Back to Journals » Drug Design, Development and Therapy » Volume 10
Treatment with a Ginkgo biloba extract, EGb 761, inhibits excitotoxicity in an animal model of spinocerebellar ataxia type 17
Authors Huang D, Lin H, Lee-Chen G, Hsieh H, Wu C ![]() , Lin J
, Lin J
Received 13 October 2015
Accepted for publication 23 December 2015
Published 18 February 2016 Volume 2016:10 Pages 723—731
DOI https://doi.org/10.2147/DDDT.S98156
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Ding-Siang Huang,1,* Hsuan-Yuan Lin,1,2,* Guey-Jen Lee-Chen,1 Hsiu-Mei Hsieh-Li,1 Chung-Hsin Wu,1 Jung-Yaw Lin1,2
1Department of Life Science, National Taiwan Normal University, 2Institute of Biochemistry and Molecular Biology, National Taiwan University, Taipei City, Taiwan, Republic of China
*These authors contributed equally to this work
Abstract: Spinocerebellar ataxia type 17 (SCA 17) is a polyglutamine disease caused by the expansion of CAG/CAA repeats in the TATA box-binding protein (TBP) gene. The Ginkgo biloba extract, EGb 761, contains flavonoids and terpenoids with a potential use for the treatment of neurodegenerative diseases such as Alzheimer’s and Parkinson’s diseases. The neuroprotective effects of EGb 761 are obvious, but whether the EGb 761 has therapeutic effects in SCA 17 is still unclear. To manage our issues, we have generated TBP/79Q-expressing SH-SY5Y cells and SCA 17 transgenic mice with the mutant hTBP gene. In in vitro experiment, we observed that the EGb 761 treatment decreased the amount of sodium dodecyl sulfate-insoluble proteins in the TBP/79Q-expressing SH-SY5Y cells. We further found that the EGb 761 treatment could inhibit excitotoxicity and calcium influx and reduce the expression of apoptotic markers in glutamate-treated SH-SY5Y neuroblastoma cells. In in vivo experiment, we observed that the EGb 761 treatment (100 mg/kg intraperitoneal injection per day) could relieve the motor deficiencies of the SCA 17 transgenic mice. Our findings provide evidence that the EGb 761 treatment can be a remedy for SCA 17 via suppressing excitotoxicity and apoptosis in SCA 17 cell and animal models. Therefore, we suggest that EGb 761 may be a potential therapeutic agent for treating SCA 17.
Keywords: spinocerebellar ataxia type 17, excitotoxicity, EGb 761, polyQ diseases, apoptosis
Introduction
Spinocerebellar ataxia type 17 (SCA 17) is a polyglutamine (polyQ) disease caused by the expansion of CAG/CAA repeats (>43 repeats) in the TATA box-binding protein (TBP) gene, leading to protein aggregation in patients with SCA 17.1–3 There is increasing evidence that excitotoxicity and polyQ diseases are related to each other. The sodium dodecyl sulfate (SDS)-resistant aggregation of insoluble proteins is increased after the glutamate-induced influx of calcium, and the aggregation is reduced by the inhibition of calcium influx in a model of SCA 3.4 Moreover, the activation of calcium-dependent proteolytic calpains aggravates SCA 3 pathology by increasing protein aggregation; however, calpastatin, an inhibitor of calpains, reduces aggregation.5,6 Moreover, the intracellular calcium concentration is elevated in neurons that are sensitive to glutamate-induced cell death in Huntington’s disease.7,8 These findings suggest that disrupted calcium signaling may lead to protein aggregation and neuronal death in polyQ diseases. Thus, the inhibition of calcium overloading may be therapeutic by preventing the pathological alterations in SCA 17.
The EGb 761, a standard extract prepared from the leaves of Ginkgo biloba, has pharmacological effects in various neurodegenerative diseases.9 Pharmacological studies had shown that the G. biloba extract, EGb 761, is an antioxidant agent that alleviates ischemia, oxidative stress, and β-amyloid-induced toxicity. The EGb 761 has been used in several neurological diseases such as Alzheimer’s disease, Parkinson’s disease, and dementia.10–13 The neuroprotective effects of the EGb 761 are obvious, but whether the EGb 761 has therapeutic effects in SCA 17 is still unclear.
In the present study, we have generated TBP/79Q-expressing SH-SY5Y cells with inducible green fluorescent protein expression and SCA 17 transgenic mice with the mutant hTBP gene driven by the Purkinje-specific protein (Pcp2/L7) gene promoter.14,15 The possible effects of the EGb 761 in pathological alterations of SCA 17 were investigated in glutamate-induced excitotoxicity model, TBP-expressing cells, and SCA 17 transgenic mice as in vivo and in vitro models of SCA 17.
