Back to Journals » Drug Design, Development and Therapy » Volume 14
Treatment with a PPAR-γ Agonist Protects Against Hyperuricemic Nephropathy in a Rat Model
Authors Wang X, Deng J, Xiong C, Chen H, Zhou Q, Xia Y, Shao X, Zou H
Received 23 January 2020
Accepted for publication 15 May 2020
Published 8 June 2020 Volume 2020:14 Pages 2221—2233
DOI https://doi.org/10.2147/DDDT.S247091
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Jianbo Sun
Xin Wang,1,* Jin Deng,2,* Chongxiang Xiong,1 Haishan Chen,1 Qin Zhou,1 Yue Xia,1 Xiaofei Shao,1 Hequn Zou1
1Department of Nephrology, The Third Affiliated Hospital, Southern Medical University, Guangzhou, People’s Republic of China; 2Department of Nephrology, The First Affiliated Hospital of University of South China, Hengyang, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Hequn Zou Tel +86 020-62784391
Email [email protected]
Purpose: Hyperuricemia is an independent risk factor for renal damage and can promote the progression of chronic kidney disease (CKD). In the present study, we employ a rat model to investigate the effects of rosiglitazone (RGTZ), a peroxisome proliferator-activated receptor-gamma agonist, on the development of hyperuricemic nephropathy (HN), and we elucidate the mechanisms involved.
Methods: An HN rat model was established by oral administration of a mixture of adenine and potassium oxonate daily for 3 weeks. Twenty-four rats were divided into 4 groups: sham treatment, sham treatment plus RGTZ, HN, and HN treated with RGTZ.
Results: Administration of RGTZ effectively preserved renal function, decreased urine microalbumin, and inhibited interstitial fibrosis and macrophage infiltration in a rat HN model. RGTZ treatment also inhibited TGF-β and NF-κB pathway activation, decreased expression of fibronectin, collagen I, α-SMA, vimentin, MCP-1, RANTES, TNF-α, and IL-1β, and increased E-cadherin expression in the kidneys of HN rats. Furthermore, RGTZ treatment preserved expression of OAT1 and OAT3 in the kidney of HN rats.
Conclusion: RGTZ attenuates the progression of HN through inhibiting TGF-β signaling, suppressing epithelial-to-mesenchymal transition, reducing inflammation, and lowering serum uric acid levels by preserving expression of urate transporters.
Keywords: hyperuricemic nephropathy, peroxisome proliferator-activated receptor-gamma, rosiglitazone, renal fibrosis, epithelial-to-mesenchymal transition, inflammation
Introduction
Hyperuricemia is an independent risk factor for renal damage and can promote the progression of chronic kidney disease (CKD).1,2 Hyperuricemia is caused by increased uric acid production and decreased renal excretion. Uric acid is formed in the liver from dietary purines and endogenously synthesized purines. Uric acid is translocated across the basolateral membrane from the bloodstream into proximal tubular cells through organic anion transporters (OAT), including OAT1 (SLC22A6) and OAT3 (SLC22A8), and is then secreted into the tubular lumen.3 Decreased expression of OAT1 and OAT3 can lead to an accumulation of uric acid, leading to hyperuricemic nephropathy (HN).
Mechanisms of renal injury induced by hyperuricemia remain poorly understood. HN is characterized by uric acid kidney stones, glomerular hypertension, arteriolosclerosis, and tubulointerstitial fibrosis, and can eventually lead to chronic renal failure. Tubulointerstitial fibrosis plays an important role in the renal injury associated with HN, and excessive deposition of extracellular matrix (ECM) in kidney tissue is a major feature of tubulointerstitial fibrosis.4 Myofibroblasts are the primary cell type that secretes collagen, and excessive collagen secretion leads to ECM overaccumulation and renal interstitial fibrosis. In renal tissue, tubular epithelial-to-mesenchymal transition (EMT) is a prominent source of myofibroblasts, and plays an important role in renal fibrosis.5,6 EMT can be induced by many factors, including transforming growth factor β1 (TGF-β1) and uric acid.7,8 Furthermore, uric acid can also induce inflammation by activating the NF-κB pathway and inducing infiltration of monocytes/macrophages and production of multiple cytokines, such as interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), monocyte chemoattractant protein-1 (MCP-1), and regulated on activation, normal T cell expressed and secreted (RANTES).9,10
Peroxisome proliferator-activated receptor γ (PPAR-γ) is a ligand-dependent transcription factor belonging to the superfamily of nuclear hormone receptors, and is widely expressed in renal tissues.11,12 In addition to its glucose-lowering activity in diabetes, PPAR-γ activation also exerts anti-inflammatory and antifibrotic effects in a variety of other diseases.13–15 However, it remains unclear whether activation of the PPAR-γ pathway is capable of halting or slowing the progression of HN. In this study, we investigated the effects of PPAR-γ activation by RGTZ, a highly selective PPAR-γ agonist, on the development of HN.
Materials and Methods
Antibodies and Reagents
Antibodies to PPAR-γ were purchased from Cell Signaling Technology (Danvers, MA, USA). Antibodies to fibronectin, p-NF-κB (p65), NF-κB (p65), and CD68 were purchased from Abcam (Cambridge, MA, USA). Antibodies to collagen I and α-smooth muscle actin (α-SMA) were purchased from Affinity Biosciences (Cincinnati, OH, USA). Antibodies to TGF-β1 were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Antibodies to Smad3 and p-Smad3 were purchased from Hangzhou HuaAn Biotechnology Co., Ltd. (Hangzhou, China). Antibodies to E-cadherin and vimentin were purchased from Proteintech (Chicago, IL, USA). Antibodies to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were purchased from CWBIO (Beijing, China). RGTZ was purchased from APExBIO Technology LLC (Houston, TX, USA). Serum XOD kit was purchased from Jiancheng Bioengineering Institute (Nanjing, China). MCP-1, RANTES, TNF-α, and IL-1β ELISA kits were purchased from CUSABIO Technology LLC (Wuhan, China).
