")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 18
Treatment and Outcome of Castleman Disease: A Retrospective Report of 31 Patients
Authors Tang D, Guo Y, Tang Y, Wang H
Received 22 December 2021
Accepted for publication 15 April 2022
Published 26 April 2022 Volume 2022:18 Pages 499—509
DOI https://doi.org/10.2147/TCRM.S354130
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Deyun Wang
Dijiao Tang,1 Yuetong Guo,1 Yi Tang,2 Hongxu Wang1
1Department of Laboratory Medicine, The First Affiliated Hospital of Chongqing Medical University, Chongqing, People’s Republic of China; 2Department of Pathology, Chongqing Medical University, Chongqing, People’s Republic of China
Correspondence: Hongxu Wang, Department of Laboratory Medicine, The First Affiliated Hospital of Chongqing Medical University, No. 1 Youyi Road, Yuzhong District, Chongqing, 400016, People’s Republic of China, Tel/Fax +86 23 89012513, Email [email protected]
Background: Castleman disease (CD) is a rare and heterogeneous lymphoproliferative disorder with a spectrum of characteristic pathological abnormalities of lymph node. Furthermore, its clinical diagnosis is very challenging until pathological results are available. This study aimed to investigate the clinical presentations, treatment and prognosis of CD, thereby improving the understanding and diagnosis of CD.
Methods: This study retrospectively analyzed the clinical data of 31 patients with CD admitted to the First Hospital Affiliated Hospital of Chongqing Medical University January 2013 to December 2020. The chi-square test and the Mann–Whitney rank sum test were employed to calculate between-group differences for categorical and quantitative data, respectively.
Results: Clinically, patients with unicentric CD (UCD) usually present with lymphadenopathy. However, the clinical presentation of patients with multicentric CD (MCD) ranged from mild lymphadenopathy with B-symptoms (5/8, 62.5%) to intense inflammation, vascular leak syndrome (3/8, 37.5%), hepatosplenomegaly (3/8, 37.5%), organ insufficiency (3/8, 37.5%), and even death (2/8, 25.0%). Compared with UCD patients, patients with MCD had significantly lower levels of hemoglobin (104 (90,129) vs 137 (120,149), p=0.018) and plasma albumin (31.5 (27.0,37.0) vs 45.0 (40.0,46.5), p=0.001), but IgG levels were significantly increased. Patients with UCD were mainly treated with surgical resection alone, with a five-year survival rate of 95.65%. When siltuximab is not an option, steroid plus rituximab-based chemotherapy and specific supportive care are common options for MCD. Except for 2 deaths, the remaining MCD patients have stable disease or partial remission.
Conclusion: CD describes a heterogeneous group of disorders characterized by morphologically benign lymphoid hyperplasia. Notably, patients with MCD present varying degrees of inflammation responses, even involving multiple systems. Surgery is a direct and effective way to diagnose and treat UCD. In the absence of IL-6 antagonists, anti-inflammatory and immunosuppressive therapeutic strategies, and cytotoxic clearance of cells responsible for hypercytokinemia could be adopted.
Keywords: Castleman disease, unicentric, multicentric, heterogeneity
Introduction
Castleman disease (CD) is a rare lymphoproliferative disorder first described by Benjamin Castleman in the 1950s as benign, localized and giant follicular lymph node hyperplasia.1 The incidence of CD in the United States is only about 0.2 per 10000 people each year.2 There is currently no clear incidence data in China, but it has been included in The Guidelines for the Diagnosis and Treatment of Rare Diseases in China (2019 edition). CD comprises three distinct clinicopathological disorders: unicentric CD (UCD), idiopathic multicentric CD (iMCD), and human herpes virus-8 (HHV-8) associated MCD (HHV-8+ MCD).3,4 Typically, UCD involves a slow-growing lymph node at a single anatomical site and is usually treated with local surgical excision. MCD involves multiple lymph node regions and is characterized by systemic inflammatory symptoms and organ dysfunction.5 Previous studies have shown that CD is highly heterogeneous in terms of clinical presentation and prognosis.4,6 However, the clinical and biological behavior of CD remains poorly elucidated. Due to its rarity and clinical heterogeneity, current understanding of CD is largely through literature and reports.
Although there have been multi-center and large-scale retrospective studies on CD, the clinical understanding of CD still needs to be continuously improved due to the clinical heterogeneity of CD. Through a retrospective analysis of the clinical characteristics of CD patients diagnosed and treated in our center, we hope to enrich or supplement the understanding of the clinical manifestations of CD and help in the differential diagnosis of CD.
Patients and Methods
Patients
After approval by the institutional review board, we retrospectively collected and analyzed the case data of CD patients who underwent surgery or chemotherapy at our medical institution (Southwest of China) from January 2013 to December 2020. All patients with biopsy-proven histopathological features consistent with a CD diagnosed at Chongqing Medical University were included in this analysis. We excluded patients with the following conditions: (i) Insufficient clinical and follow-up data. (ii) Other diseases that cause lymphadenectasis and hyperinflammatory manifestations include lymphoma, IgG4-associated diseases, and patients with active systemic lupus erythematosus (SLE).7,8 Ultimately, 31 patients were eligible for this study.
Clinical and Laboratory Parameters
Blood routine results were obtained using XN1000 Hematology Analyzer (Sysmex, Japan), while albumin (Alb, g/L), lactate dehydrogenase (LDH, IU/L), and alkaline phosphatase (ALP, IU/L) were collected from the biochemical system database (Roche, Switzerland). IgA (g/L), IgG (g/L), IgM (g/L) and CRP were detected by immunoturbidimetry (Beckman, USA).
Pathology
Eligibility depends on pathological diagnosis, which was independently reviewed by two pathologists for this study.
Histopathological features consistent with the diagnosis of CD include abnormally regressed or hyperplastic germinal centers, prominent follicular dendritic cells, hypervascularization, expanded mantle zones and interfollicular plasmacytosis.7,8 According to the grading of these pathological features, CD can be classified into the following pathological types: hyaline-vascular (HV) subtype, plasmacytic (PC) subtype, and some lesions show mixed features.8,9 HHV-8 was detected in all MCD patients, using Maixin HHV-8 antibody, and stained with Leica bond-max immunohistochemical autostainer.
