Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 14
Trigger-Specific Remodeling of KCa2 Potassium Channels in Models of Atrial Fibrillation
Authors Rahm AK, Gramlich D ![]() , Wieder T, Müller ME
, Wieder T, Müller ME ![]() , Schoeffel A, El Tahry FA, Most P, Heimberger T, Sandke S, Weis T, Ullrich ND
, Schoeffel A, El Tahry FA, Most P, Heimberger T, Sandke S, Weis T, Ullrich ND ![]() , Korff T, Lugenbiel P, Katus HA, Thomas D
, Korff T, Lugenbiel P, Katus HA, Thomas D ![]()
Received 1 December 2020
Accepted for publication 6 April 2021
Published 20 May 2021 Volume 2021:14 Pages 579—590
DOI https://doi.org/10.2147/PGPM.S290291
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Ann-Kathrin Rahm,1– 3 Dominik Gramlich,1– 3 Teresa Wieder,1,2 Mara Elena Müller,1– 3 Axel Schoeffel,1,2 Fadwa A El Tahry,1 Patrick Most,1,2 Tanja Heimberger,1,3 Steffi Sandke,1,3 Tanja Weis,1,3 Nina D Ullrich,4 Thomas Korff,4,5 Patrick Lugenbiel,1– 3 Hugo A Katus,1– 3 Dierk Thomas1– 3
1Department of Cardiology, Medical University Hospital Heidelberg, Heidelberg, 69120, Germany; 2HCR (Heidelberg Center for Heart Rhythm Disorders), University Hospital Heidelberg, Heidelberg, 69120, Germany; 3DZHK (German Centre for Cardiovascular Research), Partner Site Heidelberg/Mannheim, University of Heidelberg, Heidelberg, 69120, Germany; 4Institute of Physiology and Pathophysiology, Department of Cardiovascular Physiology, Heidelberg University, Heidelberg, 69120, Germany; 5European Center for Angioscience (ECAS), Medical Faculty Mannheim, Heidelberg University, Mannheim, 68167, Germany
Correspondence: Dierk Thomas
Department of Cardiology, University of Heidelberg, Im Neuenheimer Feld 410, Heidelberg, 69120, Germany
Tel +49 6221 568855
Fax +49 6221 565514
Email [email protected]
Aim: Effective antiarrhythmic treatment of atrial fibrillation (AF) constitutes a major challenge, in particular, when concomitant heart failure (HF) is present. HF-associated atrial arrhythmogenesis is distinctly characterized by prolonged atrial refractoriness. Small-conductance, calcium-activated K+ (KCa, SK, KCNN) channels contribute to cardiac action potential repolarization and are implicated in AF susceptibility and therapy. The mechanistic impact of AF/HF-related triggers on atrial KCa channels is not known. We hypothesized that tachycardia, stretch, β-adrenergic stimulation, and hypoxia differentially determine KCa 2.1– 2.3 channel remodeling in atrial cells.
Methods: KCNN1-3 transcript levels were assessed in AF/HF patients and in a pig model of atrial tachypacing-induced AF with reduced left ventricular function. HL-1 atrial myocytes were subjected to proarrhythmic triggers to investigate the effects on Kcnn mRNA and KCa channel protein.
Results: Atrial KCNN1-3 expression was reduced in AF/HF patients. KCNN2 and KCNN3 suppression was recapitulated in the corresponding pig model. In contrast to human AF, KCNN1 remained unchanged in pigs. Channel- and stressor-specific remodeling was revealed in vitro. Lower expression levels of KCNN1/KCa 2.1 were linked to stretch and β-adrenergic stimulation. Furthermore, KCNN3/KCa 2.3 expression was suppressed upon tachypacing and hypoxia. Finally, KCNN2/KCa 2.2 abundance was specifically enhanced by hypoxia.
Conclusion: Reduction of KCa 2.1– 2.3 channel expression might contribute to the action potential prolongation in AF complicated by HF. Subtype-specific KCa 2 channel remodeling induced by tachypacing, stretch, β-adrenergic stimulation, or hypoxia is expected to differentially determine atrial remodeling, depending on patient-specific activation of each triggering factor. Stressor-dependent KCa 2 regulation in atrial myocytes provides a starting point for mechanism-based antiarrhythmic therapy.
Keywords: atrial fibrillation, calcium, KCa channel, KCNN, remodeling, SK channel
Introduction
Atrial fibrillation (AF) is the most common cardiac arrhythmia and accounts for significant morbidity and mortality. Effective antiarrhythmic treatment of AF still exhibits suboptimal effectiveness due to patient-specific characteristics that individually determine atrial arrhythmogenesis. Among these, heart failure (HF) worsens the prognosis of AF patients and poses a particular therapeutic challenge that is mechanistically attributed to a distinct atrial substrate. Atrial action potential duration (APD) and effective refractory period (AERP) are prolonged in AF complicated by HF in humans and in animal models,1 requiring tailored therapeutic approaches. During AF and HF, multiple mechanisms including tachycardia, stretch, β-adrenergic stimulation, and hypoxia are activated and may differentially affect APD through remodeling of repolarizing atrial ion channels.
Atrial APD is determined by the combined action of ionic currents. The role of small-conductance, calcium-activated K+ (KCa, SK) channels in atrial action potential repolarization has recently been delineated.2,3 KCa channels conduct the cardiac IK,Ca current and are primarily activated by intracellular Ca2+ via binding of calmodulin to the channel C-terminus, while their voltage sensitivity is negligible.4 Three KCa2 channels (KCa2.1–2.3) and their respective genes KCNN1-3 have been identified in the heart.5–9 KCa2.2 and KCa2.3 exhibit higher expression levels than KCa2.1 in human atrial tissue.7,8 KCa2.1 and KCa2.2 are expressed in human and murine atria with predominance over ventricular tissue,3,5,6 indicating a potential advantage as atrial-selective therapeutic targets. KCa2 channel inhibition induces prolongation of atrial refractoriness, APD duration, and suppression of atrial arrhythmia.5,8,10,11 The mechanistic role of KCa2 channels in AF has been suggested in genome-wide association studies (GWAS) and candidate gene-based approaches, revealing the relationship between KCNN2 and KCNN3 variants and AF risk in humans.12 Furthermore, reduced expression of KCa2.1, KCa2.2, and KCa2.3 has been detected in patients with persistent or permanent AF and preserved cardiac function when compared to sinus rhythm (SR) subjects.7,8,13 Of note, either inappropriate shortening or excessive prolongation of atrial APD may increase AF susceptibility.3,11,14,15 Thus, the therapeutic efficacy of interventions targeting KCa2 channels will depend on the functional KCa2 homeostasis, which is affected by individual patient characteristics and by environmental factors that determine specific KCa2 channel remodeling. However, the mechanistic impact of proarrhythmic triggers on KCa2 channel expression in AF and HF as basis for patient-specific antiarrhythmic therapy is poorly understood. This study was designed to assess KCa2.1–2.3 channel remodeling in patients and in a porcine model of AF with concomitant HF, and to elucidate differential effects of tachycardia, stretch, β-adrenergic stimulation, and hypoxia on KCa2.1–2.3 expression in atrial cells.