Animals and Experimental Design
The animal experiments were approved by the Southern Medical University Experimental Animal Ethics Committee. All animal research was conducted in accordance with the Guiding Opinions on the Treatment of Laboratory Animals issued and the Laboratory Animal-Guideline for Ethical Review of Animal Welfare issued by the National Standard GB/T35892-2018 of the People’s Republic of China. Male Sprague–Dawley (SD) rats of specific pathogen free grade, weighing 200–220 g, were purchased from the Experimental Animal Center of Southern Medical University. The rats were housed under standard conditions with controlled 12 h light/dark cycles, temperature, and humidity, and had unrestrained activity and free access to water and food. The rats were allowed 7 days to acclimate to the environment before experiments. The HN rat model was established by oral administration of a mixture of adenine (0.1 g/kg) and potassium oxonate (1.5 g/kg) daily for 3 weeks, as we described previously.10 Twenty-four male rats were randomized into four treatment groups: sham treatment, sham treatment plus RGTZ (5 mg/kg/d, orally), HN, and HN treated with RGTZ (5 mg/kg/d, orally). For oral administration, RGTZ was dissolved in Carboxymethylcellulose sodium.
Functional Parameters
After three weeks, blood samples were taken to measure serum creatinine, blood urea nitrogen, serum uric acid, and other biochemical indices. In addition, 24 hours urine samples were collected, and levels of urine microalbumin and urine uric acid were measured.
Histology and Immunohistochemistry
Formalin-fixed kidneys were embedded in paraffin blocks and sections 3 μm in thickness were prepared. Sections were stained with hematoxylin–eosin (HE), Masson’s trichrome, and periodic acid-Schiff (PAS) staining. Renal tissue sections were transferred into a 10 mmol/L citrate buffer solution adjusted to a pH of 6.0. The sections were then treated in a microwave for 20 min for antigen retrieval. Afterwards, endogenous peroxidase activity was inactivated by treatment with 3% hydrogen peroxide. Thereafter, primary antibodies were added dropwise to the renal tissue sections and sections were incubated at 4°C overnight. Sections were then washed with PBS, and incubated in secondary antibody for 30 min at room temperature. The sections were again washed with PBS. Subsequently, the sections were visualized with 3,3ʹ-diaminobenzidine (DAB, Golden Bridge Biotechnology Co., Beijing, China) and counterstained with hematoxylin. Masson’s Trichrome-stained sections from each kidney were graded for the presence of interstitial fibrosis according to the following scale: 0, no evidence of interstitial fibrosis; 1, <25% involvement; 2, 25 to 50% involvement and 3, >50% involvement. The score of each kidney sample was carried out as the average of at least 10 random high-power (×400) fields.16
Western Blotting Analysis
Total protein was extracted from renal tissue samples using RIPA lysis Buffer. The protein concentrations of the samples were measured using a Bicinchoninic Acid (BCA) Protein Assay Kit (Beyotime, Shanghai, China). Equal amounts of total protein were electrophoresed and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, MA, USA). The membranes were blocked with 5% BSA and incubated overnight with primary antibodies at 4°C. Membranes were subsequently incubated with corresponding secondary antibodies (1:5000, CWBIO) for 60 min at room temperature. Immunoreactive bands were detected using chemiluminescence detection reagents and the density of the immunoreactive bands was measured by a digital visualizer (Eastman Kodak Company, USA). Data were analyzed using ImageJ software version 1.46 (Wayne Rasband, National Institutes of Health, USA). All experiments were repeated three times.
RT-PCR
Total RNA was extracted from renal tissue using TRIzol reagent (TaKaRa, Dalian, China). Total mRNA was reversed transcribed into cDNA using a Transcriptor First Strand cDNA synthesis kit (TaKaRa). Quantitative real-time PCR was performed using SYBR Green PCR master mix (Applied Biosystems) on a BioRadiCycleriQ Detection System. The primer sequences used were as follows: OAT1 forward 5ʹ-GAGCTGTACCCCACAGTGATT-3ʹ, reverse 5ʹ-GAACTCTGCAGTCATACTC- ACC-3ʹ; OAT3 forward 5ʹ-AGTCCTCGGAATAGCCAACC-3ʹ, reverse 5ʹ-TGTA- CGAAGCGGAGACACTT-3ʹ; GAPDH forward 5ʹ-CCTCGTCTCATAGACAAG- ATGGT-3ʹ, reverse 5ʹ-GGGTAGAGTCATACTGGAACATG-3ʹ.
Assessment of Serum Xanthine Oxidase Activity
Serum activity of xanthine oxidase (XOD) was examined using a serum XOD kit according to the instructions provided by the manufacturer.
ELISA Analysis
Multiple renal cytokine levels were measured using appropriate ELISA kits according to the protocols provided by the manufacturer (CUSABIO Technology LLC, Wuhan, China).
Statistical Analysis
All data are presented as mean ± standard error of mean (SEM) and were analyzed using SPSS 20 software. Comparisons between groups were performed using one-way ANOVA. A P-value of p < 0.05 was considered to be statistically significant.
Results
RGTZ Induces PPAR-γ Activation in the Kidneys of Hyperuricemic Rats
To investigate the role of PPAR-γ in the development and progression of HN, we employed a rat model of HN. HN rats were treated daily with 5 mg/kg RGTZ, a PPAR-γ agonist. The expression of PPAR-γ was decreased in the kidney of HN rats, and treatment with RGTZ significantly increased PPAR-γ expression (Figure 1). This demonstrated that RGTZ induces PPAR-γ expression in the kidneys of HN rats.
 |
Figure 1 RGTZ induces PPAR-γ activation in the kidneys of HN rats. (A) A rat model of HN was established by daily oral administration of a mixture of adenine and potassium oxonate. In some rats, RGTZ was administrated. After 3 weeks, the kidneys were taken for immunoblot analysis for PPAR-γ or glyceraldehyde 3-phosphate dehydrogenase (GAPDH). (B) Expression levels of PPAR-γ were calculated by densitometry, and the ratio between PPAR-γ and GAPDH was determined. (C) Photomicrographs (original magnification, ×400) illustrate immunohistochemical PPAR-γ staining of kidney tissues. RGTZ, rosiglitazone. Data are represented as the mean ± SEM. *p < 0.05 versus sham group; #p < 0.05 versus sham + RGTZ group. **p< 0.05 versus HN group. |
PPAR-γ Activation Improves Renal Function and Alleviates Proteinuria and Renal Damage in HN Rats
Levels of serum creatinine, blood urea nitrogen, and urine microalbumin were significantly increased in the HN group (Figure 2A–C). These results are consistent with previous reports.10 Administration of RGTZ significantly reduced serum creatinine, blood urea nitrogen, and urine microalbumin levels in HN rats (Figure 2A–C). Furthermore, no marked histological changes were observed in the kidneys of sham treated rats who were fed normal chow, and not feed containing a mixture of adenine and potassium oxonate to induce HN (Figure 2D). The main histological disorders observed in the kidney sections from HN rats were severe glomerulosclerosis and tubulointerstitial damage with tubular atrophy, tubular dilatation, and interstitial fibrosis (Figure 2D). These lesions and tissue damage were noticeably reduced by treatment with RGTZ. These data demonstrate that PPAR-γ activation through RGTZ treatment exerts a protective effect and attenuates renal damage in this HN model.