Definition
Criteria for the diagnosis and classification of CD were derived from criteria established by the Castleman Disease Collaborative Network.7 First, patients with Castleman-like histopathology on lymph node biopsy but thought to resemble CD were excluded. CD (UCD versus MCD) was then staged based on clinical examination, computed tomography (CT), magnetic resonance imaging (MRI) or positron emission tomography/CT scans (PET-CT). UCD was defined as the lesion affected only one lymph node station or a single extra-nodal lesion. Meanwhile, involvement of two or more lymph nodes or regions was considered MCD. HHV-8 was detected by immunohistochemistry. Finally, HIV infection was comprehensively considered and all patients underwent HIV serology testing. TAFRO syndrome, first reported by Takai et al, is characterized by thrombocytopenia (T), anasarca (A), myelofibrosis/fever (F), renal dysfunction/ reticulin fibrosis (R), and organomegaly (O). Currently, CD can be divided into UCD and MCD, and the latter could be subdivided into HHV-8-associated MCD, POEMS syndrome-associated MCD, and idiopathic MCD (iMCD). iMCD can be further divided into 2 distinct subtypes: iMCD, not otherwise specified (iMCD-NOS) and TAFRO syndrome with iMCD (TAFRO-iMCD), which have distinct clinical presentations.3,10,11
Treatment of CD included surgery, siltuximab, rituximab, steroids, immunosuppressive agents, radiotherapy and chemotherapy.12–14 Response to treatment was divided into the following four categories: complete response (CR), defined as no residual hyperplastic lymphadenopathy and no associated clinical or biological abnormalities; partial response (PR), defined as at least 50% mass regression and significant improvement of associated symptoms; stable disease (SD) refers to a stable mass and persistent associated symptoms; progressive disease (PD) refers to enlargement of the mass or worsening of associated symptoms.15 Disease-free survival was defined as the time from surgery to tumor recurrence or the last follow-up. Overall survival was defined as the time from pathological diagnosis to death, loss to follow-up, or last follow-up.
Follow-Up and Statistical Analysis
The end date of follow-up was March 30, 2021, with a median follow-up of 41 (18–97) months.
All analyses were performed using SPSS version 23.0 software. Patients were divided into two groups: (i) UCD and (ii) MCD. Continuous variables were expressed as mean and standard deviation and compared using independent samples t-test. Categorical variables were expressed as frequencies and percentages and were analyzed using the chi-square test. Two independent samples with skewed distribution were compared using the Mann–Whitney rank-sum test. P < 0.05 was considered statistically significant.
Ethical Considerations
The data and the samples analyzed in this study were obtained in accordance with the standards and approval of the Chongqing Medical University Institutional Review Board and Biomedical Ethics Committee. The ethics committee waived the need for written informed consent provided by participants due to the retrospective nature of the study. Because all patient data were analyzed in anonymity, no additional informed consent was required.
Results
Demographic and Clinical Characteristics
A total of 31 patients with CD were enrolled in the current study. There were 17 men and 14 women with a median age of 47 years (ranges,14–69 years) at the time of diagnosis. Among them, twenty-three cases were UCD and 8 were MCD. Clinical characteristics of CD patients were shown in Table 1. There were no statistical differences in age at diagnosis, gender distribution, and history of smoking and drinking.