Materials and methods
Materials
EGb 761, containing 24% Ginkgo flavone glycosides (including quercetin, kaempferol, and isorhamnetin) and 6% terpenlactones (including ginkgolides A, B, C, and bilobalide), is a standardized extract of G. biloba (Dr Willmar Schwabe Pharmaceuticals, Karlsruhe, Germany). 3-(4,5-Dimethylthiazole-2-yl)-2,5-diphenyltetrazolum bromide (MTT), l-glutamic acid monosodium salt monohydrate, and MK-801 were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Fluo-4 AM was obtained from Thermo Fisher Scientific (Waltham, MA, USA). Trypsin-ethylenediaminetetraacetic acid (0.5%) and penicillin/streptomycin were purchased from Thermo Fisher Scientific. Fetal bovine serum (FBS) was obtained from Corning Incorporated (Corning, NY, USA). Antibodies specific for calpain 2, calbindin, cleaved caspase 3, and cleaved poly(adenosine diphosphate-ribose) polymerase (PARP) were produced by Cell Signaling Technology (Danvers, MA, USA). Antibodies against Bax and actin were purchased from EMD Millipore (Billerica, MA, USA), and antibodies against spectrin, transcription factor II D, and Bcl-2 were obtained from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). Horseradish peroxidase (HRP)-conjugated secondary goat anti-mouse and goat anti-rabbit antibodies were purchased from EMD Millipore.
Cell culture
Human neuroblastoma SH-SY5Y cells were cultured in a 1:1 mixture of Dulbecco’s Modified Eagle’s Medium and Ham’s Nutrient Mixture F-12 containing 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. SH-SY5Y cells were cultured with or without the inducible TBP/36Q and TBP/79Q expression constructs. TBP expression was induced by the addition of 10 μg/mL doxycycline. The culture medium was changed every 48 hours, and cells were grown at 37°C in the presence of 5% CO2. Cell experiments were approved by the Biological Experimental Safety Committee at the National Taiwan Normal University.
Cell viability assay
A total of 104 SH-SY5Y cells/well in a 100 μL culture medium were grown in a 96-well plate for 24 hours to reach ~60% confluence. Cells were treated with vehicle, EGb 761 (5 μg/mL, 10 μg/mL, and 20 μg/mL), or 10 μM MK-801 (N-methyl-D-aspartate [NMDA] receptor antagonist) for 1 hour, and then they were incubated in the presence of 100 mM glutamate for 24 hours. The number of viable cells was compared between the control and treated conditions. The culture medium was supplemented with MTT (0.5 mg/mL) for 3 hours, and then 10% SDS/HCl buffer was added to each well. The number of viable cells was determined by the measurement of MTT absorbance at 570 nm using a microplate reader (BioTek Instruments, Winooski, Vermont, USA).
Measurement of calcium influx
To measure the calcium concentration in viable cells, Fluo-4 AM, a green fluorescent calcium indicator, was used. SH-SY5Y cells were pretreated with vehicle, 20 μg/mL EGb 761, or 10 μM MK-801 for 1 hour, then the cells were washed twice in phosphate-buffered saline (PBS) and stained in the dark with 10 μM Fluo-4 AM for 1 hour at 37°C. Cells were washed twice in PBS and exposed to 100 mM glutamate for 1 hour in Locke’s buffer. Cells were then centrifuged at 1,000 rpm and resuspended in PBS, and the fluorescence intensity of cells, representing the cytosolic calcium concentration, was measured using the BD FACSCalibur flow cytometer (BD, Franklin Lakes, NJ, USA).
Western blotting experiment and analysis
SH-SY5Y cells were pretreated with vehicle, 20 μg/mL EGb 761, or 10 μM MK-801 for 1 hour, and then they were incubated in the presence of 100 mM glutamate for 24 hours. In TBP-expressing cells (TBP/36Q-enhanced green fluorescent protein [EGFP] or TBP/79Q-EGFP cells), the cells were pretreated with vehicle and 20 μg/mL EGb 761 for 1 hour, and 10 μg/mL doxycycline was added to induce TBP/36Q or TBP/79Q expression for 5 days. Cells were harvested and lysed in radioimmunoprecipitation assay buffer (50 mM Tris–HCl, 150 mM NaCl, 6 mM sodium deoxycholate, 1% NP-40, pH 7.4) supplemented with protease inhibitors (Hoffman-La Roche Ltd., Basel, Switzerland) on ice for 10 minutes. Cell lysates were sonicated and centrifuged at 13,000 rpm for 15 minutes at 4°C to collect the supernatants, and protein concentration was measured using the BCA Protein Assay Kit (Thermo Fisher Scientific). Proteins were separated on 12.5% or 15% SDS polyacrylamide gels, and they were transferred to polyvinylidene difluoride membranes. The antibodies used in this study were β-actin, calpain 2, Bax, Bcl-2, cleaved caspase 3, cleaved PARP, transcription factor II D, and α-spectrin (Cell Signaling Technology). Antibodies were detected by suitable HRP-conjugated secondary antibody, protein-immunoreactive bands were visualized by the enhanced chemiluminescence substrate (EMD Millipore), and the band intensities were quantified with the ImageJ analysis software (Version 1.48t; Wayne Rasnabd, Washington, DC, USA).
Animals
SCA 17 transgenic mice in the experiment were obtained from the laboratory of Professor HM Hsieh-Li (National Taiwan Normal University, Taipei City, Taiwan, Republic of China).15 They were generated with the mutant hTBP gene driven by the Purkinje-specific protein (Pcp2/L7) gene promoter. The mice with the expanded hTBP allele developed ataxia within 2–5 months. They were identified using polymerase chain reaction with a forward primer from the Pcp2/L7 promoter (L7-F: 5′-TATGGTGAGAGCAGAGATGG-3′) and a backward primer from hTBP cDNA (hTBP-3R: 5′-CTGCTGGGACGTTGACTGCTG-3′). SCA 17 transgenic mice and their wild-type (FVB/N mice) were housed in a temperature-controlled environment at 22°C±2°C with a 12-hour light/dark cycle. All mice were housed and maintained on a 12:12-hour light–dark cycle with water and food ad libitum. Two groups of three transgenic mice each and littermates received water vehicle or 100 mg/kg EGb 761 intraperitoneally every other day from the age of 4 weeks to 24 weeks. Animal experiments were approved by the Institutional Animal Care and Use Committee at the National Taiwan Normal University.