 |
Figure 2 RGTZ improves renal function and alleviates proteinuria and renal damage in HN rats. Levels of (A) serum creatinine, (B) blood urea nitrogen (BUN), and (C) urine microalbumin were examined using biochemical assays. (D) Photomicrographs illustrating hematoxylin–eosin (HE), Masson’s trichrome, and periodic acid-Schiff (PAS) staining of kidney tissue from the different groups (original magnification ×200).(E) Interstitial fibrosis score of kidney tissue from the different groups. RGTZ, rosiglitazone. Data are represented as the mean ± SEM. *p < 0.05 vs sham group; #p< 0.05 vs sham + RGTZ group. **p < 0.05 vs HN group. |
PPAR-γ Activation Attenuates the Progression of Renal Fibrosis in HN Rats
Renal interstitial fibrosis is characterized by massive accumulation of ECM proteins in interstitial areas and is a common pathological feature of all types of CKD, including HN.10,17,18 We examined ECM protein deposition and expression to explore whether RGTZ alleviated renal fibrogenesis in HN rats. Masson’s trichrome staining showed that renal interstitial fibrosis was significantly more obvious in the HN group compared with the sham and RGTZ treatment groups (Figure 2D and E). Kidneys from rats treated with RGTZ showed significantly improved morphology, with less fibrosis observed in the interstitium (Figure 2D and E). We next explored the effect of RGTZ on collagen I and fibronectin expression by Western blot and immunohistochemistry. Both collagen I and fibronectin expression were increased in the kidneys of HN rats, and administration of RGTZ significantly reduced expression of collagen I and fibronectin (Figure 3). These data demonstrate that PPAR-γ activation attenuates the progression of renal fibrosis and accumulation of ECM proteins in the kidneys of HN rats.
 |
Figure 3 RGTZ decreases fibronectin and collagen I expression in the kidneys of HN rats. (A) Kidney tissue lysates were subjected to immunoblot analysis with specific antibodies against fibronectin, collagen I, or glyceraldehyde 3-phosphate dehydrogenase (GAPDH). (B) Expression levels of fibronectin were quantified by densitometry and normalized to GAPDH. (C) Expression levels of collagen I were quantified by densitometry and normalized to GAPDH. (D) Photomicrographs (original magnification, ×400) illustrate immunohistochemical fibronectin staining of the kidney tissues. RGTZ, rosiglitazone. Data are represented as the mean ± SEM. *p < 0.05 vs sham group; #p < 0.05 vs sham + RGTZ group. **p < 0.05 vs HN group. |
PPAR-γ Activation Abrogates the TGF-β/Smad3 Signaling Pathway in the Kidneys of HN Rats
TGF-β/Smad3 signaling pathway plays a critical role in renal fibrosis,7,19,20 but whether RGTZ inhibits uric acid–induced activation of β/Smad3 signaling remains unclear. We first investigated the effects of RGTZ on levels of TGF-β1 in the kidneys of HN rats by Western blot. TGF-β1 expression was upregulated in the kidneys of HN rats, and treatment with RGTZ prevented the upregulation of TGF-β1 protein (Figure 4A and B). Smad3 is the major downstream mediator of TGF-β signaling and directly binds to specific sites in promoter regions to mediate transcription of fibrosis-associated genes, such as collagen I.21 To further elucidate the effects of RGTZ on renal TGF-β signaling in the HN model, we analyzed the expression of phosphorylated Smad3 (p-Smad3) in kidney tissue from HN rats and HN rats treated with RGTZ. Western blot analysis indicated that the expression of p-Smad3 in the kidneys of HN rat was significantly increased, and that treatment with RGTZ reduced the upregulation of p-Smad3 expression in the HN model (Figure 4C and D). Collectively, these results suggest that PPAR-γ activation can inhibit TGF-β/Smad3 signaling in HN.
