")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 11
Treatment of adult-onset Still’s disease: a review
Authors Jamilloux Y, Gerfaud-Valentin M, Henry T, Sève P
Received 3 October 2014
Accepted for publication 13 November 2014
Published 22 December 2014 Volume 2015:11 Pages 33—43
DOI https://doi.org/10.2147/TCRM.S64951
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Garry Walsh
Yvan Jamilloux,1–3,* Mathieu Gerfaud-Valentin,1,* Thomas Henry,3 Pascal Sève1
1Department of Internal Medicine, Hôpital de la Croix-Rousse, Université Claude Bernard-Lyon 1, Lyon, France; 2Department of Biochemistry, University of Lausanne, Epalinges, Switzerland; 3International Research Center on Infectiology. INSERM U1111. Université Claude Bernard-Lyon 1, Lyon, France
*These authors contributed equally to this work
Abstract: Adult-onset Still’s disease (AOSD) is a rare inflammatory disorder that has been recently classified as a polygenic autoinflammatory disorder. The former classification, based on the disease course, seems to be quite dated. Indeed, there is accumulating evidence that AOSD can be divided into two distinct phenotypes based on cytokine profile, clinical presentation, and outcome, ie, a “systemic” pattern and an “articular” pattern. The first part of this review deals with the treatments that are currently available for AOSD. We then present the different strategies based on the characteristics of the disease according to clinical presentation. To do so, we focus on the two subsets of the disease. Finally, we discuss the management of life-threatening complications of AOSD, along with the therapeutic options during pregnancy.
Keywords: adult-onset Still’s disease, treatment, anakinra, tocilizumab, canakinumab
Introduction
Adult-onset Still’s disease (AOSD) is a rare inflammatory disorder characterized by the classical triad of daily spiking fever, arthritis, and typical salmon-colored rash. It was first described in 1971 by Bywaters, who defined the disease on the basis of clinical and laboratory resemblance to juvenile Still’s disease.1 Indeed, in 1897, George Frederic Still had described 22 children with what is now called systemic-onset idiopathic juvenile arthritis (JIA).2,3 Whether AOSD and systemic-onset JIA belong to the same continuum of disease is still debated, but the evidence strongly suggests that AOSD and systemic-onset JIA are the same disease.4–6
The epidemiology, diagnostic criteria7,8 (Table 1), and classification of AOSD have been reviewed recently.9,10 The pathophysiology of AOSD remains obscure, and identification of an etiologic trigger is still lacking.
 | Table 1 Criteria for the diagnosis of adult-onset Still’s disease |
Over the last decade, one striking event was the reclassification of AOSD as a polygenic autoinflammatory disorder.11,12 This has mainly been deduced from demonstration of the pivotal role of innate immune pathways, mostly those involved in the processing of two cytokines of the interleukin (IL)-1 family (namely, IL-1β and IL-18). Other cytokines, such as IL-6 and to a lesser extent tumor necrosis factor alpha (TNF-α), are also involved in the pathogenesis of AOSD. Data from genetic and immunologic studies, together with the dramatic effect of biologic treatments, have confirmed the major role of these cytokines. Recently, there has been accumulating evidence that AOSD can be divided into two distinct phenotypes based on cytokine profile, clinical presentation, and outcome.10,13–15 These are discussed in this review.
The renewed comprehension of the disease, along with the availability of new cytokine inhibitors, has led to new therapeutic approaches. The general aim of this review is to discuss the optimal management of AOSD. The first part deals with the treatments that are currently available for AOSD. We then present the different strategies based on characteristics of the disease according to clinical presentation.
Available treatments
Given that the current information on treatment efficacy is obtained from small retrospective case series and not from prospective, double-blind, randomized trials, the treatment of AOSD remains empirical. In contrast, due to a higher prevalence, more data are available for systemic-onset JIA and will be discussed briefly. Recently, the management of AOSD has benefited from proofs of the efficacy of targeted biotherapies.
Nonsteroidal anti-inflammatory drugs and corticosteroids
Regarding available data on AOSD, the risk/benefit ratio is not favorable with regard to nonsteroidal anti-inflammatory drugs (NSAIDs). Indeed, more than 80% of AOSD patients did not achieve remission with NSAIDs and approximately 20% suffered adverse events.16,17 Nevertheless, temporary use of NSAIDs can be considered during diagnostic workup or for early relapse of the disease.17
Corticosteroids remain the first-line treatment for AOSD, regardless of the clinical presentation. Nevertheless, studies of systemic-onset JIA are providing evidence that some biologics should be used earlier in the course of the disease (see section on IL-1 antagonists).18–21 In addition, new treatment plans for systemic-onset JIA have placed methotrexate, anakinra, and tocilizumab as possible first-line treatments.22 Corticosteroids control about 60% of patients and show greater efficacy with regard to systemic symptoms than articular ones.17,23,24 Steroid dependency occurs in approximately 45% of cases. and has been associated with splenomegaly, low glycosylated ferritin, an elevated erythrocyte sedimentation rate, and young age at onset of AOSD.16,25 Thus, early addition of a steroid-sparing agent may be considered in patients who meet these criteria.
Disease-modifying antirheumatic drugs and intravenous immunoglobulin
In the event of failure of corticosteroid treatment or steroid-dependence, disease-modifying anti-rheumatic drugs (DMARDS) can be considered.16,25 Some retrospective case series and case reports have reported the efficacy of several DMARDs, such as cyclosporine A, leflunomide, azathioprine, hydroxychloroquine,23,26 D-penicillamine, and tacrolimus.17,27,28 However, positive results remain exceptional and these agents cannot be recommended unless severe complications occur and other more specific drugs have failed.27
In contrast, methotrexate has proved beneficial and remains the first-line steroid-sparing treatment in AOSD.16,17 As for systemic-onset JIA, targeted biologic therapies (such as anakinra or tocilizumab) are possible alternatives, which could be used for a steroid-sparing effect. Methotrexate can lead to complete remission in up to 70% of patients and corticosteroid weaning has also been reported in some cases.29 Liver enzyme abnormalities do not contraindicate its prescription but require close biological monitoring.
Data concerning intravenous immunoglobulin are more controversial, with two randomized open-label trials showing some efficacy when used early in the course of AOSD,30,31 whereas retrospective data do not support the efficacy of intravenous immunoglobulin in AOSD.16,25 Overall, intravenous immunoglobulin has only a suspensive effect but is well tolerated. Thus, it should be considered in the event of reactive hemophagocytic lymphohistiocytosis (RHL; formerly known as macrophage activation syndrome),32 in some life-threatening complications, or in case of AOSD flare-up occurring during pregnancy.33,34 The usual dosage is 2 g/kg body weight administered in 2–5 days.
Targeted biologic therapies
Although dramatic responses have been reported in systemic-onset JIA when given as first-line treatment, targeted biologic agents are actually reserved for refractory AOSD.19,21,22,35 Resistance to first-line corticosteroids and second-line DMARDs defines refractory AOSD, which mostly includes the polycyclic or chronic courses of the disease. Data concerning biologic agents used in AOSD are summarized in Table 2.