Rotarod performance
Rotarod performance was used to examine the motor coordination of SCA 17 transgenic mice. The mice were tested on a rotarod apparatus (Ugo Basile, Varese, Italy) starting at 5 rpm and accelerating to 15 rpm over a period of 2 minutes and then maintaining a constant speed of 15 rpm for 2 minutes. The mice were rested for 30 minutes between trials and trained at 5 rpm for 10 minutes/day for 3 days at 8 weeks of age to become acquainted with the rotarod apparatus. Then, they were tested three times a week from the age of 9 weeks to 24 weeks, and the latency period was recorded before the mice fell off the rotating rod.
Immunohistochemistry
Wild-type and SCA 17 transgenic mice were cardiac perfused first with PBS and then fixed with 4% formaldehyde (EM grade) at the age of 24 weeks. Cerebellar specimens were then embedded in paraffin, cut into sections (5 μm), and placed on adhesion microscope slides (Thermo Fisher Scientific). By using the heat-induced epitope retrieval method, the sections were separately stained at room temperature for 1 hour with antibodies of calbindin (1:500; Cell Signaling Technology) or TBP (1:1,000; Santa Cruz Biotechnology Inc.). Detection was performed by incubation with biotinylated secondary antibodies (NovolinkTM Polymer Detection System l; Leica Microsystems, Wetzlar, Germany), for 30 minutes at room temperature, followed by 30-minute incubation with avidin–biotin–HRP complex (NovolinkTM Polymer Detection System l). Visualization was performed with 3,3′-diaminobenzidine Chromogen (NovolinkTM Polymer Detection System l) and counterstained with hematoxylin (NovolinkTM Polymer Detection System l) following the supplier’s protocol.
Statistical analysis
Statistical analyses were performed with Student’s t-test or one-way analysis of variance test (using Bonferroni’s test for post hoc comparisons) where appropriate, and the results are shown as mean ± standard error of mean values. Results were considered significant at P<0.05.
Results
The effect of EGb 761 on glutamate-induced cytotoxicity
Glutamate is the major excitatory neurotransmitter in the central nervous system, but high levels of released glutamate can cause neuronal dysfunction via the activation of NMDA receptors, and then inducing excitotoxicity in the cells.16,17 To examine whether EGb 761 alleviates excitotoxicity, SH-SY5Y cells were pretreated with various concentrations of EGb 761 (0–20 μg/mL) for 1 hour followed by an incubation with glutamate (100 mM) for 24 hours, and finally the cell viability was measured using the MTT assay. Cell survival was significantly increased in the presence of 20 μg/mL EGb 761 or the NMDA receptor antagonist MK-801 (10 μM; Figure 1A).
 | Figure 1 Protective effects of EGb 761 in glutamate-induced excitotoxicity of SH-SY5Y cells. |
EGb 761 suppressed the calcium influx induced by glutamate
To assess whether the cell death is triggered by the excessive calcium influx induced by glutamate, Fluo-4 AM was used to measure the amount of intracellular calcium using flow cytometry. Calcium concentration was increased after glutamate treatment, and EGb 761 (20 μg/mL) or MK-801 (10 μM) decreased calcium concentration in the glutamate-treated cells (Figure 1B). The calcium influx induced by glutamate increased the expression of calpain 2 and the amount of its specific spectrin breakdown products (SBDPs) which are cleaved by calpains.18 Similarly, our results showed that glutamate increased the expression of calpain 2 and the amount of SBDPs; however, EGb 761 or MK-801 treatment decreased the expression of calpain 2 or the amount of SBDPs remarkably (Figure 1C). Therefore, EGb 761 ameliorated the glutamate-induced excitotoxicity through the inhibition of calcium overloading.
EGb 761 inhibits glutamate-mediated apoptosis
To examine the effect of EGb 761 on its downstream mediators related to calcium accumulation, we examined the expression of caspase 3 and PARP, which play an important role in neuronal cell death. After 24 hours of exposure to 20 μg/mL EGb 761 or 10 μM MK-801, caspase 3 decreased by 63.8% and 80.1%, whereas PARP decreased by 48.7% and 62.5%, respectively (Figure 2A and B). Then, we studied the effect of EGb 761 on Bcl-2 expression as Bcl-2 plays an important physiological role in neuronal activity, the regulation of intracellular calcium levels, and mitochondrial dynamics.19 A profound decrease was found in the expression of Bcl-2 after a treatment with glutamate, whereas MK-801 and EGb 761 pretreatment significantly reversed the decrease in Bcl-2 expression (Figure 2C). Meanwhile, the ratio of Bcl-2 and Bax was significantly increased after MK-801 or EGb 761 administration, and the cytosolic release of mitochondrial cytochrome C induced by glutamate was suppressed (Figure 2D). Therefore, the cytosolic calcium accumulation induced by glutamate triggered a caspase-dependent cell death that was rescued by MK-801 or EGb 761 through the regulation of Bcl-2 expression.
