patients at high cardiovascular risk due to dyslipidemia, the majority of current guidelines recommend LDL-C as a primary treatment target, with statins as first-line therapy.2–4 Despite significant LDL-C lowering with statin therapy, substantial residual cardiovascular risk often remains.5 This residual risk is thought to be due, in part, to inadequate reduction of LDL-C for a given level of risk, low levels of high-density lipoprotein cholesterol (HDL-C), and/or high levels of triglycerides (TG).5 Additionally, there is increasing evidence that non-high-density lipoprotein cholesterol (non-HDL-C), which is calculated for individuals by subtracting their HDL-C value from total cholesterol (including TG-rich lipoproteins [TRLs]), may be more strongly associated with atherosclerotic risk than LDL-C alone.6
Approximately 25% of US adults are estimated to have hypertriglyceridemia, defined as a TG level of ≥150 mg/dL (≥1.7 mmol/L).7 Hypertriglyceridemia often coexists with secondary disorders that are independently associated with increased plasma TG levels, such as type 2 diabetes mellitus, chronic kidney disease, metabolic syndrome, and obesity.8 Severe hypertriglyceridemia, defined by the 2014 National Lipid Association guidelines as a TG level of ≥500 mg/dL (≥5.6 mmol/L), is a well-established risk factor for acute pancreatitis,9 and moderately elevated TG levels have been shown to be independently associated with increased CVD risk, even in patients treated effectively with statins to reduce LDL-C.10,11
Plasma TG levels are known to correspond with the levels of TRLs and their remnants.2 However, hypertriglyceridemia is also often accompanied by further lipoprotein disturbances, including increased very low-density lipoproteins (VLDLs) and total apolipoprotein (apo) C-III, elevated levels of small, dense LDL-C and total LDL particles, and decreased levels of HDL-C, all of which have been shown to be associated with increased CVD risk.12 This, along with the fact that cardiovascular outcome studies of TG-lowering agents have produced inconsistent results,13,14 means that there remains some uncertainty regarding the direct causal role of TRLs in the progression of atherosclerosis and CVD.8
This review aims to provide an overview of the available genetic, epidemiological, and clinical evidence relating to the atherogenicity of TRLs and their role in the progression of CVD.
Search strategy
A search of PubMed was performed using the following search strategy: (“triglyceride-rich lipoproteins” OR “apolipoprotein C-III” OR “remnant lipoproteins” OR “intermediate-density lipoproteins”) AND (“Cardiovascular Diseases”[MeSH]).
The search was limited to English-language publications published between July 2005 and July 2015. The reference lists of articles identified using this search strategy was also searched such that widely referenced, older publications were also screened.
TRL metabolism
Cholesterol esters and TG are the two most important circulating lipids.15 Owing to their hydrophobic nature, they are combined into lipoprotein particles in association with proteins that allow them to be transported in the plasma. Cholesterol is transported by all lipoproteins and is particularly concentrated in HDL and LDL particles. In general, TGs are transported in the plasma in VLDL, chylomicrons, and their remnants created during metabolism.16 These TRLs are the largest lipoprotein particles. In addition to size, lipoproteins may also be characterized by the apos they contain, with apoB100 (also known as apoB) being associated with VLDL and LDL and apoA-I associated with HDL.17 There are several other apos involved in lipoprotein metabolism, many of which are thought to potentially contribute to a number of diseases, such as CVD, multiple sclerosis, and Alzheimer’s disease.18–20
TRLs are highly heterogeneous, differing in size, density, composition, and associated cardiovascular risk.21 They are composed of a neutral core of TG and cholesterol esters and a surface monolayer comprising phospholipids, free cholesterol, and apos, which participate in the regulation of transport and metabolism of the TRL.22 The metabolism of TRLs occurs via two principal pathways, an exogenous pathway that originates in the small intestine and an endogenous pathway regulated by the liver (Figure 1). During the exogenous pathway, TGs from dietary fat are absorbed by enterocytes following the ingestion of a meal. Here, they are incorporated into chylomicrons, which are large apoB48-containing lipoproteins with a large TG core (80%–95%).23 Newly synthesized chylomicrons are then exported via perimesenteric lymphatics before entering the circulation, where they acquire apoC-II, apoC-III, and apoE. Once in circulation, chylomicrons are quickly hydrolyzed by lipoprotein lipase (LPL) along the luminal surface of the capillaries. LPL is synthesized by myocytes and adipocytes before being transported to the lumen of the capillaries via GPIHBPI, a small glycosylphosphatidylinositol-anchored protein synthesized by the capillary endothelial cells.24,25 LPL requires activation via apoC-II,26 and its activity is highly regulated by various proteins, including apoC-III, apoA5, and angiopoietin-like proteins 3 and 4.27 The hydrolysis of chylomicrons via LPL results in the production of free fatty acids and chylomicron remnants. The free fatty acids liberated by lipolysis are oxidized by a variety of cell types, such as skeletal and myocardial myocytes, or stored in adipose tissue. The chylomicron remnants, which are rich in cholesterol esters and apoE, are removed from circulation by the liver via binding to the LDL receptor or the LDL receptor-related protein.28
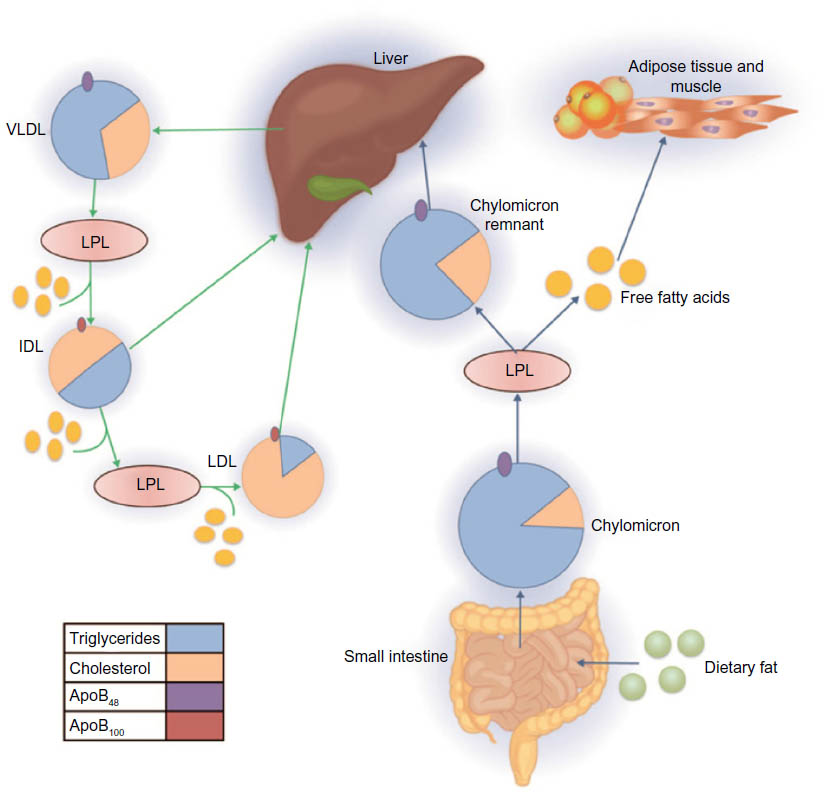 | Figure 1 Overview of triglyceride-rich lipoprotein metabolism. |
During the endogenous pathway, TGs are synthesized in hepatocytes from free fatty acids and glycerol and then incorporated into the core of apoB-containing VLDL particles. ApoC-I, apoC-II, apoC-III, and apoE are added to the surface of VLDL particles during secretion. Following secretion, VLDLs undergo LPL-mediated hydrolysis in the plasma, generating in succession progressively smaller VLDLs and then intermediate-density lipoproteins (IDLs). Some IDL particles are taken up by the liver, and some undergo further catabolism by LPL and hepatic TG lipase to produce LDL particles.28
Importantly, polymorphisms in genes encoding key components of the TRL biosynthetic pathways are known to be strongly associated with CVD risk.16,29–31
Proposed pathophysiology of TRLs in the progression of atherosclerosis
Evidence suggests that TRLs and their remnants and specific markers of TG metabolism, such as LPL and apoC-III, contribute to the progression of atherosclerosis and CVD both directly and indirectly.12,32 These proposed pathophysiological mechanisms are summarized in Figure 2.