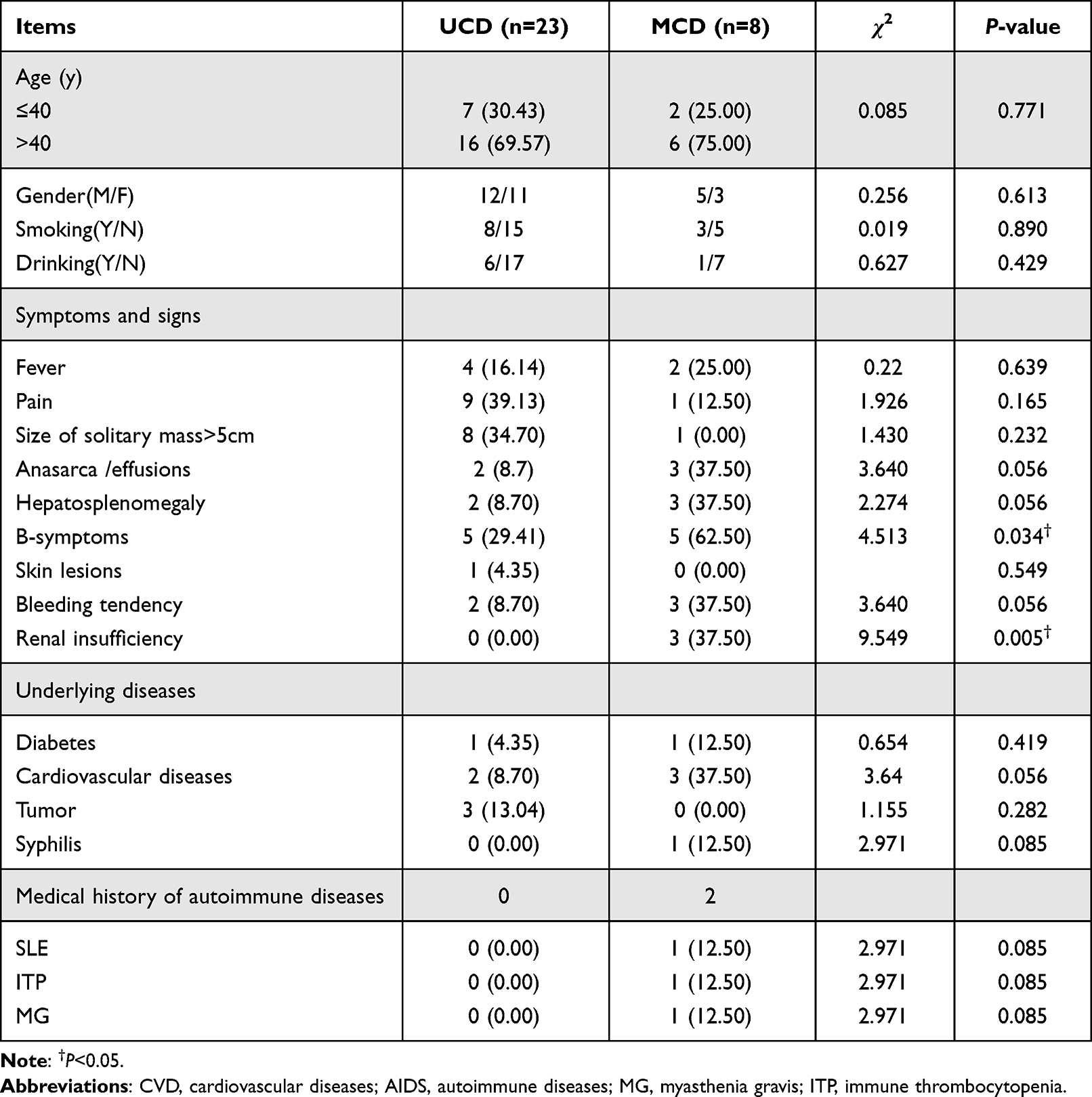 |
Table 1 Baseline Demographic and Clinical Characteristics of Patients with UCD and MCD |
47.83% of patients with UCD were identified anomalies in regular medical examinations. In addition, the main complaints of UCD patients are palpable masses or local compression symptoms due to lymphadenopathy. The symptom complaint rate of MCD patients was higher than that of UCD patients (100% vs 52.17%, p=0.015). Patients with MCD were more likely to have B-symptoms (62.50% vs 29.4%), oedema and/or pleural effusion and/or ascites (75% vs 13.05%), renal dysfunction (37.5% vs 0.00%, p=0.005) and medical history of autoimmune diseases (25% vs 0.00%, p <0.05) than those with UCD. Notably, the accompanied autoimmune diseases were myasthenia gravis (MG) and immune thrombocytopenia (ITP). All patients in this study were HIV-negative, and 1 MCD patient was infected with syphilis. Only one patient with CD was positive for HHV-8 by immunohistochemistry. Meanwhile, two patients were diagnosed with thrombocytopenia, anasarca, fever, reticular fibrosis of bone marrow, and organomegaly (TAFRO) syndrome, which was a distinct subtype of HHV-8(-) MCD.10,13
Imaging and Laboratory Findings
Imaging examinations could provide essential evidence for clinical diagnosis of UCD and MCD. The top three predominant sites of lymphadenopathy in patients with UCD were abdominal (57%), mediastinum (22%) and cervical (17%) (Figure 1). For MCD, the commonly involved locations of lymphadenopathies were inguinal (87.5%), cervical (75.0%), and axillary (62.5%) and abdominal region (50%) (Figure 2). Five cases of UCD (5/23, 21.7%) had enlarged lymph nodes with a maximum diameter greater than 5 cm.
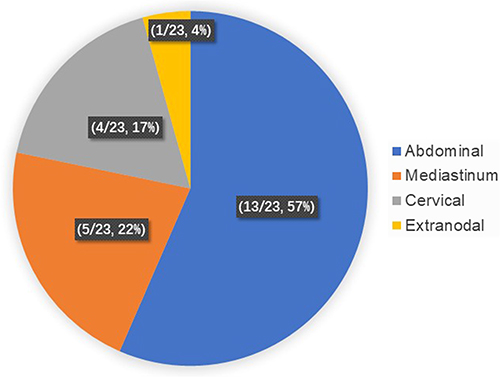 |
Figure 1 The distribution of lymphadenopathy in patients with UCD. Lymph node lesions in UCD patients were located in the abdomen (57%), mediastinum (22%), neck (17%) and extranodal (4%), with the abdomen being the most widespread. |
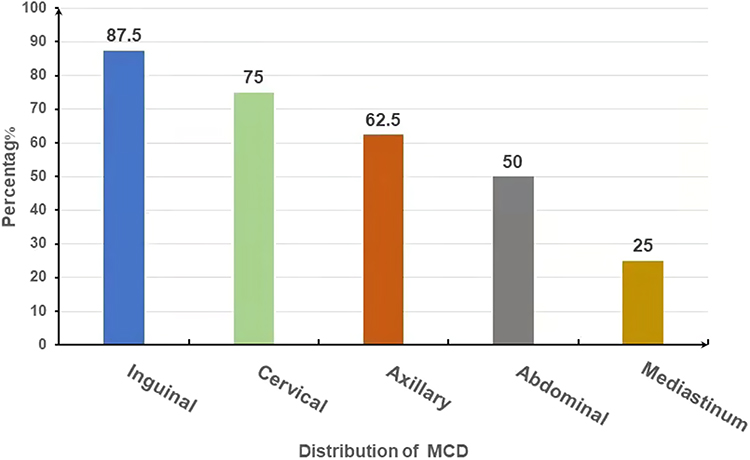 |
Figure 2 Locations of lymphadenopathies in patients with MCD. In MCD, the involved locations of lymphadenopathies were inguinal (87.5%), cervical (75.0%), and axillary (62.5%), abdominal and mediastinum region (50%). Lymphadenopathy mostly occurred in the groin area. |
Pathological examination of lymph node revealed that patients with UCD and MCD mainly demonstrated the HV and plasma/mixed type (PC/ Mix), respectively (Figures 3 and 4). There was no statistical difference in WBC (×109/L) between UCD and MCD patients. However, the Hb (g/L) of MCD patients was significantly lower than that of UCD patients (104 (90,129) vs 137 (120,149), p=0.018), and the red blood cell distribution width coefficient of variation (RDW-CV) was significantly higher (14.60 (13.5,17.1) vs 13.00 (11.8, 13.8), p=0.014). The difference in PLT distribution (×109/L) between UCD and MCD patients was statistically significant (>300,150–300 and <150, ×109/L) (p=0.016), and a higher proportion of patients had PLT (×109/L) less than 150×109/L in MCD than UCD (5/8 vs 3/23, p=0.006). Meanwhile, the albumin (ALB) level of MCD patients was lower than that of UCD patients (31.5 (27.0,37.0) vs 45.0 (40.0,46.5), p=0.001). Additionally, the level of serum ALP (IU/L) was lower in cases with UCD than MCD. Serum level of immunoglobulin (Ig, g/L) demonstrated that IgG (g/L) in the MCD group was significantly higher than that in the UCD group, while there was no significant difference in the levels of IgA (g/L) and IgM (g/L) (Table 2).
