Back to Journals » Neuropsychiatric Disease and Treatment » Volume 19
Treatment-Resistant Epilepsy and Tuberous Sclerosis Complex: Treatment, Maintenance, and Future Directions
Authors Singh A, Hadjinicolaou A ![]() , Peters JM, Salussolia CL
, Peters JM, Salussolia CL
Received 1 December 2022
Accepted for publication 22 March 2023
Published 5 April 2023 Volume 2023:19 Pages 733—748
DOI https://doi.org/10.2147/NDT.S347327
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Roger Pinder
Avantika Singh,1,* Aristides Hadjinicolaou,1,* Jurriaan M Peters,1 Catherine L Salussolia1,2
1Division of Epilepsy and Neurophysiology, Department of Neurology, Boston Children’s Hospital and Harvard Medical School, Boston, MA, USA; 2F.M. Kirby Neurobiology Center, Department of Neurology, Boston Children’s Hospital and Harvard Medical School, Boston, MA, USA
*These authors contributed equally to this work
Correspondence: Catherine L Salussolia, 3 Blackfan Circle, Center for Life Sciences 14060, Boston, MA, 02115, USA, Tel +617-355-7970, Email [email protected]
Abstract: Tuberous sclerosis complex (TSC) is a neurogenetic disorder that affects multiple organ systems, including the heart, kidneys, eyes, skin, and central nervous system. The neurologic manifestations have the highest morbidity and mortality, in particular in children. Clinically, patients with TSC often present with new-onset seizures within the first year of life. TSC-associated epilepsy is often difficult to treat and refractory to multiple antiseizure medications. Refractory TSC-associated epilepsy is associated with increased risk of neurodevelopmental comorbidities, including developmental delay, intellectual disability, autism spectrum disorder, and attention hyperactivity disorder. An increasing body of research suggests that early, effective treatment of TSC-associated epilepsy during critical neurodevelopmental periods can potentially improve cognitive outcomes. Therefore, it is important to treat TSC-associated epilepsy aggressively, whether it be with pharmacological therapy, surgical intervention, and/or neuromodulation. This review discusses current and future pharmacological treatments for TSC-associated epilepsy, as well as the importance of early surgical evaluation for refractory epilepsy in children with TSC and consideration of neuromodulatory interventions in young adults.
Keywords: neurodevelopment, infantile spasms, vigabatrin, epilepsy surgery
Introduction
Tuberous sclerosis complex (TSC) is a neurogenetic disorder that affects multiple organ systems due to an inactivating variant in either the TSC1 gene, which encodes hamartin, or the TSC2 gene, which encodes tuberin.1,2 TSC1 and TSC2 form a complex that regulates the mTOR pathway (Figure 1).3 Hyperactivation of mTOR signaling due to an inactivation of one of the TSC genes results in the disinhibition of the mTORC1 complex and leads to increased activation of the 4EBP1 and S6K1/2 downstream pathways, resulting in increased cell growth and proliferation of benign tumors or hamartomas that can affect multiple organs, including the heart, eyes, kidneys, lungs, skin, and brain.3,4 Further, dysregulated mTOR signaling results in ubiquitous subtle abnormalities within the central nervous system, including alterations in the white-matter connectivity and myelination,5–7 neuronal migration,8–12 axonal guidance,13,14 and dendritic pruning,15,16 contributing to the great phenotypic variability seen in TSC and high incidence of neurologic and neuropsychiatric morbidity and mortality.2,17,18 For more detailed information regarding the effects of dysregulated mTOR signaling on central nervous system development, please see the following reviews.2,19
 |
Figure 1 The mTOR signaling pathway and TSC disease-specific therapies. Figure depicting the mTOR signaling pathway and disease-specific treatments. Everolimus and sirolimus, mTOR inhibitors, inhibit RAPTOR, a protein in the mTORC1 complex. Evidence suggests that the ketogenic diet acts through the GATOR pathway to increase inhibition on RagAC and subsequent decreased activation of mTORC1. The exact mechanism of action of Epidiolex is unknown; however, evidence suggests it may modulate the PI3K–PDK1–Akt–mTOR pathway. Figure created with Biorender.com. |
Many patients with TSC present clinically as a newborn or young child with either new-onset seizures or dermatologic manifestations (eg, hypomelanotic macules or shagreen patch), while some patients are identified in utero through the presence of a cardiac rhabdomyoma on prenatal ultrasound. However, a subset of patients have more subtle symptoms due to variable penetrance of the inactivation of the TSC genes and may not be diagnosed until adulthood. Children often present with seizure onset within the first year of life; however, seizures can present at any point throughout the life span, with 80%–85% of TSC patients having at least one lifetime seizure.20 Focal seizures (68%) followed by infantile spasms (39%) are the most common presenting seizure semiology in children.1 Patients with a pathogenic variant in TSC2 generally have a more severe phenotype of TSC than patients with pathogenic variants in TSC1.21–23 In fact, TSC2 variants are associated with an earlier age of onset of seizures, as well as epilepsy that is more difficult to treat and often refractory to multiple antiseizure medications. Further, patients with a pathogenic variant in TSC2 are more likely to be diagnosed with infantile spasms than those with TSC1 mutations or no mutation identified.22,24
TSC-associated neuropsychiatric disorder (TAND) is present in approximately 90% of patients with TSC and encompasses a wide range of neuropsychiatric disorders and neurobehavioral symptoms, including but not limited to behavior problems, sleep disorders, autism spectrum disorder, attention deficit–hyperactivity disorder, intellectual disability, and mood disorders.25,26 The TOSCA study showed that while learning disabilities, intellectual disability, autism spectrum disorder, and attention deficit–hyperactivity disorder are relatively well recognized, the mean age of diagnosis of TAND-related symptoms in children with TSC is later than that of children without TSC.26 Further, studies have shown that children with TSC who have an earlier age of seizure onset are associated with more significant cognitive deficits and neuropsychiatric comorbidities.22,24,27 While the role of mTOR dysregulation and the underlying molecular mechanisms contributing to the presence of TAND symptoms are not clearly understood, it is evident that early seizure onset and refractory epilepsy are associated with poorer cognitive outcomes.1,24 Therefore, early diagnosis and treatment of seizures and implementation of early intervention therapy are of the utmost importance in improving neurodevelopmental outcomes and improving the patient’s quality of life.1
Diagnostic Criteria
TSC affects approximately one in 5000–6000 individuals, with variable penetrance.3,17,28 While pathogenic variants in either TSC1 or TSC2 are inherited in an autosomal-dominant manner, a majority of patients with TSC have a de novo variant that arises spontaneously early in embryogenesis. Further, up to 15% of patients with TSC do not have a definitive pathogenic variant identified (no mutation identified), despite a clinical diagnosis of TSC.2,17,29
The diagnosis of TSC requires fulfillment of either the genetic and/or clinical criteria recommended by the International Tuberous Sclerosis Complex Consensus Group.4 Identification of a pathogenic variant in either TSC1 or TSC2 is sufficient to make a definite diagnosis of tuberous sclerosis. Clinical diagnosis of TSC requires two major features or one major feature with two or more minor features. For a probable diagnosis, the presence of either one major feature or two or more minor features is sufficient.4,30
Some of the clinical features of TSC present at distinct time points throughout the patient’s lifespan.17 Cardiac rhabdomyomas, which can be detected by prenatal ultrasound as early as 20 weeks gestational age, are often the first clinical symptom of TSC and often spontaneously resolve within the first 2 years of life.31 Cortical tubers form between 7–20 weeks gestational age and can be identified on fetal MRI.8 Subependymal nodules line the lateral and third ventricles, and are present in 90% of patients with TSC, often at birth and/or infancy, and must be monitored for enlargement and transition into a subependymal giant-cell astrocytoma (SEGA; lesion >10 mm with >5 mm of growth).3 SEGAs are benign, slow-growing grade 1 astrocytomas that occur in childhood (rare in both newborns and after age 20 years) and can result in cerebrospinal fluid obstruction at the level of the foramen of Monro and subsequent obstructive hydrocephalus.8 Hypomelanotic macules are often present at birth or in early infancy, and are often a presenting clinical manifestation of TSC. Additional dermatologic manifestations, such as shagreen patches and facial angiofibromas, are often present by 5 years of age, whereas ungual fibromas do not typically appear until the second decade of life.4 Renal and hepatic angiomyolipomas can develop anytime between childhood and adulthood, whereas pulmonary lymphangiomyomatosis typically presents in women during young adulthood.4 Therefore, routine screening and surveillance in patients with TSC is necessary to assess the progression of TSC stigmata,30 as well as aid in the detection of rarer TSC manifestations, such as pancreatic neuroendocrine tumors. While the latter are not a formal major or minor criterion of TSC, they were seen in up to 4% of adult TSC patients in a single-center retrospective study.32
Synaptic Development and Epileptogenesis in TSC
Synapses are highly dynamic structures within the brain that govern normal neuronal activity, including learning and memory. The strengthening or weakening of a synapse in response to neuronal activity due to localized protein synthesis is known as activity-dependent synaptic plasticity. The mTOR pathway is a critical regulator of protein synthesis, and thus plays an integral role in the synaptic changes underlying synaptic plasticity and neuronal activity.33,34
Epileptogenesis is the development and extension of neural tissue capable of generating spontaneous seizures, including the development of an epileptic condition and/or progressive worsening of the patient’s epilepsy.35 Cortical tubers, which are histologically similar to focal cortical dysplasia type IIb, are characterized by gliosis, loss of lamination, and the presence of multinucleated giant cells with aberrant and disordered processes that are composed of both dysplastic neurons and reactive astrocytes.36 Epilepsy surgical workup utilizing scalp EEG recordings in combination with magnetoencephalography, PET, and single photon–emission computed tomography often leads to the identification of one or more specific tubers thought to be at the center of the epileptogenic zone.36 Thus, one may hypothesize that identifying the primary epileptogenic tuber(s) with subsequent tuberectomy would result in seizure freedom; however, this occurs in only 60%–80% of patients.37–40 A recent retrospective study by Alexander et al showed that quantitative EEG analysis of stereoelectroencephalography (sEEG) recordings of four TSC patients, there was a change in global and intertuberal connectivity in the beta and gamma frequencies in the epileptogenic tubers.41 Therefore, there must be other mechanisms mediating epileptogenesis in TSC, including the possibility of an epileptogenic network including multiple tubers and/or surrounding tissue, given that perituberal tissue contains subtle structural and cellular abnormalities on a microscopic level that are not appreciated on MRI.36,41 This is supported by the fact that multiple mouse models of tuberous sclerosis in which Tsc1 is conditionally knocked out in either glial cells42,43 or neurons5 have progressive seizures without any focal abnormalities or tubers present within the mouse brain.