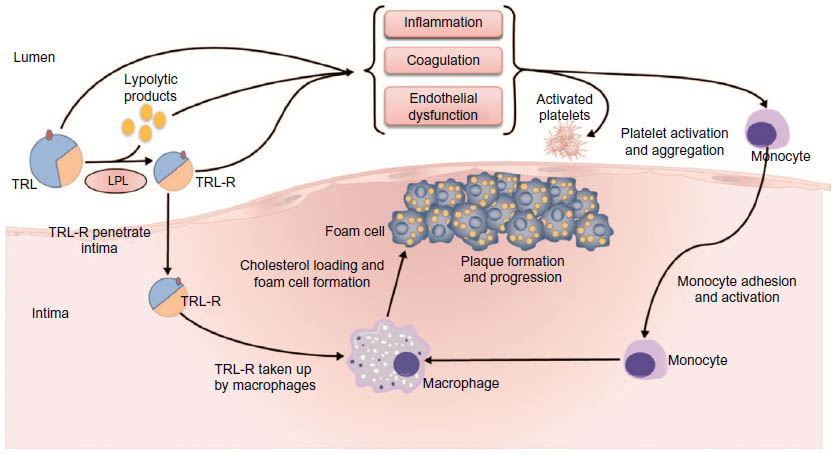 | Figure 2 Proposed pathophysiology of triglyceride-rich lipoproteins in the progression of atherosclerosis. |
It has been suggested that it is primarily the cholesterol content of TRL remnants that directly contributes to the progression of atherosclerosis, rather than the TGs themselves.15 Like LDL, cholesterol-enriched, TG-depleted TRL remnants are able to penetrate the arterial intima, where they become selectively bound to the connective tissue matrix. Once entrapped in the subendothelial space, TRLs can be scavenged by resident macrophages, thereby contributing to macrophage foam cell formation as well as plaque formation and progression.12,33–36 TRLs are thought to be equally or more atherogenic than LDL. In contrast to LDL, TRL remnants can be taken up directly by arterial macrophages without oxidative modification37–39 and, due to their larger size, carry more cholesterol per particle than LDL.12 TRL remnants have also been shown to promote endothelial dysfunction, which potentiates atherogenesis.40
LPL-mediated TRL hydrolysis results in a high concentration of lipolytic products, such as oxidized free fatty acids, along the vascular endothelium or within the arterial intima. These lipolytic products, along with TRLs themselves, are also known to activate a number of proinflammatory, procoagulant, and proapoptotic signaling pathways that play a fundamental role in the pathogenesis of atherosclerosis.12 Oxidized free fatty acids are known to increase the expression of inflammatory interleukins and cytokines, leading to endothelial inflammation,41–43 while TRL remnants have been shown to upregulate the endothelial expression of intercellular adhesion molecule-1 and vascular cell adhesion molecule-1.44,45 These proatherogenic adhesion molecules facilitate the transendothelial migration of leukocytes to sites of inflammation.46 Consequently, their TRL remnant-mediated activation leads to endothelial monocyte adhesion and an enhanced inflammatory response.44,45 TRL remnants have also been shown to induce early monocyte and neutrophil activation.47
Additionally, TRL remnants are known to increase the production of reactive oxygen species, which can increase vascular endothelial permeability, promote leukocyte adhesion, and, at high concentrations, cause cellular injury and death.43,48 TRL remnants have also been shown to induce endothelial cell apoptosis via increased secretion of the proapoptotic cytokines, tumor necrosis factor-α, and interleukin-1β, a process that is known to contribute to vascular injury and atherosclerosis.49 TRLs and their remnants enhance platelet aggregation and clot formation and amplify the coagulation cascade by 1) supporting the assembly of the prothombinase complex and 2) upregulating the expression of plasminogen activator inhibitor-1 and plasminogen activator inhibitor-1 antigen.50 They have also been shown to upregulate the endothelial expression of tissue factor, a key initiator of the coagulation cascade.44,45,51 Finally, TRL remnants suppress the atheroprotective and anti-inflammatory effects of HDL52 and have been shown to significantly correlate with impairment of coronary vasodilation.53
Key components of TRL metabolism have also been shown to be associated with CVD, in particular apoC-III, a key contributor to hypertriglyceridemia due to its inhibitory effects on LPL.30,31,54 ApoC-III is thought to contribute to the progression of atherosclerosis via a number of mechanisms:54 it can impair VLDL binding to cellular receptors, resulting in small, dense LDL particle formation,12 and has been shown to induce the expression of proinflammatory mediators and to stimulate monocyte activation and the adhesion of monocytes to endothelial cells.55 It is also thought to induce apoptosis56,57 and may accumulate on atheroprotective HDL particles on secretion, thus rendering them dysfunctional.58
Genetic evidence
The fact that monogenic disorders of TG metabolism, such as hyperlipoproteinemia type 3, predispose individuals to CVD suggests that raised TG and remnant cholesterol levels contribute to this process.29 Conversely, a recent Mendelian randomization study based on data from 10,208 individuals included in the Copenhagen City Heart Study found that subjects with genetically confirmed reduction in nonfasting plasma TG levels had reduced all-cause mortality.31 An additional meta-analysis involving 188,578 genotyped individuals with 185 different single nucleotide polymorphisms found that the strength of a variant’s effect on TG levels strongly correlated with the magnitude of its effect on coronary artery disease, even after adjustment for effects on LDL-C and HDL-C. These results support the hypothesis that TRLs causally influence cardiovascular risk.16
Sequence variants in several key genes involved in the metabolism of TRLs, such as those encoding LPL and the proteins that regulate it, appear to be strongly associated with CVD risk.30,31 For example, apoC-III, an apolipoprotein playing a central role in TG metabolism by inhibiting LPL, is overexpressed in hypertriglyceridemia and is significantly associated with cardiovascular risk.54 In one study, which evaluated 18,666 genes in 3,734 participants, four loss-of-function mutations were identified in APOC3, the gene encoding apoC-III. Heterozygous carriers of any of these mutations had 46% lower circulating levels of apoC-III, corresponding to 39% lower plasma TG levels, and a 40% lower risk of coronary heart disease than noncarriers.59 Similarly, a second study, which analyzed data from 75,725 participants, found that heterozygosity of loss-of-function mutations in APOC3 were associated with a mean reduction in nonfasting TG levels of 44% and a corresponding 41% decrease in the incidence of ischemic vascular disease compared with wild-type individuals.60 In addition, mutations in the gene encoding apoAV (APOA5), an activator of LPL, have also been shown to be associated with CVD risk. Carriers of nonsynonymous APOA5 mutations have higher plasma TG levels, lower HDL-C levels, but similar overall cholesterol levels, compared with noncarriers. Carriers of these mutations were shown to have a 2.2-fold higher risk of myocardial infarction (MI) and coronary artery disease compared with noncarriers.20 Furthermore, polymorphisms in the APOA5 promoter region, which led to decreased APOA5 expression, were strongly related to increased plasma TG levels and a concordant increase in coronary heart disease risk.61 Finally, loss-of-function variants in the gene encoding angiopoietin-like protein 4 (ANGPTL4), an inhibitor of LPL, were found to be associated with substantially decreased TG levels and decreased coronary heart disease risk.62
Genetically elevated levels of TG and remnant lipoprotein cholesterol are also associated with increased low-grade inflammation, marked by elevated C-reactive protein levels. As this association was not observed for genetically elevated LDL-C, this suggests that the inflammatory component of atherosclerosis may be driven by elevated TRLs and remnant cholesterol.63 Taken together, these findings are consistent with the hypothesis that elevated TG levels, and consequently TRLs, are causally associated with CVD.