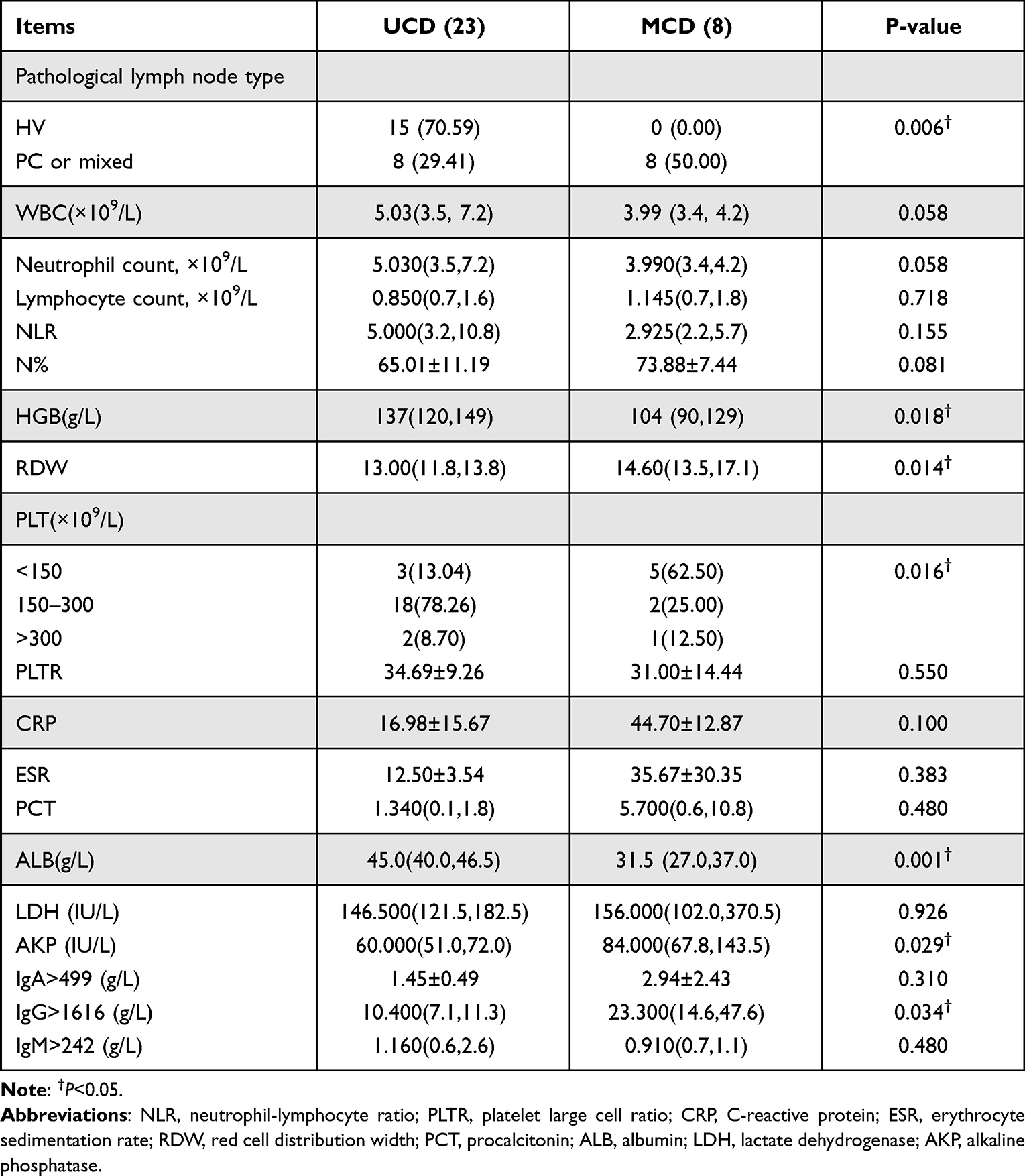 |
Table 2 Comparison of Laboratory Results Between UCD and MCD |
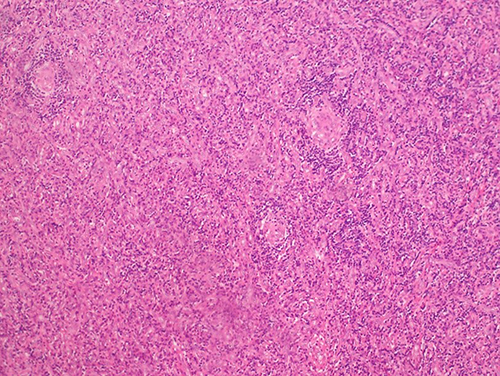 |
Figure 3 Typical pathological image of the hyaline-vascular (HV) subtype. The picture shows a patient with UCD who underwent lymph node biopsy and was diagnosed as HV subtype of CD by H&E staining. |
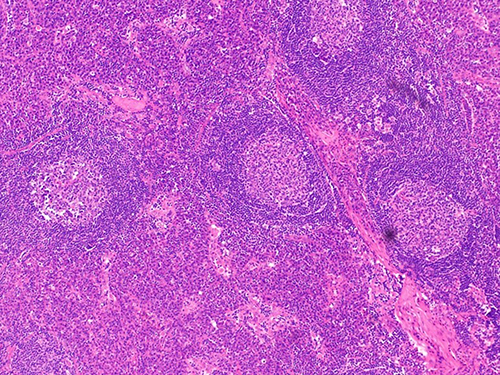 |
Figure 4 Typical pathological image of plasma cell (PC) subtype. The picture shows a patient with MCD who underwent pathological puncture of lymph nodes and was diagnosed as PC subtype of CD by H&E staining. |
Treatment and Prognosis of Patients with UCD
A total of 23 patients (22/23, 95.7%) received therapeutic surgical resection, including twenty-two first-line therapy and one acute abdomen after watch-and-wait period. A middle-aged woman was identified with cervical lymphadenopathy 9 years after left mastectomy because of left breast cancer. She received a complete resection of left supraclavicular lymph nodes and the biopsy specimen exhibited classical HV features. However, PET-CT still showed increased radioactive uptake and the SUV (standard uptake value) max was 3.1 after complete removal of left supraclavicular lymph nodes. Continuing lymph node dissection carries a high risk, in which case clinicians are concerned about both late breast cancer recurrence and UCD recurrence and decide to administer local radiotherapy. After a median follow-up of 37 months (ranges from 17–78 months), twenty-one patients (21/23, 91.3%) were in CR. One patient with lymph node pathology of PC had recurrence in the same lymph node region 22 months after lymphadenectomy, and the pathological results of recurrence were the same as before. Notably, an elderly man with subtype of PC died of respiratory failure caused by pulmonary infection without any evidence of association with UCD recurrence. None of the patients developed lymphoma at the last evaluation.