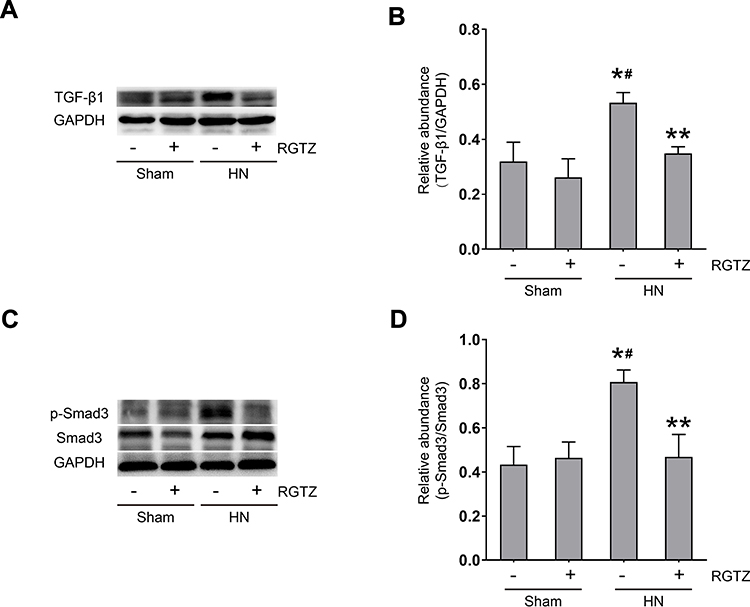 |
Figure 4 RGTZ abrogates TGF-β-Smad3 signaling in the kidneys of HN rats. (A) Kidney tissue lysates were subjected to immunoblot analysis with specific antibodies against TGF-β1 or glyceraldehyde 3-phosphate dehydrogenase (GAPDH). (B) Expression levels of TGF-β1 were quantified by densitometry and normalized to GAPDH. (C) The kidney tissue lysates were subjected to immunoblot analysis with specific antibodies against p-Smad3, Smad3, or GAPDH. (D) Expression levels of p- Smad3 were quantified by densitometry and normalized to Smad3. RGTZ, rosiglitazone. Data are represented as the mean ± SEM. *p < 0.05 vs sham group; #p< 0.05 vs sham + RGTZ group. **p < 0.05 vs HN group. |
PPAR-γ Activation Inhibits Renal Tubular Epithelial-to-Mesenchymal Transition in HN Rats
Renal tubular epithelial-to-mesenchymal transition (EMT) is defined as the process during which renal tubular epithelial cells lose their epithelial phenotype and acquire characteristic mesenchymal features, which leads to the secretion of ECM proteins.6 Renal tubular EMT plays an important role in the pathogenesis of renal interstitial fibrosis in all types of CKD, including HN.4,6,8 To investigate whether RGTZ inhibits renal tubular EMT in HN rats, we examined expression of E-cadherin, α-SMA, and vimentin in the kidneys of HN rats by Western blot and immunohistochemistry. Western blot of renal tissue showed that RGTZ treatment prevented decreases in E-cadherin, and prevented increases in α-SMA and vimentin expression induced by uric acid (Figure 5A–D). Immunohistochemistry staining revealed that expression of the epithelial marker E-cadherin was evident both in the cell membranes and cytoplasm in normal rat kidneys (Figure 5E), while kidneys of HN rats had reduced E-cadherin expression at 3 weeks (Figure 5E). Meanwhile, some tubules in kidneys of HN rats exhibited de novo staining of α-SMA, a marker of mesenchymal cells (Figure 5E). Treatment with RGTZ significantly increased E-cadherin expression and decreased α-SMA expression in kidney tissue of HN rats. These findings suggest that renal tubular EMT occurs in the kidneys of HN rats, and that PPAR-γ activation inhibits renal tubular EMT in the kidneys of HN animals.
 |
Figure 5 RGTZ inhibits renal tubular epithelial-to-mesenchymal transition in the kidneys of HN rats. (A) Kidney tissue lysates were subjected to immunoblot analysis with specific antibodies against E-cadherin, vimentin, α-SMA, or glyceraldehyde 3- phosphate dehydrogenase (GAPDH). (B) Expression levels of E-cadherin were quantified by densitometry and normalized to GAPDH. (C) Expression levels of vimentin were quantified by densitometry and normalized to GAPDH. (D) Expression levels of α-SMA were quantified by densitometry and normalized to GAPDH. (E) Photomicrographs (original magnification, ×400) illustrate immunohistochemical staining of E-cadherin and α-SMA in the kidney tissues. RGTZ, rosiglitazone. Data are represented as the mean ± SEM. *p < 0.05 vs sham group; #p < 0.05 vs sham + RGTZ group. **p < 0.05 vs HN group. |
PPAR-γ Activation Inhibits NF-κB Pathway Activation and Macrophage Infiltration in the Kidneys of HN Rats
NF-κB is a transcription factor and regulator of many genes, and is involved in the inflammatory response.22 NF-κB activation increases leukocyte infiltration and cytokine and chemokine expression.23 Expression of p-NF-κB (p65) was increased in the kidneys of HN rats, and this increase was significantly downregulated by RGTZ (Figure 6A and B). Inflammatory cell infiltration in the renal interstitium is a common pathologic feature of CKD, including HN.10,24,25 To assess whether RGTZ inhibited macrophage infiltration in HN, CD68, which is a marker of active macrophages, was examined by immunohistochemistry. The number of infiltrated macrophages was increased in the kidneys of HN rats, and was significantly decreased by treatment with RGTZ (Figure 6C). These results show that PPAR-γ activation can inhibit the NF-κB signaling pathway and macrophage infiltration in the kidneys of HN rats.
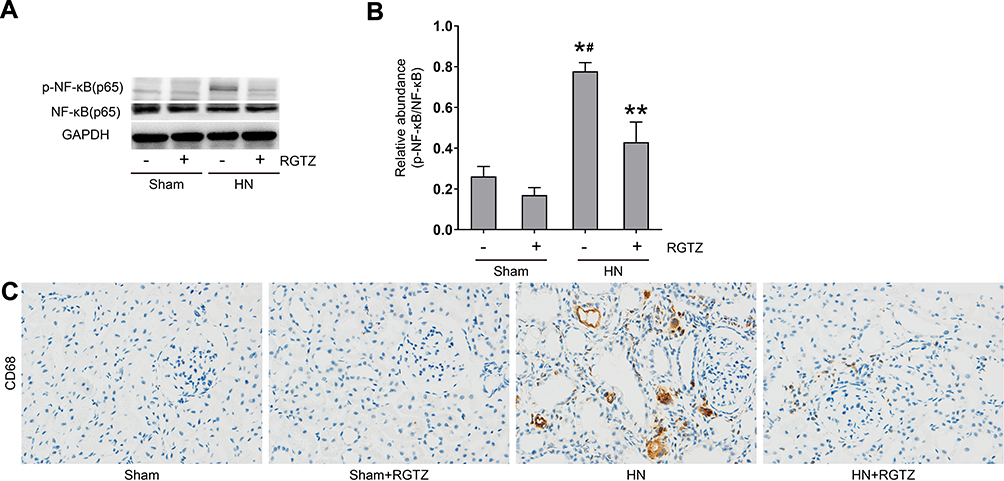 |
Figure 6 RGTZ mediates NF-κB pathway activation and inhibits macrophage infiltration in the kidneys of HN rats. (A) Kidney tissue lysates were subjected to immunoblot analysis with specific antibodies against p-NF-κB (p65), NF-κB (p65), or glyceraldehyde 3-phosphate dehydrogenase (GAPDH). (B) Expression levels of p-NF- κB (p65) were quantified by densitometry and normalized to NF-κB (p65). (C) Photomicrographs (original magnification, ×400) illustrate immunohistochemical staining for CD68 in kidney tissue. RGTZ, rosiglitazone. Data are represented as the mean ± SEM. *p < 0.05 vs sham group; #p < 0.05 vs sham + RGTZ group. **p < 0.05vs. HN group. |
PPAR-γ Activation Inhibits the Release of Cytokines/Chemokines in the Kidney of HN Rats
The production of proinflammatory cytokines/chemokines is a key initiating step in renal fibrogenesis.26 Therefore, we examined the expression of some proinflammatory cytokines/chemokines, including MCP-1, RANTES, TNF-α, and IL-1β, in the kidney by enzyme-linked immunosorbent assay (ELISA). The results showed that the levels of these molecules were significantly increased in the kidney of HN rats. Administration of RGTZ to HN rats resulted in decreased MCP-1, ICAM-1, TNF-α, and IL-1β levels (Figure 7). These data demonstrate that PPAR-γ activation suppresses the expression of proinflammatory cytokines/chemokines in the HN rat model.