Epidemiological evidence
In the Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis In Myocardial Infarction 22 (PROVE IT-TIMI 22) trial, it was demonstrated that, among patients receiving statin therapy following acute coronary syndrome, an on-treatment fasting TG level <150 mg/dL (<1.7 mmol/L) was associated with a reduction in recurrent coronary heart disease risk versus higher TG levels (Table 1), even after adjustment for HDL-C and LDL-C levels (hazard ratio 0.8; P=0.025).10 A wealth of epidemiological evidence exists, demonstrating that both fasting and nonfasting TG levels are significant predictors of cardiovascular events, even in individuals who have already achieved guideline-recommended LDL-C levels with lipid-lowering therapy.64–68
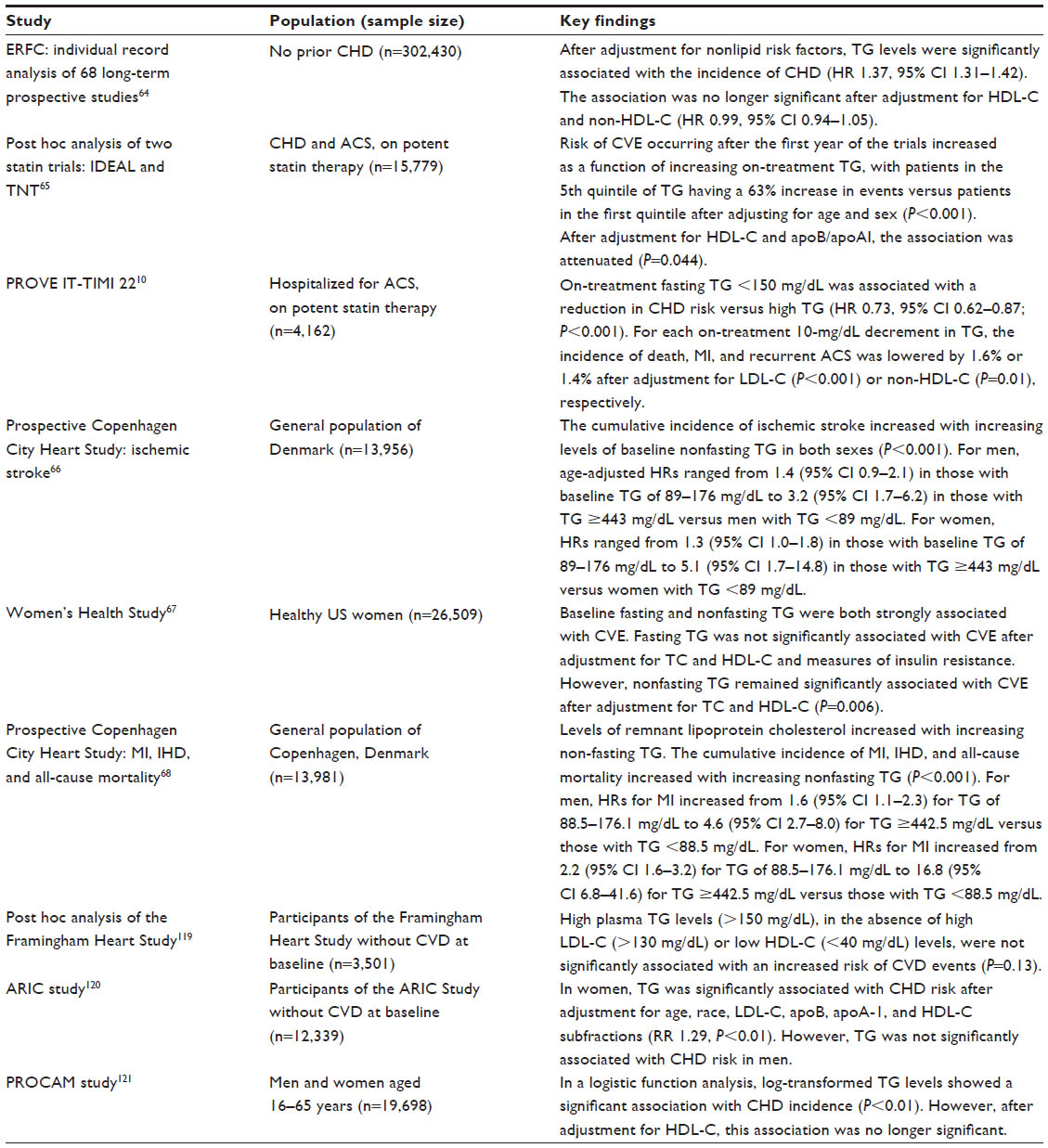 | Table 1 Key studies investigating the association between triglycerides and cardiovascular disease |
However, nonfasting TG levels are thought to be a much stronger predictor of cardiovascular events than fasting TG levels. The Women’s Health Study (n=26,509) showed that both fasting and nonfasting TG levels were strongly associated with an increased risk of cardiovascular events, independent of baseline cardiovascular risk factors (age, blood pressure, smoking, and use of hormone therapy). However, after adjustment for TC and HDL-C levels and indicators of insulin resistance, the association between fasting TG levels and the risk of cardiovascular events was no longer significant (P=0.90). In contrast, the association between nonfasting TG levels and cardiovascular risk remained strong even after adjustment for other lipid levels and markers of insulin resistance (P=0.006).67 Likewise, two prospective cohort studies using data from the Copenhagen City Heart Study found that the cumulative incidence of cardiovascular events (ischemic stroke, MI, ischemic heart disease) and all-cause mortality were strongly associated with increasing nonfasting TG levels (all P<0.001) (Table 1). That being said, these associations were not adjusted for other lipid parameters.66,68
A number of studies have found that the association between plasma TG levels (both fasting and nonfasting) and cardiovascular risk is often attenuated once adjusted for other lipid parameters, including HDL-C and non-HDL-C. An analysis conducted by the Emerging Risk Factors Collaboration, which included data from 302,430 individuals from 68 long-term prospective studies, demonstrated that there was a significant and stepwise association between fasting and nonfasting TG levels and CVD risk. However, this association was no longer significant after adjustment for HDL-C and non-HDL-C (Table 1).64 Likewise, in a combined analysis of the Incremental Decrease in End Points through Aggressive Lipid Lowering (IDEAL) and Treating to New Targets (TNT) trials in patients achieving low LDL-C (<70 mg/dL [1.8 mmol/L]), CVD risk increased incrementally with increasing on-treatment fasting TG level, with patients in the highest quintile experiencing a 63% higher rate of cardiovascular events than those in the lowest quintile (P<0.001). However, this association was also attenuated (P=0.044) after adjustment for HDL-C and apoB/apoA1 (Table 1).65 Elevated TG levels are closely associated with higher levels of non-HDL-C and apoB and low levels of HDL-C,2 and this may explain why this association is weakened after adjustment for these parameters.
Elevated remnant cholesterol levels, which directly correlate with elevated levels of TRLs, have also been shown to be associated with CVD. Using data from 73,513 subjects, Varbo et al30 found that every 88.6 mg/dL (1 mmol/L) increase in remnant cholesterol was associated with a 2.8-fold increase in CVD risk, independent of low HDL-C.