Presentations of 8 Patients with MCD
To deepen the cognition of MCD, features of all 8 patients with MCD were comprehensively summarized (Table 3). Siltuximab, the only approved and established first-line treatment for iMCD by the US Food and Drug Administration,16 has not been approved for marketing in China during the study period. In this study, the primary therapies for 8 patients with MCD were steroids and chemotherapy with or without rituximab.
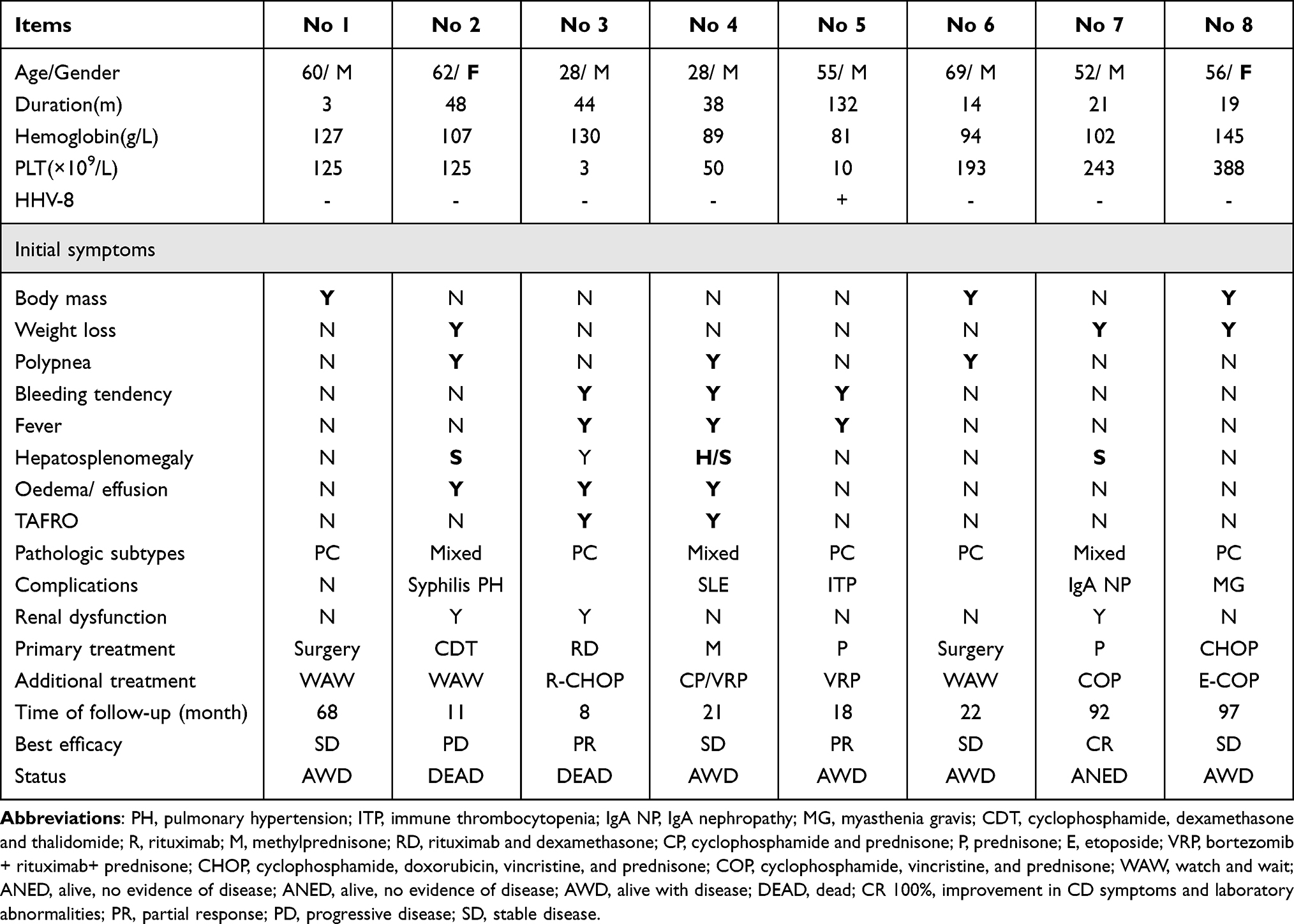 |
Table 3 Efficacy of Drug Treatments for 8 MCD |
Two patients (No. 3 and No. 4) presented features consistent with TAFRO syndrome, and both patients had grade 3 of TAFRO syndrome severity on admission.11,14 Patient No. 3 had fever, lymphadenopathy, fatigue, pleural effusion, thrombocytopenia and bleeding tendency. The patient rapidly developed severe dyspnea and was treated with imipenem plus teicoplanin for a week, but his symptoms and inflammatory markers did not improve. The patient subsequently developed abdominal distension, peritoneal effusion, and incomplete bowel obstruction. Physicians considered that tuberculosis and Crohn’s disease could not be ruled out. Because the patient refused PET-CT and lymph node biopsy antituberculosis quadruple therapy was administered. During antituberculosis therapy, the patient underwent a lymph node biopsy, resulting in of the PC type of CD. Subsequently, the patient received RD (rituximab and dexamethasone) and 3 courses of sequential R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) chemotherapy, and PLT recovered to normal range. However, persistent diarrhea and dyspnea worsened. Cultures of blood, stool, pleural fluid, and ascites fluid were negative during hospitalization. After that, the patient was discharged from the hospital with persistent Castleman and finally died of respiratory failure. The time from onset to death was 8 months.
A total of 3 patients with MCD (No.3, 4, and 5) had thrombocytopenia (PLT≤50×109/L), of which case No.5 was CD complicated with ITP. After RBT (rituximab-based therapy rituximab), the PLT of patients No. 3 and 4 gradually increased and returned to normal, but the PLT of patient No. 5 remained unchanged, at around 20×109/L.