Studies have shown that perilesional or non-tuber areas of the cortex can contribute to the epileptogenic zone and/or potential within the cortex.44 One study showed that resection of normal-appearing tuber-free neuronal tissue resulted in seizure freedom in a TSC1 patient with refractory epilepsy.44 Molecular analysis of surgically resected cortical tubers has demonstrated changes at both the mRNA and protein level in both excitatory, ionotropic glutamatergic receptors and inhibitory GABAergic receptors.45–49 Aberrant expression of the calcium-binding proteins calbindin and parvalbumin have also been observed in the dysplastic cortex of cortical tubers.50 Therefore, subtle abnormalities at the cellular and molecular level of the perilesional tissue may be contributing to the excitatory/inhibitory imbalance contributing to seizure generation in TSC patients.
Epilepsy in TSC: Heterogeneous Presentation
Epilepsy among TSC patients is often the most challenging manifestation and has the greatest effect on both the patient’s and family’s quality of life. Children often present with seizure onset within the first year of life; however, seizures can present at any point throughout the life span, with 80%–85% of TSC patients having at least one lifetime seizure.20 Further, the likelihood of developing epilepsy after a single seizure is nearly 100% in patients with TSC.24 While focal seizures (67.5%) followed by infantile spasms (38.9%) are the most common presenting seizure semiology in children with TSC,1 patients can present with almost all seizure semiologies, including focal to bilateral tonic–clonic, atonic, tonic, atypical absences, and myoclonic seizures.24,51 Epilepsy is truly heterogeneous among TSC patients, and seizure semiology can change throughout a patient’s lifetime.
Focal seizures are the most frequent seizure type noted in TSC patients, with a mean age at diagnosis of 2.7 years.1 Given the early onset of focal seizures in children with TSC, focal seizures often precede or occur concomitantly with the presentation of infantile spasms.52 However, focal seizures can also evolve into infantile spasms in up to a third of patients with TSC.1,53 Therefore, early treatment of new-onset seizures, even after one clinical seizure, in a patient with a diagnosis of TSC can have significant implications on the patient’s neurodevelopment and overall morbidity and mortality.1,20 Given the dynamic nature of TSC-associated epilepsy during critical periods of neurodevelopment, early seizure control via pharmacological and/or surgical interventions, albeit even if only temporizing, is essential in maximizing the patient’s developmental trajectory.
Among the 2216 patients enrolled in the international TOSCA study, 720 (38.9%) presented with infantile spasms, with a mean age at diagnosis of 4–5 months (0.4 years), which is slightly lower than the mean age of onset of infantile spasms of all etiologies (6.1 months).1,54 Rarely, late-onset epileptic spasms in TSC patients older than 2 years may be seen (2%–6%). The occurrence of spasms has been strongly associated with increased cortical tuber count.53,55 Clinically, the presentation of infantile spasms in patients with TSC can appear to be more subtle than classic flexor or extensor spasms, as well as be asymmetric, similar to those noted in other cortical malformations.53,56,57 Further, electrophysiologically, up to 70% of TSC patients with infantile spasms do not have interictal hypsarrhythmia present on EEG.53,56 Infantile spasms are a risk factor of autism spectrum disorder and poor cognitive outcomes in patients with TSC.58,59 However, effective seizure control has been shown to result in improved cognitive and developmental outcomes in TSC patients with infantile spasms.55,60,61 Finally, acute management of status epilepticus does not differ in patients with TSC, and institutional, regional, or international guidelines should be applied in order to ensure a safe and timely response to these neurological emergencies.
EEG Abnormalities as a Biomarker and Early Preventive Treatment
Given the significant neurologic and neuropsychiatric comorbidities associated with TSC, it is important to identify a biomarker that can monitor for patients at risk of developing seizures and allow for early pharmacological intervention. Several studies have shown that patients with TSC have abnormal EEGs with the presence of epileptiform activity prior to the onset of clinical seizures and/or infantile spasms.62,63 Wu et al monitored infants with TSC with serial EEGs and identified epileptiform activity on average at 4.2 months of age (range 1.2–9.0 months), which preceded seizure onset by a median of 1.9 months (average age at seizure onset 6.7±4.1 months).64 Serial routine EEGs were found to be a feasible strategy to identify individuals at high risk of developing seizures and epilepsy. These early studies formed the basis for current studies aimed at determining whether early pharmacological intervention at the onset of epileptiform activity and prior to onset of seizures can change the natural history of epilepsy due to TSC.65,66
Vigabatrin is the recommended first-line monotherapy for both TSC-associated infantile spasms and focal seizures in TSC patients <1 year of age in Europe and for infantile spasms in the US.67 In 2011, an open-label study of 45 patients monitored infants with EEG every 6 weeks to assess for epileptiform activity.66 Patients with epileptiform activity on EEG either received preventive vigabatrin prior to onset of clinical seizures or standard treatment with initiation of vigabatrin after clinical seizure onset. At 2 years of age, the preventive group had a significantly higher rate of seizure freedom (93% vs 35%) and lower incidence of drug-resistant epilepsy (7% vs 42%), and there were fewer patients requiring multidrug therapy (21% vs 55%) than the standard-treatment arm. Additionally, the standard-treatment group had a significantly higher prevalence of intellectual disability (48% vs 14%) and lower mean IQ scores (68.7 vs 92.3) than the preventive arm. As such, this study showed that preventive vigabatrin treatment markedly improves neurodevelopmental and epilepsy outcomes at 2 years of age.66
More recently, the multicenter EPISTOP clinical trial (NCT02098759) utilized serial EEG as a biomarker of epileptogenesis in infants with TSC <4 months of age and compared the efficacy of either preventive or standard vigabatrin treatment on clinical outcomes. The trial initially followed 94 infants with a definite TSC diagnosis with monthly video EEG. A total of 55 patients were included for analysis of primary outcomes of epilepsy and received either preventive vigabatrin in the setting of an abnormal EEG prior to the onset of seizures or standard vigabatrin treatment after the onset of clinical or electrographic seizures. The time to first clinical seizure was significantly longer with preventive than conventional treatment. Compared to the standard treatment group, preventive vigabatrin treatment decreased the risk of clinical seizures (OR 0.21, p=0.032), drug-resistant epilepsy (OR 0.23, p=0.022), and infantile spasms (OR 0, p<0.001) at 24 months of age.65 Further, the incidence of neurodevelopmental delay by 2 years of age was lower in the preventive vigabatrin arm; however, this difference was not significant compared to the standard-treatment arm.65
While the authors stated that the results of the EPISTOP trial suggested that early intervention with vigabatrin at the onset of epileptiform activity on EEG prior to onset of clinical or electrographic seizures may modify the natural history of TSC-associated epilepsy and improve cognitive outcomes, there are limitations to this study that dampen this interpretation. Ideally, the trial would have been designed as a double-blind, placebo-controlled study to assess the efficacy of vigabatrin on seizure control and neurodevelopmental outcomes; however, it did not include a placebo arm and only six of ten of the sites that participated in the study randomized patients to either preventive or conventional vigabatrin treatment. The remaining four sites were designated open-label, with treatment according to local clinical practice, two offering preventive treatment and two conventional treatment. Further, the study sample was relatively small, with 94 patients undergoing EEG surveillance to determine eligibility, and had a high dropout rate (>10%). Only 54 patients were deemed eligible for the primary end-point analysis, with 27 patients participating in each of the randomized and open-label arms. Of the 27 patients that were in the randomized control arm, only 13 were randomized to the preventive arm and 14 to the conventional arm, with one patient dropping out. To be eligible for randomization, a patient must have had epileptiform activity prior to the onset of clinical or electrographic seizures; however, epileptiform activity was defined as unifocal discharges present in >10% of the recording, which is a high standard requiring at least one focal spike to be present on each page on average. Therefore, while vigabatrin is approved in Europe as a preventive treatment for TSC patients with an abnormal EEG, further studies are needed to address these weaknesses.
The PREVeNT trial (NCT02849457) is an ongoing phase IIb prospective, multicenter, randomized, placebo-controlled, double-blind clinical trial that seeks to determine the cognitive and neurodevelopmental effects of early preventive vigabatrin treatment in infants with TSC who have not yet developed clinical seizures at 24 months of age.68 This trial has finished enrollment and is currently slated to complete data collection for primary outcome analysis by May 2023. Infants <6 months of age underwent monthly surveillance EEG, and once epileptiform activity was detected the infants were randomized to vigabatrin or placebo. The primary outcome measure utilizes the cognitive assessment scores on the Bayley Scales of Infant and Toddler Development at 24 months of age to determine the developmental impact of early versus delayed treatment with vigabatrin. The secondary measures will assess the number of patients who develop seizures despite preventive vigabatrin treatment, time to first clinical seizure, prevalence of drug-resistant epilepsy, and neurodevelopmental impact of early versus late treatment with vigabatrin evaluated by the Vineland II and Autism Diagnostic Observation Schedule second edition (ADOS 2). Additional secondary measures will include assessing the safety profile and adverse events of vigabatrin, as well as the feasibility of using a routine 1-hour video EEG as a biomarker for the risk of developing epilepsy.