Available treatment options to reduce TG levels and their potential impact on cardiovascular outcomes
Although the clinical definition of the severity of hypertriglyceridemia differs among guidelines,2,7,28,69–71 the majority of guidelines define severe hypertriglyceridemia as a TG level of ≥500 mg/dL (≥5.6 mmol/L).28,70 In such cases, guidelines recommend the initiation of TG-lowering therapy to reduce the risk of pancreatitis.3,4,70 Guidelines also acknowledge that a TG level of <150 mg/dL (<1.7 mmol/L) is desirable, and if elevated TG or non-HDL-C levels remain following lifestyle intervention and statin therapy, a number of guidelines recommend the use of TG-lowering agents, primarily fibrates, niacin, or omega-3 fatty acids (Table 2).2,4,8,70
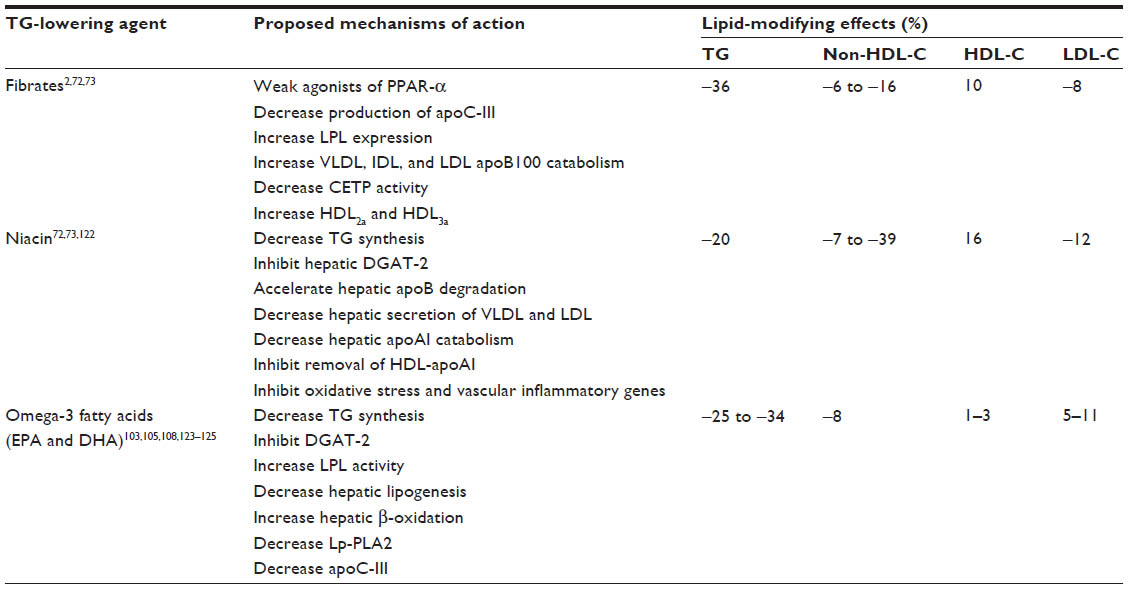 | Table 2 Summary of available triglyceride-lowering therapies |
Fibrates
Fibrates decrease TG levels by ~36%, non-HDL-C levels by ~6%–16%, and LDL-C levels by ~8% and increase HDLC levels by ~10%.72,73 Of note, however, fibrate-induced increases in LDL-C may occur in patients with severe hypertriglyceridemia.74,75 To date, cardiovascular outcome studies of fibrates have produced varied results, with some studies suggesting a small benefit, particularly in patients with other factors besides hypertriglyceridemia, such as low HDL-C or metabolic syndrome, and others showing no benefit.76–79 A meta-analysis including data from 45,058 participants from 18 clinical trials showed that fibrate therapy was associated with a significant decrease in major cardiovascular events (relative risk [RR] reduction 10%; P=0.048), although this did not translate into a benefit for all-cause mortality (RR reduction 0%; P=0.92).14 In another meta-analysis of 7,389 patients with high TG levels (>200 mg/dL [2.3 mmol/L]), fibrate therapy was associated with a 25% decrease in vascular events, and in 5,068 patients with both high TG and low HDL-C levels (<40 mg/dL [1 mmol/L]), a 29% decrease in vascular events was observed.80
Fibrate therapy is associated with a number of adverse effects, including increases in creatinine levels, myopathy, and, in rare cases, rhabdomyolysis, especially when used in combination with other lipid-lowering therapies.79,81,82 Gemfibrozil, in particular, has been shown to increase exposure to and reduce the renal clearance of statins by inhibiting their glucuronidation,83 potentially leading to severe side effects.82 However, fenofibrate appears to be better tolerated than other fibrates, with no cases of rhabdomyolysis observed with fenofibrate–statin combination therapy in two large-scale clinical trials.79,84 Fenofibrate is often recommended for use in combination with statin therapy in patients requiring additional non-HDL-C lowering.2,4,69 Studies have so far failed to demonstrate any significant reduction in cardiovascular risk with fenofibrate–simvastatin combination therapy, compared with simvastatin monotherapy. However, in a subgroup of patients with a TG level in the upper third (≥204 mg/dL [≥2.30 mmol/L]) and an HDL-C level in the lower third (≤34 mg/dL [≤0.88 mmol/L]), there was a nonsignificant trend (P=0.06) for reduction in cardiovascular risk.84
Niacin
Prescription strength niacin is indicated to reduce elevated total cholesterol, LDL-C, apoB, and TG levels and to increase HDL-C in patients with primary hyperlipidemia, severe hypertriglyceridemia, and mixed dyslipidemia. Niacin has been shown to decrease TG levels by 20%, LDL-C levels by 12%, and non-HDL-C levels by 7%–39% and to increase HDL-C levels by an average of 16%.72,73 Despite improvements in coronary atherosclerosis and carotid intima-media thickness, niacin does not appear to impact risk for cardiovascular events when added to statin therapy.85–89 That being said, one post hoc analysis showed that among patients with TG >200 mg/dL (>2.3 mmol/L) and HDL-C <32 mg/dL (<0.8 mmol/L), niacin may reduce cardiovascular events by 37% (P<0.05).90
The use of niacin is often limited due to the high incidence of associated adverse effects.91 The most common adverse effect, cutaneous vasodilatation or “flushing”, reportedly occurs in up to 70% of patients receiving niacin therapy72 and often leads to treatment discontinuation.4,91 Although attempts have been made to reduce the incidence of niacin-induced flushing using laropiprant, a specific antagonist of the prostaglandin D2 receptor, this combination therapy does not significantly reduce the risk of major cardiovascular events and has been shown to actually increase the incidence of adverse effects.88 Additional adverse effects, including hyperglycemia, insulin resistance, hyperuricemia, myopathy, pruritus, and elevations in liver enzymes, are also associated with niacin monotherapy or statin–niacin combination therapy.88,89
Omega-3 fatty acids
Three prescription omega-3 fatty acid formulations are currently approved in the US, including formulations comprising omega-3 carboxylic acids, a mixture of long-chain omega-3 fatty acids in free fatty acid form, primarily eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA), and docosapentaenoic acid (Epanova®);92 omega-3 fatty acid ethyl esters, a mixture of long-chain omega-3 fatty acid ethyl esters, primarily EPA and DHA (Lovaza®, Omtryg®, and some generics);93,94 and icosapent ethyl (EPA ethyl esters) (Vascepa®).95 These prescription omega-3 fatty acids have been shown to reduce plasma TG levels by 25%–45%, VLDL-C levels by 20%–42%, and non-HDL-C levels by 8%–14% in patients with severe hypertriglyceridemia.92,93,95 Increases in HDL-C levels of 5%–9% have also been observed with DHA-containing formulations.92,93 Higher doses of each formulation and higher baseline TG levels are associated with greater TG reductions.96 Of note, DHA-containing formulations are thought to increase LPL expression, leading to increased TG removal from circulating VLDL and chylomicron particles. This results in the increased production of IDL particles, some of which undergo further catabolism by LPL to produce LDL particles.28 Consequently, DHA-containing formulations have been shown to significantly increase LDL-C levels in patients with severe hypertriglyceridemia by up to 45%.92,93 However, these increases are accompanied by reductions in non-HDL-C. This may be of particular importance, as non-HDL-C levels are more strongly associated with the risk of cardiovascular events than LDL-C,6 particularly in patients with hypertriglyceridemia.70
Like fibrates, cardiovascular outcome studies of omega-3 fatty acids have also produced inconsistent results.97–100 A meta-analysis including data from 63,030 individuals from 20 clinical trials demonstrated that omega-3 fatty acid therapy did not have an impact on a composite cardiovascular end point or total mortality (P=0.24 and P=0.28, respectively) but was associated with a significantly decreased rate of vascular death (RR 0.86; 95% confidence interval, 0.75–0.99; P=0.03).13 In subgroup analysis, protection against the composite cardiovascular end point with omega-3 fatty acid use was observed in those trials that enrolled patients with high baseline TG levels (≥150 mg/dL [≥1.7 mmol/L]; RR 0.82; 95% confidence interval 0.74–0.91) versus those with lower baseline TG levels (P=0.006).13 Notably, many of the clinical trials included in the meta-analysis did not use recommended prescription doses of omega-3 fatty acids. The Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardio (GISSI)-Prevenzione Trial showed that in post-MI patients, EPA and DHA significantly reduced the risk of reinfarction and death over a 3-year follow-up period.97 Additionally, in the Japan EPA Lipid Intervention Study (JELIS), the addition of 1.8 mg EPA provided an incremental 18% reduction in major coronary events. In patients with TG >200 mg/dL (>2.3 mmol/L) and HDL-C <40 mg/dL (<1.0 mmol/L), risk reduction was 53% compared with statin monotherapy.98
Heterogeneity in results observed in cardiovascular outcome studies of TG-lowering agents may be due, in part, to the inclusion of subjects with normal baseline TG levels (<150 mg/dL [<1.7 mmol/L]). Therefore, additional large-scale cardiovascular outcome studies in patients with clinically defined hypertriglyceridemia may be beneficial. In this regard, the Reduction of Cardiovascular Events with EPA – Intervention Trial (REDUCE-IT; NCT01492361)101 and the STatin Residual risk reduction with EpaNova in hiGh cardiovascular risk paTients with Hypertriglyceridemia (STRENGTH; NCT02104817) trial102 are currently under way. These studies should provide valuable information on the utility of omega-3 fatty acids in combination with statin therapy in high-risk patients with TG levels of 200–500 mg/dL.