Totally, three patients with MCD (No.2, 3 and 4) developed multiple serous effusions, involving the thoracic cavity, abdominal cavity, pelvis, and pericardium. Routine and biochemical examinations of effusion revealed 2 cases of exudate and 1 case of transudate. Chemotherapy, diuretics and drainage were applied to relieve the compression. Patient No. 4 was in remission and patient of No. 3 was partially relieved. Nevertheless, patient of No.2 failed to respond to primary treatment of CDT (cyclophosphamide, dexamethasone and thalidomide), and she discontinued additional therapy for financial reasons. Eleven months after the onset of the disease, the patient’s multiple serosal effusion continued to worsen and died of infection and electrolyte disturbance.
Discussion
Current knowledge of CD is largely based on retrospective literature studies and case reports.4,17–21 Meanwhile, due to the high rarity and clinical heterogeneity of CD, continued exploration is required. To improve understanding of CD, this study summarized the clinical characteristics of patients with CD treated in our hospital.
Usually, UCD manifests as enlargement of single or single regional lymph nodes, and local compressive symptoms caused by the enlarged lymph nodes.22 The onset of UCD is often insidious due to the frequent absence of systemic symptoms, so patients with UCD are more likely to have a single lymph node enlargement of more than 5cm.23 Moreover, 3 patients with UCD had a history of tumor in this study. Likewise, UCD needs to be differentiated from the tumor at the corresponding site.22,24 Most patients with UCD have normal in laboratory examinations, and only a few patients have mild anemia, hyperglobulinemia, and accelerated ESR. Complete surgical resection is often curative, if possible, and is therefore the first-line treatment for UCD.22 Most clinical symptoms and laboratory abnormalities gradually recover after surgical resection.18,25 All patients with UCD underwent curative therapeutic surgical resection. One patient relapsed in the same lymph node area, and the biopsy pathology still showed type of PC. Rituximab or radiation therapy26,27 could be effective for UCD patients which are refractory to surgical resection or inoperable.22
Although there were only 8 cases of MCD in this study, and 2 of them were diagnosed with TAFRO syndrome,28 it also reflected the clinical heterogeneity of MCD. Of note, patients with MCD present with multiple or multi-regional lymphadenopathy, but not every enlarged lymph node has CD changes. To improve the pathological detection rate of MCD, it is rather crucial for clinicians to select biopsy targets in multiple enlarged lymph nodes. Based on the size and blood supply of lymph nodes reflected by imaging findings, biopsy results showed reactive hyperplasia in 2 patients. A PET-CT scan was then performed,24,29 taking into account the metabolic activity of the lymph nodes, and a second lymph node biopsy is performed. The final diagnosis is made with a complete lymph node biopsy. Therefore, the size, blood supply and metabolic activity of enlarged lymph nodes should be comprehensively considered, and complete lymph node biopsy should be performed to improve the detection rate of MCD.
Compared with UCD, B-symptoms were more common in MCD patients, and MCD accompanied with B-symptoms was easily confused with tuberculosis, so careful differential diagnosis was required. Two patients initially disagreed with invasive lymph node biopsy and were administrated to a week of anti-tuberculosis therapy. They received lymph node biopsy and were diagnosed with MCD after poor anti-tuberculosis efficacy.
In addition, thrombocytopenia was common in MCD patients.4 A total of 3 patients with MCD had thrombocytopenia (PLT≤50×109/L), of which 2 patients were due to CD-induced thrombocytopenia. One case of MCD patients with ITP, we considered that ITP occurred as part of CD, due to CD-induced anti-platelet antibodies. CD-induced thrombocytopenia was recovered after rituximab-based chemotherapy. For MCD patients with ITP, other symptoms related to MCD were relieved after treatment of MCD, but thrombocytopenia was not alleviated due to the persistence of anti-platelet antibodies. Therefore, the production of platelet antibodies could be a poor prognostic factor.
The results of this study showed that MCD patients had lower Hb and higher RDW than UCD patients. Increased RDW is mostly associated with systemic inflammation and critical illness.30 In addition, malnutrition and anemia can lead to increased RDW, but we believe that this is not the main reason for increased RDW in MCD patients. We speculate that high levels of RDW reflect an inflammatory state and are associated with poor clinical outcomes.31 Although the exact mechanism by which increased RDW is associated with poor prognosis is not known, multiple factors may contribute to this association. These include disturbances of erythrocyte metabolism, abnormal iron release from reticuloendothelial macrophages, increased erythrocyte vulnerability by free histone release and binding, and inflammatory factors that inhibit erythrocyte maturation in the bone marrow, such as tumor necrosis alpha (TNF-ɑ) and interleukins (IL-6).30,32 In addition, a study on RDW and dementia showed that the locus was strongly associated with an increase in RDW.33 In conclusion, there are few reports of RDW in Castleman, and whether RDW is associated with the prognosis of MCD also deserves further attention.
Currently, there is no unified standard for the treatment of MCD.34–37 Siltuximab was tested in iMCD randomized trials38 and became the first iMCD treatment approved by the US Food and Drug Administration in 2014. Siltuximab with or without glucocorticoids is the first-line treatment of choice for iMCD, with a 34% durable response rate.11 Additionally, rituximab-based therapy significantly improved the 5-year OS of HHV8-MCD from 33% to 90%.39 When Siltuximab is not an option, other drugs that can be used for the treatment of MCD include corticosteroids, rituximab, thalidomide, lenalidomide, bortezomib, cyclosporine, sirolimus, interferon etc.3,34 Hematopoietic stem cell transplantation is an option for patients with relapsed and refractory MCD.34 In this study, MCD patients had different therapeutic regimens. Two patients with MCD had a good prognosis and stable disease after resection of the mainly enlarged lymph nodes without other treatment due to financial reasons. While some patients with MCD did not respond well and even died despite aggressive treatment. This indicated that the prognosis of MCD patients varies greatly. Therefore, exploring possible prognostic risk factors is crucial for the stratified diagnosis and treatment of MCD patients. Studies have suggested that advanced age, splenomegaly and hypoproteinemia are independent prognostic factors for MCD.4 Importantly, MCD patients 2 and 3 died in this study. Among them, case No. 2 did not respond to the initial treatment of CDT (cyclophosphamide, dexamethasone, and thalidomide), and later died of disease progression after discontinuation of subsequent treatment for economic reasons. While case 3 received RD (rituximab and dexamethasone) and 3 courses of sequential R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone). After chemotherapy, platelets returned to normal, but his persistent diarrhea and dyspnea worsened. With no better treatment options, the patient and his family discontinued follow-up treatment and were discharged. The intractable diarrhea of patient No. 3 is a key factor affecting its prognosis. Regarding the cause of diarrhea, although infectious factors were excluded, the cause could not be revealed because the patient failed to undergo colonoscopy and bowel biopsy. However, studies have shown that this may be related to MCD-related intestinal mucosal thrombotic microangiopathy40 and intestinal amyloidosis,41 and there are reports of Castleman disease and Crohn’s disease.42 For some patients with a dangerous clinical presentation, plasma exchange can be performed in the acute phase, and a patient with Castleman’s disease with diarrhea was relieved by six courses of rituximab, cyclophosphamide, and dexamethasone.40 Although Siltuximab was only launched in the country in December 2021, the drug was not an option at the time. However, the recovery of platelets in patient No. 3 indicates that the treatment is effective, which may be related to the insufficient treatment course. The number of cases in this study was too small for prognostic correlation analysis. Further investigations are expected to explore clinical stratification of MCD in order to instruct hierarchical therapeutic regimen and prognosis.