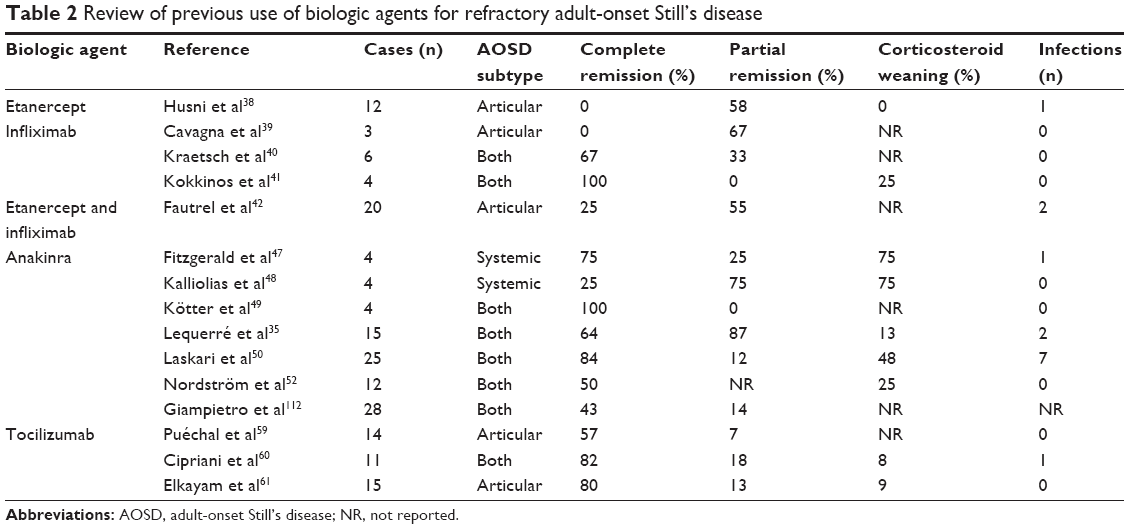 | Table 2 Review of previous use of biologic agents for refractory adult-onset Still’s disease |
Anti-TNF-α
Historically, anti-TNF therapy was the first biologic therapy that was used to treat refractory AOSD. The administration schemes were based on those used in the treatment of rheumatoid arthritis. The three anti-TNF-α agents (ie, infliximab, etanercept, and adalimumab) have been used to treat refractory AOSD but data on adalimumab are limited to a few cases.36 Data from case reports, retrospective case series, and one prospective open-label prospective trial are available. Complete remissions have been observed37–41 but anti-TNF therapy was rather disappointing, mostly for systemic symptoms.10,42 Switching from one molecule to another had no additional effect.16,43 Moreover, two patients who were started on etanercept and adalimumab developed RHL that could be linked with initiation of the treatment.44,45 Overall, TNF-α blockers seem to be of interest only for the treatment of chronic polyarticular disease.16,17,23,25 In that case, based on our experience and on analysis of the literature, infliximab should be the preferred molecule because of better efficacy. However, no prospective evidence sustains this preferred option, and each case should be carefully evaluated by AOSD experts before therapeutic changes.
IL-1 antagonists
Three IL-1 antagonists are presently available, ie, a recombinant antagonist of the IL-1 receptor (IL-1Ra, anakinra), a human monoclonal antibody directed against IL-1β (canakinumab), and a soluble IL-1 trap fusion protein (rilonacept). Anakinra has been more frequently reported in the treatment of AOSD.35,46–52
The efficacy of anakinra has been reported in retrospective case series35,46–51 and in one prospective, randomized, open-label trial.52 Anakinra is particularly efficient in the rapid relief of systemic symptoms. Its effect on articular symptoms is less frequently reported. In almost all cases, inflammatory markers reverted to normal within about 2 weeks and corticosteroids could be tapered and discontinued. However, the effect of anakinra was suspensive, and relapses occurred frequently as soon as the treatment was stopped. In some cases, progressive dose reduction has enabled the weaning of anakinra. In the event of an insufficient response to anakinra, rilonacept and canakinumab can be considered because they have longer half-lives and can be administered every week or every 8 weeks, respectively. Indeed, both agents have been reported to be efficient in AOSD.53–56
More data are available in the setting of systemic-onset JIA.19,20,55–58 Swart et al reviewed the literature and reported on 140 children with systemic-onset JIA treated with anakinra.57 Systemic symptoms resolved in 98% of the patients who had such symptoms. Fatigue and well-being improved in 93% of cases. Arthritis improved in 66% of the cases within a longer time (few months).
Complete disease remission was mostly observed in patients with systemic symptoms, less arthritis, and a shorter duration of disease. Adverse events were most frequently pain and erythema at the injection site. More serious adverse events were rare (occurring in eight cases). Quartier et al confirmed the efficacy of anakinra in systemic-onset JIA by performing a multicenter, randomized, placebo-controlled clinical trial.58 The study included 24 patients with a systemic-onset JIA duration of more than 6 months and steroid dependency. After one month, anakinra had been effective in 8/12 patients (versus one in the placebo group) who reached the modified American College of Rheumatology Pediatric 30 score. At month 2, 9/10 patients in the placebo group who had been switched to anakinra were also responders. However, during the following open-label treatment period, a loss of response was observed over time and no patient achieved inactive disease.
Nigrovic et al reported on 46 patients who received anakinra as first-line treatment.19 Rapid resolution of systemic symptoms was observed in more than 95% of cases, along with an additional preventive effect on refractory arthritis in almost 90% of the patients. Notably, 60% of the patients achieved inactive disease. Based on these results, Nigrovic postulated that there could be a “window of opportunity” for IL-1 blockade if given at the very beginning of the disease (ie, within 6 months after onset).18 In Nigrovic’s model, systemic-onset JIA/AOSD would have a biphasic pattern. The first phase, being febrile and inflammatory, involves innate immunity, and evolves to an afebrile phase with chronic destructive arthritis involving adaptive immunity. This hypothesis has been somewhat reinforced by the results reported by Vastert et al in 2014.20 Indeed, they reported on a prospective series of 20 patients who received anakinra as first-line therapy. Eighty-five percent of the patients showed an American College of Rheumatology Pediatric 90 score response or had inactive disease within 3 months. Overall, 75% of the patients treated with anakinra alone achieved inactive disease. Although it is difficult to evaluate to what extent children with new-onset systemic-onset JIA enrolled in that study would have underwent monocyclic course (ie disease that never relapses), these dramatic results clearly indicate that IL-1 blockade has an early place in the treatment strategy.
Tocilizumab
Tocilizumab is a humanized monoclonal antibody directed against the IL-6 receptor that has shown positive results in patients with refractory AOSD from several case series.59–62 Results from randomized placebo-controlled trials are available in systemic-onset JIA but not in AOSD.63,64 Generally, the patients responded rapidly and experienced a sustained clinical remission over time. Moreover, the effect of tocilizumab persisted for ≥6 months after its discontinuation. Tocilizumab seems to have a similar beneficial effect on the systemic and articular features of AOSD, has a marked corticosteroid-sparing effect, and has a good safety and tolerance profile.59–61,63 Indeed, in a recent retrospective series, only two cases among 34 tocilizumab-treated patients with AOSD had to withdraw from treatment due to severe infections.62 Other side effects were mild leukopenia or neutropenia, elevation of hepatic enzymes, hypercholesterolemia, and headache associated with tocilizumab infusion.
Optimal management of AOSD
Since the management of AOSD varies widely depending on the various clinical presentations of the disease, the optimal management is discussed in the second part of this review. A management algorithm is provided in Figure 1.