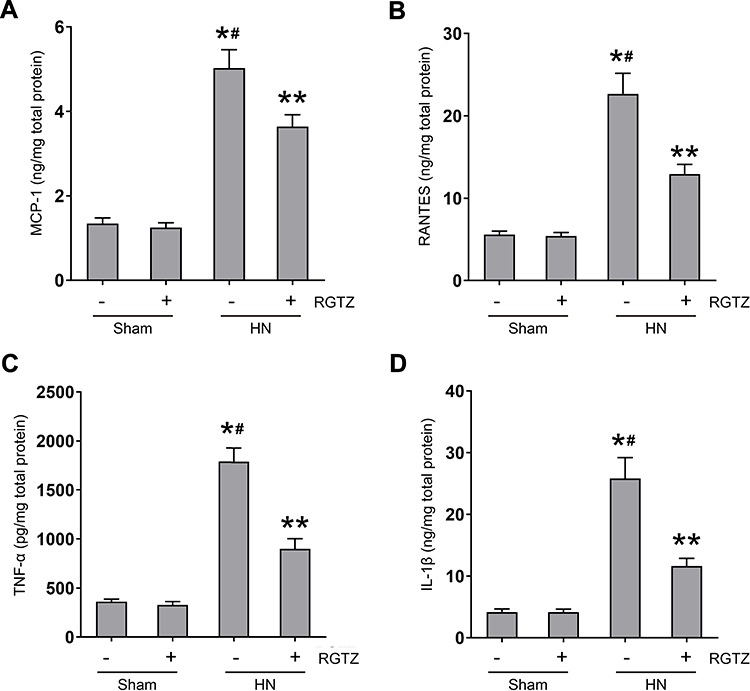 |
Figure 7 RGTZ inhibits the release of cytokines/chemokines in the kidney of HN rats. Protein was extracted from kidney and subjected to ELISA. Protein expression levels of (A) MCP-1, (B) RANTES, (C) TNF-α, and (D) IL-1β are indicated. RGTZ, rosiglitazone. Data are represented as the mean ± SEM. *p < 0.05 vs sham group; #p< 0.05 vs sham + RGTZ group. **p < 0.05 vs HN group. |
PPAR-γ Activation Decreases Serum Uric Acid and Preserves Expression of Urate Transporters Expression in the Kidneys of HN Rats
After three weeks of daily feeding with a mixture of adenine and potassium oxonate, serum uric acid levels were significantly increased, and urinary uric acid levels were significantly decreased (Figure 8A and B). RGTZ treatment dramatically reduced serum uric acid levels and increased urinary uric acid, which is consistent with the inhibitory effects of RGTZ on renal dysfunction. Urate transporters, including OAT1 and OAT3, promote uric acid excretion and play a critical role in uric acid homeostasis.27 Therefore, we assessed the effects of RGTZ on expression of OAT1 and OAT3 in the kidneys of HN rats. The expression of OAT1 and OAT3 were significantly reduced in the kidneys of HN, and administration of RGTZ partially rescued expression of OAT1 and OAT3 (Figure 8C and D). Increased uric acid production is also associated with hyperuricemia, and is commonly associated with upregulation of serum XOD activity. Therefore, we assessed the effects of RGTZ on serum XOD activity in HN rats. The activity of serum XOD was increased in the kidneys of HN rats, and RGTZ administration was ineffective in reducing the increase in activity of serum XOD in the HN model (Figure 8E). These data demonstrated that PPAR-γ activation decreases serum uric acid and promotes uric acid excretion through preservation of OAT1 and OAT3 expression.