Medical Therapies
Vigabatrin
Vigabatrin is an irreversible blocker of GABA transaminase and prevents the breakdown of synaptic GABA, the major inhibitory neurotransmitter in the central nervous system, thus increasing GABA levels in the cerebrospinal fluid several 100-fold.69 Vigabatrin is recommended as the first-line treatment for both TSC-associated infantile spasms and/or focal seizures in children <1 year of age (Table 1).52,67 Recently, the EPISTOP trial established a potential role for use of vigabatrin (100–150 mg/kg/day) in the prevention of clinical seizures, drug-resistant epilepsy and infantile spasms among patients with TSC.65 While further studies must be performed to determine the ideal recommended dosing for preventive vigabatrin treatment, given the concerns for possible retinal toxicity and risk of irreversible loss of peripheral vision with prolonged and higher doses of vigabatrin treatment, we recommend a dosing of 100 mg/kg/day for preventive therapy. However, if clinical seizures — either infantile spasms or focal seizures — do occur, it is our experience that increased dosing to 150 mg/kg/day or possibly higher is effective.52,70,71 Additionally, there is evidence that higher doses of vigabatrin are associated with lower relapse rates.71 While irreversible vision loss associated with vigabatrin can occur at any point throughout treatment, thereby warranting close monitoring with routine ophthalmologic exams every 3 months while on treatment, studies have shown that short treatment courses of vigabatrin of 6 months or less can result in recurrence of infantile spasms.70,72 Of note, previous studies in TSC animal models suggested that taurine deficiency may underlie vigabatrin-related retinal toxicity;73,74 however, a clinical study did not find any evidence for this hypothesis.75 Therefore, while there are no formal recommendations for taurine supplementation for children receiving vigabatrin, some centers do provide supplementation, as there are no known contraindications to taurine supplementation. Therefore, it is recommended vigabatrin treatment should be continued for longer treatment courses — up to 2 years — while balancing the additive risk of prolonged vigabatrin treatment.
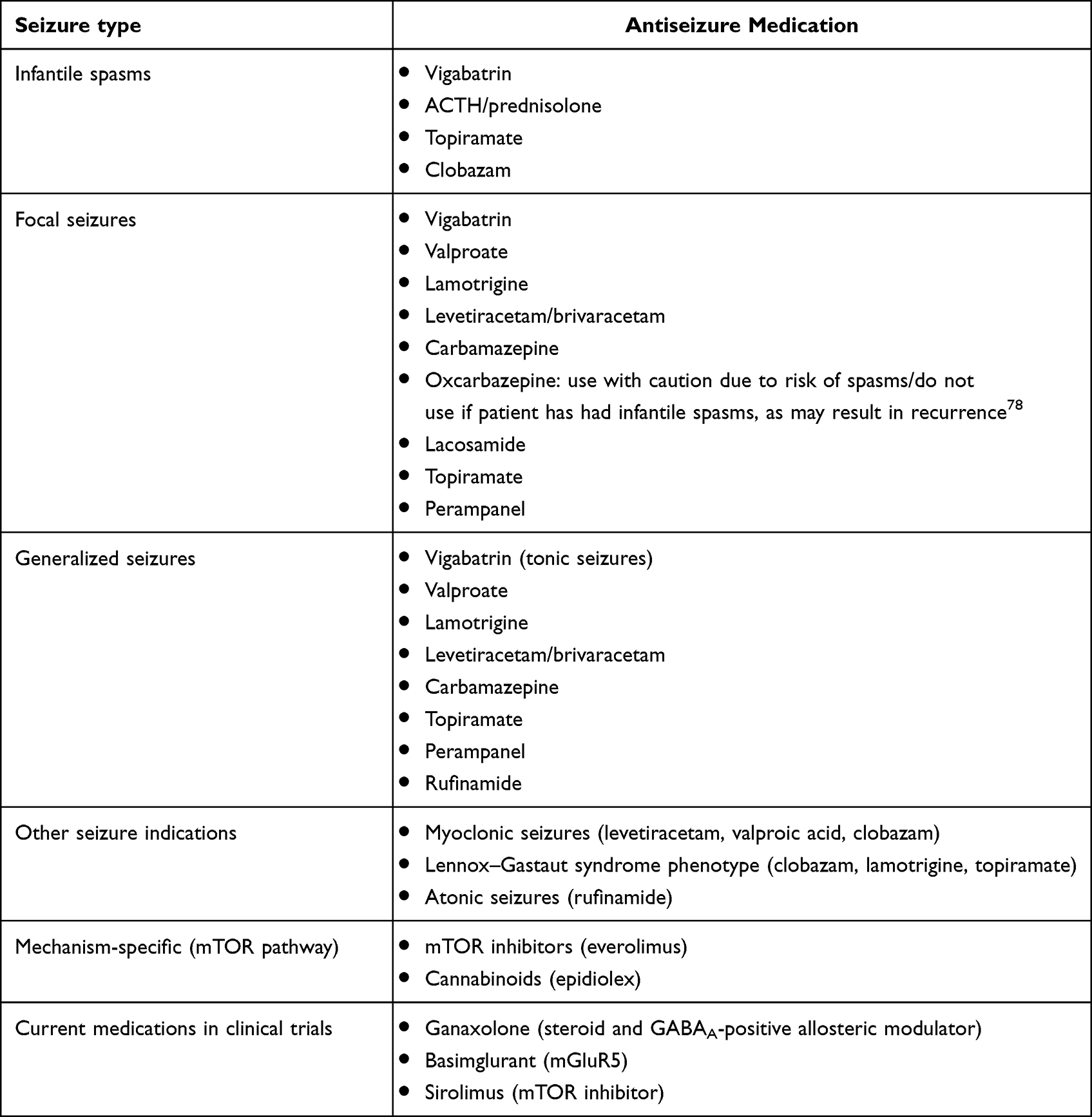 |
Table 1 Medication management options for TSC-associated seizures by indication.53,65,129–132 |
If the patient’s epilepsy is refractory to vigabatrin monotherapy, second-line treatment for TSC-associated infantile spasms includes corticosteroids/hormonal treatment with ACTH and/or prednisolone, as well as topiramate or clobazam.52,76,77 Traditional antiepileptic medications used in combination and/or as replacement therapy can be used for refractory TSC-associated focal seizures. However, caution must be used when using oxcarbazepine in children <2 years of age, given concerns that oxcarbazepine could result in the recurrence of infantile spasms.78
Traditional Antiseizure Medications
Traditional antiseizure medications continue to play a role in management of seizures in TSC (Table 1). As with all other patients, their selection is based on seizure type, medication interactions, side-effect profile, and tolerability. As previously mentioned, there are many options for focal seizures, including valproate, lamotrigine, levetiracetam, brivaracetam, carbamazepine, oxcarbazepine, topiramate, zonisamide, lacosamide, and perampanel.70,79–81 Rufinamide has been shown to be effective in treating TSC-associated tonic seizures and/or drop attacks.67 In addition, clobazam, which enhances GABAergic tone within the brain by binding to the GABAA receptor and upregulation of GABA transporters, is used as an adjunctive treatment in treating refractory epilepsy in TSC.77
mTOR Inhibitors
Given that TSC is the result of aberrant mTOR signaling, mTOR inhibitors such as everolimus and sirolimus, analogs of rapamycin, offer targeted, disease-specific treatment in TSC (Table 1). The EXIST-1 and EXIST-3 trials have shown that everolimus is effective in decreasing the size and progression of SEGAs, as well as maintaining a sustained decrease in seizure frequency in children older than 2 years.82,83 In 2018, based on the results from the EXIST-3 trial, the FDA approved the use of everolimus as an adjunctive treatment for refractory TSC-associated focal seizures, joining the European Medicinal Agency, which had approved its use in 2016. The other two FDA-approved indications for everolimus treatment in TSC are for TSC-associated SEGA and TSC-associated renal angiomyolipoma.
In 2016, Overwater et al conducted a randomized control study that assessed the efficacy of sirolimus treatment in modulating seizure frequency in children aged 2–12 years with TSC. They found that 6 months of treatment decreased seizure frequency by 61%; however, the result was not significant (p=0.06) due to the sample size.84 However, the study provided supportive data and the basis for two current clinical trials that assess its efficacy in younger children: STOP2 (NCT04595513) and TSC-STEPS (NCT05104983). STOP2 is a phase I/II open label clinical trial that verified the safety and dosing of sirolimus in infants. The STOP2 trial has finished enrolling and the data are expected to be published soon, but were favorable and allowed for the TSC-STEPS trial to ensue. TSC-STEPS is a randomized, double-blind, placebo-controlled trial that is enrolling infants <6 months of age with definite TSC who have not yet developed seizures to evaluate the safety and efficacy of early sirolimus treatment in preventing and/or delaying seizure onset in infants with TSC at 12 months of age. Secondary outcomes will assess neurodevelopmental outcomes at 12 and 24 months of age, quality-of-life measures, EEG and MRI measures of neuronal connectivity and validate the feasibility of sirolimus dosing in infants.