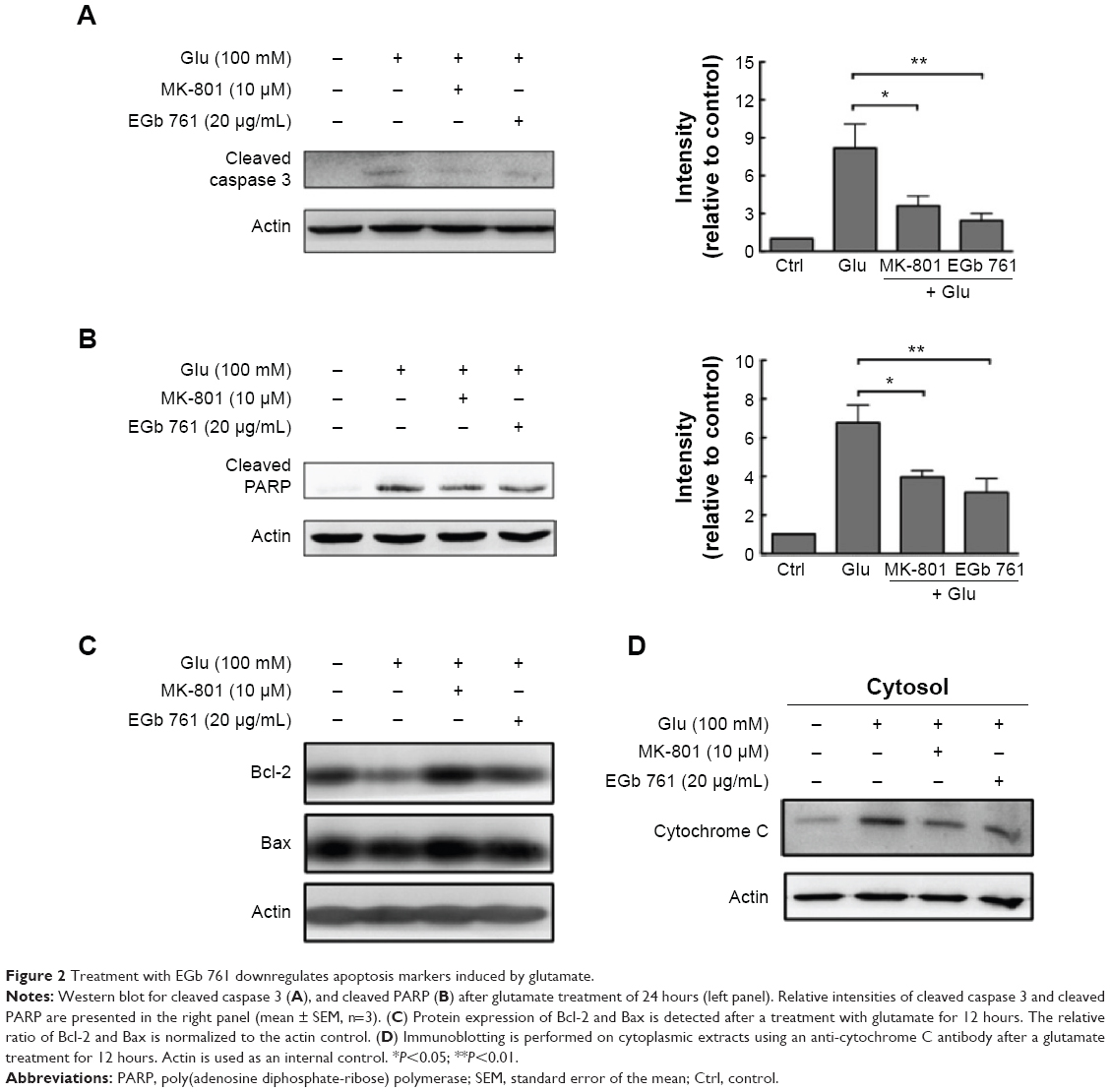 | Figure 2 Treatment with EGb 761 downregulates apoptosis markers induced by glutamate. |
EGb 761 attenuates TBP/79Q aggregation and apoptosis in the TBP/79Q-EGFP cells
To determine the neuroprotective effects of EGb 761 in the cellular model of SCA 17, the expression of TBP containing 36 or 79 glutamines fused to EGFP (TBP/36Q-EGFP and TBP/79Q-EGFP cells) was induced by doxycycline in SH-SY5Y cells. The levels of cleaved caspase 3 and PARP were increased in the TBP/79Q-EGFP cells, but not in the TBP/36Q-EGFP-expressing cells. EGb 761 decreased the expression of cleaved caspase-3 and PARP by 65.5% and 41.2%, respectively, after the induction of TBP/79Q-EGFP cells (Figure 3A and B). Pretreatment with EGb 761 inhibited TBP aggregation by 59.9% in the TBP/79Q-EGFP cells, and no green fluorescent protein aggregation was visible in the TBP/36Q-EGFP cells (Figure 4A). Furthermore, an SDS-insoluble aggregation was detected by the dot blot filter retardation assay stained with a TBP-specific antibody; EGb 761 reduced TBP aggregation by 58.5% in the SDS-insoluble materials of TBP/79Q-EGFP cells (Figure 4B). Overall, these results suggest that apoptosis is activated by TBP/79Q aggregation, and EGb 761 decreases apoptosis by inhibiting TBP/79Q aggregation.