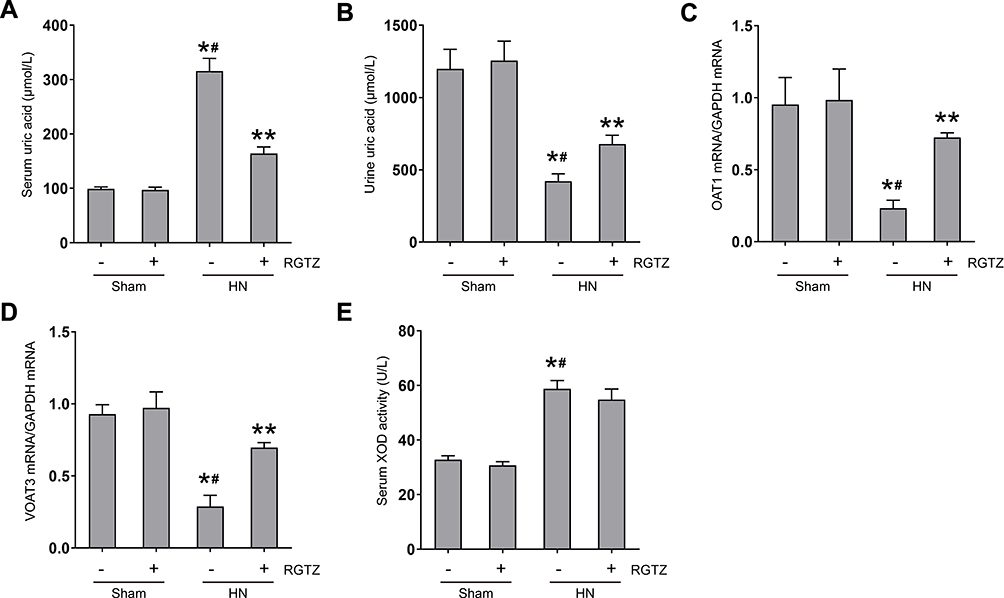 |
Figure 8 RGTZ decreases serum uric acid and preserves expression of OAT1 and OAT3 in the kidneys of HN rats. Levels of uric acid in the (A) serum and (B) urine were examined using an automatic biochemical assay. (C) OAT1 and (D) OAT3 mRNA expression in renal tissue from the different groups (normalized to GAPDH). (E) Serum XOD activity was examined using a XOD kit. RGTZ, rosiglitazone. Data are represented as the mean ± SEM. *p < 0.05 vs sham group; #p < 0.05 vs sham + RGTZ group. **p < 0.05 vs HN group. |
Discussion
In the present study, we investigated the effects of PPAR-γ activation on the development and pathological mechanisms of HN. We demonstrated that treatment with RGTZ, a highly selective PPAR-γ agonist, improves renal function, decreases urine microalbumin levels, and inhibits interstitial fibrosis and macrophage infiltration in kidney tissue in a rat model of HN. Additionally, we report that RGTZ treatment reduces TGF-β expression and NF-κB pathway activation, decreases expression of fibronectin, collagen I, α-SMA, vimentin, MCP-1, RANTES, TNF-α, and IL-1β, and increases E-cadherin expression in the kidneys of HN rats. Furthermore, we showed that RGTZ treatment decreases serum uric acid and promotes renal excretion of uric acid through preservation of OAT1 and OAT3 expression.
Interstitial fibrosis is a reliable prognostic indicator and a major determinant of renal insufficiency, and is the ultimate pathological outcome of most kinds of CKD, including HN.28 TGF-β has an central role in glomerulosclerosis and interstitial fibrosis, and influences increased matrix protein synthesis, suppression of matrix degradation, and changing cell-cell interactions in various kidney diseases.18,29
Abnormal and excessive accumulation of ECM in glomeruli and in renal tubulointerstitial regions exacerbates the seriousness of kidney injury, and are typical characteristics of renal fibrosis.30,31 In a study by Choi et al, PPAR-γ activation inhibits dimethylnitrosamine-induced hepatic fibrosis by inhibiting the TGF-β1/Smad pathway.32 Jiang et al found that PPAR-γ agonist treatment significantly attenuated renal interstitial fibrosis by reducing the expression of TGF-β, collagen IV, and fibronectin in kidneys in a rat model of unilateral ureteral obstruction.33 In our study, we observed enhanced TGF-β activation, diffuse interstitial fibrosis, and increased expression of collagen I and fibronectin in kidneys in a rat model of HN. Moreover, administration of RGTZ dramatically suppressed TGF-β activation, alleviated interstitial fibrosis, and decreased expression of collagen I and fibronectin in renal tissue of HN rats.
Renal tubular EMT is a prominent source of fibroblasts and an important event in the pathogenesis of renal interstitial fibrosis in CKD.5,6 A report by Lee et al suggested that high glucose-induced EMT was ameliorated by the PPAR-γ agonist troglitazone in a primary culture model of renal tubular epithelial cells.34 PPAR-γ activation reduced renal fibrosis induced by perfluorooctanesulfonate by regulating EMT.35 In our study, we report that a significant reduction of E-cadherin expression and an increase of α-SMA and vimentin expression in the kidneys of HN rats. These findings suggested that renal tubular EMT may participate in the progression of HN in rats. Our study demonstrated that RGTZ can attenuate renal tubular EMT in HN rats, evidenced by increased E-cadherin expression and decreased α-SMA and vimentin expression in kidney tissue. The suppression of renal EMT may represent the mechanism by which RGTZ attenuates renal fibrosis in HN.
Hyperuricemia has been shown to induce inflammatory responses, leading to kidney injury.36,37 Activation of the NF-κB pathway plays a crucial role in the hyperuricemia-induced inflammatory response, and increases expression of pro-inflammatory cytokines and chemokines.38,39 In a study by Zhu et al, PPAR-γ activation induced an anti-inflammatory effect by inhibiting the NF-κB pathway in chronic asthma models.40 It has been also reported that RGTZ suppresses LPS-mediated inflammatory responses, indicating that PPAR-γ agonists may be effective protection against pulmonary inflammation in rats.41 Moreover, our previous study in a chronic renal allograft dysfunction model demonstrated that PPAR-γ agonists inhibit NF-κB activation and infiltration of inflammatory cells into the interstitium.42 In the present study, we showed that activation of PPAR-γ with RGTZ inhibits NF-κB activation, macrophage infiltration, and reduces expression of MCP-1, RANTES, TNF-α, and IL-1β induced by hyperuricemia. These data suggest that inhibition of the inflammatory response may be another mechanism by which RGTZ attenuates the renal fibrosis and the development of HN.
Hyperuricemia is an independent risk factor for renal damage and can exacerbate the progression of kidney fibrosis and progressive CKD.1 Therapies that lower uric acid may retard the progression of CKD.43,44 In the present study, we found that RGTZ dramatically reduced serum uric acid levels in HN rats, which was consistent with the inhibitory effect of RGTZ on renal dysfunction. Previous studies in patients with type 2 diabetes mellitus demonstrated that RGTZ treatment decreases levels of serum urate.45 However, the mechanism by which RGTZ decreases serum uric acid levels remains unclear. Serum uric acid levels are largely determined by uric acid production and renal excretion. OAT1 and OAT3 are two primary organic anion transporters, that play a critical role in the excretion of uric acid, and aberrant expression of OAT1 and OAT3 causes excessive uric acid accumulation leading to hyperuricemia.3 In the present study, we demonstrate that expression of OAT1 and OAT3 is decreased in the kidney tissue of HN rats, and we show that OAT1 and OAT3 expression is partially restored by RGTZ treatment. We also examined the effects of RGTZ on the activity of serum XOD, a fundamental enzyme that promotes the production of serum uric acid. Our data indicated that the activity of serum XOD is significantly increased in HN rats. However, RGTZ administration was ineffective in reducing the activity of serum XOD in HN rats. These results suggest that RGTZ decreases serum uric acid and promotes uric acid excretion through preservation of OAT1 and OAT3 expression, which may constitute a mechanism by which RGTZ attenuates hyperuricemia-associated renal injury.