Conclusion
The clinical manifestations of MCD are diverse and lack specificity, and the prognosis varies greatly. Castleman disease, especially MCD, is still a huge challenge for clinicians.
Data Sharing Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Compliance with Ethics Guidelines
This study was performed in accordance with the Helsinki Declaration of 1964, and its later amendments. This retrospective study was reviewed and approved by the Ethics Committee of The First Affiliated Hospital of Chongqing Medical University. For this type of study formal consent is not required.
Funding
There is no funding to report.
Disclosure
The authors declare that they have no competing interests.
References
1. Castleman B, Iverson L, Menendez VP. Localized mediastinal lymphnode hyperplasia resembling thymoma. Cancer. 1956;9(4):822–830. doi:10.1002/1097-0142(195607/08)9:4<822:AID-CNCR2820090430>3.0.CO;2-4
2. Munshi N, Mehra M, van de Velde H, Desai A, Potluri R, Vermeulen J. Use of a claims database to characterize and estimate the incidence rate for Castleman disease. Leuk Lymphoma. 2015;56(5):1252–1260. doi:10.3109/10428194.2014.953145
3. Dispenzieri A, Fajgenbaum DC. Overview of Castleman disease. Blood. 2020;135(16):1353–1364. doi:10.1182/blood.2019000931
4. Yu L, Tu M, Cortes J, et al. Clinical and pathological characteristics of HIV- and HHV-8–negative Castleman disease. Blood. 2017;129(12):1658–1668. doi:10.1182/blood-2016-11-748855
5. van Rhee F, Voorhees P, Dispenzieri A, et al. International, evidence-based consensus treatment guidelines for idiopathic multicentric Castleman disease. Blood. 2018;132(20):2115–2124. doi:10.1182/blood-2018-07-862334
6. Fajgenbaum DC, van Rhee F, Nabel CS. HHV-8-negative, idiopathic multicentric Castleman disease: novel insights into biology, pathogenesis, and therapy. Blood. 2014;123(19):2924–2933. doi:10.1182/blood-2013-12-545087
7. Abramson JS. Diagnosis and management of Castleman disease. J Natl Compr Canc Netw. 2019;17:1417–1419. doi:10.6004/jnccn.2019.5037
8. Fajgenbaum DC, Uldrick TS, Bagg A, et al. International, evidence-based consensus diagnostic criteria for HHV-8-negative/idiopathic multicentric Castleman disease. Blood. 2017;129(12):1646–1657. doi:10.1182/blood-2016-10-746933
9. Keller AR, Hochholzer L, Castleman B. Hyaline-vascular and plasma-cell types of giant lymph node hyperplasia of the mediastinum and other locations. Cancer. 1972;29(3):670–683.
10. Fujimoto S, Sakai T, Kawabata H, et al. Is TAFRO syndrome a subtype of idiopathic multicentric Castleman disease? Am J Hematol. 2019;94(9):975–983. doi:10.1002/ajh.25554
11. Masaki Y, Kawabata H, Takai K, et al. Proposed diagnostic criteria, disease severity classification, and treatment strategy for a novel disorder; TAFRO syndrome. Rinsho Ketsueki. 2016;57(10):2029–2037. doi:10.11406/rinketsu.57.2029
12. Kawabata H, Takai K, Kojima M, et al. Castleman-Kojima disease (TAFRO syndrome): a novel systemic inflammatory disease characterized by a constellation of symptoms, namely, thrombocytopenia, ascites (anasarca), microcytic anemia, myelofibrosis, renal dysfunction, and organomegaly: a status report and summary of Fukushima (6 June, 2012) and Nagoya meetings (22 September, 2012). J Clin Exp Hematop. 2013;53(1):57–61. doi:10.3960/jslrt.53.57
13. Iwaki N, Fajgenbaum DC, Nabel CS, et al. Clinicopathologic analysis of TAFRO syndrome demonstrates a distinct subtype of HHV-8-negative multicentric Castleman disease. Am J Hematol. 2016;91(2):220–226. doi:10.1002/ajh.24242
14. Masaki Y, Kawabata H, Takai K, et al. 2019 Updated diagnostic criteria and disease severity classification for TAFRO syndrome. Int J Hematol. 2020;111(1):155–158. doi:10.1007/s12185-019-02780-1
15. Hillman SL, An MW, O’Connell MJ, et al. Evaluation of the optimal number of lesions needed for tumor evaluation using the response evaluation criteria in solid tumors: a north central cancer treatment group investigation. J Clin Oncol. 2009;27(19):3205–3210. doi:10.1200/JCO.2008.18.3269
16. Mukherjee S, Martin R, Sande B, Paige JS, Fajgenbaum DC. Epidemiology and treatment patterns of idiopathic multicentric Castleman disease in the era of IL-6-directed therapy. Blood Adv. 2022;6(2):359–367. doi:10.1182/bloodadvances.2021004441
17. Tang JQ, Chen HK, Wang X, et al. Retrospective analysis of 45 cases of localized retroperitoneal Castleman disease from a single center. Hepatobiliary Surg Nutr. 2020;9(3):304–311. doi:10.21037/hbsn.2019.05.05
18. Zhang X, Rao H, Xu X, et al. Clinical characteristics and outcomes of Castleman disease: a multicenter study of 185 Chinese patients. Cancer Sci. 2018;109(1):199–206. doi:10.1111/cas.13439
19. Oksenhendler E, Boutboul D, Fajgenbaum D, et al. The full spectrum of Castleman disease: 273 patients studied over 20 years. Br J Haematol. 2018;180(2):206–216. doi:10.1111/bjh.15019