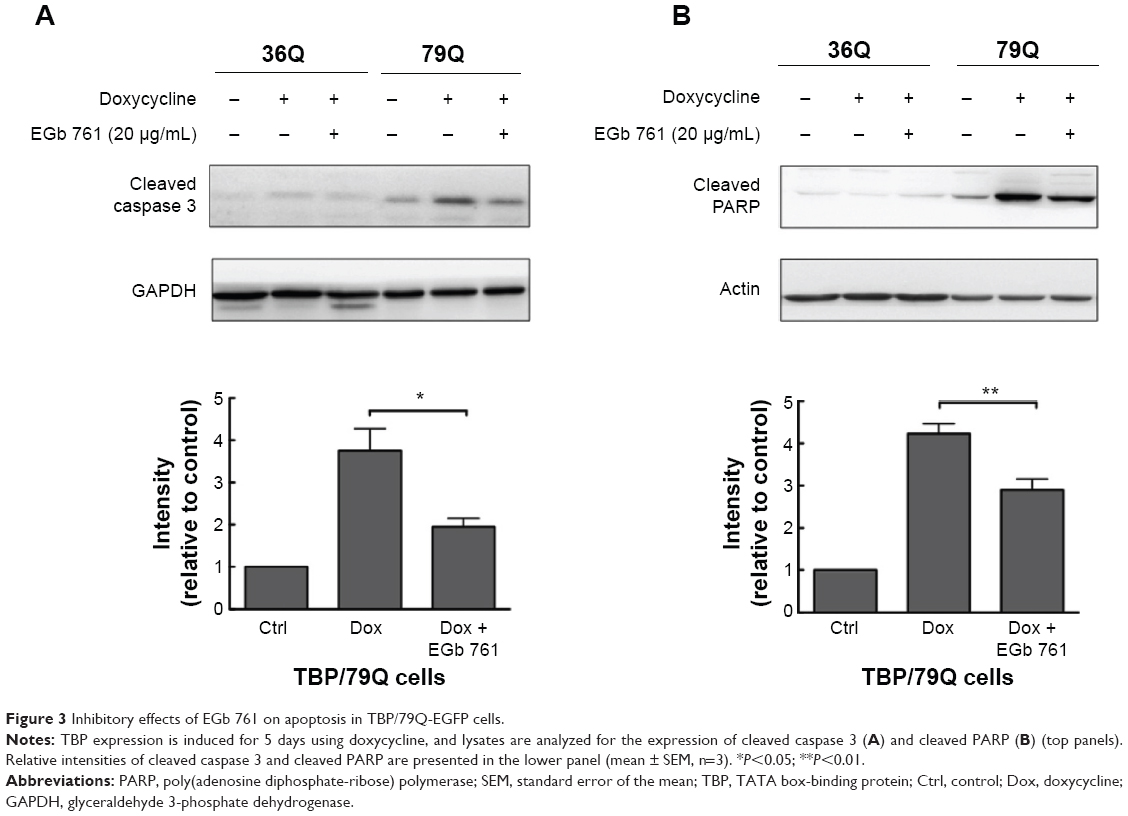 | Figure 3 Inhibitory effects of EGb 761 on apoptosis in TBP/79Q-EGFP cells. |
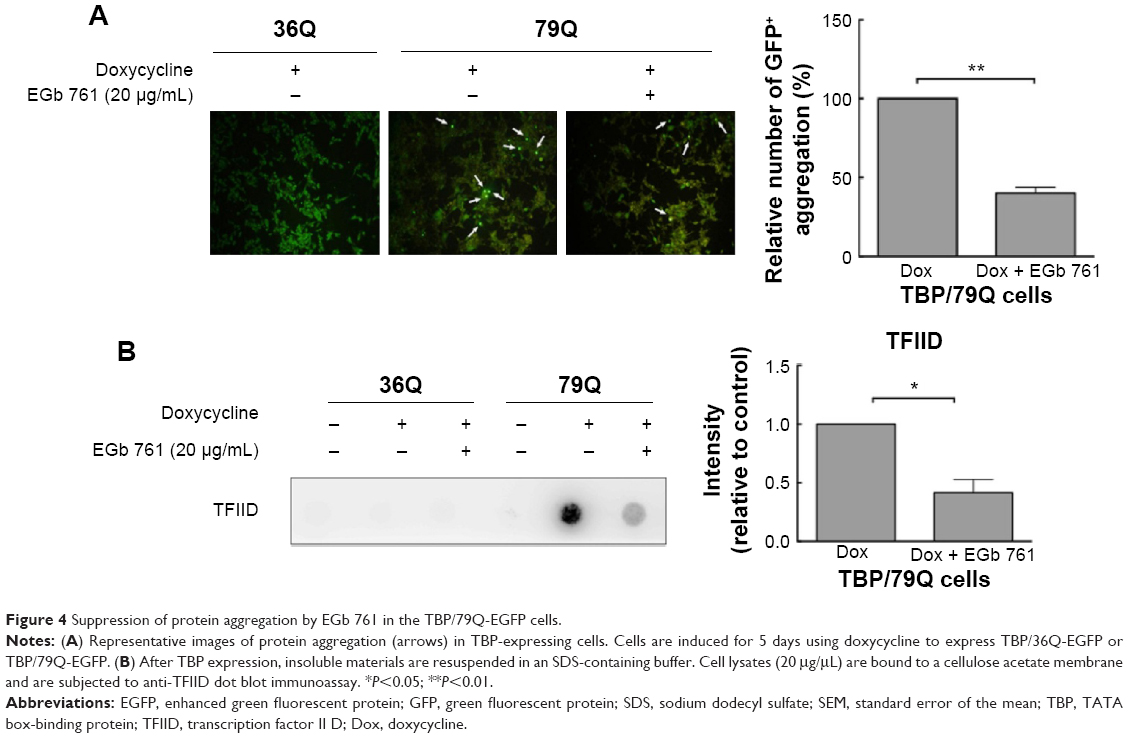 | Figure 4 Suppression of protein aggregation by EGb 761 in the TBP/79Q-EGFP cells. |
EGb 761 ameliorated the motor function of the SCA 17 transgenic mice
Cerebellum plays an important role in motor control, and it is the brain region most affected by SCAs. In the present study, 100 mg/kg EGb 761 was administrated intraperitoneally that there was no observed effect on embryo–fetal development in mice20 and was also used for in vivo study of neurodegenerative disease.21,22 To examine whether EGb 761 improves motor function in the SCA 17 transgenic mice, we examined the highly abundant cerebellar Purkinje cells using calbindin staining at the age of 24 weeks.23,24 The Purkinje cells of the wild-type mice formed a compact layer, but the alignment of Purkinje cells was disrupted in the SCA 17 transgenic mice. The disrupted stratification was restored by the EGb 761 treatment in the SCA 17 transgenic mice (Figure 5A). Furthermore, EGb 761 inhibited TBP aggregation in the SCA 17 transgenic mice (Figure 5B). The impairment of motor functions in the SCA 17 transgenic mice was observed at the age of 11 weeks as demonstrated by the rotarod experiments that analyze motor coordination (Figure 5C). For 20 weeks, 100 mg/kg EGb 761 was administered intraperitoneally every other day to the SCA 17 transgenic mice from the age of 4 weeks. We observed that the EGb 761-treated SCA 17 transgenic mice showed increased rotarod performance by 64.8% and 54.7% compared with the untreated SCA 17 transgenic mice at the age of 15 weeks and 17 weeks, respectively (Figure 5C).
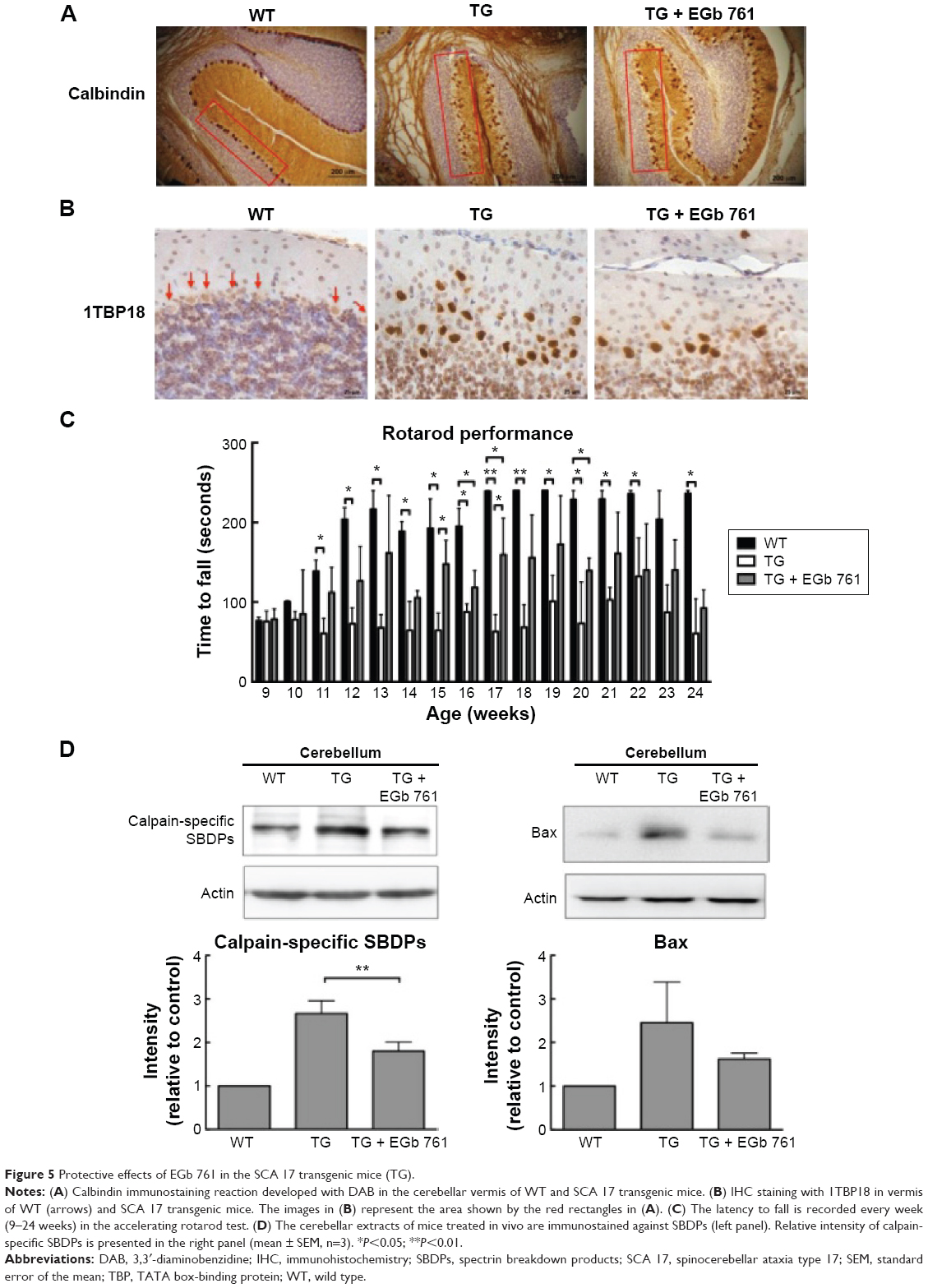 | Figure 5 Protective effects of EGb 761 in the SCA 17 transgenic mice (TG). |