While both everolimus and sirolimus are associated with side effects including mucositis/stomatitis, gastrointestinal upset, and increased infection, they are generally well tolerated. For stomatitis/mucositis, one can use dexamethasone swish and spit at the start of treatment to reduce the incidence of oral ulcers85,86 and/or one can add lysine supplementation to decrease the incidence of recurrent ulcers.87 Prior to starting an mTOR inhibitor, one should obtain screening labs that include fasting lipid panel, liver-function tests, and hemoglobin A1c, given concerns for hyperlipidemia, elevated transaminases, and hyperglycemia.84
Epidiolex
Epidiolex is a synthetic cannabinoid with a 50:1 ratio of cannabidiol to tetrahydrocannabinol (Table 1). Cannabidiol is the pharmacologically active component of epidiolex, and does not cause the psychoactive features associated with tetrahydrocannabinol in cannabis. While the exact mechanism of action of epidiolex is not clear, prior studies suggest multiple potential anticonvulsive mechanisms, including inhibition of adenylate cyclase and voltage-gated calcium channels with subsequent decreased intracellular calcium, resulting in decreased presynaptic glutamate release and decreased synaptic excitability,88 but there are several studies that suggest that cannabidiol may mediate the mTOR pathway.89–91 Although Barnett et al showed that therapeutic doses of epidiolex did not reduce the volume of TSC-associated SEGAs or renal angiomyolipomas, in contrast to mTOR inhibitors,92 recent studies have shown that cannabidiol decreases seizure frequency.93,94 In 2020, Thiele et al published the findings of the GWPCARE6 study (NCT02544763), a randomized, placebo-controlled study that showed the efficacy of cannabidiol as an adjunctive treatment in TSC, as well as providing guidance with regard to optimal dosing (25 mg/kg/day), leading to FDA approval for TSC for patients >1 years of age and >2 years of age by the European Medicinal Agency.94 Additionally, longer-term adjunctive cannabidiol treatment was studied in an open-label extension to the GWPCARE6 trial and demonstrated maintenance of seizure reduction through 48 weeks, as well as 87% of patients and caregivers reporting subjective global improvement in quality of life and epilepsy control.95 The major adverse effects of epidiolex are mild to moderate gastrointestinal upset (diarrhea and decreased appetite) and somnolence, present in 91% of patients on <25 mg/kg/day and nearly all patients on >25 mg/kg/day.95 However, one must also consider the patient’s other antiseizure medications when prescribing epidiolex and often adjust the concurrent medication doses, particularly if they are on valproic acid or clobazam, as epidiolex affects the metabolism of several medications and can exacerbate their side effects.96,97
Ketogenic Diet
The ketogenic diet is an effective nonpharmacological, low-carbohydrate, high-fat diet for drug-resistant epilepsy of various etiologies that mimics the physiological state of fasting or starvation to use ketones as the body’s main energy source.98 Preclinical studies suggest a role for modulation of the mTOR pathway by the ketogenic diet, providing the basis for clinical studies (Table 2).70,99,100 Recent studies aimed at determining the efficacy of the ketogenic diet specifically in TSC and mTORopathies have shown that it results in seizure reduction, often within the first few months of initiation of the diet, but it does not guarantee long-term efficacy.101,102 However, due to its known efficacy in drug-resistant epilepsy and tolerability in children, both the European consensus TSC guidelines and the International Ketogenic Diet Study Group recommend early consideration of the ketogenic diet in infants and young children with TSC when surgery is not an option.52,103
 |
Table 2 Alternative (nonpharmacological) management options |
Surgical Options
The multilesional nature of cortical tubers in TSC poses significant challenges in the assessment of a patient’s surgical options (Table 2). There are many additional challenges in assessing the surgical candidacy of TSC patients, including the timing of surgery (ie, children <2–3 years of age may not have sufficient skull thickness to allow for sEEG electrode placement versus early surgery to improve developmental outcomes), the presence of generalized ictal or interictal epileptiform activity on EEG, and determining the goals of the surgery, ie, sustained seizure freedom with a single large intervention versus repeated smaller surgeries with temporary or incremental seizure control (serial minimally invasive surgeries could target specific seizure types and gain quality of life or improve learning during critical periods of neurodevelopment). TSC presurgical workup utilizing EEG recordings, in combination with magnetoencephalography, PET, and single photon–emission computed tomography often identifies one or more specific tubers thought to be at the center of the epileptogenic zone.36 However, tuberectomy of the dominant cortical tuber results in seizure freedom in only 60%–80% of patients,37–40 suggesting that perilesional or non-tuber areas of the cortex can contribute to the epileptogenic zone and/or potential within the cortex.44 In recent years, clinical and technological advances (eg, sEEG, laser interstitial thermal therapy, diffusion tensor imaging, α-[11C]methyl-
Resective Surgery
Presurgical evaluation, identification/localization of the epileptogenic zone, and operative management of patients with TSC differs considerably across institutions. In a prospective, observational study by Grayson et al, single-stage open resections guided by intraoperative electrocorticography were the most common procedure, followed by subdural grids with or without depth electrodes with extraoperative monitoring and rarely sEEG with extraoperative monitoring.104 However, certain centers exclusively proceed with multistage or even bilateral invasive monitoring strategies.37 When invasive monitoring is required, a tendency toward lower surgical success rates has been seen, which may reflect the inherently greater complexity of the underlying epilepsy, rather than technical failure.104 Whether a minimally invasive intratuberal ablation versus a large perituberal resection is preferred remains unresolved, as children with TSC have thus far only been included within larger case series.3 Traditionally, resection volume has been considered a major predictor of surgical success, suggesting larger resections encompassing both the tuber and perituberal rim,105 but this is not necessarily the case in TSC, and patients may actually require a multistaged approach with multiple smaller surgeries to achieve seizure freedom/good surgical outcomes. The advent of sEEG and magnetic resonance–guided laser interstitial thermal therapy has increased the ability to perform smaller lesional ablations or highly targeted focal resections that can improve surgical outcomes.41,106 However, overall outcomes appear similar among the different surgical approaches, which lends one to postulate that familiarity, expertise, and experience with TSC epilepsy surgery may play a more important role than the surgical technique used.37,107
Increased seizure burden in early development in TSC has been repeatedly demonstrated to result in significant cognitive delays, as well as higher rates of autism spectrum disorder.27,108 This was further confirmed by the recent TSC Natural History study, where data from 1657 children demonstrated that epilepsy onset prior to 2 years of age was associated with increased severity and frequency of intellectual disability.109 However, future studies are needed to determine if early, effective pharmacological treatment of epilepsy can prevent intellectual delays, as evidence suggests that even relative seizure control during critical developmental periods may mitigate seizure-related developmental injury.104 Grayson et al and the TACERN Study Group showed that early epilepsy surgery in children with TSC <2 years of age resulted in developmental gains within language domains — receptive greater than expressive language — despite patients rarely achieving full seizure freedom.104 Interestingly, Grayson et al did not find an association between resection size and postoperative neurodevelopmental outcomes, whereas Fallah et al reported a significant association between a greater extent of resection and probability of seizure freedom.104,105
Good surgical outcomes have been shown to directly promote neurodevelopment and cognition, with early and aggressive surgery potentially increasing developmental gains, as well as improving the patient’s quality of life.1,110–112 However, even if the patient experiences only partial or temporary improvement in their seizure frequency during critical developmental periods following surgery, this could result in a significant improvement in the patient’s quality of life and overall development. Surgical success rates may be higher in young children; however, systematic assessment of surgical and cognitive outcomes in this population are lacking.105,107 In the TACERN surgical pilot study, 19 patients with TSC underwent epilepsy surgery prior to 2 years of age with excellent (Engel I, n=10) and favorable (Engel II, n=2) outcomes, similar to those reported in older children, with surgical complications occurring in 11/19 (58%) patients, most of which were minor: subgaleal or subdural fluid collections, local edema, or hemorrhage.1,104,107,110,113 However, even transient deficits can play a detrimental role in neurodevelopment and impact assessment, highlighting the importance of long-term follow-up to evaluate for lasting effects.
Therefore, in order to properly understand the effects of early aggressive surgery in refractory TSC-associated epilepsy in children, better methods are required to serially monitor developmental outcomes and efficacy on seizure frequency. Ideally, a multicenter, prospective, observational study is needed to examine the predictive value of each presurgical workup modality, the location and number of implanted electrodes, the different types of surgeries, and long-term results on both seizure freedom and/or amelioration of the targeted seizure type, as well as on neurodevelopment.
Neuromodulation
Given the refractory nature of TSC-associated epilepsy, for patients that are not surgical candidates for resective surgery, they may benefit from neuomodulation with vagus nerve stimulation (VNS), deep brain stimulation (DBS), or brain-responsive neurostimulation (RNS) (Table 2). Following FDA approval of VNS in 1997 for drug-resistant focal epilepsy in individuals 12 years of age and older, tens of thousands of stimulators have been implanted worldwide. The first study published on the safety and efficacy of VNS in TSC was in 2001. In the open-label, retrospective, multicenter study by Parain et al, they reported that nine of ten of children with TSC experienced >50% reduction in seizure frequency and five had >90% reduction in seizures.114 Several studies since have confirmed both safety and efficacy, in addition to potential improvements in depressed mood, although seizure freedom is a rare occurrence.115–117
Recently, a small retrospective review of five adult TSC patients who were treated with direct RNS (RNS System, NeuroPace) reported that RNS is safe and effective in treating TSC-associated drug-resistant epilepsy.118 The RNS System targets one or two seizure foci in response to epileptiform activity detected by the system. Patients were followed postimplantation, and all five had ≥50% reduction in seizures at last follow-up (average length of follow-up was 20 months). Additionally, three of the five patients experienced some period of seizure freedom ranging from 3 months to >1 year. This study highlights the potential promise of RNS as a treatment option for TSC patients with refractory focal epilepsy who are not resective surgical candidates.
DBS of the anterior nucleus of the thalamus has been shown to be an effective treatment for patients with refractory epilepsy. Recently, Zheng et al reported a single case report of a 22-year-old male patient with refractory TSC-associated epilepsy who received bilateral DBS of anterior thalamic nucleus.119 Following placement of the bilateral DBS, the patient experienced a 90% reduction in seizure frequency and achieved a satisfactory response on quality-of-life measures at 15-month follow-up.119 This suggests that anterior thalamus DBS may be an alternative option to other neuromodulatory interventions in TSC patients.