In conclusion, our results demonstrate that RGTZ attenuates the progression of HN in a rat model through multiple mechanisms, including inhibition of TGF-β signaling, inhibiting renal tubular EMT and inflammation, and lowering serum uric acid levels by preserving expression of urate transporters.
Acknowledgments
This work was supported by Risk factors and prediction model of chronic kidney disease caused by metabolic syndrome: A multicentric prospective cohort study clinical trial training project of Southern Medical University (LC2016PY047, 2016), the Science and Technique Program of Guangzhou (201604020015, 2015), the South Wisdom Valley Innovative Research Team Program (CXTD-004, 2014), and The National Natural Science Foundation of China (81873620).
Disclosure
Xin Wang and Jin Deng are co-first authors. The authors report no conflicts of interest in this work.
References
1. Srivastava A, Kaze AD, McMullan CJ, Isakova T, Waikar SS. Uric acid and the risks of kidney failure and death in individuals with CKD. Am J Kidney Dis. 2018;71(3):362–370. doi:10.1053/j.ajkd.2017.08.017
2. Johnson RJ, Bakris GL, Borghi C, et al. Hyperuricemia, acute and chronic kidney disease, hypertension, and cardiovascular disease: report of a scientific workshop organized by the national kidney foundation. Am J Kidney Dis. 2018;71(6):851–865. doi:10.1053/j.ajkd.2017.12.009
3. Otani N, Ouchi M, Hayashi K, Jutabha P, Anzai N. Roles of organic anion transporters(OATs) in renal proximal tubules and their localization. Anat Sci Int. 2017;92(2):200–206.
4. Huijuan W, Xiaoxu C, Rui S, Xinghui L, Beibei T, Jianchun M. Qi-Zhu-Xie-Zhuo-Fang reduces serum uric acid levels and ameliorates renal fibrosis in hyperuricemic nephropathy rats. Biomed Pharmacother. 2017;91:358–365. doi:10.1016/j.biopha.2017.04.031
5. Lovisa S, LeBleu VS, Tampe B, Sugimoto H, Vadnagara K, Carstens JL. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med. 2015;21(9):998–1009. doi:10.1038/nm.3902
6. Stone RC, Pastar I, Ojeh N, et al. Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell Tissue Res. 2016;365(3):495–506. doi:10.1007/s00441-016-2464-0
7. Meng XM, Tang PM, Li J, Lan HY. TGF-beta/Smad signaling in renal fibrosis. Front Physiol. 2015;6:82. doi:10.3389/fphys.2015.00082
8. Ryu ES, Kim MJ, Shin HS, et al. Uric acid-induced phenotypic transition of renal tubular cells as a novel mechanism of chronic kidney disease. Am J Physiol Renal Physiol. 2013;304(5):F471–480. doi:10.1152/ajprenal.00560.2012
9. Mulay SR, Evan A, Anders HJ. Molecular mechanisms of crystal-related kidney inflammation and injury. Implications for cholesterol embolism, crystalline nephropathies and kidney stone disease. Nephrol Dial Transplant. 2014;29(3):507–514. doi:10.1093/ndt/gft248
10. Liu N, Wang L, Yang T, et al. EGF receptor inhibition alleviates hyperuricemic nephropathy. J Am Soc Nephrol. 2015;26(11):2716–2729. doi:10.1681/ASN.2014080793
11. Lefterova MI, Haakonsson AK, Lazar MA, Mandrup S. PPARγ and the global map of adipogenesis and beyond. Trends Endocrinol Metab. 2014;25(6):293–302. doi:10.1016/j.tem.2014.04.001
12. Wang S, Dougherty EJ, Danner RL. PPARγ signaling and emerging opportunities for improved therapeutics. Pharmacol Res. 2016;111:76–85. doi:10.1016/j.phrs.2016.02.028
13. Kadam L, Gomez-Lopez N, Mial TN, Kohan-Ghadr HR, Drewlo S. Rosiglitazone regulates TLR4 and rescues HO-1 and NRF2 expression in myometrial and decidual macrophages in inflammation-induced preterm birth. Reprod Sci (Thousand Oaks, Calif). 2017;24(12):1590–1599. doi:10.1177/1933719117697128
14. Gutting T, Weber CA, Weidner P, et al. PPARgamma-activation increases intestinal M1 macrophages and mitigates formation of serrated adenomas in mutant KRAS mice. Oncoimmunology. 2018;7(5):e1423168. doi:10.1080/2162402X.2017.1423168
15. Sarafidis PA, Bakris GL. Protection of the kidney by thiazolidinediones: an assessment from bench to bedside. Kidney Int. 2006;70(7):1223–1233. doi:10.1038/sj.ki.5001620
16. Wang P, Luo ML, Song E, et al. Long noncoding RNA lnc-TSI inhibits renal fibrogenesis by negatively regulating the TGF-β/Smad3 pathway. Sci Transl Med. 2018;10(462):eaat2039. doi:10.1126/scitranslmed.aat2039
17. Eddy AA. Overview of the cellular and molecular basis of kidney fibrosis. Kidney Int Suppl. 2014;4(1):2–8. doi:10.1038/kisup.2014.2
18. Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-beta: the master regulator of fibrosis. Nat Rev Nephrol. 2016;12(6):325–338. doi:10.1038/nrneph.2016.48
19. Ding Y, Kim S, Lee SY, Koo JK, Wang Z, Choi ME. Autophagy regulates TGF-beta expression and suppresses kidney fibrosis induced by unilateral ureteral obstruction. J Am Soc Nephrol. 2014;25(12):2835–2846. doi:10.1681/ASN.2013101068
20. Lan HY. Diverse roles of TGF-beta/Smads in renal fibrosis and inflammation. Int J Biol Sci. 2011;7(7):1056–1067. doi:10.7150/ijbs.7.1056
21. Zhang Y, Huang XR, Wei LH, Chung AC, Yu CM, Lan HY. miR-29b as a therapeutic agent for angiotensin II-induced cardiac fibrosis by targeting TGF-beta/Smad3 signaling. Mol Ther. 2014;22(5):974–985. doi:10.1038/mt.2014.25
22. Mitchell JP, Carmody RJ. NF-kappaB and the transcriptional control of inflammation. Int Rev Cell Mol Biol. 2018;335:41–84.