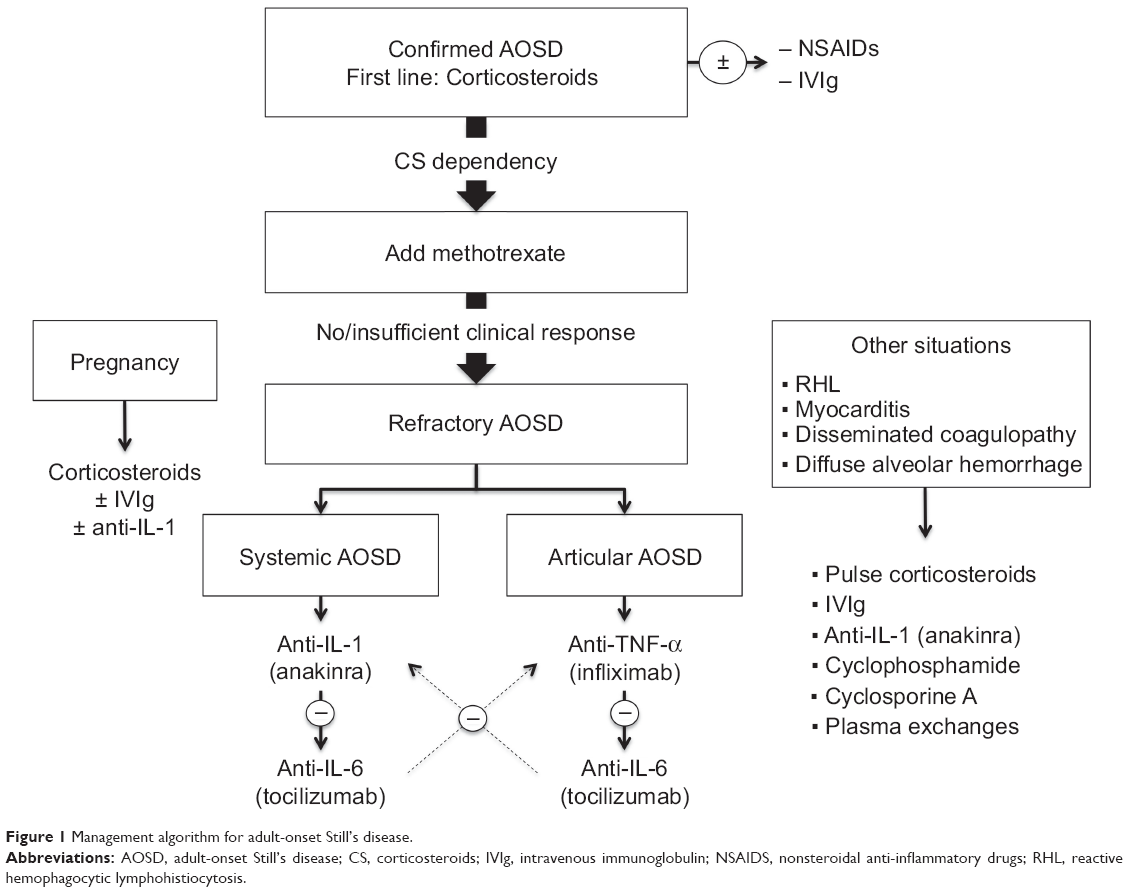 | Figure 1 Management algorithm for adult-onset Still’s disease. |
Nonrefractory AOSD
According to the abovementioned definition, nonrefractory AOSD corresponds to disease patterns that can be cured by using first-line corticosteroids ± methotrexate. This mostly includes the monocyclic and polycyclic courses of the disease, when corticosteroid dependency can be overcome with a first-line steroid-sparing agent. NSAIDs can be used during the diagnostic workup, in case of a monocyclic course of AOSD without major systemic or articular involvement or in case of isolated mild articular involvement. In such cases, the preferred NSAID is high-dose indomethacin (150–200 mg/day).26,65,66
Corticosteroids should be started promptly once the diagnosis is confirmed. Usually, corticosteroid therapy starts at a dosage of 0.5–1 mg/kg/day, but an intravenous pulse of high-dose methylprednisolone can be used, particularly where there is severe visceral involvement.67,68 Higher dosages seem to be more efficient in controlling the disease, achieving remission earlier, and decreasing the number of relapses.10 Fractionated daily intakes of corticosteroids have also provided interesting results in insufficiently controlled patients.26,69 The response to corticosteroids should be obtained within hours or days.70 Usually, the tapering of corticosteroids can start after 4–6 weeks, when symptoms have resolved and biological parameters have returned to normal.
If patients show early signs of corticosteroid dependency or have the previously mentioned risk factors (splenomegaly, low glycosylated ferritin, elevated erythrocyte sedimentation rate, or young age at AOSD onset16,25), methotrexate could be considered early for its steroid-sparing effect. Usually, methotrexate (7.5–20 mg/week) enables complete remission of the disease (70%) or at least a significant reduction of daily corticosteroid intake.29 Methotrexate has a similar effect on systemic and arthritic AOSD.17,29 Blood count, renal function, and liver enzymes should be monitored before initiation of methotrexate and then every 1–2 months.71 If methotrexate fails to control the disease, other DMARDs could be considered. However, growing evidence suggests using targeted biologic therapies as early second-line treatment.
Refractory AOSD
It is becoming increasingly apparent that AOSD patients fall into two distinct subsets, ie, those presenting with systemic features and those presenting with prominent arthritis.10,13–15 These findings are supported by molecular evidence, cytokine profiles, clinical course, and response to treatment (Figure 2). In AOSD, such as in systemic-onset JIA, it remains unclear whether the two subsets of the disease are temporally related (biphasic disease)18 or present since the disease onset.