20. Dong Y, Wang M, Nong L, et al. Clinical and laboratory characterization of 114 cases of Castleman disease patients from a single centre: paraneoplastic pemphigus is an unfavourable prognostic factor. Br J Haematol. 2015;169(6):834–842. doi:10.1111/bjh.13378
21. Borocco C, Ballot-Schmit C, Ackermann O, et al. The French paediatric cohort of Castleman disease: a retrospective report of 23 patients. Orphanet J Rare Dis. 2020;15(1):95. doi:10.1186/s13023-020-1345-5
22. van Rhee F, Oksenhendler E, Srkalovic G, et al. International evidence-based consensus diagnostic and treatment guidelines for unicentric Castleman disease. Blood Adv. 2020;4(23):6039–6050. doi:10.1182/bloodadvances.2020003334
23. Wang W, Dong D, Wen J, Li H. A 10-year observational single-center study of retroperitoneal unicentric Castleman disease. Medicine. 2021;100(10):e25088. doi:10.1097/MD.0000000000025088
24. Jiang Y, Hou G, Zhu Z, Huo L, Cheng W, Li F. The value of multiparameter (18) F-FDG PET/CT imaging in differentiating retroperitoneal paragangliomas from unicentric Castleman disease. Sci Rep. 2020;10(1):12887. doi:10.1038/s41598-020-69854-7
25. Boutboul D, Fadlallah J, Chawki S, et al. Treatment and outcome of unicentric Castleman disease: a retrospective analysis of 71 cases. Br J Haematol. 2019;186(2):269–273. doi:10.1111/bjh.15921
26. Neuhof D, Debus J. Outcome and late complications of radiotherapy in patients with unicentric Castleman disease. Acta Oncol. 2006;45(8):1126–1131. doi:10.1080/02841860600812701
27. Chronowski GM, Ha CS, Wilder RB, Cabanillas F, Manning J, Cox JD. Treatment of unicentric and multicentric Castleman disease and the role of radiotherapy. Cancer. 2001;92(3):670–676. doi:10.1002/1097-0142(20010801)92:3<670:AID-CNCR1369>3.0.CO;2-Q
28. Zhang Y, Suo SS, Yang HJ, et al. Clinical features and treatment of 7 Chinese TAFRO syndromes from 96 de novo Castleman diseases: a 10-year retrospective study. J Cancer Res Clin Oncol. 2020;146(2):357–365. doi:10.1007/s00432-019-03120-w
29. Han EJ, JH O, Jung SE, et al. FDG PET/CT findings of Castleman disease assessed by histologic subtypes and compared with laboratory findings. Diagnostics. 2020;10:12.
30. Tonelli M, Sacks F, Arnold M, et al. Relation between red blood cell distribution width and cardiovascular event rate in people with coronary disease. Circulation. 2008;117(2):163–168. doi:10.1161/CIRCULATIONAHA.107.727545
31. Ridker PM, Rifai N, Clearfield M, et al. Measurement of C-reactive protein for the targeting of statin therapy in the primary prevention of acute coronary events. N Engl J Med. 2001;344(26):1959–1965. doi:10.1056/NEJM200106283442601
32. Baba Y, Saito B, Shimada S, et al. Association of red cell distribution width with clinical outcomes in myelodysplastic syndrome. Leuk Res. 2018;67:56–59. doi:10.1016/j.leukres.2018.02.004
33. Winchester LM, Powell J, Lovestone S, Nevado-Holgado AJ. Red blood cell indices and anaemia as causative factors for cognitive function deficits and for Alzheimer’s disease. Genome Med. 2018;10(1):51. doi:10.1186/s13073-018-0556-z
34. Fajgenbaum DC. Novel insights and therapeutic approaches in idiopathic multicentric Castleman disease. Blood. 2018;132(22):2323–2330. doi:10.1182/blood-2018-05-848671
35. Zhang L, Zhao AL, Duan MH, et al. Phase 2 study using oral thalidomide-cyclophosphamide-prednisone for idiopathic multicentric Castleman disease. Blood. 2019;133(16):1720–1728. doi:10.1182/blood-2018-11-884577
36. van Rhee F, Casper C, Voorhees PM, et al. Long-term safety of siltuximab in patients with idiopathic multicentric Castleman disease: a prespecified, open-label, extension analysis of two trials. Lancet Haematol. 2020;7(3):e209–e217. doi:10.1016/S2352-3026(19)30257-1
37. Deisseroth A, Ko CW, Nie L, et al. FDA approval: siltuximab for the treatment of patients with multicentric Castleman disease. Clin Cancer Res. 2015;21(5):950–954. doi:10.1158/1078-0432.CCR-14-1678
38. van Rhee F, Wong RS, Munshi N, et al. Siltuximab for multicentric Castleman’s disease: a randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2014;15(9):966–974. doi:10.1016/S1470-2045(14)70319-5
39. Bower M, Newsom-Davis T, Naresh K, et al. Clinical features and outcome in HIV-associated multicentric Castleman’s disease. J Clin Oncol. 2011;29(18):2481–2486. doi:10.1200/JCO.2010.34.1909
40. Flahault A, Vignon M, Rabant M, et al. Case report and literature review: glomerular and neurologic thrombotic microangiopathy as a primary manifestation of multicentric Castleman disease. Medicine. 2016;95(41):e5047. doi:10.1097/MD.0000000000005047
41. Miura A, Sato I, Suzuki C. Fatal diarrhea in a patient with Castleman’s disease associated with intestinal amyloidosis. Intern Med. 1995;34(11):1106–1109. doi:10.2169/internalmedicine.34.1106
42. Yavasoglu I, Kadikoylu G, Meteoglu I, Bolaman Z. Hyaline-vascular type of Castleman's disease associated with Crohn’s disease. Int J Colorectal Dis. 2009;24(1):123–124. doi:10.1007/s00384-008-0545-6
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.