The most common adverse effects associated with omega-3 fatty acids are gastrointestinal (such as nausea and diarrhea).103,104 The rate of treatment discontinuation observed in clinical trials is similar between omega-3 fatty acid groups and placebo groups.81,105–107 Moreover, omega-3 fatty acids do not affect liver function and do not exhibit drug–drug interactions with other lipid-lowering agents, including statins.108,109 They are metabolized by mitochondrial beta-oxidation.
Emerging TG-lowering therapies
As studies further elucidate the proatherogenic mechanisms of TRLs, new treatment targets are beginning to emerge, and a number of novel therapeutic agents are currently in clinical development. These agents include antisense apoC-III inhibitors and LPL gene replacement therapy. Volanesorsen (formerly ISIS-APOCIIIRx) is an antisense apoC-III inhibitor currently undergoing Phase III clinical trials. It inhibits hepatic apoC-III synthesis by binding to apoC-III messenger RNA, thereby promoting its degradation. Phase II clinical trials conducted in patients with LPL deficiency demonstrated that Volanesorsen successfully reduced levels of apoC-III, TG, and non-HDL-C.110 However, concerns have been raised that the inhibition of hepatic apoC-III synthesis could lead to hepatic lipid accumulation.111
Another agent with promise in reducing TG levels is alipogene tiparvovec (AAV1-LPLS447X), a nonreplicating and nonreplacing adeno-associated viral vector that delivers copies of the human LPL gene to muscle tissue. It has been approved in Europe for adult patients diagnosed with familial LPL deficiency, a disorder characterized by severe hypertriglyceridemia and increased risk of pancreatitis. However, it has not yet been approved in the US. Intramuscular administration of alipogene tiparvovec has been shown to be generally well tolerated and is associated with clinical improvement and reduced incidence and severity of acute pancreatitis.112
Mipomersen and lomitapide are both approved by the US Food and Drug Administration for the treatment of homozygous familial hypercholesterolemia. However, as both agents interfere with TRL synthesis in the liver, along with substantial reductions in LDL-C, they have also been shown to diminish plasma TG levels.113,114 Mipomersen is an antisense oligonucleotide that blocks the translation of the apoB-100 gene, reducing its synthesis and consequently decreasing the circulation of atherogenic apoB-100-containing lipoproteins, including VLDL, IDL, and LDL.113 Lomitapide is a microsomal TG transfer protein inhibitor that blocks the microsomal TG transfer protein-mediated transfer of lipids to apoB, and therefore, results in significant reductions in VLDL, LDL-C, TG, and non-HDL-C.114 Currently, the sole indication for both mipomersen and lomitapide is homozygous familial hypercholesterolemia. However, in the future they may be found to have a place in the treatment of hypertriglyceridemia once their effect on TRLs has been fully defined.
Additional treatment considerations
Several guidelines recommend evaluating TG levels in the fasting state.4,69,115 However, nonfasting TG levels significantly correlate with increased levels of remnant lipoprotein cholesterol, are associated with a stepwise increase in the incidence of cardiovascular events,66,68 and have been shown to be a superior predictor of cardiovascular risk compared with fasting TG levels.67,68 Therefore, the measurement of nonfasting TG levels in the absence of a high-fat meal is now suggested by the American Heart Association and the European Atherosclerosis Society.2,28
Additionally, although a number of guidelines recommend LDL-C as a primary treatment target, there is increasing evidence that non-HDL-C, the sum of the total cholesterol carried by all atherogenic lipoproteins (including LDL, IDL, and VLDL and chylomicrons and their remnants) provides a better indication of cardiovascular risk than LDL-C.6 Therefore, the use of non-HDL-C as a treatment target is now advocated by several guidelines, particularly in patients with hypertriglyceridemia.8,69,70 It is also worth noting that LDL-C levels are ordinarily estimated by the Friedewald equation.116 The Friedewald equation is inapplicable for patients with fasting TG >400 mg/dL; however, it has also been shown to underestimate LDL-C levels in the presence of TG levels ≥150 mg/dL, particularly in patients with low LDL-C. Therefore, alternative evaluation is warranted in high-risk patients with elevated TG levels.117,118
Conclusion
Although some studies have failed to report a link between raised TG levels and CVD when adjusting for other lipid parameters, there is now a large body of evidence indicating that elevated TG levels are independently associated with an increased incidence of cardiovascular events. Genetic studies have further clarified the causal association between TRLs and CVD, with variants in several key genes involved in TRL metabolism, such as LPL and its regulators, shown to be strongly associated with cardiovascular risk. Moreover, it has been shown that the strength of a variant’s effect on TG levels correlates with the magnitude of the variant’s effect on CVD. TRLs are known to contribute to the progression of atherosclerosis and CVD via a number of direct and indirect mechanisms. For example, they directly contribute to intimal cholesterol deposition and are also involved in the activation and enhancement of several proinflammatory, proapoptotic, and procoagulant pathways. Consequently, a number of guidelines now recommend the use of TG-lowering agents, primarily fibrates, niacin, or omega-3 fatty acids, if elevated TG or non-HDL-C levels remain following lifestyle intervention and statin therapy. Although cardiovascular outcome studies of TG-lowering agents have produced inconsistent results, post hoc analyses of large-scale clinical trials have shown a clinical benefit of TG-lowering agents in patients with hypertriglyceridemia, those with atherogenic dyslipidemia, and patients with features of metabolic syndrome. However, further large-scale clinical trials conducted in patients with clinically defined hypertriglyceridemia are required to further clarify this clinical benefit.
Acknowledgments
Medical writing support was provided by Alex Mellors of Prime Medica Ltd, Knutsford, Cheshire, UK, and funded by AstraZeneca. The opinions, conclusion, and interpretation of the data are the responsibility of the author.
Disclosures
Doctor Toth received no compensation and was involved in all stages of manuscript development. Doctor Toth is a member of speakers bureaus for Amarin, Amgen, AstraZeneca, GSK, Kowa, and Merck & Co., Inc. and is a consultant/advisory board member for Amgen, AstraZeneca, Merck & Co., Inc. Novartis, Sanofi, Regeneron, and Kowa. The author reports no other conflicts of interest in this work.