Methods
Ethics Statement
The study involving human tissue samples was conducted in accordance with the Declaration of Helsinki, and the study protocol was approved by the University of Heidelberg Ethics Committee (Germany; institutional approval number S-390/2011). Written informed consent was obtained from all patients. Animal experiments have been carried out in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the US National Institutes of Health (NIH publication No. 86–23, revised 1985) and with EU Directive 2010/63/EU, and the current version of the German Law on the Protection of Animals was followed. Experiments involving pigs (institutional approval numbers G-106/10 and G-165/12) have been approved by the local animal welfare authority (Regierungspräsidium Karlsruhe, Karlsruhe, Germany).
Patients and Human Tissue Handling
The study comprised a total of 30 patients (66.7% male, mean age 51.0±12.2 years) with sinus rhythm (SR; n = 10), paroxysmal (p)AF (n = 10), and chronic (c)AF (ie, persistent, long-standing persistent or permanent AF; n = 10) undergoing heart transplantation were included (Supplementary Table 1). The patient cohort with detailed characteristics has been reported previously.1,16–18 Atrial tissue samples were obtained from the Heidelberg CardioBiobank (Department of Cardiology, University Hospital Heidelberg, Heidelberg, Germany) and quality controlled by the tissue bank of the National Center for Tumor Diseases (NCT, Heidelberg, Germany) in accordance with the regulations of the tissue bank. Cardiac tissue samples were dissected immediately following explantation of the recipient’s heart in the operating room. The atrial tissue sections were shock-frozen in liquid nitrogen and stored at −80°C.
AF Pig Model
Remodeling of study proteins was evaluated using an established porcine AF model.1 AF was induced in domestic swine by right atrial burst pacing via an implanted cardiac pacemaker. High-rate atrial pacing and AF with rapid ventricular rate response resulted in reduced left ventricular function. Animals receiving inactive pacemakers served as controls. Cardiac tissue was obtained from previously reported pigs 7 days (n = 5)19 or 14 days (n = 5)1 after the initiation of atrial burst pacing or from corresponding control pigs not subjected to AF induction (n = 5 each). Animal characteristics were previously described in detail.1,19
HL-1 Cell Culture
HL-1 cells (kindly provided by Dr. William Claycomb, Louisiana State University Health Science Center, New Orleans, LA, USA) originate from atrial tumor cells of transgenic mice and exhibit cardiomyocyte-specific ion channel expression.20,21 KCa2 channel expression in HL-1 cells has been shown previously.9,16 Cells were cultured in a supplemented Claycomb medium (Sigma-Aldrich, Steinheim, Germany) containing 0.1 mM norepinephrine.20 A total of 4–5×106 HL-1 cells per well were seeded on gelatin-/fibronectin-coated 6-well dishes and subjected to experimental procedures, unless stated otherwise. For ß-adrenergic stimulation 50 µM isoproterenol was added to the culture medium for 24 h. Similar doses of isoprenaline have been employed previously in experimental studies.22–24 For HL-1 cell authentication, Short Tandem Repeat (STR) profiling of HL-1 cells used in this study (obtained directly from Dr. Claycomb) and of commercially available HL-1 cells (Sigma-Aldrich, Steinheim, Germany) for comparison was performed by American Type Culture Collection (ATCC) cell-line authentication service (Wesel, Germany). This approach confirmed a high degree of similarity between cell lines (Supplementary Table 2). In addition, human and/or African green monkey has not been detected in these samples.
Rapid Electrical Stimulation of HL-1 Cells
Cells were subjected to electrical stimulation as reported using the C-Pace EP system (IonOptix, Westwood, MA, USA).25 The cells were stimulated with 10 V/10 ms pulses at 5 Hz stimulation rate. Following rapid electrical stimulation for 24 h, cell viability was visually assessed by microscopic examination and TUNEL assays. Cells were harvested and subjected to RNA and protein isolation. Control cells not subjected to stimulation were otherwise maintained and handled similarly.
HL-1 Cardiomyocyte Stretch Experiments
Prior to stretching, amino-coated FlexCell dishes or amino-coated membranes in 6-well tissue culture plates were coated for 24 h with gelatin/fibronectin, and 1×107 cells were seeded into each well. HL-1 cells were stretched for 24 h with 13% stretch at 1 Hz using the FlexCell Tension system (FlexCell International Corporation, Burlington, NC, USA). Control cells plated on similar control plates were maintained at identical conditions (without stretch) for the same duration.
Hypoxia
Hypoxia was induced by maintaining HL-1 cells in an incubator with 1% oxygen for 24 h (HERAcell150i, ThermoFisher Scientific, Waltham, MA, USA). Cells receiving 21% oxygen served as controls.
RNA Isolation and Quantitative RT-PCR
Total RNA was isolated from tissue samples and from HL-1 cells with QiaZol-Reagent (Qiagen, Hilden, Germany), followed by chloroform extraction, isopropanol precipitation, and assessment of quantity and integrity using a NanoDrop2000 (Thermo Fisher Scientific, Waltham, MA, USA). Genomic DNA was digested using TurboDNase-Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. Complementary DNA was synthesized using the Maxima First Strand cDNA Synthesis Kit for RT-qPCR (Thermo Fisher Scientific) using 3 µg of total RNA. Quantitative real-time PCR (qRT-qPCR) was performed with 7500 Fast Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s protocol. Ninety-six-well optical detection plates (Applied Biosystems) were loaded to a total volume of 10 μL per well, consisting of 0.5 μL cDNA, 5 μL TaqMan Fast Universal Master Mix (Applied Biosystems), and 6-carboxyfluorescein (FAM)-labeled TaqMan probes and primers (TaqMan Gene Expression Assays; Applied Biosystems) detecting human, porcine, and murine KCNN1-3 (Supplementary Table 3). Primers and probes detecting glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA or ribosomal protein L32 (Rpl32) mRNA were used for normalization. All qRT-PCR reactions were performed in duplicates or higher replicates, and non-template control (NTC) and dilution series were included on each plate for quantification. Data are expressed as averages of replicates. Data analyses were performed using the second derivative method. All measurements were adjusted using a standard probe, and quantification was corrected for efficiency calculated with the standard curves.