To determine whether calcium homeostasis is disrupted in the SCA 17 transgenic mice, the cerebellar extracts were examined for SBDPs generated by calpains. The amount of calpain-specific SBDPs was increased in the cerebellum of the SCA 17 transgenic mice, but it was decreased in the EGb 761-treated SCA 17 transgenic animals (Figure 5D). To conclude, we observed the impairment of motor function and the activation of intracellular calcium signaling in the cerebellum of the SCA 17 transgenic mice that were attenuated by EGb 761, suggesting that EGb 761 ameliorates motor dysfunction by inhibiting the abnormal calcium signaling and TBP aggregation in the SCA 17 mice.
Discussion
In the present study, we examined the molecular mechanisms of EGb 761 treatment in decreasing cell death caused by excitotoxicity. Our results show that EGb 761 inhibits the glutamate-induced apoptosis in SH-SY5Y cells through the suppression of excessive calcium influx and subsequent inhibition of excitotoxicity and cell death demonstrated by the decreased expression of cleaved caspase 3 and PARP. Excessive influx of calcium is implied in neurodegenerative diseases and results in excitotoxicity and subsequent neuronal injury. Furthermore, EGb 761 was protective against cell death induced by calcium overloading in vitro (Figures 1 and 2) and in vivo. EGb 761 decreased the amount of calpain-specific SBDPs in the cerebellum of the SCA 17 transgenic mice (Figure 5B). This observation suggests that EGb 761 may decrease calcium overloading in SCA 17 and rescue neuronal cells from injury.
Patients with SCA 17 develop a variety of neuroanatomical abnormalities, including the formation of neuronal inclusions and neuronal loss affecting particularly the Purkinje cells of the cerebellum, and subsequent cerebellar atrophy. A previous study also indicated that quality control may be inefficient for polyQ proteins, and