References
Cholesterol Treatment Trialists’ (CTT) Collaboration, Fulcher J, O’Connell R, et al. Efficacy and safety of LDL-lowering therapy among men and women: meta-analysis of individual data from 174,000 participants in 27 randomised trials. Lancet. 2015;385(9976):1397–1405. | |
Chapman MJ, Ginsberg HN, Amarenco P, et al. Triglyceride-rich lipoproteins and high-density lipoprotein cholesterol in patients at high risk of cardiovascular disease: evidence and guidance for management. Eur Heart J. 2011;32(11):1345–1361. | |
Jellinger PS, Smith DA, Mehta AE, et al. American Association of Clinical Endocrinologists’ guidelines for management of dyslipidemia and prevention of atherosclerosis. Endocr Pract. 2012;18(suppl 1):1–78. | |
Catapano AL, Reiner Z, De Backer G, et al. ESC/EAS guidelines for the management of dyslipidaemias. The task force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS). Atherosclerosis. 2011;217(1):3–46. | |
Sampson UK, Fazio S, Linton MF. Residual cardiovascular risk despite optimal LDL cholesterol reduction with statins: the evidence, etiology, and therapeutic challenges. Curr Atheroscler Rep. 2012;14(1):1–10. | |
Boekholdt SM, Arsenault BJ, Mora S, et al. Association of LDL cholesterol, non-HDL cholesterol, and apolipoprotein B levels with risk of cardiovascular events among patients treated with statins: a meta-analysis. JAMA. 2012;307(12):1302–1309. | |
Carroll M, Kit B, Lacher D. Trends in elevated triglyceride in adults: United States, 2001–2012. NCHS Data Brief. 2015;198:198. | |
Hegele RA, Ginsberg HN, Chapman MJ, et al. The polygenic nature of hypertriglyceridaemia: implications for definition, diagnosis, and management. Lancet Diabetes Endocrinol. 2014;2(8):655–666. | |
Brunzell JD, Schrott HG. The interaction of familial and secondary causes of hypertriglyceridemia: role in pancreatitis. J Clin Lipidol. 2012;6(5):409–412. | |
Miller M, Cannon CP, Murphy SA, et al; PROVE IT-TIMI 22 Investigators. Impact of triglyceride levels beyond low-density lipoprotein cholesterol after acute coronary syndrome in the PROVE IT-TIMI 22 trial. J Am Coll Cardiol. 2008;51(7):724–730. | |
Schwartz GG, Abt M, Bao W, et al. Fasting triglycerides predict recurrent ischemic events in patients with acute coronary syndrome treated with statins. J Am Coll Cardiol. 2015;65(21):2267–2275. | |
Rosenson RS, Davidson MH, Hirsh BJ, Kathiresan S, Gaudet D. Genetics and causality of triglyceride-rich lipoproteins in atherosclerotic cardiovascular disease. J Am Coll Cardiol. 2014;64(23):2525–2540. | |
Kotwal S, Jun M, Sullivan D, Perkovic V, Neal B. Omega 3 fatty acids and cardiovascular outcomes: systematic review and meta-analysis. Circ Cardiovasc Qual Outcomes. 2012;5(6):808–818. | |
Jun M, Foote C, Lv J, et al. Effects of fibrates on cardiovascular outcomes: a systematic review and meta-analysis. Lancet. 2010; 375(9729):1875–1884. | |
Nordestgaard BG, Varbo A. Triglycerides and cardiovascular disease. Lancet. 2014;384(9943):626–635. | |
Do R, Willer CJ, Schmidt EM, et al. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat Genet. 2013;45(11):1345–1352. | |
Zheng C. Updates on apolipoprotein CIII: fulfilling promise as a therapeutic target for hypertriglyceridemia and cardiovascular disease. Curr Opin Lipidol. 2014;25(1):35–39. | |
Fazekas F, Enzinger C, Ropele S, Schmidt H, Schmidt R, Strasser-Fuchs S. The impact of our genes: consequences of the apolipoprotein E polymorphism in Alzheimer disease and multiple sclerosis. J Neurol Sci. 2006;245(1–2):35–39. | |
Mahley RW, Huang Y, Weisgraber KH. Detrimental effects of apolipoprotein E4: potential therapeutic targets in Alzheimer’s disease. Curr Alzheimer Res. 2007;4(5):537–540. | |
Do R, Stitziel NO, Won HH, et al. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature. 2015;518(7537):102–106. | |
Ginsberg HN. New perspectives on atherogenesis: role of abnormal triglyceride-rich lipoprotein metabolism. Circulation. 2002;106(16):2137–2142. | |
Xiao C, Hsieh J, Adeli K, Lewis GF. Gut-liver interaction in triglyceride-rich lipoprotein metabolism. Am J Physiol Endocrinol Metab. 2011; 301(3):E429–E446. | |
Ginsberg HN. Lipoprotein physiology. Endocrinol Metab Clin North Am. 1998;27(3):503–519. | |
Beigneux AP, Davies BS, Bensadoun A, Fong LG, Young SG. GPIHBP1, a GPI-anchored protein required for the lipolytic processing of triglyceride-rich lipoproteins. J Lipid Res. 2009;50(suppl):S57–S62. | |
Davies BS, Beigneux AP, Barnes RH 2nd, et al. GPIHBP1 is responsible for the entry of lipoprotein lipase into capillaries. Cell Metab. 2010;12(1):42–52. | |
Goldberg IJ, Scheraldi CA, Yacoub LK, Saxena U, Bisgaier CL. Lipoprotein ApoC-II activation of lipoprotein lipase. Modulation by apolipoprotein A-IV. J Biol Chem. 1990;265(8):4266–4272. | |
Johansen CT, Kathiresan S, Hegele RA. Genetic determinants of plasma triglycerides. J Lipid Res. 2011;52(2):189–206. | |
Miller M, Stone NJ, Ballantyne C, et al. Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2011;123(20):2292–2333. | |
Brahm A, Hegele RA. Hypertriglyceridemia. Nutrients. 2013;5(3):981–1001. | |
Varbo A, Benn M, Tybjaerg-Hansen A, Jorgensen AB, Frikke- Schmidt R, Nordestgaard BG. Remnant cholesterol as a causal risk factor for ischemic heart disease. J Am Coll Cardiol. 2013;61(4):427–436. | |
Thomsen M, Varbo A, Tybjaerg-Hansen A, Nordestgaard BG. Low nonfasting triglycerides and reduced all-cause mortality: a mendelian randomization study. Clin Chem. 2014;60(5):737–746. | |
Hodis HN. Triglyceride-rich lipoprotein remnant particles and risk of atherosclerosis. Circulation. 1999;99(22):2852–2854. | |
Nordestgaard BG, Stender S, Kjeldsen K. Reduced atherogenesis in cholesterol-fed diabetic rabbits. Giant lipoproteins do not enter the arterial wall. Arteriosclerosis. 1988;8(4):421–428. | |
Rapp JH, Lespine A, Hamilton RL, et al. Triglyceride-rich lipoproteins isolated by selected-affinity anti-apolipoprotein B immunosorption from human atherosclerotic plaque. Arterioscler Thromb. 1994;14(11):1767–1774. | |
Nordestgaard BG, Wootton R, Lewis B. Selective retention of VLDL, IDL, and LDL in the arterial intima of genetically hyperlipidemic rabbits in vivo. Molecular size as a determinant of fractional loss from the intima-inner media. Arterioscler Thromb Vasc Biol. 1995; 15(4):534–542. | |
Yla-Herttuala S, Jaakkola O, Ehnholm C, et al. Characterization of two lipoproteins containing apolipoproteins B and E from lesion-free human aortic intima. J Lipid Res. 1988;29(5):563–572. | |
Goldstein JL, Ho YK, Brown MS, Innerarity TL, Mahley RW. Cholesteryl ester accumulation in macrophages resulting from receptor-mediated uptake and degradation of hypercholesterolemic canine beta-very low density lipoproteins. J Biol Chem. 1980;255(5):1839–1848. | |
Pitas RE, Innerarity TL, Mahley RW. Foam cells in explants of atherosclerotic rabbit aortas have receptors for beta-very low density lipoproteins and modified low density lipoproteins. Arteriosclerosis. 1983; 3(1):2–12. | |
Batt KV, Patel L, Botham KM, Suckling KE. Chylomicron remnants and oxidised low density lipoprotein have differential effects on the expression of mRNA for genes involved in human macrophage foam cell formation. J Mol Med (Berl). 2004;82(7):449–458. | |
Aung HH, Lame MW, Gohil K, An CI, Wilson DW, Rutledge JC. Induction of ATF3 gene network by triglyceride-rich lipoprotein lipolysis products increases vascular apoptosis and inflammation. Arterioscler Thromb Vasc Biol. 2013;33(9):2088–2096. | |
Sun C, Alkhoury K, Wang YI, et al. IRF-1 and miRNA126 modulate VCAM-1 expression in response to a high-fat meal. Circ Res. 2012;111(8):1054–1064. | |
Gower RM, Wu H, Foster GA, et al. CD11c/CD18 expression is upregulated on blood monocytes during hypertriglyceridemia and enhances adhesion to vascular cell adhesion molecule-1. Arterioscler Thromb Vasc Biol. 2011;31(1):160–166. | |
Wang L, Gill R, Pedersen TL, Higgins LJ, Newman JW, Rutledge JC. Triglyceride-rich lipoprotein lipolysis releases neutral and oxidized FFAs that induce endothelial cell inflammation. J Lipid Res. 2009; 50(2):204–213. | |