Protein Isolation and Western Blotting
HL-1 cells were lysed in a radioimmunoprecipitation (RIPA) buffer consisting of 20 mM Tris-HCl, 0.5% NP-40, 0.5% sodium-deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 mM Na3VO4, 1 mM NaF and inhibitors of proteases (CompleteMini, Roche Applied Science, Indianapolis, IN, USA). After centrifugation of homogenates for 30 min at 14,000 g supernatants were collected and the protein concentration was determined using the bicinchoninic acid (BCA) protein assay (Thermo Scientific, Rockford, IL, USA) and proteins were diluted to equal concentrations with sterile water. Protein immunodetection was performed using sodium dodecyl sulfate (SDS) gel electrophoresis and Western blotting as described previously.25 Equal amounts of protein were separated on 10% SDS polyacrylamide gels and transferred onto polyvinylidene difluoride membranes. Membranes were blocked with 5% milk in PBS-T for 2 hours at room temperature and developed using primary antibodies directed against KCa2.1 (1:1000, APC-039), KCa2.2 (1:1000, APC-028) and KCa2.3 (1:1000, APC-025, all from Alomone Lab, Jerusalem, Israel). Primary antibodies were incubated overnight at 4°C. Horseradish peroxidase (HRP)-conjugated donkey anti-rabbit (ab6802; Abcam, Cambridge, UK) secondary antibody was used. Signals were developed using an enhanced chemiluminescence assay (ECL Western Blotting Reagents, GE Healthcare, Buckinghamshire, UK). After removal of primary and secondary antibodies (ReBlot Strong Stripping Solution, Merck, Germany), the membranes were re-probed with anti-GAPDH (1:10.000, ab181602, Abcam) or anti-β-actin (1:1000, ab 8227, Abcam) antibody and corresponding secondary antibodies (ab6802; Abcam). Protein content was normalized to GAPDH or β-actin for quantification of optical density with ImageJ 1.50i Software (National Institutes of Health, Bethesda, MD, USA).
TUNEL Staining
HL-1 cells were seeded on glass coverslips before treatment with tachypacing or hypoxia, respectively. Apoptosis was detected by TUNEL (terminal deoxyribonucleotide transferase-mediated dUTP nick end labeling) according to the manufacturer’s instructions (TMR red In Situ Cell Death Detection Kit, Roche Applied Science). Negative controls (omitting terminal transferase) and positive controls (using recombinant DNAse I) were included. Cells were mounted with Fluoroshield Mounting Medium with DAPI (ab10433, Abcam) and sealed on object slides with nail polish. TUNEL-positive cells were counted using a fluorescence microscope (Zeiss, Oberkochen, Germany), and ImageJ 1.41 software was used to calculate the total cell number in 3×3 tiles. The percentage of TUNEL-positive cells was calculated by dividing TUNEL-positive cells by total cell count.
Statistics
Patient data are presented as mean ± standard deviation (SD) or number and percentage. Experimental data are presented as box plots with dots representing raw data. Statistical differences of continuous variables were determined using unpaired Student’s t tests (two-sided tests). Categorical data were analyzed using the chi-square test. Multiple comparisons were performed using ANOVA. Statistical analyses were performed with Origin 2020 (OriginLab, Northampton, MA, USA). P<0.05 was considered statistically significant.
Results
AF-Related KCa2 Channel Remodeling in HF Patients
KCa2 channel mRNA expression was analyzed in the right atrium of patients with severe heart failure (mean left ventricular ejection fraction, 17%-21%) and SR, pAF, or cAF, respectively (Figure 1A and B). AF and concomitant HF were associated with macroscopic atrial alteration, reflected by increased LA size that was numerically more pronounced in pAF and cAF subjects compared with SR patients (Figure 1A). Paroxysmal AF was associated with reduced expression of KCNN1 (−73%, n = 10, P = 0.010), KCNN2 (−62%, n = 10, P = 0.010), and KCNN3 (−50%, n = 10, P = 0.003) compared to HF patients with SR (n = 10) (Figure 1C), as reported earlier.16,18 In HF patients with cAF, KCNN expression was similarly suppressed by 78% (KCNN1, n = 10, P < 0.0001), 55% (KCNN2, n = 10, P = 0.010), and 48% (KCNN3, n = 10, P= 0.037), respectively (Figure 1C).16,18
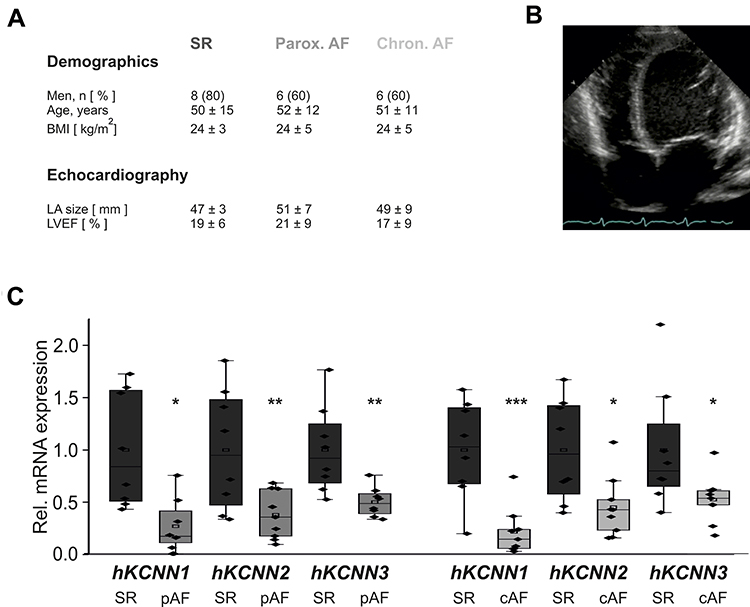 |
Figure 1 Remodeling of KCa2 channels in atrial fibrillation (AF) patients with concomitant heart failure. (A) Key characteristics of study patients (LA, left atrium; LVEF, left ventricular ejection fraction; see Supplementary Table 1 for details). Please note that patient characteristics have been published previously.1,16–18 (B) Representative apical 4-chamber echocardiography view of a patient with severely reduced LVEF and sinus rhythm (SR). (C) KCNN mRNA levels normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and respective SR control, obtained from patients with SR, paroxysmal (pAF), or chronic AF (cAF). KCNN118 and KCNN2/316 mRNA expression data were reported previously and are shown here for reference. Data are presented as box plots with underlying dots representing raw data; *P < 0.05, **P < 0.01, ***P < 0.001 versus SR controls. |