Novel Therapeutics and Future Directions
Ganaxolone
Ganaxolone is a neuroactive steroid and positive allosteric modulator of GABAA receptors that was recently FDA-approved to treat epilepsy in CDKL5-deficiency disorder.120 Given its mechanism of action and modulation of GABAergic signaling, ganaxolone is being investigated as a possible therapy for TSC (Table 1). Koenig et al recently presented preliminary findings from an open-label phase II clinical trial in which ganaxolone was evaluated as an adjunctive therapy for refractory TSC-associated epilepsy (more than eight seizures during 4-week baseline) in 23 patients aged 2–65 years (median age 11 years).121 The primary outcome was the median percentage change from baseline in frequency of TSC-associated seizures over the course of the 12-week treatment period (including a 4-week titration) and the secondary outcome responder rate (50% reduction in seizure frequency). Preliminary results showed a 16.6% reduction in TSC-associated seizures after 28 days, with 30% of patients achieving at least a 50% reduction in seizure frequency. Overall, the trial showed a median of 25% reduction in focal seizures. Ganaxolone was generally well tolerated; however, four patients (17.4%) withdrew from the trial due to adverse events, which most often was somnolence (43.5%).121 These findings served as the basis for TrustTSC (NCT05323734), a phase III, double-blind, randomized, placebo-controlled trial aimed at further evaluating the efficacy of ganaxolone in reducing the frequency of TSC-associated seizures. This trial is currently enrolling, and is predicted to end in the winter of 2025.
mGLUR5
mGluR5 is located both presynaptically on excitatory glutamatergic neurons and present in the postsynaptic membrane, lending itself as a promising therapeutic target for neurodevelopmental disorders (Table 1). Evidence has shown that modulation of mGluR5-mediated signaling with a negative allosteric modulator (NAM) ameliorates long-term memory deficits, excessive repetitive behaviors, motor stereotypies, and abnormal social interactions in different mouse models of autism.122–126 In addition, mGluR5 NAMs have anticonvulsant effects, as seen in Tsc2 mouse models in which MPEP, an mGluR5 NAM, reduced ictal bursting in acute slice preparations and acute treatment with MPEP decreased seizure frequency and total seizure time.127,128 Further, acute administration of a positive allosteric modulator of mGluR5 resulted in increased seizure frequency and total seizure time in a Tsc2 conditional knockout mouse model.127 Together, these studies suggest that inhibition of mGluR5-mediated signaling is a promising therapeutic target for TSC-associated seizures. Basimglurant in Children, Adolescents, and Young Adults with TSC (NCT05059327) is a phase II, randomized, double-blind, placebo-controlled study that is currently enrolling patients to determine the effectiveness and optimal dose of basimglurant, an mGluR5 inhibitor, in reducing seizure frequency in patients with TSC (5–30 years of age). This study is anticipated to be completed in the summer of 2024.
Conclusions
TSC-associated epilepsy can be challenging to treat; however, with advances in the understanding of the epileptogenesis in TSC, mTOR signaling including the role of the GATOR complex, and the advent of mTOR inhibitors, we have entered a new era in precision therapy for TSC. The ongoing technological and investigational advances in invasive EEG monitoring, particularly in sEEG methodology, offer great promise in the treatment of TSC patients with refractory seizures. Preliminary evidence suggests that early treatment with vigabatrin for infantile spasms and perhaps focal seizures in infants and young children can improve neurodevelopmental outcomes during critical developmental periods; however, further studies are needed. If TSC-associated epilepsy remains pharmacoresistant, patients should undergo early evaluation for surgical intervention, even prior to 2 years of age, to consider both curative and/or palliative options, as this may provide not only potential improvement in seizure outcomes but also improvement in cognition and quality of life for patients and caregivers. A patient’s age or the presence of generalized interictal epileptiform activity on EEG should not preclude them from consideration for possible resective or targeted laser ablation surgery. The advent of disease-specific therapies that target mTOR signaling with everolimus and sirolimus, as well as the promise of novel ways to enhance GABAergic signaling via ganaxolone and new therapeutic targets with mGluR5, may offer patients even more precise therapies aimed at treating TSC-associated seizures in the near future.
Acknowledgments
CLS was funded by the CH/BIDMC/Harvard Medical School Neurology Resident Research Education Program, NIH (R25NS070682) and is funded by the National Institutes of Neurological Disorders and Stroke (5K12NS098482).
Disclosure
Dr Jurriaan Peters is the site PI on the Marinus TrustTSC Trial, a phase III study of ganaxolone in refractory epilepsy in TSC. He is a speaker and consultant for Jazz Pharmaceuticals, SK Life Sciences, and Neurelis. The authors report no other conflicts of interest in this work.
References
1. Nabbout R, Belousova E, Benedik MP, et al. Epilepsy in tuberous sclerosis complex: findings from the TOSCA Study. Epilepsia Open. 2019;4(1):73–84. doi:10.1002/epi4.12286
2. Salussolia CL, Klonowska K, Kwiatkowski DJ, Sahin M. Genetic Etiologies, Diagnosis, and Treatment of Tuberous Sclerosis Complex. Annu Rev Genomics Hum Genet. 2019;20:217–240. doi:10.1146/annurev-genom-083118-015354
3. Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355(13):1345–1356. doi:10.1056/NEJMra055323
4. Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus G. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 international tuberous sclerosis complex consensus conference. Pediatr Neurol. 2013;49(4):243–254. doi:10.1016/j.pediatrneurol.2013.08.001
5. Meikle L, Talos DM, Onda H, et al. A mouse model of tuberous sclerosis: neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced myelination, seizure activity, and limited survival. J Neurosci. 2007;27(21):5546–5558. doi:10.1523/JNEUROSCI.5540-06.2007
6. Ercan E, Han JM, Di Nardo A, et al. Neuronal CTGF/CCN2 negatively regulates myelination in a mouse model of tuberous sclerosis complex. J Exp Med. 2017;214(3):681–697. doi:10.1084/jem.20160446
7. Lebrun-Julien F, Bachmann L, Norrmén C, et al. Balanced mTORC1 activity in oligodendrocytes is required for accurate CNS myelination. J Neurosci. 2014;34(25):8432–8448. doi:10.1523/JNEUROSCI.1105-14.2014
8. Feliciano DM, Lin TV, Hartman NW, et al. A circuitry and biochemical basis for tuberous sclerosis symptoms: from epilepsy to neurocognitive deficits. Int J Dev Neurosci. 2013;31(7):667–678. doi:10.1016/j.ijdevneu.2013.02.008
9. Feliciano DM, Quon JL, Su T, Taylor MM, Bordey A. Postnatal neurogenesis generates heterotopias, olfactory micronodules and cortical infiltration following single-cell Tsc1 deletion. Hum Mol Genet. 2012;21(4):799–810. doi:10.1093/hmg/ddr511
10. Lin TV, Hsieh L, Kimura T, Malone TJ, Bordey A. Normalizing translation through 4E-BP prevents mTOR-driven cortical mislamination and ameliorates aberrant neuron integration. Proc Natl Acad Sci U S A. 2016;113(40):11330–11335. doi:10.1073/pnas.1605740113
11. Goto J, Talos DM, Klein P, et al. Regulable neural progenitor-specific Tsc1 loss yields giant cells with organellar dysfunction in a model of tuberous sclerosis complex. Proc Natl Acad Sci U S A. 2011;108(45):E1070–9. doi:10.1073/pnas.1106454108
12. Zhou J, Shrikhande G, Xu J, et al. Tsc1 mutant neural stem/progenitor cells exhibit migration deficits and give rise to subependymal lesions in the lateral ventricle. Genes Dev. 2011;25(15):1595–1600. doi:10.1101/gad.16750211
13. Gong X, Zhang L, Huang T, et al. Activating the translational repressor 4E-BP or reducing S6K-GSK3β activity prevents accelerated axon growth induced by hyperactive mTOR in vivo. Hum Mol Genet. 2015;24(20):5746–5758. doi:10.1093/hmg/ddv295
14. Choi YJ, Di Nardo A, Kramvis I, et al. Tuberous sclerosis complex proteins control axon formation. Genes Dev. 2008;22(18):2485–2495. doi:10.1101/gad.1685008
15. Wong RO, Ghosh A. Activity-dependent regulation of dendritic growth and patterning. Nat Rev Neurosci. 2002;3(10):803–812. doi:10.1038/nrn941
16. LiCausi F, Hartman NW. Role of mTOR Complexes in Neurogenesis. Int J Mol Sci. 2018;19(5):1544. doi:10.3390/ijms19051544
17. Zöllner JP, Franz DN, Hertzberg C, et al. A systematic review on the burden of illness in individuals with tuberous sclerosis complex (TSC). Orphanet J Rare Dis. 2020;15(1):23. doi:10.1186/s13023-019-1258-3
18. Farach LS, Richard MA, Lupo PJ, et al. Epilepsy risk prediction model for patients with tuberous sclerosis complex. Pediatr Neurol. 2020;113:46–50. doi:10.1016/j.pediatrneurol.2020.07.015
19. Winden KD, Ebrahimi-Fakhari D, Sahin M. Abnormal mTOR activation in autism. Annu Rev Neurosci. 2018;41:1–23. doi:10.1146/annurev-neuro-080317-061747
20. Ihnen SKZ, Capal JK, Horn PS, et al. Epilepsy Is heterogeneous in early-life tuberous sclerosis complex. Pediatr Neurol. 2021;123:1–9. doi:10.1016/j.pediatrneurol.2021.06.012
21. Avgeris S, Fostira F, Vagena A, et al. Mutational analysis of TSC1 and TSC2 genes in Tuberous Sclerosis Complex patients from Greece. Sci Rep. 2017;7(1):16697. doi:10.1038/s41598-017-16988-w
22. Curatolo P, Moavero R, Roberto D, Graziola F. Genotype/phenotype correlations in tuberous sclerosis complex. Semin Pediatr Neurol. 2015;22(4):259–273. doi:10.1016/j.spen.2015.10.002
23. Dabora SL, Jozwiak S, Franz DN, et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am J Hum Genet. 2001;68(1):64–80. doi:10.1086/316951