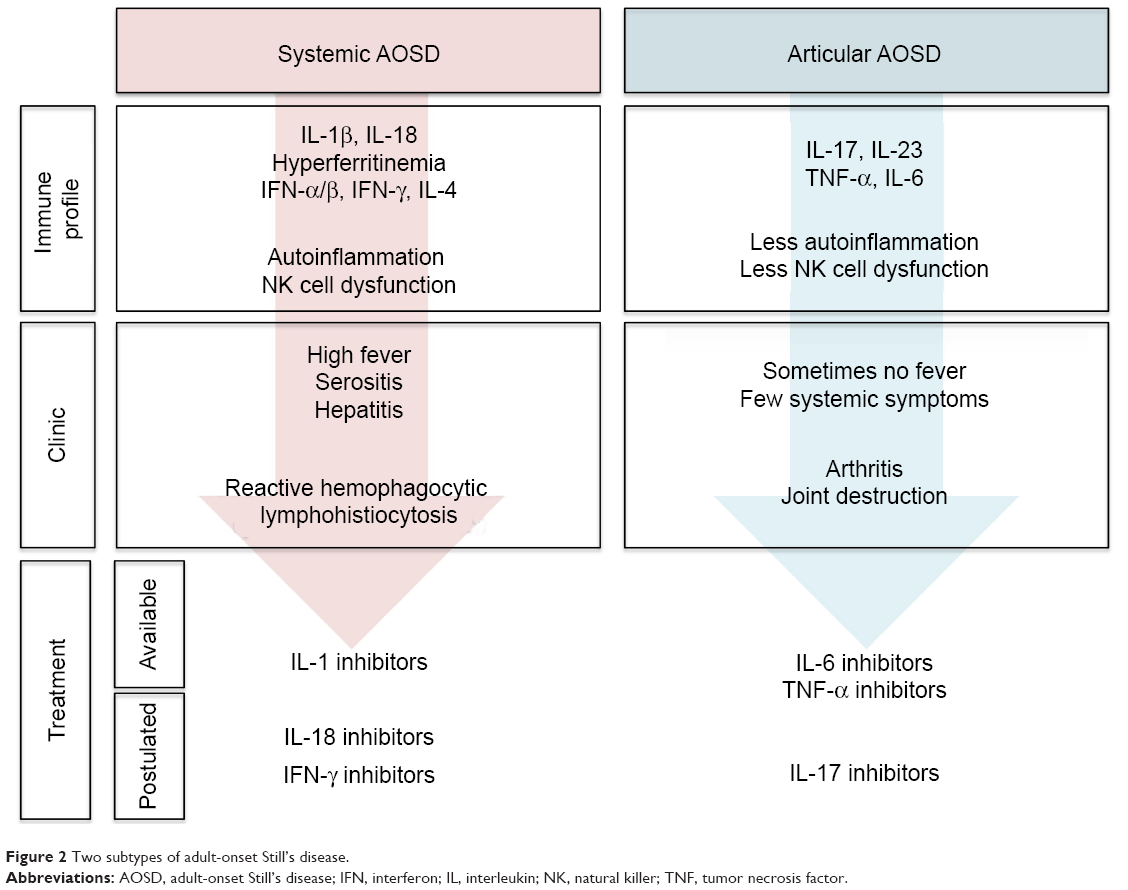 | Figure 2 Two subtypes of adult-onset Still’s disease. |
Predictive factors for a prominent articular pattern include female sex, proximal arthritis at disease onset, thrombocytosis, and corticosteroid dependency, whereas high fever, high levels of liver enzymes, or high acute phase reactants are more likely to be associated with a systemic pattern of AOSD.72–74 Other clues to identify the systemic subtype of AOSD are the following: thrombocytopenia, RHL, and hyperferritinemia. Although the cytokine dosage is not performed in routine care, IL-18, interferon-γ, IL-10, and IL-4 are associated with systemic AOSD whereas IL-6, IL-17, and IL-23 are associated with arthritic AOSD.15,74,75 This dichotomy may be of the utmost importance for the management of AOSD patients, as patients falling into one of the two categories should benefit from different treatments.10,13
Patients with systemic AOSD are more likely to be responders to first-line corticosteroid therapy. In the case of refractory systemic AOSD, IL-1 antagonists (mostly anakinra) should be considered first as they have proved to be dramatically more efficient for systemic symptoms than for articular features.16,17,35,76 Anakinra is used at 100 mg/day via subcutaneous injection. Once the disease is controlled and the biological parameters have returned to normal, subcutaneous injections can be spaced. The most commonly reported adverse event with anakinra has been self-limiting erythema at the injection site. Unlike anakinra, anti-TNF-α agents usually have a less sustained effect on systemic symptoms.
Tocilizumab should be considered as an alternative to IL-1 antagonists, particularly when articular signs such as joint erosion accompany systemic symptoms.10,13,77 Nevertheless, tocilizumab has been less widely used in treating AOSD and should actually be used only if anti-IL-1 treatments fail to control the disease or if relapses occur during weaning of anti-IL-1. Tocilizumab is given at a dosage of 5–8 mg/kg body weight every 2–4 weeks. Nevertheless, larger randomized studies are still needed to further determine the optimal therapeutic scheme for tocilizumab.
In patients with articular refractory AOSD, anti-TNF-α should be the preferred treatment.10,13,77,78 Infliximab, given with a therapeutic scheme similar to that used for rheumatoid arthritis (ie, 3–5 mg/kg at weeks 0, 2, and 6, and then once every 8 weeks), seems to have better efficacy than etanercept, but larger studies are required to compare these two molecules. Since available data on the efficacy of adalimumab in AOSD are lacking, it cannot be recommended so far. In the case of failure of anti-TNF-α, tocilizumab should be considered first as it has proven efficient for both the articular and systemic features of AOSD.10,13 If tocilizumab fails, anakinra may be tried.
Finally, it should be noted that, in an effort to standardize therapeutic management and evaluate comparative effectiveness in an observational setting, the Childhood Arthritis and Rheumatology Research Alliance has developed four consensus treatment plans for systemic-onset JIA.22 These plans include a glucocorticoid plan, a methotrexate plan, an anakinra plan, and a tocilizumab plan. As no guidelines are available for AOSD, this consensus plan should be of help for physicians dealing with new-onset AOSD.
Life-threatening complications of AOSD
Reactive hemophagocytic lymphohistiocytosis
RHL corresponds to an uncontrollable activation of the reticuloendothelial system leading to phagocytosis of hematopoietic cells by activated tissue macrophages. In AOSD, RHL has been reported to have an incidence of 12%–17%.79–81 Evidence for occult RHL in a substantial proportion of patients with AOSD (up to 50%) supports the possibility that RHL and AOSD could represent the same disease within a continuum of severity.82,83 Considering all the underlying causes, the mortality rate for RHL ranges between 10% and 22%.84–86 The clinical picture and diagnostic criteria are presented in Table 3.81 Although RHL features resemble those of AOSD, differences exist and may help the early recognition of RHL during AOSD:83 the fever pattern is mostly non-remitting in RHL; there are less central nervous system involvement and hepatosplenomegaly during AOSD flare-ups than during RHL; and neutrophil and platelet counts are elevated in AOSD whereas they are low in RHL. Notably, a rapid decrease in leukocyte count or the rapid appearance of an hypertriglyceridemia may alert the physician on the onset of RHL complicating previously diagnosed AOSD. Hypofibrinogenemia is also one of the most important clues indicating RHL, since AOSD patients are more likely to have hyperfibrinogenemia due to the underlying inflammatory state. Hyperferritinemia is difficult to interpret in an AOSD patient because an acute increase in total and glycosylated serum ferritin can indicate a flare-up of AOSD or RHL. Bone marrow aspiration is the gold standard for diagnosis of RHL. It is not to be performed systematically but can be required in atypical cases and when there is a diagnostic dilemma.
 | Table 3 Diagnostic criteria for hemophagocytic lymphohistiocytosis |
Treatment of RHL in AOSD patients has mostly been empirical and has consisted of intravenous pulsed corticosteroids or intravenous immunoglobulin.83 Several immunosuppressive drugs have also been used, including cyclosporine A, mycophenolate mofetil, or etoposide.83,87 More recently, several authors have reported on the successful management of RHL complicating adult or pediatric Still’s disease using IL-1 antagonists.88–91 It is possible that higher doses of anakinra are required to treat Still’s disease-associated RHL (ie, 100 mg/6 hours).91 On the contrary, anti-TNF-α has proven ineffective or even harmful in treating RHL. Yet, several cases of RHL associated with etanercept have been reported in patients with AOSD.88,92 Finally, tocilizumab has been reported to be effective in the management of AOSD-related RHL, but these preliminary results still need to be confirmed.93
Disseminated intravascular coagulopathy
Disseminated intravascular coagulopathy (DIC) is characterized by an unregulated overactivation of the coagulation system. Fewer than ten cases have been reported in the setting of AOSD.83 However, the diagnosis can be challenging since the clinical picture may mimic sepsis and DIC may complicate AOSD-related RHL.94–96 The reported efficient treatments of AOSD-associated DIC include prednisolone,97 cyclosporine A,96,98 anakinra,49 and tocilizumab.99 So far, since only few data are available, it is not possible to recommend one or another molecule. Nevertheless, taking into account the morbidity of DIC, it seems reasonable to propose early therapy with anti-IL-1 or anti-IL-6 to AOSD patients who do not respond rapidly to corticosteroids.96,98
Myocarditis
We recently described four previously unreported cases of AOSD-related myocarditis and reviewed 20 other cases from the literature (Gerfaud-Valentin et al Medicine100). Myocarditis was an early life-threatening complication of AOSD occurring within the first year after the disease onset and affecting younger patients, mostly males. All the patients were given high-dose corticosteroids (either intravenous pulses or 1 mg/kg body weight). Intravenous immunoglobulins were added in 6/24 patients, methotrexate in 5/24, and anti-TNF-α in 3/24. In addition, one patient received cyclophosphamide and another received anakinra (100 mg/day). All but one had a favorable outcome, giving a fatality rate of about 4%. Thus, AOSD-associated myocarditis can be life-threatening but has a good prognosis when recognized early and efficiently treated. High-dose corticosteroids are the mainstay of treatment. Whether the other treatments are useful cannot be determined on the basis of this retrospective study, but in our experience, intravenous immunoglobulin seems to be useful and methotrexate steroid-sparing.