KCa2 Channel Remodeling in a Porcine AF/HF Model
To assess KCa2 channel expression changes in an animal model, previously described domestic pigs subjected to induction of atrial fibrillation for 7 days (n = 5)19 and 14-day duration (n=5)1 by pacemaker-induced right atrial burst stimulation were studied (Figure 2A). Control pigs exhibiting sinus rhythm (SR) during 14 days (n = 5)1 served as controls. The pigs exhibited a reduction in LV function and atrial dilation due to rapid ventricular rate response during AF.1 We observed a trend towards reduced expression of the right atrial KCNN2 mRNA levels 7 days (−30%, P = 0.105) and 14 days (−34%, P = 0.063) after AF induction compared to SR control animals, while KCNN1 mRNA abundance did not differ between the study groups (Figure 2B and C). KCNN3 transcript levels showed trends towards reduced abundance after 14 days AF (−32%, P = 0.22) and towards enhanced expression after 7 days AF (+19%, P = 0.39), respectively (Figure 2D). Previous protein analyses were confined to animals studied after 14 days atrial burst pacing due to limited sample availability and indicated a tendency towards decreased KCa2.1 protein expression in RA tissue.18 Furthermore, reduced expression of the right atrial KCa2.2 and KCa2.3 protein after 14 days AF in pigs has been reported previously.16
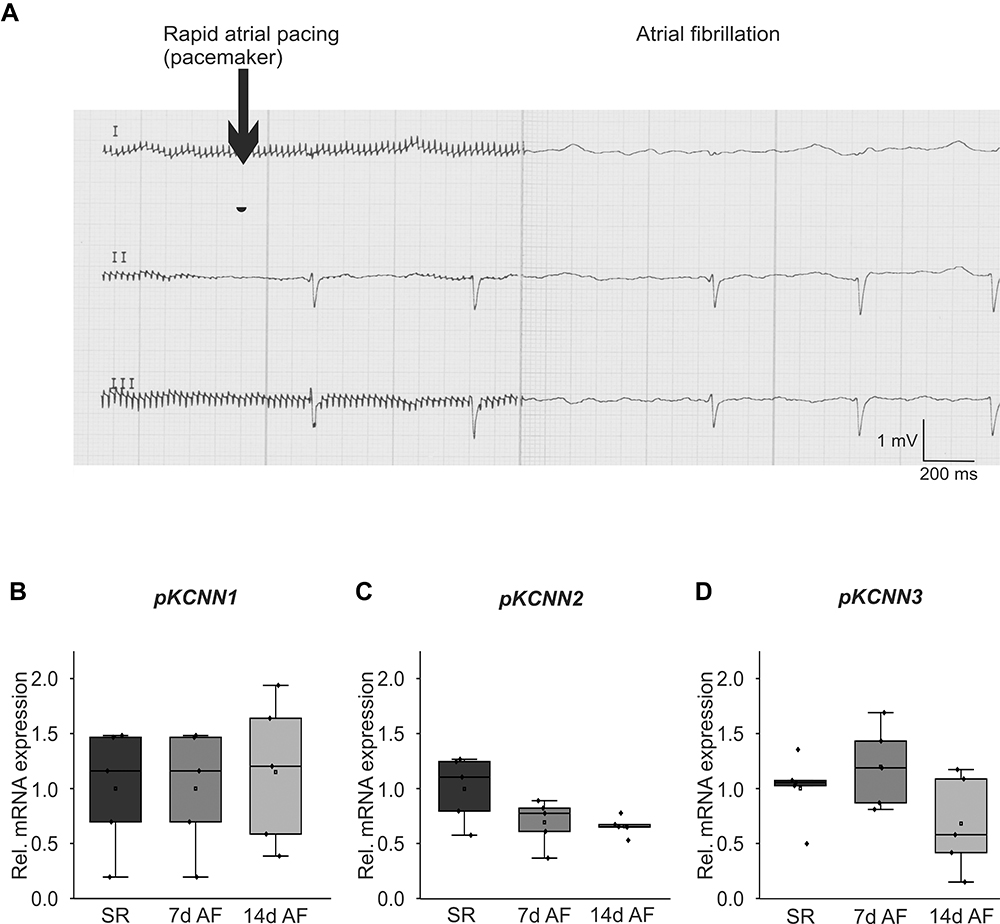 |
Figure 2 Remodeling of KCa2 channels in a porcine atrial fibrillation (AF)/heart failure model. (A) Representative ECG recordings illustrate AF (right) induced by rapid atrial pacing (left). (B-D) Relative right atrial mRNA expression levels of KCNN1 (B), KCNN2 (C), and KCNN3 (D) are provided for AF pigs after 7 and 14 days of atrial burst pacing compared to sinus rhythm (SR) controls. |
Tachypacing-Related Remodeling of KCa2 Channels in vitro
To test the hypothesis that potential proarrhythmic triggers (ie high-rate electrical activity, myocardial wall stress, β-adrenergic activation, hypoxia) differentially affect KCa2 channel expression, we next analyzed their effects in vitro using immortalized murine atrial cardiomyocytes (HL-1 cells). Rapid electrical pacing of cultured cells provides an established model of cardiac tachyarrhythmia that may induce electrophysiological remodeling similar to findings obtained in humans or animal models. HL-1 cells were subjected to tachypacing (TP; 24 h) by electric field stimulation (Figure 3A). TP resulted in a significant reduction of KCa2.3 at mRNA (−56%, P = 0.029, n = 6; Figure 3B) and protein levels (−63%, P = 0.024, n = 6; Figure 3C and D). KCa2.1 or KCa2.2 expression was not significantly affected by TP (Figure 3C and 4). To exclude tachypacing-related apoptosis as an unspecific mechanism underlying KCa2.3 expression changes, apoptosis was quantified using TUNEL staining (Figure 3E). Apoptosis rates were low and not significantly different between HL-1 cells following TP (3.7 ± 0.3%) or maintained under control conditions (3.2 ± 0.4%, P=0.294, n = 3; Figure 3F).
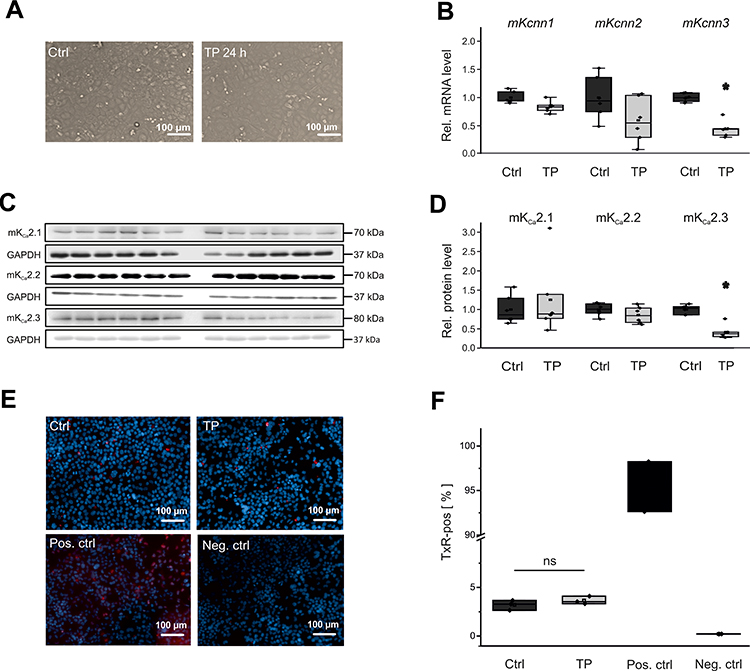 |
Figure 3 Effects of electrical tachypacing on KCa2 channel expression and viability in HL-1 atrial cells. (A) Representative microscopic images under control conditions (Ctrl) and after 24 h tachypacing (TP), respectively (scale bar, 100 µm). (B) Kcnn1 (n = 6), Kcnn2 (n = 6), and Kcnn3 mRNA expression (n = 6) calculated relative to untreated controls (n = 6 each). (C, D) Remodeling of KCa2.1 (n = 6), KCa2.2 (n = 6), and KCa2.3 protein (n = 6), evaluated using Western blot analyses and compared to controls (n = 6 each). (E, F) Apoptosis of HL-1 cells subjected to tachypacing (n = 3), including untreated (Ctrl; n = 3), positive (Pos. ctrl; n = 2), and negative controls (Neg. ctrl; n = 2). (E) Representative fluorescence microphotographs corresponding to terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assays (scale bar, 100 µm). Red nuclear fluorescence reflects endonucleolytic DNA degradation and apoptosis. (F) Mean apoptosis rates. TUNEL (Texas Red, TxR)-positive cells are expressed in relation to the total number of cells. Data are provided as box plots with underlying dots representing original data; *P < 0.05 versus HL-1 cells not subjected to tachypacing, ns= non significant. |
Effects of Cell Stretch on KCa2 Channel Expression
Membrane stretch is another key factor in pathologic atrial remodeling following pressure and volume overload. To evaluate its effects on electrical remodeling, HL-1 cells were exposed to 24 h membrane stretch mimicking atrial dilatation (Figure 4A). Contrary to effects of TP on KCa2.3 channel expression, stretch reduced KCa2.1 channel abundance without affecting KCa2.2 or KCa2.3 levels (Figure 4B–D). Kcnn1 mRNA was lowered by 38% (P = 0.015, n = 6) compared to untreated controls (n = 6), while KCa2.1 protein was reduced by 51% (P = 0.019, n = 6) compared with controls (n = 6).