24. Chu-Shore CJ, Major P, Camposano S, Muzykewicz D, Thiele EA. The natural history of epilepsy in tuberous sclerosis complex. Epilepsia. 2010;51(7):1236–1241. doi:10.1111/j.1528-1167.2009.02474.x
25. de Vries PJ, Whittemore VH, Leclezio L, et al. Tuberous sclerosis associated neuropsychiatric disorders (TAND) and the TAND Checklist. Pediatr Neurol. 2015;52(1):25–35. doi:10.1016/j.pediatrneurol.2014.10.004
26. de Vries PJ, Belousova E, Benedik MP, et al. TSC-associated neuropsychiatric disorders (TAND): findings from the TOSCA natural history study. Orphanet J Rare Dis. 2018;13(1):157. doi:10.1186/s13023-018-0901-8
27. Capal JK, Bernardino-Cuesta B, Horn PS, et al. Influence of seizures on early development in tuberous sclerosis complex. Epilepsy Behav. 2017;70(Pt A):245–252. doi:10.1016/j.yebeh.2017.02.007
28. O’Callaghan FJ, Shiell AW, Osborne JP, Martyn CN. Prevalence of tuberous sclerosis estimated by capture-recapture analysis. Lancet. 1998;351(9114):1490. doi:10.1016/S0140-6736(05)78872-3
29. Sancak O, Nellist M, Goedbloed M, et al. Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype--phenotype correlations and comparison of diagnostic DNA techniques in Tuberous Sclerosis Complex. Eur J Hum Genet. 2005;13(6):731–741. doi:10.1038/sj.ejhg.5201402
30. Northrup H, Aronow ME, Bebin EM, et al. Updated international tuberous sclerosis complex diagnostic criteria and surveillance and management recommendations. Pediatr Neurol. 2021;123:50–66. doi:10.1016/j.pediatrneurol.2021.07.011
31. Hinton RB, Prakash A, Romp RL, Krueger DA, Knilans TK; Group ITSC. Cardiovascular manifestations of tuberous sclerosis complex and summary of the revised diagnostic criteria and surveillance and management recommendations from the International Tuberous Sclerosis Consensus Group. J Am Heart Assoc. 2014;3(6):e001493. doi:10.1161/JAHA.114.001493
32. Larson AM, Hedgire SS, Deshpande V, et al. Pancreatic neuroendocrine tumors in patients with tuberous sclerosis complex. Clin Genet. 2012;82(6):558–563. doi:10.1111/j.1399-0004.2011.01805.x
33. Tang SJ, Reis G, Kang H, Gingras AC, Sonenberg N, Schuman EM. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci U S A. 2002;99(1):467–472. doi:10.1073/pnas.012605299
34. Tsokas P, Grace EA, Chan P, et al. Local protein synthesis mediates a rapid increase in dendritic elongation factor 1A after induction of late long-term potentiation. J Neurosci. 2005;25(24):5833–5843. doi:10.1523/JNEUROSCI.0599-05.2005
35. Pitkänen A. Therapeutic approaches to epileptogenesis--hope on the horizon. Epilepsia. 2010;51(Suppl 3):2–17. doi:10.1111/j.1528-1167.2010.02602.x
36. Wong M. Mechanisms of epileptogenesis in tuberous sclerosis complex and related malformations of cortical development with abnormal glioneuronal proliferation. Epilepsia. 2008;49(1):8–21. doi:10.1111/j.1528-1167.2007.01270.x
37. Weiner HL, Carlson C, Ridgway EB, et al. Epilepsy surgery in young children with tuberous sclerosis: results of a novel approach. Pediatrics. 2006;117(5):1494–1502. doi:10.1542/peds.2005-1206
38. Aronica E, Leenstra S, van Veelen CW, et al. Glioneuronal tumors and medically intractable epilepsy: a clinical study with long-term follow-up of seizure outcome after surgery. Epilepsy Res. 2001;43(3):179–191. doi:10.1016/s0920-1211(00)00208-4
39. Tassi L, Colombo N, Garbelli R, et al. Focal cortical dysplasia: neuropathological subtypes, EEG, neuroimaging and surgical outcome. Brain. 2002;125(Pt 8):1719–1732. doi:10.1093/brain/awf175
40. Neal A, Ostrowsky-Coste K, Jung J, et al. Epileptogenicity in tuberous sclerosis complex: a stereoelectroencephalographic study. Epilepsia. 2020;61(1):81–95. doi:10.1111/epi.16410
41. Alexander H, Govindan RB, Anwar T, et al. Global and intertuberal epileptic networks in tuberous sclerosis based on stereoelectroencephalographic (sEEG) findings: a quantitative EEG analysis in pediatric subjects and surgical implications. Childs Nerv Syst. 2022;38(2):407–419. doi:10.1007/s00381-021-05342-1
42. Uhlmann EJ, Wong M, Baldwin RL, et al. Astrocyte-specific TSC1 conditional knockout mice exhibit abnormal neuronal organization and seizures. Ann Neurol. 2002;52(3):285–296. doi:10.1002/ana.10283
43. Erbayat-Altay E, Zeng LH, Xu L, Gutmann DH, Wong M. The natural history and treatment of epilepsy in a murine model of tuberous sclerosis. Epilepsia. 2007;48(8):1470–1476. doi:10.1111/j.1528-1167.2007.01110.x
44. Wang Y, Greenwood JS, Calcagnotto ME, Kirsch HE, Barbaro NM, Baraban SC. Neocortical hyperexcitability in a human case of tuberous sclerosis complex and mice lacking neuronal expression of TSC1. Ann Neurol. 2007;61(2):139–152. doi:10.1002/ana.21058
45. Talos DM, Sun H, Kosaras B, et al. Altered inhibition in tuberous sclerosis and type IIb cortical dysplasia. Ann Neurol. 2012;71(4):539–551. doi:10.1002/ana.22696
46. Talos DM, Kwiatkowski DJ, Cordero K, Black PM, Jensen FE. Cell-specific alterations of glutamate receptor expression in tuberous sclerosis complex cortical tubers. Ann Neurol. 2008;63(4):454–465. doi:10.1002/ana.21342
47. White R, Hua Y, Scheithauer B, Lynch DR, Henske EP, Crino PB. Selective alterations in glutamate and GABA receptor subunit mRNA expression in dysplastic neurons and giant cells of cortical tubers. Ann Neurol. 2001;49(1):67–78. doi:10.1002/1531-8249(200101)49:1<67::aid-ana10>3.0.co;2-l
48. Mori K, Mori T, Toda Y, et al. Decreased benzodiazepine receptor and increased GABA level in cortical tubers in tuberous sclerosis complex. Brain Dev. 2012;34(6):478–486. doi:10.1016/j.braindev.2011.09.001
49. Boer K, Crino PB, Gorter JA, et al. Gene expression analysis of tuberous sclerosis complex cortical tubers reveals increased expression of adhesion and inflammatory factors. Brain Pathol. 2010;20(4):704–719. doi:10.1111/j.1750-3639.2009.00341.x
50. Valencia I, Legido A, Yelin K, Khurana D, Kothare SV, Katsetos CD. Anomalous inhibitory circuits in cortical tubers of human tuberous sclerosis complex associated with refractory epilepsy: aberrant expression of parvalbumin and calbindin-D28k in dysplastic cortex. J Child Neurol. 2006;21(12):1058–1063. doi:10.1177/7010.2006.00242
51. van der Poest Clement E, Jansen FE, Braun KPJ, Peters JM. Update on drug management of refractory epilepsy in tuberous sclerosis complex. Paediatr Drugs. 2020;22(1):73–84. doi:10.1007/s40272-019-00376-0
52. Curatolo P, Nabbout R, Lagae L, et al. Management of epilepsy associated with tuberous sclerosis complex: updated clinical recommendations. Eur J Paediatr Neurol. 2018;22(5):738–748. doi:10.1016/j.ejpn.2018.05.006
53. Thiele EA. Managing epilepsy in tuberous sclerosis complex. J Child Neurol. 2004;19(9):680–686. doi:10.1177/08830738040190090801
54. Berg AT, Chakravorty S, Koh S, et al. Why West? Comparisons of clinical, genetic and molecular features of infants with and without spasms. PLoS One. 2018;13(3):e0193599. doi:10.1371/journal.pone.0193599
55. Hamano S, Tanaka M, Mochizuki M, Sugiyama N, Eto Y. Long-term follow-up study of west syndrome: differences of outcome among symptomatic etiologies. J Pediatr. 2003;143(2):231–235. doi:10.1067/s0022-3476(03)00323-8
56. Curatolo P, Verdecchia M, Bombardieri R. Vigabatrin for tuberous sclerosis complex. Brain Dev. 2001;23(7):649–653. doi:10.1016/s0387-7604(01)00290-x
57. Curatolo P, Verdecchia M, Bombardieri R. Tuberous sclerosis complex: a review of neurological aspects. Eur J Paediatr Neurol. 2002;6(1):15–23. doi:10.1053/ejpn.2001.0538
58. Bolton PF, Park RJ, Higgins JN, Griffiths PD, Pickles A. Neuro-epileptic determinants of autism spectrum disorders in tuberous sclerosis complex. Brain. 2002;125(Pt 6):1247–1255. doi:10.1093/brain/awf124
59. Asano E, Chugani DC, Muzik O, et al. Autism in tuberous sclerosis complex is related to both cortical and subcortical dysfunction. Neurology. 2001;57(7):1269–1277. doi:10.1212/wnl.57.7.1269
60. Fukushima K, Inoue Y, Fujiwara T, Yagi K. Long-term follow-up study of West syndrome associated with tuberous sclerosis. Brain Dev. 2001;23(7):698–704. doi:10.1016/s0387-7604(01)00275-3
61. Jambaqué I, Chiron C, Dumas C, Mumford J, Dulac O. Mental and behavioural outcome of infantile epilepsy treated by vigabatrin in tuberous sclerosis patients. Epilepsy Res. 2000;38(2–3):151–160. doi:10.1016/s0920-1211(99)00082-0
62. Domanska-Pakiela D, Kaczorowska M, Jurkiewicz E, Kotulska K, Dunin-Wasowicz D, Jozwiak S. EEG abnormalities preceding the epilepsy onset in tuberous sclerosis complex patients - A prospective study of 5 patients. Eur J Paediatr Neurol. 2014;18(4):458–468. doi:10.1016/j.ejpn.2013.12.006
63. Davis PE, Kapur K, Filip-Dhima R, et al. Increased electroencephalography connectivity precedes epileptic spasm onset in infants with tuberous sclerosis complex. Epilepsia. 2019;60(8):1721–1732. doi:10.1111/epi.16284
64. Wu JY, Peters JM, Goyal M, et al. Clinical electroencephalographic biomarker for impending epilepsy in asymptomatic tuberous sclerosis complex infants. Pediatr Neurol. 2016;54:29–34. doi:10.1016/j.pediatrneurol.2015.09.013
65. Kotulska K, Kwiatkowski DJ, Curatolo P, et al. Prevention of epilepsy in infants with tuberous sclerosis complex in the EPISTOP trial. Ann Neurol. 2021;89(2):304–314. doi:10.1002/ana.25956
66. Jozwiak S, Kotulska K, Domanska-Pakiela D, et al. Antiepileptic treatment before the onset of seizures reduces epilepsy severity and risk of mental retardation in infants with tuberous sclerosis complex. Eur J Paediatr Neurol. 2011;15(5):424–431. doi:10.1016/j.ejpn.2011.03.010
67. Curatolo P, Jóźwiak S, Nabbout R; Management TCMfSaE. Management of epilepsy associated with tuberous sclerosis complex (TSC): clinical recommendations. Eur J Paediatr Neurol. 2012;16(6):582–586. doi:10.1016/j.ejpn.2012.05.004
68. Preventing epilepsy using vigabatrin in infants with tuberous sclerosis complex; 2022. Available from: https://clinicaltrials.gov/ct2/show/NCT02849457.