Thrombotic thrombocytopenia purpura
About ten cases of AOSD-related thrombotic thrombocytopenic purpura (TTP) have been reported.83 The pathophysiologic link between TTP and AOSD remains unknown. The clinical sign that should alert the clinician is acute visual loss. Early detection of TTP is required because it is associated with high morbidity and mortality.101 Delayed treatment of TTP may lead to renal failure, brain edema, and death.102,103 Thrombocytopenia and unexplained microangiopathic hemolytic anemia are arguments for a diagnosis of TTP during AOSD.104 Plasma exchange is always required in the event of AOSD-associated TTP. Additional drugs have been used with success, such as corticosteroids, intravenous immunoglobulin, azathioprine, cyclophosphamide, or rituximab.101,103 Only one author has reported on successful treatment of AOSD-related TTP using tocilizumab, which seemed to be efficient in treating both TTP and the underlying refractory AOSD.105
Diffuse alveolar hemorrhage
Diffuse alveolar hemorrhage is a rare life-threatening complication of AOSD characterized by accumulation of erythrocytes in the lung alveolar spaces. Whether diffuse alveolar hemorrhage is a specific complication of AOSD or is coincidental remains unclear. Clinicians should think about diffuse alveolar hemorrhage when AOSD patients present with hemoptysis, cough, and dyspnea progressing to respiratory distress, together with falling hematocrit and hemorrhagic bronchoalveolar lavage fluid. Therapeutic options include an intravenous pulse of high-dose methylprednisolone (1 g/day for 5 days), plasma exchange, or intravenous cyclophosphamide.106,107
AOSD and pregnancy
We recently reviewed the link between AOSD and pregnancy.34 In the subset of women without previously known AOSD, the disease occurred mainly during the first or second trimester. Due to flares, some cases were complicated by oligohydramnios or prematurity. No life-threatening complications were reported. In the second subset of women with known AOSD, flares were less frequent and more likely to occur during the second trimester and the postpartum period. Of note, data concerning preventive treatment are controversial and do not allow guidelines or recommendations.34,108 Treatment of AOSD flares during pregnancy always included prednisone at a dosage of 0.5–1 mg/kg body weight. The safety of corticosteroids has been established for usual dosages, but their effect at higher dosages remains unclear.109 In our previous article, it was not possible to state whether obstetric complications were due to the treatment or to the underlying disease.34 Thus, multidisciplinary expertise is required when corticosteroids are required. In some cases, intravenous immunoglobulins has been added but their effect on the outcome cannot be clearly determined.33,34 Although NSAIDs have been used, they are not recommended during pregnancy, particularly after week 24. Anakinra was used during three pregnancies in patients with AOSD. Both the outcome of the AOSD and the pregnancies were positive. The disease was controlled in all three cases and the children were born at term and healthy.110,111 Finally, close multidisciplinary monitoring before, during, and after pregnancy is required for women with AOSD.
Conclusion
Adult-onset Still’s disease is a complex disease with a polymorphic clinical presentation. In some cases, AOSD is as simple as a unique flare easily cured by NSAIDs or short-course corticosteroid therapy. On another hand, AOSD can present with stormy systemic features and lead to life-threatening complications (such as RHL) or as a chronic articular disease that may be either indolent or destructive. Recent advances in our understanding of the pathophysiology of AOSD and the availability of anti-cytokine-targeted treatments have given rise to more personalized treatments. In the near future, understanding of AOSD will probably benefit further from wide genetic analyses. Targeted biologic therapies seem to have a dramatic effect when given as first-line treatment in systemic-onset JIA. Results from ongoing observational studies and future prospective trials may lead to the recommendation of earlier use of these treatments in AOSD. Finally, the management of AOSD will also benefit from the release of new targeted biotherapies, such as anti-IL-18 or anti-IL-17.
Acknowledgments
YJ acknowledges the Foundation for the Development of Internal Medicine in Europe, the Société Nationale Française de Médecine Interne, Groupama Fundation, and Genzyme for their help in funding his PhD research project. YJ is supported by a “poste d’accueil” at INSERM.
Disclosure
The authors report no conflicts of interest in this work.
References
Bywaters EG. Still’s disease in the adult. Ann Rheum Dis. 1971;30(2):121–133. | ||
Still GF. On a form of chronic joint disease in children. Med Chir Trans. 1897;80:47–60.9. | ||
Efthimiou P, Paik PK, Bielory L. Diagnosis and management of adult onset Still’s disease. Ann Rheum Dis. 2006;65(5):564–572. | ||
Tanaka S, Matsumoto Y, Ohnishi H, et al. [Comparison of clinical features of childhood and adult onset Still’s disease]. Ryūmachi Rheum. 1991;31(5):511–518. Japanese. | ||
Uppal SS, Pande IR, Kumar A, et al. Adult onset Still’s disease in northern India: comparison with juvenile onset Still’s disease. Br J Rheumatol. 1995;34(5):429–434. | ||
Luthi F, Zufferey P, Hofer MF, So AK. “Adolescent-onset Still’s disease”: characteristics and outcome in comparison with adult-onset Still’s disease. Clin Exp Rheumatol. 2002;20(3):427–430. | ||
Yamaguchi M, Ohta A, Tsunematsu T, et al. Preliminary criteria for classification of adult Still’s disease. J Rheumatol. 1992;19(3):424–430. | ||
Fautrel B, Zing E, Golmard J-L, et al. Proposal for a new set of classification criteria for adult-onset Still disease. Medicine. 2002;81(3):194–200. | ||
Mahroum N, Mahagna H, Amital H. Diagnosis and classification of adult Still’s disease. J Autoimmun. 2014;48–49:34–37. | ||
Gerfaud-Valentin M, Jamilloux Y, Iwaz J, Sève P. Adult-onset Still’s disease. Autoimmun Rev. 2014;13(7):708–722. | ||
McGonagle D, McDermott MF. A proposed classification of the immunological diseases. PLoS Med. 2006;3(8):e297. | ||
Kastner DL, Aksentijevich I, Goldbach-Mansky R. Autoinflammatory disease reloaded: a clinical perspective. Cell. 2010;140(6):784–790. | ||