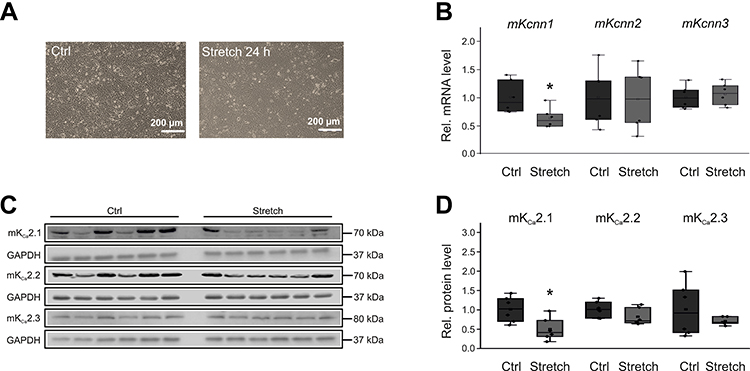 |
Figure 4 Stretch-related KCa2 channel remodeling in HL-1 cells. (A) Representative microphotographs under control conditions (Ctrl) and after 24 h cell stretch, respectively (scale bar, 200 µm). (B) Kcnn1 (n = 6), Kcnn2 (n = 6), and Kcnn3 mRNA expression (n = 6) relative to untreated controls (n = 6 each). (C, D) Expression of KCa2.1 (n = 6), KCa2.2 (n = 6), and KCa2.3 protein (n = 6) compared with controls (n = 6 each). Data are shown as box plots with underlying dots representing original data; *P < 0.05 versus HL-1 cells not subjected to stretch. |
KCa2 Channel Remodeling Upon β-Adrenergic Stimulation
We next assessed the potential effect of β-adrenergic stress as a proarrhythmic mechanism during AF/HF. To this end, HL-1 cells were maintained for 24 h in culture medium supplemented with 50 µM isoproterenol (Figure 5A). Adrenergic stimulation reduced Kcnn1 mRNA levels by 38% (P < 0.001, n = 6) compared to non-paced control cells (n = 6) (Figure 5B). KCa2.1 protein was not significantly affected by isoproterenol (Figure 5C and D). Furthermore, β-adrenergic stimulation did not change KCa2.2 and KCa2.3 transcript and protein levels, respectively (Figure 5B–D).
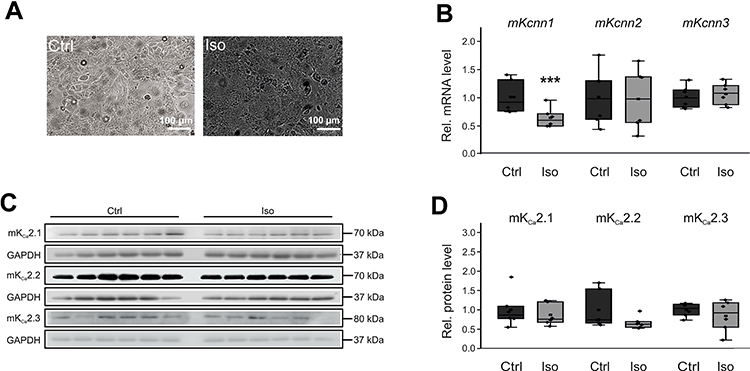 |
Figure 5 Effects of β-adrenergic stimulation on KCa2 channel expression in HL-1 cells. (A) Representative microphotographs under control conditions (Ctrl) and after incubation with 50 µM isoproterenol for 24 h, respectively (scale bar, 100 µm). (B) Kcnn1 (n = 6), Kcnn2 (n = 6), and Kcnn3 mRNA expression (n = 6) relative to untreated controls (n = 6 each). (C, D) Relative KCa2.1 (n = 6), KCa2.2 (n = 6), and KCa2.3 protein levels (n = 6) compared with controls (n = 6 each). Data are shown as box plots with underlying dots representing original data; ***P < 0.001 versus HL-1 cells not subjected to isoproterenol administration. |
Effects of Hypoxia on KCa2 Channel Expression
High rates of electrical activity and HF may be associated with regional myocardial hypoxia. To examine the impact of hypoxia on KCa2 expression, HL-1 cells were maintained at 1% oxygen for 24 h (Figure 6A). Hypoxia increased Kcnn2 mRNA (+46%, P = 0.013, n = 6; Figure 3B) and KCa2.2 protein (+66%, P = 0.059, n = 6; Figure 3C and D) compared with controls receiving 21% oxygen (n = 6 each). By contrast, hypoxia resulted in reduction of KCa2.3 expression at mRNA (−42%, P < 0.001, n =6; Figure 6B) and protein levels (−63%, P = 0.001, n = 6; Figure 6C and D) compared with controls (n = 6 each). KCa2.1 expression was not significantly altered upon hypoxia. Finally, we observed low apoptosis rates that did not differ between HL-1 cells maintained at 1% oxygen (1.9 ± 0.3%, n = 3) or 21% oxygen (1.1 ± 0.1%, P = 0.064, n = 3), indicating that KCa2 channel expression changes were not triggered by potential hypoxia-related apoptosis (Figure 6E and F).