one of the therapeutic strategies suggested is to assist the correct folding of polyQ proteins to reduce aggregation.2,25,26 The aggregation of misfolded proteins has been documented in polyQ diseases such as Huntington’s disease, Alzheimer’s disease, and SCA 3. The present study demonstrates that EGb 761 inhibits the SDS-insoluble TBP aggregation in the TBP/79Q cells (Figures 3 and 4). These data suggest that EGb 761 alleviates cell toxicity by inhibiting the TBP/79Q aggregation.
The rotarod performance is used to evaluate motor coordination in animal models. In this study, motor coordination declined in the SCA 17 transgenic mice compared with the wild-type mice, and the intraperitoneal administration of EGb 761 improved the rotarod performance of the SCA 17 transgenic mice at the age of 15 weeks and 17 weeks (Figure 5C), but rotarod performance was not significantly increased from the age of 18 weeks to 24 weeks. These data suggest that the motor dysfunction is delayed by the administration of EGb 761 in the SCA 17 transgenic mice.
Conclusion
In conclusion, in vitro application of EGb 761 alleviates excitotoxicity and the subsequent apoptosis by inhibiting the apoptotic pathways induced by the aggregation of TBP/79Q. Moreover, in vivo EGb 761 delays the motor deficit and neuropathological alterations found in the SCA 17 transgenic mice. These data suggest that EGb 761 may be a potential therapeutic agent for SCA 17.
Acknowledgments
This study was supported by the grants NSC 100-2325-B-002-064, NSC 101-2325-B-002-057, NSC 102-2325-B-002-055, MOST 103-2325-B-003-002, and MOST 104-2325-B-003-002 and from the Ministry of Science and Technology, and 103T3040B00, 104T3040C0, 104T3040D0, and 105T3040D0 from the National Taiwan Normal University, Taipei, Taiwan, Republic of China.
Disclosure
The authors report no conflicts of interest in this work.