Doi H, Kugiyama K, Oka H, et al. Remnant lipoproteins induce proatherothrombogenic molecules in endothelial cells through a redox-sensitive mechanism. Circulation. 2000;102(6):670–676. | |
Wang YI, Bettaieb A, Sun C, et al. Triglyceride-rich lipoprotein modulates endothelial vascular cell adhesion molecule (VCAM)-1 expression via differential regulation of endoplasmic reticulum stress. PLoS One. 2013;8(10):e78322. | |
Hua S. Targeting sites of inflammation: intercellular adhesion molecule-1 as a target for novel inflammatory therapies. Front Pharmacol. 2013;4:127. | |
Alipour A, van Oostrom AJ, Izraeljan A, et al. Leukocyte activation by triglyceride-rich lipoproteins. Arterioscler Thromb Vasc Biol. 2008;28(4):792–797. | |
Hadi HA, Carr CS, Al Suwaidi J. Endothelial dysfunction: cardiovascular risk factors, therapy, and outcome. Vasc Health Risk Manag. 2005;1(3):183–198. | |
Shin HK, Kim YK, Kim KY, Lee JH, Hong KW. Remnant lipoprotein particles induce apoptosis in endothelial cells by NAD(P)H oxidase-mediated production of superoxide and cytokines via lectin-like oxidized low-density lipoprotein receptor-1 activation: prevention by cilostazol. Circulation. 2004;109(8):1022–1028. | |
Olufadi R, Byrne CD. Effects of VLDL and remnant particles on platelets. Pathophysiol Haemost Thromb. 2006;35(3–4):281–291. | |
Steffel J, Luscher TF, Tanner FC. Tissue factor in cardiovascular diseases: molecular mechanisms and clinical implications. Circulation. 2006;113(5):722–731. | |
Patel S, Puranik R, Nakhla S, et al. Acute hypertriglyceridaemia in humans increases the triglyceride content and decreases the anti-inflammatory capacity of high density lipoproteins. Atherosclerosis. 2009;204(2):424–428. | |
Zheng XY, Liu L. Remnant-like lipoprotein particles impair endothelial function: direct and indirect effects on nitric oxide synthase. J Lipid Res. 2007;48(8):1673–1680. | |
Wyler von Ballmoos MC, Haring B, Sacks FM. The risk of cardiovascular events with increased apoplipoprotein CIII: a systematic review and meta-analysis. J Clin Lipidol. 2015;9:498–510. | |
Kawakami A, Aikawa M, Alcaide P, Luscinskas FW, Libby P, Sacks FM. Apolipoprotein CIII induces expression of vascular cell adhesion molecule-1 in vascular endothelial cells and increases adhesion of monocytic cells. Circulation. 2006;114(7):681–687. | |
Juntti-Berggren L, Refai E, Appelskog I, et al. Apolipoprotein CIII promotes Ca2+-dependent beta cell death in type 1 diabetes. Proc Natl Acad Sci U S A. 2004;101(27):10090–10094. | |
Sol EM, Sundsten T, Bergsten P. Role of MAPK in apolipoprotein CIII-induced apoptosis in INS-1E cells. Lipids Health Dis. 2009;8:3. | |
Kones R. Molecular sources of residual cardiovascular risk, clinical signals, and innovative solutions: relationship with subclinical disease, undertreatment, and poor adherence: implications of new evidence upon optimizing cardiovascular patient outcomes. Vasc Health Risk Manag. 2013;9:617–670. | |
Crosby J, Peloso GM, Auer PL, et al. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med. 2014;371(1):22–31. | |
Jorgensen AB, Frikke-Schmidt R, Nordestgaard BG, Tybjaerg-Hansen A. Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N Engl J Med. 2014;371(1):32–41. | |
Triglyceride Coronary Disease Genestics Consortium and Emerging Risk Factors Collaboration, Sarwar N, Sandhu MS, et al. Triglyceride-mediated pathways and coronary disease: collaborative analysis of 101 studies. Lancet. 2010;375(9726):1634–1639. | |
Folsom AR, Peacock JM, Demerath E, Boerwinkle E. Variation in ANGPTL4 and risk of coronary heart disease: the Atherosclerosis Risk in Communities Study. Metabolism. 2008;57(11):1591–1596. | |
Varbo A, Benn M, Tybjaerg-Hansen A, Nordestgaard BG. Elevated remnant cholesterol causes both low-grade inflammation and ischemic heart disease, whereas elevated low-density lipoprotein cholesterol causes ischemic heart disease without inflammation. Circulation. 2013;128(12):1298–1309. | |
Di Angelantonio E, Sarwar N, Perry P, et al. Major lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009;302(18):1993–2000. | |
Faergeman O, Holme I, Fayyad R, et al. Plasma triglycerides and cardiovascular events in the treating to new targets and incremental decrease in end-points through aggressive lipid lowering trials of statins in patients with coronary artery disease. Am J Cardiol. 2009;104(4):459–463. | |
Freiberg JJ, Tybjaerg-Hansen A, Jensen JS, Nordestgaard BG. Nonfasting triglycerides and risk of ischemic stroke in the general population. JAMA. 2008;300(18):2142–2152. | |
Bansal S, Buring JE, Rifai N, Mora S, Sacks FM, Ridker PM. Fasting compared with nonfasting triglycerides and risk of cardiovascular events in women. JAMA. 2007;298(3):309–316. | |
Nordestgaard BG, Benn M, Schnohr P, Tybjaerg-Hansen A. Nonfasting triglycerides and risk of myocardial infarction, ischemic heart disease, and death in men and women. JAMA. 2007;298(3):299–308. | |
Berglund L, Brunzell JD, Goldberg AC, et al. Evaluation and treatment of hypertriglyceridemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2012;97(9):2969–2989. | |
Jacobson TA, Ito MK, Maki KC, et al. National Lipid Association recommendations for patient-centered management of dyslipidemia: part 1 – executive summary. J Clin Lipidol. 2014;8(5):473–488. | |
Stone NJ, Robinson JG, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63(25 pt B):2889–2934. | |
Birjmohun RS, Hutten BA, Kastelein JJ, Stroes ES. Efficacy and safety of high-density lipoprotein cholesterol-increasing compounds: a meta-analysis of randomized controlled trials. J Am Coll Cardiol. 2005;45(2):185–197. | |
Robinson JG, Wang S, Smith BJ, Jacobson TA. Meta-analysis of the relationship between non-high-density lipoprotein cholesterol reduction and coronary heart disease risk. J Am Coll Cardiol. 2009;53(4):316–322. | |
Davidson MH, Bays HE, Stein E, et al; TRIMS Investigators. Effects of fenofibrate on atherogenic dyslipidemia in hypertriglyceridemic subjects. Clin Cardiol. 2006;29(6):268–273. | |
Farnier M. Update on the clinical utility of fenofibrate in mixed dyslipidemias: mechanisms of action and rational prescribing. Vasc Health Risk Manag. 2008;4(5):991–1000. | |
Frick MH, Elo O, Haapa K, et al. Helsinki Heart Study: primary-prevention trial with gemfibrozil in middle-aged men with dyslipidemia. Safety of treatment, changes in risk factors, and incidence of coronary heart disease. N Engl J Med. 1987;317(20):1237–1245. | |
Rubins HB, Robins SJ, Collins D, et al. Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial Study Group. N Engl J Med. 1999;341(6):410–418. | |
The BIP Study Group. Secondary prevention by raising HDL cholesterol and reducing triglycerides in patients with coronary artery disease. The Bezafibrate Infarction Prevention (BIP) study. Circulation. 2000; 102(1):21–27. | |
Keech A, Simes RJ, Barter P, et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet. 2005;366(9500):1849–1861. | |
Lee M, Saver JL, Towfighi A, Chow J, Ovbiagele B. Efficacy of fibrates for cardiovascular risk reduction in persons with atherogenic dyslipidemia: a meta-analysis. Atherosclerosis. 2011;217(2):492–498. | |
Davidson MH, Armani A, McKenney JM, Jacobson TA. Safety considerations with fibrate therapy. Am J Cardiol. 2007;99:3C–18C. | |
Chang JT, Staffa JA, Parks M, Green L. Rhabdomyolysis with HMG-CoA reductase inhibitors and gemfibrozil combination therapy. Pharmacoepidemiol Drug Saf. 2004;13(7):417–426. | |
Kyrklund C, Backman JT, Neuvonen M, Neuvonen PJ. Gemfibrozil increases plasma pravastatin concentrations and reduces pravastatin renal clearance. Clin Pharmacol Ther. 2003;73(6):538–544. | |
Ginsberg HN, Elam MB, Lovato LC, et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. 2010; 362(17):1563–1574. | |
Bruckert E, Labreuche J, Amarenco P. Meta-analysis of the effect of nicotinic acid alone or in combination on cardiovascular events and atherosclerosis. Atherosclerosis. 2010;210(2):353–361. | |
Lavigne PM, Karas RH. The current state of niacin in cardiovascular disease prevention: a systematic review and meta-regression. J Am Coll Cardiol. 2013;61(4):440–446. | |