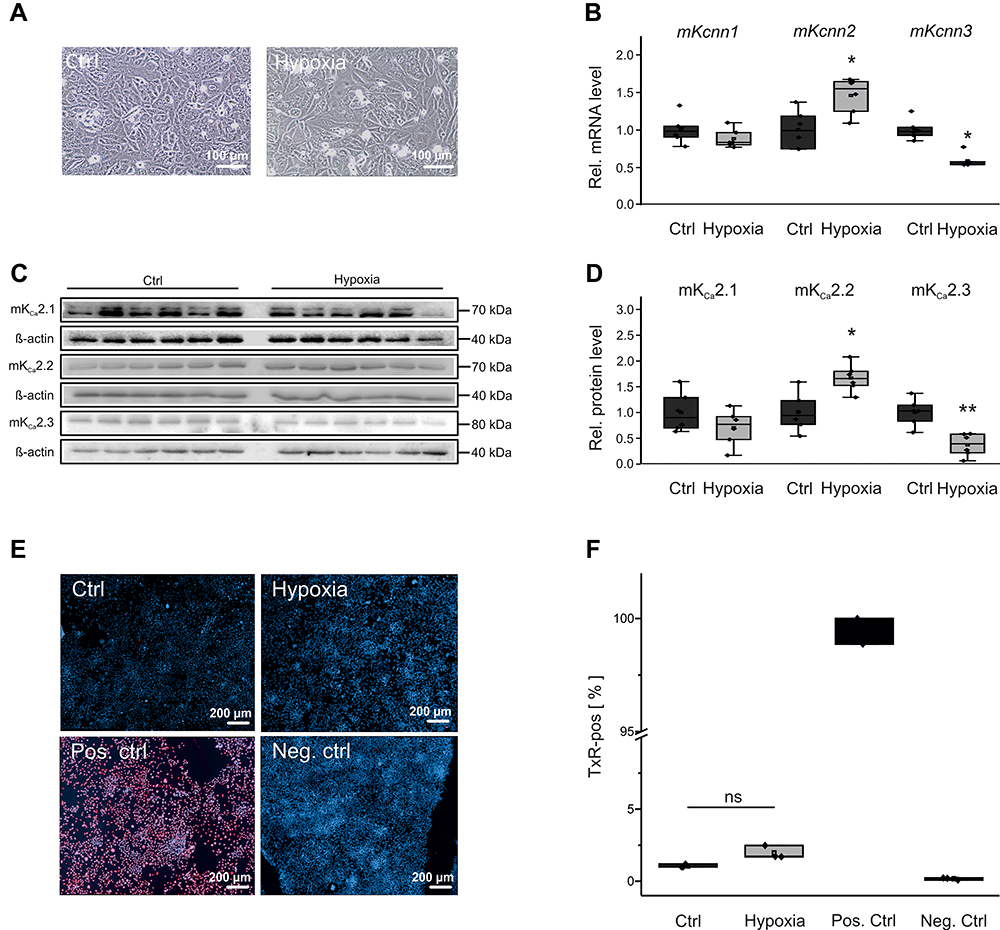 |
Figure 6 KCa2 channel expression and viability of HL-1 atrial cells during hypoxia. (A) Representative microscopic images under control conditions (Ctrl, 21% O2) and after 24 h hypoxia (1% O2), respectively (scale bar, 100 µm). (B) Kcnn1 (n = 6), Kcnn2 (n = 6), and Kcnn3 mRNA expression (n = 6) calculated relative to untreated controls (n = 6). (C, D) Relative KCa2.1 (n = 6), KCa2.2 (n = 6), and KCa2.3 protein expression (n = 6) during hypoxia and under control conditions (n = 6 each). (E, F) Apoptosis of HL-1 cells subjected to hypoxia, including untreated (Ctrl), positive (Pos. ctrl), and negative controls (Neg. ctrl). (E) Representative fluorescence microphotographs corresponding to TUNEL assays (scale bar, 200 µm). (F) Mean apoptosis ratios. TUNEL (Texas Red, TxR)-positive cells are provided in relation to the total number of cells. Data are given as box plots with underlying dots representing original data; *P < 0.05, **P < 0.01 versus HL-1 cells not exposed to hypoxia. |
Discussion
Remodeling of KCa2.1, KCa2.2, and KCa2.3 Channels During AF Complicated by HF
Paroxysmal and chronic atrial fibrillation was associated with reduced atrial KCa2.1, KCa2.2, and KCa2.3 transcript levels in patients with severe HF. In pigs subjected to AF induction for a limited time (ie, 7 days and 14 days, respectively), suppression of KCa2 channel expression occurred in time- and channel-dependent manner. While KCa2.1 mRNA levels were not significantly affected, KCa2.2 transcripts were reduced after 7 and 14 days. KCa2.3 mRNA exhibited an intermediate phenotype characterized by reduction requiring 14 days of AF, whereas expression was not changed after 7 days AF. These observations suggest that KCa2 channel suppression may be more pronounced in cases with long AF duration. Reduced expression of KCa2.1, KCa2.2, and KCa2.3 in patients with preserved cardiac function and persistent or permanent AF has previously been reported.7,8,13 The present work extends AF-related KCa2 downregulation to patients with severely impaired left ventricular function, highlighting a broader mechanism of atrial electrophysiological remodeling. While these results suggest uniform KCa2 remodeling during AF, a more complex picture with species- and mechanism-based remodeling of the channels is supported by data obtained from a canine model of AF induced by 7 days atrial tachypacing.11 In dogs, left atrial KCa2.1 channel protein was increased without changes in the corresponding mRNA. This observation may correspond to a tendency towards higher KCNN1 mRNA in the present pig model that did not reach statistical significance (Figure 2B). Importantly, there was no remodeling of KCa2.2 or KCa2.3 in the canine model. In case of KCa2.3 this may be due to the duration of AF that was reduced only after 14 days AF in pigs. In addition, species-dependent differences could explain the lack of KCa2.2 remodeling in dogs vs pigs. Considering that right atrial tissue was analyzed in the present work, whereas Qi et al11 obtained left atrial samples for analyses, KCa2 channel remodeling may exhibit side-specific components. Finally, different degrees of proarrhythmic trigger activity may account for differential KCa2 channel remodeling and were investigated further in this study.
Differential Mechanistic Effects of AF/HF-Related Triggers on KCa2 Channel Abundance
The aim of the present approach was to dissect individual mechanisms leading to electrical remodeling in AF. Potential proarrhythmic triggers that may be differentially activated during AF and/or HF comprise high-rate electrical activity, myocardial wall stress, β-adrenergic activation, and hypoxia. In HL-1 atrial myocytes, KCa2.1 channel suppression was mediated by cell stretch and β-adrenergic stress, whereas KCa2.3 expression was reduced by electrical tachypacing and hypoxia. Thus, either of these mechanisms (or a combination thereof) may contribute to the downregulation of both channels observed in humans and in pigs. Considering the lack of KCa2.3 reduction after 7 days AF in the porcine model, we may speculate that high-rate electrical activity and hypoxia play a less important role in short-term AF in these animals. In contrast, KCa2.2 channel reduction was not detected under any of these conditions, suggesting additional triggers of KCa2.2 downregulation in human AF and in pigs. However, KCa2.2 was enhanced by hypoxia, representing a previously unrecognized regulatory mechanism in cardiomyocytes, with no apparent impact on the present patient subgroup or on the animal model studied here. Of note, upregulation or activation of KCa2.2 is associated with reduction of reactive oxygen species and neuroprotection or reduction of arrhythmias in neuronal and cardiac cell models, respectively.26–28
In summary, this work provides a comprehensive analysis of the effects of different AF triggers on Kcnn1-3 mRNA expression in a cellular model. We acknowledge that this approach cannot yet yield a complete assessment of KCNN remodeling observed in AF patients or in animal models. Rather, it provides initially novel insights into the mechanistic effects of AF-related triggers on atrial KCa channels. Further studies are required to integrate these individual mechanisms and to assess their functional, in vivo significance in more detail. To further delineate the specific impact of individual proarrhythmic triggers in humans, future studies that include larger patient cohorts are required to establish significant associations between clinical characteristics, such as heart rate, left atrial size, or catecholamine levels and KCa2 channel expression, and to elucidate underlying signal transduction pathways.