Maria AT, Le Quellec A, Jorgensen C, Touitou I, Rivière S, Guilpain P. Adult onset Still’s disease (AOSD) in the era of biologic therapies: dichotomous view for cytokine and clinical expressions. Autoimmun Rev. August 27, 2014. [Epub ahead of print]. | ||
Canna SW. Editorial. Interferon-γ: friend or foe in systemic juvenile idiopathic arthritis and adult Still’s disease. Arthritis Rheumatol. 2014; 66(5):1072–1076. | ||
Shimizu M, Nakagishi Y, Yachie A. Distinct subsets of patients with systemic juvenile idiopathic arthritis based on their cytokine profiles. Cytokine. 2013;61(2):345–348. | ||
Gerfaud-Valentin M, Maucort-Boulch D, Hot A, et al. Adult-onset Still disease: manifestations, treatments, outcome, and prognostic factors in 57 patients. Medicine (Baltimore). 2014;93(2):91–99. | ||
Franchini S, Dagna L, Salvo F, Aiello P, Baldissera E, Sabbadini MG. Efficacy of traditional and biologic agents in different clinical phenotypes of adult-onset Still’s disease. Arthritis Rheum. 2010;62(8):2530–2535. | ||
Nigrovic PA. Review: is there a window of opportunity for treatment of systemic juvenile idiopathic arthritis? Arthritis Rheumatol. 2014; 66(6):1405–1413. | ||
Nigrovic PA, Mannion M, Prince FH, et al. Anakinra as first-line disease-modifying therapy in systemic juvenile idiopathic arthritis: report of forty-six patients from an international multicenter series. Arthritis Rheum. 2011;63(2):545–555. | ||
Vastert SJ, de Jager W, Noordman BJ, et al. Effectiveness of first-line treatment with recombinant interleukin-1 receptor antagonist in steroid-naive patients with new-onset systemic juvenile idiopathic arthritis: results of a prospective cohort study. Arthritis Rheumatol. 2014;66(4):1034–1043. | ||
Moulis G, Sailler L, Astudillo L, Pugnet G, Arlet P. May anakinra be used earlier in adult onset Still disease? Clin Rheumatol. 2010; 29(10):1199–1200. | ||
DeWitt EM, Kimura Y, Beukelman T, et al. Consensus treatment plans for new-onset systemic juvenile idiopathic arthritis. Arthritis Care Res. 2012;64(7):1001–1010. | ||
Pay S, Türkçapar N, Kalyoncu M, et al. A multicenter study of patients with adult-onset Still’s disease compared with systemic juvenile idiopathic arthritis. Clin Rheumatol. 2006;25(5):639–644. | ||
Nagashima T, Iwamoto M, Matsumoto K, Minota S. Interleukin-18 in adult-onset Still’s disease: treatment target or disease activity indicator? Intern Med. 2012;51(4):449. | ||
Kim H-A, Sung J-M, Suh C-H. Therapeutic responses and prognosis in adult-onset Still’s disease. Rheumatol Int. 2012;32(5):1291–1298. | ||
Pouchot J, Sampalis JS, Beaudet F, et al. Adult Still’s disease: manifestations, disease course, and outcome in 62 patients. Medicine (Baltimore). 1991;70(2):118–136. | ||
Mitamura M, Tada Y, Koarada S, et al. Cyclosporin A treatment for Japanese patients with severe adult-onset Still’s disease. Mod Rheumatol. 2009;19(1):57–63. | ||
Nakamura H, Odani T, Shimizu Y, Takeda T, Kikuchi H. Usefulness of tacrolimus for refractory adult-onset Still’s disease: report of six cases. Mod Rheumatol. 2014;18:1–5. | ||
Fautrel B, Borget C, Rozenberg S, et al. Corticosteroid sparing effect of low dose methotrexate treatment in adult Still’s disease. J Rheumatol. 1999;26(2):373–378. | ||
Vignes S, Wechsler B, Amoura Z, et al. Intravenous immunoglobulin in adult Still’s disease refractory to non-steroidal anti-inflammatory drugs. Clin Exp Rheumatol. 1998;16(3):295–298. | ||
Permal S, Wechsler B, Cabane J, Perrot S, Blum L, Imbert JC. [Treatment of Still’s disease in adults with intravenous immunoglobulins]. Rev Med Interne. 1995;16(4):250–254. French. | ||
Emmenegger U, Frey U, Reimers A, et al. Hyperferritinemia as indicator for intravenous immunoglobulin treatment in reactive macrophage activation syndromes. Am J Hematol. 2001;68(1):4–10. | ||
Liozon E, Ly K, Aubard Y, Vidal E. Intravenous immunoglobulins for adult Still’s disease and pregnancy. Rheumatology. 1999;38(10):1024–1025. | ||
Gerfaud-Valentin M, Hot A, Huissoud C, Durieu I, Broussolle C, Seve P. Adult-onset Still’s disease and pregnancy: about ten cases and review of the literature. Rheumatol Int. 2014;34(6):867–871. | ||
Lequerré T, Quartier P, Rosellini D, et al. Interleukin-1 receptor antagonist (anakinra) treatment in patients with systemic-onset juvenile idiopathic arthritis or adult onset Still disease: preliminary experience in France. Ann Rheum Dis. 2008;67(3):302–308. | ||
Benucci M, Li GF, Del Rosso A, Manfredi M. Adalimumab (anti-TNF-alpha) therapy to improve the clinical course of adult-onset Still’s disease: the first case report. Clin Exp Rheumatol. 2005;23(5):733. | ||
Felson DT, Anderson JJ, Boers M, et al. American College of Rheumatology. Preliminary definition of improvement in rheumatoid arthritis. Arthritis Rheum. 1995;38(6):727–735. | ||
Husni ME, Maier AL, Mease PJ, et al. Etanercept in the treatment of adult patients with Still’s disease. Arthritis Rheum. 2002;46(5):1171–1176. | ||
Cavagna L, Caporali R, Epis O, Bobbio-Pallavicini F, Montecucco C. Infliximab in the treatment of adult Still’s disease refractory to conventional therapy. Clin Exp Rheumatol. 2001;19(3):329–332. | ||
Kraetsch HG, Antoni C, Kalden JR, Manger B. Successful treatment of a small cohort of patients with adult onset of Still’s disease with infliximab: first experiences. Ann Rheum Dis. 2001;60 Suppl 3:iii55–iii57. | ||
Kokkinos A, Iliopoulos A, Greka P, Efthymiou A, Katsilambros N, Sfikakis PP. Successful treatment of refractory adult-onset Still’s disease with infliximab. A prospective, non-comparative series of four patients. Clin Rheumatol. 2004;23(1):45–49. | ||
Fautrel B, Sibilia J, Mariette X, Combe B, Club Rhumatismes et Inflammation. Tumour necrosis factor alpha blocking agents in refractory adult Still’s disease: an observational study of 20 cases. Ann Rheum Dis. 2005;64(2):262–266. | ||
Aikawa NE, Ribeiro AC, Saad CG, et al. Is anti-TNF switching in refractory Still’s disease safe and effective? Clin Rheumatol. 2011; 30(8):1129–1134. | ||
Kaneko K, Kaburaki M, Muraoka S, et al. Exacerbation of adult-onset Still’s disease, possibly related to elevation of serum tumor necrosis factor-alpha after etanercept administration. Int J Rheum Dis. 2010; 13(4):e67–e69. | ||
Agarwal S, Moodley J, Ajani Goel G, Theil KS, Mahmood SS, Lang RS. A rare trigger for macrophage activation syndrome. Rheumatol Int. 2011; 31(3):405–407. | ||
Vasques Godinho FM, Parreira Santos MJ, Canas da Silva J. Refractory adult onset Still’s disease successfully treated with anakinra. Ann Rheum Dis. 2005;64(4):647–648. | ||