Canner PL, Berge KG, Wenger NK, et al. Fifteen year mortality in coronary drug project patients: long-term benefit with niacin. J Am Coll Cardiol. 1986;8(6):1245–1255. | |
HPS2-THRIVE Collaborative Group. HPS2-THRIVE randomized placebo-controlled trial in 25 673 high-risk patients of ER niacin/laropiprant: trial design, pre-specified muscle and liver outcomes, and reasons for stopping study treatment. Eur Heart J. 2013;34(17):1279–1291. | |
Boden WE, Probstfield JL, Anderson T, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365(24):2255–2267. | |
Guyton JR, Slee AE, Anderson T, et al. Relationship of lipoproteins to cardiovascular events: the AIM-HIGH Trial (atherothrombosis intervention in metabolic syndrome with low HDL/high triglycerides and impact on global health outcomes). J Am Coll Cardiol. 2013;62(17):1580–1584. | |
Guyton JR, Bays HE. Safety considerations with niacin therapy. Am J Cardiol. 2007;99(6A):22C–31C. | |
AstraZeneca. Epanova Prescribing Information. 2014. Available from: http://www.astrazeneca-us.com/pi/epanova.pdf. Accessed August 26, 2015. | |
GlaxoSmithKline [webpage on the Internet]. Lovaza Prescribing Information. 2014. Available from: https://www.gsksource.com/gskprm/htdocs/documents/LOVAZA-PI-PIL.PDF. Accessed August 26, 2015. | |
Trygg Pharma [webpage on the Internet]. Omtryg Prescribing Information. 2014. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/204977s000lbl.pdf. Accessed August 26, 2015. | |
Amarin Corporation [webpage on the Internet]. Vascepa Prescibing Information. 2014. Available from: www.vascepa.com/full-prescribing- information.pdf. Accessed August 26, 2015. | |
Harris WS. n-3 fatty acids and serum lipoproteins: human studies. Am J Clin Nutr. 1997;65(5 suppl):1645S–1654S. | |
GISSI-Prevenzione Investigators. Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: results of the GISSI-Prevenzione trial. Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto miocardico. Lancet. 1999;354(9177):447–455. | |
Yokoyama M, Origasa H, Matsuzaki M, et al. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): a randomised open-label, blinded endpoint analysis. Lancet. 2007;369(9567):1090–1098. | |
Kromhout D, Giltay EJ, Geleijnse JM, Alpha Omega Trial Group. n-3 fatty acids and cardiovascular events after myocardial infarction. N Engl J Med. 2010;363(21):2015–2026. | |
Rauch B, Schiele R, Schneider S, et al. OMEGA, a randomized, placebo-controlled trial to test the effect of highly purified omega-3 fatty acids on top of modern guideline-adjusted therapy after myocardial infarction. Circulation. 2010;122(21):2152–2159. | |
Clinicaltrials.gov. [webpage on the Internet]. A Study of AMR101 to Evaluate Its Ability to Reduce Cardiovascular Events in High Risk Patients with Hypertriglyceridemia and on Statin. The Primary Objective is to Evaluate the Effect of 4 g/Day AMR101 for Preventing the Occurrence of a First Major Cardiovascular Event. (REDUCE-IT). Clinicaltrials. gov; 2014. Available from: https://clinicaltrials.gov/ct2/show/NCT01492361. Accessed August 20, 2015. | |
Clinicaltrials.gov. [webpage on the Internet]. Outcomes Study to Assess Statin Residual Risk Reduction with Epanova in High CV Risk Patients with Hypertriglyceridemia (STRENGTH). 2015. Available from: https://clinicaltrials.gov/ct2/show/NCT02104817. Accessed August 20, 2015. | |
Kastelein JJ, Maki KC, Susekov A, et al. Omega-3 free fatty acids for the treatment of severe hypertriglyceridemia: the Epanova for lowering very high triglycerides (EVOLVE) trial. J Clin Lipidol. 2014;8(1):94–106. | |
Wang C, Harris WS, Chung M, et al. n-3 Fatty acids from fish or fish-oil supplements, but not alpha-linolenic acid, benefit cardiovascular disease outcomes in primary- and secondary-prevention studies: a systematic review. Am J Clin Nutr. 2006;84(1):5–17. | |
Bays HE, Ballantyne CM, Kastelein JJ, Isaacsohn JL, Braeckman RA, Soni PN. Eicosapentaenoic acid ethyl ester (AMR101) therapy in patients with very high triglyceride levels (from the multi-center, placebo-controlled, randomized, double-blind, 12-week study with an open-label extension [MARINE] trial). Am J Cardiol. 2011;108(5):682–690. | |
Ballantyne CM, Bays HE, Kastelein JJ, et al. Efficacy and safety of eicosapentaenoic acid ethyl ester (AMR101) therapy in statin-treated patients with persistent high triglycerides (from the ANCHOR study). Am J Cardiol. 2012;110(7):984–992. | |
Filion KB, El Khoury F, Bielinski M, Schiller I, Dendukuri N, Brophy JM. Omega-3 fatty acids in high-risk cardiovascular patients: a meta-analysis of randomized controlled trials. BMC Cardiovasc Disord. 2010;10:24. | |
Harris WS, Ginsberg HN, Arunakul N, et al. Safety and efficacy of Omacor in severe hypertriglyceridemia. J Cardiovasc Risk. 1997;4(5–6):385–391. | |
Kostapanos MS, Milionis HJ, Elisaf MS. Rosuvastatin-associated adverse effects and drug-drug interactions in the clinical setting of dyslipidemia. Am J Cardiovasc Drugs. 2010;10(1):11–28. | |
Gaudet D, Alexander VJ, Diane B, et al. An antisense inhibitor of apolipoprotein C-III substantially decreases fasting apolipoprotein C-III and triglyceride levels in LPL deficiency. J Clin Lipidol. 2014; 8(3):353–354. | |
Huff MW, Hegele RA. Apolipoprotein C-III: going back to the future for a lipid drug target. Circ Res. 2013;112(11):1405–1408. | |
Gaudet D, Methot J, Kastelein J. Gene therapy for lipoprotein lipase deficiency. Curr Opin Lipidol. 2012;23(4):310–320. | |
Toth PP. Emerging LDL therapies: mipomersen-antisense oligonucleotide therapy in the management of hypercholesterolemia. J Clin Lipidol. 2013;7(3 suppl):S6–S10. | |
Toth PP, Shah PK, Wilkinson MJ, Davidson MH, McCullough PA. Use of microsomal triglyceride transfer protein inhibitors in patients with homozygous familial hypercholesterolemia: translating clinical trial experience into clinical practice. Rev Cardiovasc Med. 2014;15(1):1–10. | |
National Cholesterol Education Program (NCEP) Expert Panel on Detection Evaluation and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation. 2002;106(25):3143–3421. | |
Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem. 1972;18(6):499–502. | |
Hermans MP, Ahn SA, Rousseau MF. Novel unbiased equations to calculate triglyceride-rich lipoprotein cholesterol from routine non-fasting lipids. Cardiovasc Diabetol. 2014;13:56. | |
Martin SS, Blaha MJ, Elshazly MB, et al. Friedewald-estimated versus directly measured low-density lipoprotein cholesterol and treatment implications. J Am Coll Cardiol. 2013;62(8):732–739. | |
Andersson C, Lyass A, Vasan RS, Massaro JM, D’Agostino RB Sr, Robins SJ. Long-term risk of cardiovascular events across a spectrum of adverse major plasma lipid combinations in the Framingham Heart Study. Am Heart J. 2014;168(6):878–883. | |
Sharrett AR, Ballantyne CM, Coady SA, et al. Coronary heart disease prediction from lipoprotein cholesterol levels, triglycerides, lipoprotein(a), apolipoproteins A-I and B, and HDL density subfractions: the Atherosclerosis Risk in Communities (ARIC) Study. Circulation. 2001;104(10):1108–1113. | |
Assmann G, Schulte H. The importance of triglycerides: results from the Prospective Cardiovascular Munster (PROCAM) Study. Eur J Epidemiol. 1992;8(suppl 1):99–103. | |
Kamanna VS, Kashyap ML. Mechanism of action of niacin. Am J Cardiol. 2008;101(8A):20B–26B. | |
Balk EM, Lichtenstein AH, Chung M, Kupelnick B, Chew P, Lau J. Effects of omega-3 fatty acids on serum markers of cardiovascular disease risk: a systematic review. Atherosclerosis. 2006;189(1):19–30. | |
Morton A, Furtado J, Amerine W, Kling D, Davidson M. The effect of omega-3 carboxylic acids on apolipoprotein CIII containing lipoproteins in moderate to severe hypertriglyceridemia. Circulation. 2014;130(suppl 1):A16864. | |
Weintraub HS. Overview of prescription omega-3 fatty acid products for hypertriglyceridemia. Postgrad Med. 2014;126(7):7–18. | |
Watts GF, Ooi EM, Chan DC. Demystifying the management of hypertriglyceridaemia. Nat Rev Cardiol. 2013;10(11):648–661. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.