Clinical Implications, Limitations, and Future Directions
We specifically investigated potentially proarrhythmic mechanisms in models of a clinically relevant subgroup of AF patients with concomitant HF. Atrial APD prolongation and increased AERPs due to distinct changes in ion channel expression are characteristic of HF patients with reduced left ventricular ejection fraction (LVEF).16 The porcine model employed in this work is characterized by a reduction in LV function and prolonged AERP, thus partially resembling findings in human AF/HF patients.16 Antiarrhythmic strategies that specifically target electrical remodeling to improve and personalize AF treatment are limited. Downregulation of repolarizing KCa2 potassium channels observed here is expected to contribute to prolonged atrial refractoriness. Reversal of KCa2 channel remodeling by channel activation or overexpression could represent an individualized strategy for rhythm control in this specific AF patient entity. Trigger-specific remodeling mechanisms reflect the need for channel-specific pharmacological modulation. Unfortunately, no subtype-specific modulators of KCa2.1–2.3 channels have been identified to date, preventing further evaluation of the channels’ individual contributions to the atrial action potential and of their potential role in antiarrhythmic therapy. Furthermore, current KCa2 channel blockers exhibit off-target effects on sodium channels.2 It cannot be expected that a single antiarrhythmic intervention will be sufficient to treat all types of AF, as both shortening and prolongation of atrial APD have been suggested to increase AF susceptibility.3,11,14,15 Rather, the mechanism-based understanding that is advanced by the present work suggests that tailored, individualized antiarrhythmic strategies are required based on patient-specific mechanisms. Inhibition of KCa2 channels and prolongation of atrial refractoriness by the compound AP30663 is currently evaluated in a Phase II clinical trial for termination of AF (EudraCT Number: 2018–004445-17).29 While this intervention may be effective in a subset of patients, we suggest that the therapeutic efficacy of interventions targeting KCa2 channels may require channel activation in other subjects, depending on the individual KCa2 expression and function.
We acknowledge that small sample sizes due to the large animal model and limitations in human tissue acquisition resulted in relatively low statistical power. In addition, this study was designed to specifically address KCa2 channel expression upon different conditions. Functional analyses were not performed due to pharmacological limitations mentioned above and require future studies when specific inhibitors are available. Additional effects of comorbidities with potential effects on KCa2 channel remodeling such as diabetes9 were beyond the scope of the present work and therefore need to be addressed in separate approaches.
Conclusions
This study advances the understanding of the role of KCa2 channels in AF by providing evidence for trigger- and channel-specific remodeling. Suppression of KCa2.1, KCa2.2, and KCa2.3 channel expression in human AF and in pigs is expected to facilitate action potential prolongation that is characteristic of AF with concomitant HF. Differential effects of proarrhythmic triggers resulting in a reduction of KCa2.1 (stretch, β-adrenergic activation) and KCa2.3 (electrical tachypacing, hypoxia) or in an enhanced expression of KCa2.2 (hypoxia) contribute to complex KCa2 channel remodeling. Trigger-dependent KCa2 regulation in atrial myocytes that is determined by individual patient characteristics and by environmental factors provides a new basis for personalized antiarrhythmic therapy.
Data Sharing Statement
The data underlying this article are available in the article and supplementary material.
Acknowledgments
We thank Teresa Caspari, Nic Geßner, Xenia Kramp, Emili Manolova, and Nadine Weiberg for their excellent technical assistance, and we are grateful to the operating room team at the Department of Cardiac Surgery of Heidelberg University for supporting our work. We gratefully acknowledge the discussions on methodology with Michael Bartolf-Kopp (Ullrich Lab, Department of Physiology and Pathophysiology, Heidelberg University, Germany).
Funding
This work was supported in part by research grants from the University of Heidelberg, Faculty of Medicine (Postdoctoral Fellowships to P.L. and to A.K.R.), from the German Cardiac Society (Fellowships to A.K.R. and to P.L., Otto-Hess-Promotionsstipendium to D.G.), from the German Internal Medicine Society (DGIM clinician-scientist program to A.K.R), from the Elisabeth und Rudolf-Hirsch Stiftung für Medizinische Forschung (to A.K.R), from the Ernst und Berta Grimmke-Stiftung (to P.L.), from the German Heart Foundation/German Foundation of Heart Research (Fellowship to A.K.R., Kaltenbach Scholarship to D.G., Project F/08/14 to D.T.), from the Joachim Siebeneicher Foundation (to D.T.), from the Deutsche Forschungsgemeinschaft (German Research Foundation; TH 1120/7-1 and TH 1120/8-1 to D.T.), and from the Ministry of Science, Research and the Arts Baden-Wuerttemberg (Sonderlinie Medizin to D.T.). D.G., T.W., and M.E.M. were supported by the Cardiology Career Program of the Department of Cardiology, University of Heidelberg, and D.G. received a scholarship from the German Academic Scholarship Foundation. The funding sources had no involvement in study design; in the collection, analysis and interpretation of data; and in the decision to submit the article for publication.
Disclosure
A.K.R. report grants from Heidelberg University, fellowships from German Cardiac Society, clinician-scientist program from German Internal Medicine Society, and fellowship from German Heart Foundation/German Foundation of Heart Research, during the conduct of the study; and educational support from Boston Scientific, Johnson & Johnson, Abbott and Medtronic. Mara Elena Müller reports grants from Heidelberg University Medical Hospital, during the conduct of the study; personal fees from Heidelberg University Medical Hospital, outside the submitted work. P.L. reports receiving lecture fees from Bayer Vital and Pfizer Pharma and educational support from Boston Scientific and Johnson & Johnson. Hugo A Katus reports personal fees from Astra Zeneca, personal fees from Daiichi Sankyo, personal fees from Bayer Vital, personal fees from Roche Diagnostics, personal fees from Novo Nordisk, and personal fees from Boehringer Ingelheim, outside the submitted work. D.T. reports lecture fees/honoraria from Bayer Vital, lecture fees/honoraria from Boehringer Ingelheim Pharma, lecture fees/honoraria from Bristol-Myers Squibb, lecture fees/honoraria and study support from Daiichi Sankyo, lecture fees/honoraria from Medtronic, lecture fees/honoraria from Pfizer Pharma, personal fees from Sanofi-Aventis, lecture fees/honoraria from St. Jude Medical, lecture fees/honoraria from ZOLL CMS, grants from German Heart Foundation/German Foundation of Heart Research, research award from Joachim Siebeneicher Foundation, grants from German Research Foundation, grant from Ministry of Science, Research and the Arts Baden-Wuerttemberg, during the conduct of the study. The authors reported no other potential conflicts of interest for this work. The remaining authors have reported that they have no relationships relevant to the content of this paper to disclose.