Fitzgerald AA, Leclercq SA, Yan A, Homik JE, Dinarello CA. Rapid responses to anakinra in patients with refractory adult-onset Still’s disease. Arthritis Rheum. 2005;52(6):1794–1803. | ||
Kalliolias GD, Georgiou PE, Antonopoulos IA, Andonopoulos AP, Liossis SN. Anakinra treatment in patients with adult-onset Still’s disease is fast, effective, safe and steroid sparing: experience from an uncontrolled trial. Ann Rheum Dis. 2007;66(6):842–843. | ||
Kötter I, Wacker A, Koch S, et al. Anakinra in patients with treatment-resistant adult-onset Still’s disease: four case reports with serial cytokine measurements and a review of the literature. Semin Arthritis Rheum. 2007;37(3):189–197. | ||
Laskari K, Tzioufas AG, Moutsopoulos HM. Efficacy and long-term follow-up of IL-1R inhibitor anakinra in adults with Still’s disease: a case-series study. Arthritis Res Ther. 2011;13(3):R91. | ||
Rudinskaya A, Trock DH. Successful treatment of a patient with refractory adult-onset Still disease with anakinra. J Clin Rheumatol. 2003; 9(5):330–332. | ||
Nordström D, Knight A, Luukkainen R, et al. Beneficial effect of interleukin 1 inhibition with anakinra in adult-onset Still’s disease. An open, randomized, multicenter study. J Rheumatol. 2012;39(10):2008–2011. | ||
Lo Gullo A, Caruso A, Pipitone N, Macchioni P, Pazzola G, Salvarani C. Canakinumab in a case of adult onset Still’s disease: efficacy only on systemic manifestations. Joint Bone Spine. 2014;81(4):376–377. | ||
Kontzias A, Efthimiou P. The use of canakinumab, a novel IL-1β long-acting inhibitor, in refractory adult-onset Still’s disease. Semin Arthritis Rheum. 2012;42(2):201–205. | ||
Ruperto N, Quartier P, Wulffraat N, et al. A phase II, multicenter, open-label study evaluating dosing and preliminary safety and efficacy of canakinumab in systemic juvenile idiopathic arthritis with active systemic features. Arthritis Rheum. 2012;64(2):557–567. | ||
Ruperto N, Brunner HI, Quartier P, et al. Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis. N Engl J Med. 2012;367(25):2396–2406. | ||
Swart JF, Barug D, Möhlmann M, Wulffraat NM. The efficacy and safety of interleukin-1-receptor antagonist anakinra in the treatment of systemic juvenile idiopathic arthritis. Expert Opin Biol Ther. 2010;10(12):1743–1752. | ||
Quartier P, Allantaz F, Cimaz R, et al. A multicentre, randomised, double-blind, placebo-controlled trial with the interleukin-1 receptor antagonist anakinra in patients with systemic-onset juvenile idiopathic arthritis (ANAJIS trial). Ann Rheum Dis. 2011;70(5):747–754. | ||
Puéchal X, DeBandt M, Berthelot J-M, et al. Tocilizumab in refractory adult Still’s disease. Arthritis Care Res. 2011;63(1):155–159. | ||
Cipriani P, Ruscitti P, Carubbi F, et al. Tocilizumab for the treatment of adult-onset Still’s disease: results from a case series. Clin Rheumatol. 2014;33(1):49–55. | ||
Elkayam O, Jiries N, Dranitzki Z, et al. Tocilizumab in adult-onset Still’s disease: the Israeli experience. J Rheumatol. 2014;41(2):244–247. | ||
Ortiz-Sanjuán F, Blanco R, Calvo-Rio V, et al. Efficacy of tocilizumab in conventional treatment-refractory adult-onset Still’s disease: multicenter retrospective open-label study of thirty-four patients. Arthritis Rheumatol. 2014;66(6):1659–1665. | ||
De Benedetti F, Brunner HI, Ruperto N, et al. Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis. N Engl J Med. 2012;367(25):2385–2395. | ||
Yokota S, Imagawa T, Mori M, et al. Efficacy and safety of tocilizumab in patients with systemic-onset juvenile idiopathic arthritis: a randomised, double-blind, placebo-controlled, withdrawal phase III trial. Lancet. 2008;371(9617):998–1006. | ||
Cush JJ, Medsger TA Jr, Christy WC, Herbert DC, Cooperstein LA. Adult-onset Still’s disease. Clinical course and outcome. Arthritis Rheum. 1987;30(2):186–194. | ||
Wouters JM, van de Putte LB. Adult-onset Still’s disease; clinical and laboratory features, treatment and progress of 45 cases. Q J Med. 1986;61(235):1055–1065. | ||
Kong X-D, Xu D, Zhang W, Zhao Y, Zeng X, Zhang F. Clinical features and prognosis in adult-onset Still’s disease: a study of 104 cases. Clin Rheumatol. 2010;29(9):1015–1019. | ||
Kim YJ, Koo BS, Kim Y-G, Lee C-K, Yoo B. Clinical features and prognosis in 82 patients with adult-onset Still’s disease. Clin Exp Rheumatol. 2014;32(1):28–33. | ||
Koizumi R, Tsukada Y, Ideura H, Ueki K, Maezawa A, Nojima Y. Treatment of adult Still’s disease with dexamethasone, an alternative to prednisolone. Scand J Rheumatol. 2000;29(6):396–398. | ||
Hot A, Toh M-L, Coppéré B, et al. Reactive hemophagocytic syndrome in adult-onset Still disease: clinical features and long-term outcome: a case-control study of 8 patients. Medicine (Baltimore). 2010;89(1):37–46. | ||
Pavy S, Constantin A, Pham T, et al. Methotrexate therapy for rheumatoid arthritis: clinical practice guidelines based on published evidence and expert opinion. Joint Bone Spine. 2006;73(4):388–395. | ||
Chen D-Y, Lan J-L, Lin F-J, Hsieh T-Y. Proinflammatory cytokine profiles in sera and pathological tissues of patients with active untreated adult onset Still’s disease. J Rheumatol. 2004;31(11):2189–2198. | ||
Fujii T, Nojima T, Yasuoka H, et al. Cytokine and immunogenetic profiles in Japanese patients with adult Still’s disease. Association with chronic articular disease. Rheumatology. 2001;40(12):1398–1404. | ||
Ichida H, Kawaguchi Y, Sugiura T, et al. Clinical manifestations of adult-onset Still’s disease presenting with erosive arthritis: association with low levels of ferritin and IL-18. Arthritis Care Res (Hoboken). October 7, 2013. [Epub ahead of print]. | ||
Shimizu M, Yokoyama T, Yamada K, et al. Distinct cytokine profiles of systemic-onset juvenile idiopathic arthritis-associated macrophage activation syndrome with particular emphasis on the role of interleukin-18 in its pathogenesis. Rheumatology. 2010;49(9):1645–1653. | ||
Gattorno M, Piccini A, Lasigliè D, et al. The pattern of response to anti-interleukin-1 treatment distinguishes two subsets of patients with systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2008; 58(5):1505–1515. | ||
Pouchot J, Arlet J-B. Biological treatment in adult-onset Still’s disease. Best Pract Res Clin Rheumatol. 2012;26(4):477–487. | ||
Lin Y-T, Wang C-T, Gershwin ME, Chiang B-L. The pathogenesis of oligoarticular/polyarticular vs systemic juvenile idiopathic arthritis. Autoimmun Rev. 2011;10(8):482–489. | ||