References
Fujigasaki H, Martin JJ, DeDeyn PP, et al. CAG repeat expansion in the TATA box-binding protein gene causes autosomal dominant cerebellar ataxia. Brain. 2001;124:1939–1947. | ||
Nakamura K, Jeong SY, Uchihara T, et al. SCA 17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum Mol Genet. 2001;10(14):1441–1448. | ||
Zühlke C, Hellenbroich Y, Dalski A, et al. Different types of repeat expansion in the TATA-binding protein gene are associated with a new form of inherited ataxia. Eur J Hum Genet. 2001;9(3):160–164. | ||
Koch P, Breuer P, Peitz M, et al. Excitation-induced ataxin-3 aggregation in neurons from patients with Machado-Joseph disease. Nature. 2011;480(7378):543–546. | ||
Hübener J, Weber JJ, Richter C, et al. Calpain-mediated ataxin-3 cleavage in the molecular pathogenesis of spinocerebellar ataxia type 3 (SCA3). Hum Mol Genet. 2013;22(3):508–518. | ||
Simoes AT, Goncalves N, Koeppen A, et al. Calpastatin-mediated inhibition of calpains in the mouse brain prevents mutant ataxin 3 proteolysis, nuclear localization and aggregation, relieving Machado-Joseph disease. Brain. 2012;135(pt 8):2428–2439. | ||
Tang TS, Tu H, Chan EY, et al. Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1. Neuron. 2003;39(2):227–239. | ||
Tang TS, Slow E, Lupu V, et al. Disturbed Ca2+ signaling and apoptosis of medium spiny neurons in Huntington’s disease. Proc Natl Acad Sci U S A. 2005;102(7):2602–2607. | ||
Ramassamy C. Emerging role of polyphenolic compounds in the treatment of neurodegenerative diseases: a review of their intracellular targets. Eur J Pharmacol. 2006;545(1):51–64. | ||
Chandrasekaran K, Mehrabian Z, Spinnewyn B, Drieu K, Fiskum G. Neuroprotective effects of bilobalide, a component of the Ginkgo biloba extract (EGb 761), in gerbil global brain ischemia. Brain Res. 2001;922(2):282–292. | ||
Oyama Y, Chikahisa L, Ueha T, Kanemaru K, Noda K. Ginkgo biloba extract protects brain neurons against oxidative stress induced by hydrogen peroxide. Brain Res. 1996;712(2):349–352. | ||
Bastianetto S, Ramassamy C, Doré S, Christen Y, Poirier J, Quirion R. The Ginkgo biloba extract (EGb 761) protects hippocampal neurons against cell death induced by beta-amyloid. Eur J Neurosci. 2000;12(6):1882–1890. | ||
Diamond BJ, Shiflett SC, Feiwel N, et al. Ginkgo biloba extract: mechanisms and clinical indications. Arch Phys Med Rehabil. 2000;81(5):668–678. | ||
Kung PJ, Tao YC, Hsu HC, et al. Indole and synthetic derivative activate chaperone expression to reduce polyQ aggregation in SCA 17 neuronal cell and slice culture models. Drug Des Devel Ther. 2014;8:1929–1939. | ||
Chang YC, Lin CY, Hsu CM, et al. Neuroprotective effects of granulocyte-colony stimulating factor in a novel transgenic mouse model of SCA 17. J Neurochem. 2011;118(2):288–303. | ||
Rothman SM, Olney JW. Excitotoxicity and the NMDA receptor – still lethal after eight years. Trends Neurosci. 1995;18(2):57–58. | ||
Sun ZW, Zhang L, Zhu SJ, Chen WC, Mei B. Excitotoxicity effects of glutamate on human neuroblastoma SH-SY5Y cells via oxidative damage. Neurosci Bull. 2010;26(1):8–16. | ||
Miao Y, Dong LD, Chen J, Hu XC, Yang XL, Wang Z. Involvement of calpain/p35-p25/Cdk5/NMDAR signaling pathway in glutamate-induced neurotoxicity in cultured rat retinal neurons. PLoS One. 2012;7(8):e42318. | ||
Hardwick JM, Soane L. Multiple functions of BCL-2 family proteins. Cold Spring Harb Perspect Biol. 2013;5(2):a008722. | ||
Koch E, Nöldner M, Leuschner J. Reproductive and developmental toxicity of the Ginkgo biloba special extract EGb 761 in mice. Phytomedicine. 2013;12(1):90–97. | ||
Mdzinarishvili A, Sumbria R, Lang D, Klein J. Ginkgo extract EGb 761 confers neuroprotection by reduction of glutamate release in ischemic brain. J Pharm Pharm Sci. 2012;15(1):94–102. | ||
Rojas P, Serrano-García N, Mares-Sámano JJ, Medina-Campos ON, Pedraza-Chaverri J, Ogren SO. EGb761 protects against nigrostriatal dopaminergic neurotoxicity in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinsonism in mice: role of oxidative stress. Eur J Neurosci. 2008;28(1):41–50. | ||
Barski JJ, Hartmann J, Rose CR, et al. Calbindin in cerebellar Purkinje cells is a critical determinant of the precision of motor coordination. J Neurosci. 2003;23(8):3469–3477. | ||
Whitney ER, Kemper TL, Rosene DL, Bauman ML, Blatt GJ. Calbindin-D28k is a more reliable marker of human Purkinje cells than standard Nissl stains: a stereological experiment. J Neurosci Methods. 2008;168(1):42–47. | ||
Park SH, Kukushkin Y, Gupta R, et al. PolyQ proteins interfere with nuclear degradation of cytosolic proteins by sequestering the Sis1p chaperone. Cell. 2013;154(1):134–145. | ||
Ross C, Poirier M. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10(suppl):10–17. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.