References
1. Lugenbiel P, Wenz F, Syren P, et al. TREK-1 (K2P2.1) K+ channels are suppressed in patients with atrial fibrillation and heart failure and provide therapeutic targets for rhythm control. Basic Res Cardiol. 2017;112:8.
2. Skibsbye L, Wang X, Axelsen LN, et al. Antiarrhythmic mechanisms of SK channel inhibition in the rat atrium. J Cardiovasc Pharmacol. 2015;66165–66176.
3. Zhang XD, Lieu DK, Chiamvimonvat N. Small-conductance Ca2+ -activated K+ channels and cardiac arrhythmias. Heart Rhythm. 2015;12:1845–1851.
4. Maylie J, Bond CT, Herson PS, Lee WS, Adelman JP. Small conductance Ca2+-activated K+ channels and calmodulin. J Physiol. 2004;554:255–261.
5. Xu Y, Tuteja D, Zhang Z, et al. Molecular identification and functional roles of a Ca2+-activated K+ channel in human and mouse hearts. J Biol Chem. 2003;278:49085–49094.
6. Tuteja D, Xu D, Timofeyev V, et al. Differential expression of small-conductance Ca2+-activated K+ channels SK1, SK2, and SK3 in mouse atrial and ventricular myocytes. Am J Physiol Heart Circ Physiol. 2005;289:H2714–23.
7. Yu T, Deng C, Wu R, et al. Decreased expression of small-conductance Ca2+-activated K+ channels SK1 and SK2 in human chronic atrial fibrillation. Life Sci. 2012;90:219–227.
8. Skibsbye L, Poulet C, Diness JG, et al. Small-conductance calcium-activated potassium (SK) channels contribute to action potential repolarization in human atria. Cardiovasc Res. 2014;103:156–167.
9. Yi F, Ling TY, Lu T, et al. Down-regulation of the small conductance calcium-activated potassium channels in diabetic mouse atria. J Biol Chem. 2015;290:7016–7026.
10. Diness JG, Sørensen US, Nissen JD, et al. Inhibition of small-conductance Ca2+-activated K+ channels terminates and protects against atrial fibrillation. Circ Arrhythm Electrophysiol. 2010;3:380–390.
11. Qi XY, Diness JG, Brundel BJ, et al. Role of small-conductance calcium-activated potassium channels in atrial electrophysiology and fibrillation in the dog. Circulation. 2014;129:430–440.
12. Christophersen IE, Rienstra M, Roselli C, et al. Large-scale analyses of common and rare variants identify 12 new loci associated with atrial fibrillation. Nat Genet. 2017;49:946–952.
13. Fan X, Yu Y, Lan H, et al. Ca2+/calmodulin-dependent protein kinase II (CaMKII) increases small-conductance Ca2+-activated K+ current in patients with chronic atrial fibrillation. Med Sci Monit. 2018;24:3011–3023.
14. Li N, Timofeyev V, Tuteja D, et al. Ablation of a Ca2+-activated K+ channel (SK2 channel) results in action potential prolongation in atrial myocytes and atrial fibrillation. J Physiol. 2009;587:1087–1100.
15. Zhang XD, Timofeyev V, Li N, et al. Critical roles of a small conductance Ca2⁺-activated K⁺ channel (SK3) in the repolarization process of atrial myocytes. Cardiovasc Res. 2014;101:317–325.
16. Rahm AK, Wieder T, Gramlich D, et al. HDAC2-dependent remodeling of KCa2.2 (KCNN2) and KCa2.3 (KCNN3) K+ channels in atrial fibrillation with concomitant heart failure. Life Sci. 2021;266:118892.
17. Lugenbiel P, Govorov K, Syren P, et al. Epigenetic regulation of cardiac electrophysiology in atrial fibrillation: HDAC2 determines action potential duration and suppresses NRSF in cardiomyocytes. Basic Res Cardiol. 2021;116:13.
18. Rahm AK, Wieder T, Gramlich D, et al. Differential regulation of KCa2.1 (KCNN1) K+ channel expression by histone deacetylases in atrial fibrillation with concomitant heart failure. Physiol Rep. 2021. doi:10.14814/phy2.14835
19. Lugenbiel P, Wenz F, Govorov K, Schweizer PA, Katus HA, Thomas D. Atrial fibrillation complicated by heart failure induces distinct remodeling of calcium cycling proteins. PLoS One. 2015;10:e0116395.
20. Claycomb WC, Lanson NA
21. White SM, Constantin PE, Claycomb WC. Cardiac physiology at the cellular level: use of cultured HL-1 cardiomyocytes for studies of cardiac muscle cell structure and function. Am J Physiol Heart Circ Physiol. 2004;286:H823–9.
22. Gizurarson S, Shao Y, Miljanovic A, et al. Electrophysiological effects of lysophosphatidylcholine on HL-1 cardiomyocytes assessed with a microelectrode array system. Cell Physiol Biochem. 2012;30:477–488.
23. Kao YH, Chen YC, Chung CC, et al. Heart failure and angiotensin II modulate atrial Pitx2c promotor methylation. Clin Exp Pharmacol Physiol. 2013;40:379–384.
24. Shao Y, Redfors B, Ståhlman M, et al. A mouse model reveals an important role for catecholamine-induced lipotoxicity in the pathogenesis of stress-induced cardiomyopathy. Eur J Heart Fail. 2013;15:9–22.
25. Lugenbiel P, Govorov K, Rahm AK, et al. Inhibition of histone deacetylases induces K+ channel remodeling and action potential prolongation in HL-1 atrial cardiomyocytes. Cell Physiol Biochem. 2018;49:65–77.
26. Honrath B, Matschke L, Meyer T, et al. SK2 channels regulate mitochondrial respiration and mitochondrial Ca2+ uptake. Cell Death Differ. 2017;24:761–773.
27. Richter M, Nickel C, Apel L, et al. SK channel activation modulates mitochondrial respiration and attenuates neuronal HT-22 cell damage induced by H2O2. Neurochem Int. 2015;81:63–67.
28. Kim TY, Terentyeva R, Roder KH, et al. SK channel enhancers attenuate Ca2+-dependent arrhythmia in hypertrophic hearts by regulating mito-ROS-dependent oxidation and activity of RyR. Cardiovasc Res. 2017;113:343–353.
29. Diness JG, Kirchhoff JE, Speerschneider T, et al. The KCa2 channel inhibitor AP30663 selectively increases atrial refractoriness, converts vernakalant-resistant atrial fibrillation and prevents its reinduction in conscious pigs. Front Pharmacol. 2020;11:159.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.