Mellins ED, Macaubas C, Grom AA. Pathogenesis of systemic juvenile idiopathic arthritis: some answers, more questions. Nat Rev Rheumatol. 2011;7(7):416–426. | ||
Arlet J-B, Le TH, Marinho A, et al. Reactive haemophagocytic syndrome in adult-onset Still’s disease: a report of six patients and a review of the literature. Ann Rheum Dis. 2006;65(12):1596–1601. | ||
Ravelli A, Grom AA, Behrens EM, Cron RQ. Macrophage activation syndrome as part of systemic juvenile idiopathic arthritis: diagnosis, genetics, pathophysiology and treatment. Genes Immun. 2012;13(4):289–298. | ||
Behrens EM, Beukelman T, Paessler M, Cron RQ. Occult macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis. J Rheumatol. 2007;34(5):1133–1138. | ||
Efthimiou P, Kadavath S, Mehta B. Life-threatening complications of adult-onset Still’s disease. Clin Rheumatol. 2014;33(3):305–314. | ||
Silva CA, Silva CH, Robazzi TC, et al. [Macrophage activation syndrome associated with systemic juvenile idiopathic arthritis]. J Pediatr (Rio J). 2004;80(6):517–522. Portuguese. | ||
Ramanan AV, Schneider R. Macrophage activation syndrome following initiation of etanercept in a child with systemic onset juvenile rheumatoid arthritis. J Rheumatol. 2003;30(2):401–403. | ||
Tristano AG. Macrophage activation syndrome: a frequent but under-diagnosed complication associated with rheumatic diseases. Med Sci Monit. 2008;14(3):RA27–RA36. | ||
Mizrahi M, Ben-Chetrit E. Relapsing macrophage activating syndrome in a 15-year-old girl with Still’s disease: a case report. J Med Case Rep. 2009;3:138. | ||
Pamuk ON, Pamuk GE, Usta U, Cakir N. Hemophagocytic syndrome in one patient with adult-onset Still’s disease. Presentation with febrile neutropenia. Clin Rheumatol. 2007;26(5):797–800. | ||
Kelly A, Ramanan AV. A case of macrophage activation syndrome successfully treated with anakinra. Nat Clin Pract Rheumatol. 2008;4(11):615–620. | ||
Bruck N, Suttorp M, Kabus M, Heubner G, Gahr M, Pessler F. Rapid and sustained remission of systemic juvenile idiopathic arthritis-associated macrophage activation syndrome through treatment with anakinra and corticosteroids. J Clin Rheumatol. 2011;17(1):23–27. | ||
Record JL, Beukelman T, Cron RQ. Combination therapy of abatacept and anakinra in children with refractory systemic juvenile idiopathic arthritis: a retrospective case series. J Rheumatol. 2011;38(1):180–181. | ||
Stern A, Riley R, Buckley L. Worsening of macrophage activation syndrome in a patient with adult onset Still’s disease after initiation of etanercept therapy. J Clin Rheumatol. 2001;7(4):252–256. | ||
Kobayashi M, Takahashi Y, Yamashita H, Kaneko H, Mimori A. Benefit and a possible risk of tocilizumab therapy for adult-onset Still’s disease accompanied by macrophage-activation syndrome. Mod Rheumatol. 2011;21(1):92–96. | ||
Aellen P, Raccaud O, Waldburger M, Chamot AM, Gerster JC. [Still’s disease in adults with disseminated intravascular coagulation]. Schweiz Rundsch Med Prax. 1991;80(15):376–378. German. | ||
Arai Y, Handa T, Mitani K. [Adult-onset Still disease presenting with disseminated intravascular coagulation]. Rinsho Ketsueki. 2004;45(4):316–318. Japanese. | ||
Park J-H, Bae JH, Choi Y-S, et al. Adult-onset Still’s disease with disseminated intravascular coagulation and multiple organ dysfunctions dramatically treated with cyclosporine A. J Korean Med Sci. 2004;19(1):137–141. | ||
Yokoyama M, Suwa A, Shinozawa T, et al. [A case of adult onset Still’s disease complicated with adult respiratory distress syndrome and disseminated intravascular coagulation]. Nihon Rinsho Meneki Gakkai Kaishi. 1995;18(2):207–214. Japanese. | ||
Mori T, Tanigawa M, Iwasaki E, et al. [Cyclosporine therapy of adult onset Still’s disease with disseminated intravascular coagulation]. Nihon Rinsho Meneki Gakkai Kaishi. 1993;34(2):147–152. Japanese. | ||
Matsumoto K, Nagashima T, Takatori S, et al. Glucocorticoid and cyclosporine refractory adult onset Still’s disease successfully treated with tocilizumab. Clin Rheumatol. 2009;28(4):485–487. | ||
Gerfaud-Valentin M, Sève P, Iwaz J, et al. Myocarditis in adult-onset Still disease. Medicine. (Baltimore). 2014; 93(17):280–289. | ||
Perez MG, Rodwig FR Jr. Chronic relapsing thrombotic thrombocytopenic purpura in adult onset Still’s disease. South Med J. 2003;96(1):46–49. | ||
Masuyama A, Kobayashi H, Kobayashi Y, et al. A case of adult-onset Still’s disease complicated by thrombotic thrombocytopenic purpura with retinal microangiopathy and rapidly fatal cerebral edema. Mod Rheumatol. 2013;23(2):379–385. | ||
Sayarlioglu M, Sayarlioglu H, Ozkaya M, Balakan O, Ucar MA. Thrombotic thrombocytopenic purpura-hemolytic uremic syndrome and adult onset Still’s disease: case report and review of the literature. Mod Rheumatol. 2008;18(4):403–406. | ||
Onundarson PT, Rowe JM, Heal JM, Francis CW. Response to plasma exchange and splenectomy in thrombotic thrombocytopenic purpura. A 10-year experience at a single institution. Arch Intern Med. 1992;152(4):791–796. | ||
Sumida K, Ubara Y, Hoshino J, et al. Etanercept-refractory adult-onset Still’s disease with thrombotic thrombocytopenic purpura successfully treated with tocilizumab. Clin Rheumatol. 2010;29(10):1191–1194. | ||
Cheema GS, Quismorio FP. Pulmonary involvement in adult-onset Still’s disease. Curr Opin Pulm Med. 1999;5(5):305–309. | ||
Sari I, Birlik M, Binicier O, et al. A case of adult-onset Still’s disease complicated with diffuse alveolar hemorrhage. J Korean Med Sci. 2009;24(1):155–157. | ||
Le Loët X, Daragon A, Duval C, Thomine E, Lauret P, Humbert G. Adult onset Still’s disease and pregnancy. J Rheumatol. 1993;20(7):1158–1161. | ||
Lockshin MD, Sammaritano LR. Corticosteroids during pregnancy. Scand J Rheumatol Suppl. 1998;107:136–138. | ||
Fischer-Betz R, Specker C, Schneider M. Successful outcome of two pregnancies in patients with adult-onset Still’s disease treated with IL-1 receptor antagonist (anakinra). Clin Exp Rheumatol. 2011;29(6):1021–1023. | ||
Berger CT, Recher M, Steiner U, Hauser TM. A patient’s wish: anakinra in pregnancy. Ann Rheum Dis. 2009;68(11):1794–1795. | ||
Giampietro C, Ridene M, Lequerre T, et al. Anakinra in adult-onset Still’s disease: long-term treatment in patients resistant to conventional therapy. Arthritis Care Res. 2013;65(5):822–826. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.