69. Ben-Menachem E. Mechanism of action of vigabatrin: correcting misperceptions. Acta Neurol Scand Suppl. 2011;192:5–15. doi:10.1111/j.1600-0404.2011.01596.x
70. Canevini MP, Kotulska-Jozwiak K, Curatolo P, et al. Current concepts on epilepsy management in tuberous sclerosis complex. Am J Med Genet C Semin Med Genet. 2018;178(3):299–308. doi:10.1002/ajmg.c.31652
71. Hussain SA, Schmid E, Peters JM, et al. High vigabatrin dosage is associated with lower risk of infantile spasms relapse among children with tuberous sclerosis complex. Epilepsy Res. 2018;148:1–7. doi:10.1016/j.eplepsyres.2018.09.016
72. Humphrey A, Neville BG, Clarke A, Bolton PF. Autistic regression associated with seizure onset in an infant with tuberous sclerosis. Dev Med Child Neurol. 2006;48(7):609–611. doi:10.1017/S0012162206001277
73. Jammoul F, Wang Q, Nabbout R, et al. Taurine deficiency is a cause of vigabatrin-induced retinal phototoxicity. Ann Neurol. 2009;65(1):98–107. doi:10.1002/ana.21526
74. Froger N, Jammoul F, Gaucher D, et al. Taurine is a crucial factor to preserve retinal ganglion cell survival. Adv Exp Med Biol. 2013;775:69–83. doi:10.1007/978-1-4614-6130-2_6
75. Spelbrink EM, Mabud TS, Reimer R, Porter BE. Plasma taurine levels are not affected by vigabatrin in pediatric patients. Epilepsia. 2016;57(8):e168–72. doi:10.1111/epi.13447
76. Nomura S, Shimakawa S, Tanabe T, Fukui M, Kashiwagi M, Tamai H. Topiramateがてんかん性スパズムの再発例に有効であった結節性硬化症の3例. [Efficacy of topiramate for relapsed epileptic spasms with tuberous sclerosis: report of three cases]. No to Hattatsu. 2011;43(6):476–481. Japanese.
77. Jennesson M, van Eeghen AM, Caruso PA, Paolini JL, Thiele EA. Clobazam therapy of refractory epilepsy in tuberous sclerosis complex. Epilepsy Res. 2013;104(3):269–274. doi:10.1016/j.eplepsyres.2012.10.010
78. Hussain SA, Heesch J, Weng J, Rajaraman RR, Numis AL, Sankar R. Potential induction of epileptic spasms by nonselective voltage-gated sodium channel blockade: interaction with etiology. Epilepsy Behav. 2021;115:107624. doi:10.1016/j.yebeh.2020.107624
79. Franz DN, Tudor C, Leonard J, et al. Lamotrigine therapy of epilepsy in tuberous sclerosis. Epilepsia. 2001;42(7):935–940. doi:10.1046/j.1528-1157.2001.042007935.x
80. Collins JJ, Tudor C, Leonard JM, Chuck G, Franz DN. Levetiracetam as adjunctive antiepileptic therapy for patients with tuberous sclerosis complex: a retrospective open-label trial. J Child Neurol. 2006;21(1):53–57. doi:10.1177/08830738060210011201
81. Geffrey AL, Belt OD, Paolini JL, Thiele EA. Lacosamide use in the treatment of refractory epilepsy in tuberous sclerosis complex. Epilepsy Res. 2015;112:72–75. doi:10.1016/j.eplepsyres.2015.02.008
82. Curatolo P, Franz DN, Lawson JA, et al. Adjunctive everolimus for children and adolescents with treatment-refractory seizures associated with tuberous sclerosis complex: post-hoc analysis of the phase 3 EXIST-3 trial. Lancet Child Adolesc Health. 2018;2(7):495–504. doi:10.1016/S2352-4642(18)30099-3
83. Franz DN, Belousova E, Sparagana S, et al. Long-Term use of everolimus in patients with tuberous sclerosis complex: final results from the EXIST-1 study. PLoS One. 2016;11(6):e0158476. doi:10.1371/journal.pone.0158476
84. Overwater IE, Rietman AB, Bindels-de Heus K, et al. Sirolimus for epilepsy in children with tuberous sclerosis complex: a randomized controlled trial. Neurology. 2016;87(10):1011–1018. doi:10.1212/WNL.0000000000003077
85. Arena C, Troiano G, Zhurakivska K, Nocini R, Lo Muzio L. Stomatitis and everolimus: a review of current literature on 8201 patients. Onco Targets Ther. 2019;12:9669–9683. doi:10.2147/OTT.S195121
86. Rugo HS, Seneviratne L, Beck JT, et al. Prevention of everolimus-related stomatitis in women with hormone receptor-positive, HER2-negative metastatic breast cancer using dexamethasone mouthwash (SWISH): a single-arm, phase 2 trial. Lancet Oncol. 2017;18(5):654–662. doi:10.1016/S1470-2045(17)30109-2
87. Peterson ME. Management of adverse events in patients with hormone receptor-positive breast cancer treated with everolimus: observations from a phase III clinical trial. Support Care Cancer. 2013;21(8):2341–2349. doi:10.1007/s00520-013-1826-3
88. Morano A, Fanella M, Albini M, et al. Cannabinoids in the treatment of epilepsy: current status and future prospects. Neuropsychiatr Dis Treat. 2020;16:381–396. doi:10.2147/NDT.S203782
89. Gobira PH, Vilela LR, Gonçalves BD, et al. Cannabidiol, a Cannabis sativa constituent, inhibits cocaine-induced seizures in mice: possible role of the mTOR pathway and reduction in glutamate release. Neurotoxicology. 2015;50:116–121. doi:10.1016/j.neuro.2015.08.007
90. Renard J, Loureiro M, Rosen LG, et al. Cannabidiol counteracts amphetamine-induced neuronal and behavioral sensitization of the mesolimbic dopamine pathway through a novel mTOR/p70S6 kinase signaling pathway. J Neurosci. 2016;36(18):5160–5169. doi:10.1523/JNEUROSCI.3387-15.2016
91. Giacoppo S, Pollastro F, Grassi G, Bramanti P, Mazzon E. Target regulation of PI3K/Akt/mTOR pathway by cannabidiol in treatment of experimental multiple sclerosis. Fitoterapia. 2017;116:77–84. doi:10.1016/j.fitote.2016.11.010
92. Barnett JR, Grinspoon RA, Harisinghani M, Caruso PA, Thiele EA. The efficacy of cannabidiol on renal angiomyolipoma and subependymal giant cell tumor volume in tuberous sclerosis complex. J Clin Neurosci. 2020;77:85–88. doi:10.1016/j.jocn.2020.05.030
93. Hess EJ, Moody KA, Geffrey AL, et al. Cannabidiol as a new treatment for drug-resistant epilepsy in tuberous sclerosis complex. Epilepsia. 2016;57(10):1617–1624. doi:10.1111/epi.13499
94. Thiele EA, Bebin EM, Bhathal H, et al. Add-on cannabidiol treatment for drug-resistant seizures in tuberous sclerosis complex: a placebo-controlled randomized clinical trial. JAMA Neurol. 2021;78(3):285–292. doi:10.1001/jamaneurol.2020.4607
95. Thiele EA, Bebin EM, Filloux F, et al. Long-term cannabidiol treatment for seizures in patients with tuberous sclerosis complex: an open-label extension trial. Epilepsia. 2022;63(2):426–439. doi:10.1111/epi.17150
96. Geffrey AL, Pollack SF, Bruno PL, Thiele EA. Drug-drug interaction between clobazam and cannabidiol in children with refractory epilepsy. Epilepsia. 2015;56(8):1246–1251. doi:10.1111/epi.13060
97. Gaston TE, Bebin EM, Cutter GR, Liu Y, Szaflarski JP; Program UC. Interactions between cannabidiol and commonly used antiepileptic drugs. Epilepsia. 2017;58(9):1586–1592. doi:10.1111/epi.13852
98. Thiele EA. Assessing the efficacy of antiepileptic treatments: the ketogenic diet. Epilepsia. 2003;44(Suppl 7):26–29. doi:10.1046/j.1528-1157.44.s7.4.x
99. Yuskaitis CJ, Modasia JB, Schrötter S, et al. DEPDC5-dependent mTORC1 signaling mechanisms are critical for the anti-seizure effects of acute fasting. Cell Rep. 2022;40(9):111278. doi:10.1016/j.celrep.2022.111278
100. McDaniel SS, Rensing NR, Thio LL, Yamada KA, Wong M. The ketogenic diet inhibits the mammalian target of rapamycin (mTOR) pathway. Epilepsia. 2011;52(3):e7–11. doi:10.1111/j.1528-1167.2011.02981.x
101. Youn SE, Park S, Kim SH, Lee JS, Kim HD, Kang HC. Long-term outcomes of ketogenic diet in patients with tuberous sclerosis complex-derived epilepsy. Epilepsy Res. 2020;164:106348. doi:10.1016/j.eplepsyres.2020.106348