23. Huang H, Liu Y, Daniluk J, et al. Activation of nuclear factor-kappaB in acinar cells increases the severity of pancreatitis in mice. Gastroenterology. 2013;144(1):202–210. doi:10.1053/j.gastro.2012.09.059
24. Meng XM, Nikolic-Paterson DJ, Lan HY. Inflammatory processes in renal fibrosis. Nat Rev Nephrol. 2014;10(9):493–503. doi:10.1038/nrneph.2014.114
25. Liu N, Xu L, Shi Y, et al. Pharmacologic targeting ERK1/2 attenuates the development and progression of hyperuricemic nephropathy in rats. Oncotarget. 2017;8(20):33807–33826. doi:10.18632/oncotarget.16995
26. Black LM, Lever JM, Agarwal A. Renal inflammation and fibrosis: a double-edged sword. J Histochem Cytochem. 2019;67(9):663–681. doi:10.1369/0022155419852932
27. Wu W, Bush KT, Nigam SK. Key role for the organic anion transporters, OAT1 and OAT3, in the in vivo handling of uremic toxins and solutes. Sci Rep. 2017;7(1):4939. doi:10.1038/s41598-017-04949-2
28. Liu Y. Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol. 2011;7(12):684–696. doi:10.1038/nrneph.2011.149
29. Isaka Y. Targeting TGF-β signaling in kidney fibrosis. Int J Mol Sci. 2018;19(9):2532. doi:10.3390/ijms19092532
30. Toba H, Lindsey ML. Extracellular matrix roles in cardiorenal fibrosis: potential therapeutic targets for CVD and CKD in the elderly. Pharmacol Ther. 2019;193:99–120. doi:10.1016/j.pharmthera.2018.08.014
31. Sun YBY, Qu X, Caruana G, Li J. The origin of renal fibroblasts/myofibroblasts and the signals that trigger fibrosis. Differentiation. 2016;92(3):102–107. doi:10.1016/j.diff.2016.05.008
32. Choi JH, Jin SW, Choi CY, et al. Capsaicin inhibits dimethylnitrosamine-induced hepatic fibrosis by inhibiting the TGF-β1/Smad pathway via peroxisome proliferator-activated receptor gamma activation. J Agric Food Chem. 2017;65(2):317–326. doi:10.1021/acs.jafc.6b04805
33. Jiang L, Chen XP, Long YB, et al. The potential signaling pathway between peroxisome proliferator-activated receptor gamma and retinoic acid receptor alpha in renal interstitial fibrosis disease. J Recept Signal Transduct Res. 2015;35(4):258–268. doi:10.3109/10799893.2014.975249
34. Lee YJ, Han HJ. Troglitazone ameliorates high glucose-induced EMT and dysfunction of SGLTs through PI3K/Akt, GSK-3β, Snail1, and β-catenin in renal proximal tubule cells. Am J Physiol Renal Physiol. 2010;298(5):F1263–1275. doi:10.1152/ajprenal.00475.2009
35. Chou HC, Wen LL, Chang CC, Lin CY, Jin L, Juan SH. From the cover: l-carnitine via pparγ- and sirt1-dependent mechanisms attenuates epithelial-mesenchymal transition and renal fibrosis caused by perfluorooctanesulfonate. Toxicol Sci. 2017;160(2):217–229. doi:10.1093/toxsci/kfx183
36. Jalal DI, Chonchol M, Chen W, Targher G. Uric acid as a target of therapy in CKD. Am J Kidney Dis. 2013;61(1):134–146. doi:10.1053/j.ajkd.2012.07.021
37. Spiga R, Marini MA, Mancuso E, et al. Uric acid is associated with inflammatory biomarkers and induces inflammation via activating the NF-kappaB signaling pathway in HepG2 cells. Arterioscler Thromb Vasc Biol. 2017;37(6):1241–1249. doi:10.1161/ATVBAHA.117.309128
38. Zhou Y, Fang L, Jiang L, et al. Uric acid induces renal inflammation via activating tubular NF-kappaB signaling pathway. PLoS One. 2012;7(6):e39738. doi:10.1371/journal.pone.0039738
39. Liu H, Xiong J, He T, et al. high uric acid-induced epithelial-mesenchymal transition of renal tubular epithelial cells via the TLR4/NF-kB signaling pathway. Am J Nephrol. 2017;46(4):333–342. doi:10.1159/000481668
40. Zhu T, Chen Z, Chen G, et al. Curcumin attenuates asthmatic airway inflammation and mucus hypersecretion involving a PPAR-dependent NF-B signaling pathway in vivo and in vitro. Mediators Inflamm. 2019;2019:4927430. doi:10.1155/2019/4927430
41. Cho RL, Yang CC, Tseng HC, Hsiao LD, Lin CC, Yang CM. Haem oxygenase-1 up-regulation by rosiglitazone via ROS-dependent Nrf2-antioxidant response elements axis or PPARγ attenuates LPS-mediated lung inflammation. Br J Pharmacol. 2018;175(20):3928–3946. doi:10.1111/bph.14465
42. Deng J, Xia Y, Zhou Q, et al. Protective effect of rosiglitazone on chronic renal allograft dysfunction in rats. Transpl Immunol. 2019;54:20–28. doi:10.1016/j.trim.2019.01.002
43. Bose B, Badve SV, Hiremath SS, et al. Effects of uric acid-lowering therapy on renal outcomes: a systematic review and meta-analysis. Nephrol Dial Transplant. 2014;29(2):406–413. doi:10.1093/ndt/gft378
44. Sampson AL, Singer RF, Walters GD. Uric acid lowering therapies for preventing or delaying the progression of chronic kidney disease. Cochrane Database Syst Rev. 2017;10:Cd009460.
45. Bao Y, Zhao T, Wang X, et al. Metabonomic variations in the drug-treated type 2 diabetes mellitus patients and healthy volunteers. J Proteome Res. 2009;8(4):1623–1630. doi:10.1021/pr800643w
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.