102. Ko A, Sim NS, Choi HS, et al. Efficacy of the ketogenic diet for pediatric epilepsy according to the presence of detectable somatic mTOR pathway mutations in the brain. J Clin Neurol. 2022;18(1):71–78. doi:10.3988/jcn.2022.18.1.71
103. Kossoff EH, Zupec-Kania BA, Auvin S, et al. Optimal clinical management of children receiving dietary therapies for epilepsy: updated recommendations of the International Ketogenic Diet Study Group. Epilepsia Open. 2018;3(2):175–192. doi:10.1002/epi4.12225
104. Grayson LE, Peters JM, McPherson T, et al. Pilot study of neurodevelopmental impact of early epilepsy surgery in tuberous sclerosis complex. Pediatr Neurol. 2020;109:39–46. doi:10.1016/j.pediatrneurol.2020.04.002
105. Fallah A, Rodgers SD, Weil AG, et al. Resective epilepsy surgery for tuberous sclerosis in children: determining predictors of seizure outcomes in a multicenter retrospective cohort study. Neurosurgery. 2015;77(4):517–524; discussion 524. doi:10.1227/NEU.0000000000000875
106. Boom M, SCurry J, Weiner DJ, Peters HL, Jurriaan M. Technological advances in pediatric epilepsy surgery: implications for tuberous sclerosis complex. Future Neurol. 2017;12:101–115. doi:10.2217/fnl-2017-0005
107. Wu JY, Salamon N, Kirsch HE, et al. Noninvasive testing, early surgery, and seizure freedom in tuberous sclerosis complex. Neurology. 2010;74(5):392–398. doi:10.1212/WNL.0b013e3181ce5d9e
108. Bombardieri R, Pinci M, Moavero R, Cerminara C, Curatolo P. Early control of seizures improves long-term outcome in children with tuberous sclerosis complex. Eur J Paediatr Neurol. 2010;14(2):146–149. doi:10.1016/j.ejpn.2009.03.003
109. Gupta A, de Bruyn G, Tousseyn S, et al. Epilepsy and neurodevelopmental comorbidities in tuberous sclerosis complex: a natural history study. Pediatr Neurol. 2020;106:10–16. doi:10.1016/j.pediatrneurol.2019.12.016
110. Arya R, Tenney JR, Horn PS, et al. Long-term outcomes of resective epilepsy surgery after invasive presurgical evaluation in children with tuberous sclerosis complex and bilateral multiple lesions. J Neurosurg Pediatr. 2015;15(1):26–33. doi:10.3171/2014.10.PEDS14107
111. Liang S, Zhang J, Yang Z, et al. Long-term outcomes of epilepsy surgery in tuberous sclerosis complex. J Neurol. 2017;264(6):1146–1154. doi:10.1007/s00415-017-8507-y
112. Zaroff CM, Morrison C, Ferraris N, Weiner HL, Miles DK, Devinsky O. Developmental outcome of epilepsy surgery in tuberous sclerosis complex. Epileptic Disord. 2005;7(4):321–326.
113. Bollo RJ, Kalhorn SP, Carlson C, Haegeli V, Devinsky O, Weiner HL. Epilepsy surgery and tuberous sclerosis complex: special considerations. Neurosurg Focus. 2008;25(3):E13. doi:10.3171/FOC/2008/25/9/E13
114. Parain D, Penniello MJ, Berquen P, Delangre T, Billard C, Murphy JV. Vagal nerve stimulation in tuberous sclerosis complex patients. Pediatr Neurol. 2001;25(3):213–216. doi:10.1016/s0887-8994(01)00312-5
115. Tong X, Wang X, Qin L, et al. Vagus nerve stimulation for drug-resistant epilepsy induced by tuberous sclerosis complex. Epilepsy Behav. 2022;126:108431. doi:10.1016/j.yebeh.2021.108431
116. Major P, Thiele EA. Vagus nerve stimulation for intractable epilepsy in tuberous sclerosis complex. Epilepsy Behav. 2008;13(2):357–360. doi:10.1016/j.yebeh.2008.04.001
117. Elliott RE, Carlson C, Kalhorn SP, et al. Refractory epilepsy in tuberous sclerosis: vagus nerve stimulation with or without subsequent resective surgery. Epilepsy Behav. 2009;16(3):454–460. doi:10.1016/j.yebeh.2009.08.018
118. McDermott DS, Mirro EA, Fetrow K, et al. Brain-responsive neurostimulation for the treatment of adults with epilepsy in tuberous sclerosis complex: a case series. Epilepsia Open. 2021;6(2):419–424. doi:10.1002/epi4.12481
119. Zheng H, Chengcheng W, Bin J, et al. Deep brain stimulation of anterior thalamic nucleus for treatment of patient with tuberous sclerosis-related refractory epilepsy. World Neurosurg. 2020;138:141–144. doi:10.1016/j.wneu.2020.03.010
120. Knight EMP, Amin S, Bahi-Buisson N, et al. Safety and efficacy of ganaxolone in patients with CDKL5 deficiency disorder: results from the double-blind phase of a randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2022;21(5):417–427. doi:10.1016/S1474-4422(22)00077-1
121. Koenig MK, Mahalingam R, Peters J, et al. Phase 2 open-label clinical study evaluating oral ganaxolone for the treatment of seizures associated with tuberous sclerosis complex (P12-8.002). Neurology. 2022;98:1.
122. Seese RR, Maske AR, Lynch G, Gall CM. Long-term memory deficits are associated with elevated synaptic ERK1/2 activation and reversed by mGluR5 antagonism in an animal model of autism. Neuropsychopharmacology. 2014;39(7):1664–1673. doi:10.1038/npp.2014.13
123. Silverman JL, Smith DG, Rizzo SJ, et al. Negative allosteric modulation of the mGluR5 receptor reduces repetitive behaviors and rescues social deficits in mouse models of autism. Sci Transl Med. 2012;4(131):131ra51. doi:10.1126/scitranslmed.3003501
124. Tian D, Stoppel LJ, Heynen AJ, et al. Contribution of mGluR5 to pathophysiology in a mouse model of human chromosome 16p11.2 microdeletion. Nat Neurosci. 2015;18(2):182–184. doi:10.1038/nn.3911
125. Mares P, Mikulecká A, Tichá K, Lojková-Janecková D, Kubová H. Metabotropic glutamate receptors as a target for anticonvulsant and anxiolytic action in immature rats. Epilepsia. 2010;51(Suppl 3):24–26. doi:10.1111/j.1528-1167.2010.02604.x
126. Spooren W, Gasparini F. mGlu5 receptor antagonists: a novel class of anxiolytics? Drug News Perspect. 2004;17(4):251–257. doi:10.1358/dnp.2004.17.4.829052
127. Kelly E, Schaeffer SM, Dhamne SC, et al. mGluR5 modulation of behavioral and epileptic phenotypes in a mouse model of tuberous sclerosis complex. Neuropsychopharmacology. 2018;43(6):1457–1465. doi:10.1038/npp.2017.295
128. Potter WB, Basu T, O’Riordan KJ, et al. Reduced juvenile long-term depression in tuberous sclerosis complex is mitigated in adults by compensatory recruitment of mGluR5 and Erk signaling. PLoS Biol. 2013;11(8):e1001627. doi:10.1371/journal.pbio.1001627
129. van der Poest Clement EA, Sahin M, Peters JM. Vigabatrin for epileptic spasms and tonic seizures in tuberous sclerosis complex. J Child Neurol. 2018;33(8):519–524. doi:10.1177/0883073818768309
130. Camposano SE, Major P, Halpern E, Thiele EA. Vigabatrin in the treatment of childhood epilepsy: a retrospective chart review of efficacy and safety profile. Epilepsia. 2008;49(7):1186–1191. doi:10.1111/j.1528-1167.2008.01589.x
131. Schubert-Bast S, Strzelczyk A. Review of the treatment options for epilepsy in tuberous sclerosis complex: towards precision medicine. Ther Adv Neurol Disord. 2021;14:17562864211031100. doi:10.1177/17562864211031100
132. Wang S, Fallah A. Optimal management of seizures associated with tuberous sclerosis complex: current and emerging options. Neuropsychiatr Dis Treat. 2014;10:2021–2030. doi:10.2147/NDT.S51789
133. Peters JM, Sahin M. Tuberous sclerosis complex. In: Cataltepe O, Jallo GI, editors. Pediatric Epilepsy Surgery: Preoperative Assessment and Surgical Treatment.
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.