Back to Journals » Cancer Management and Research » Volume 6
Treating advanced melanoma: current insights and opportunities
Authors Tronnier M, Mitteldorf C
Received 2 June 2014
Accepted for publication 5 July 2014
Published 10 September 2014 Volume 2014:6 Pages 349—356
DOI https://doi.org/10.2147/CMAR.S49494
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Michael Tronnier, Christina Mitteldorf
Department of Dermatology, Klinikum Hildesheim GmbH, Hildesheim, Germany
Abstract: Whereas thin melanomas have an excellent prognosis after sufficient surgical treatment, melanoma disease in advanced stages is still a therapeutic challenge. After decades of frustrating studies, new therapeutic strategies have come up in the past few years. On the one hand, increasing insights into the molecular aberrations in melanoma have led to specific "targeted" therapies to affect only the mutated tumor cells, as in many other types of cancers. Today there are few "targeted" substances which are already approved and successfully used for single or combination therapy, but many others are under development. While on the other hand, nonpersonalized strategy substances have been developed successfully inducing an immunologic tumor response. Both kinds of therapy have been found to result in an improvement not only of the response rate, but also of the overall survival in metastatic disease, which represents a milestone in melanoma therapy. However, using these therapies there is still much to learn regarding the effects, the side effects, and the limitations of these promising substances.
Keywords: melanoma, treatment, targeted therapy, immunotherapy, BRAF, CTLA-4
Introduction
In countries with a fair-skinned population, the incidence of melanoma is increasing faster than in any other type of cancer – a fact, which has led to the use of the term melanoma “epidemic”.1,2 The incidence rate per year is rising by 2%–7% annually – thus it doubles around every 10 years.3,4 This impressive rise in incidence may be due to several factors, including behavioral changes, better early detection by screening instruments, changes in diagnostic criteria in histopathology, and perhaps also the change in the medico-legal climate.2,5
Whereas surgical procedures are usually the treatment of choice with primary tumors, regional disease, and single metastases, an inoperable tumor manifestation requires a systemic therapy. For many years various chemotherapeutic regimen have been applied, either as monochemotherapy or as polychemotherapy, which in general did not result in an improvement of progression-free or overall survival, but sometimes in severe toxicities.6
The development of new substances, targeted therapies, and immunologic substances has completely changed the former treatment guidelines for metastatic disease in melanoma.2,7 This review focuses on the new development in therapy and future perspectives. Nevertheless, the chemotherapeutical procedures still remain an option in treating metastatic melanoma.
Targeted therapy
The number of mutations found in melanoma is high compared to the number in other metastatic tumors. This may be due to the fact that ultraviolet (UV) light is involved in the pathogenesis of melanoma.7 The analysis of the mutational status of melanoma disease clearly shows that the various clinical manifestations of melanoma also differ in their molecular changes, which subsequently will be of importance for therapies directed against tumors bearing a distinct mutation. According to the findings of molecular studies in melanoma, the unifying concept of one melanoma disease which is mainly based on dermatopathologic criteria is outdated.8 However, the knowledge of the mutational landscape in melanoma alone does not help in the development of therapeutic strategies. Concerning the high rate of mutations it must be differentiated which mutation is causative in the disease (driver mutation) and which is only a bystander mutation (passenger mutation).8 Approval of single substances directed against mutated proteins has dramatically changed the options available in melanoma therapy. The most important task for the future will be to overcome primary and, even more important, secondary resistance to the targeted therapeutics. In addition, the understanding and management of the frequent side effects of these new therapies have to be improved.
BRAF
BRAF is a key member of the rat sarcoma (RAS) mitogen-activated protein kinase (MAPK) pathway which regulates cell growth and proliferation. Mutation of BRAF has been reported in about 50% of all melanomas and in most of the melanocytic nevi, which implies that the mutation per se is not responsible for malignancy in melanocytic proliferations. However, due to the fact that in BRAF (and NRAS) wild-type melanoma five times more mutations are observed (or needed) it may be speculated that the relative specificity of BRAF/NRAS mutations for the disease is quite high.9 Most of the mutations of BRAF are found in exon 15, at codon 600 (V600).10 In about 75% of the mutations in that area valine is substituted by glutamic acid (V600E). Other substitutions include valine by lysine (V600K) (about 20%) and valine by arginine (V600R). For the available diagnostic tests for the detection of BRAF mutation it is of great importance not to overlook the abovementioned mutations beside V600E, which account for about one quarter of all mutated cases. BRAF mutation, and in part also the special type of substitution, correlates with age, localization of the primary tumor, sun damage, and, in part, geographic region. In primary melanoma, the presence or absence of a mutant BRAF gene has no impact on the disease-free interval or overall survival.11 For the detection of a BRAF mutation in melanoma tissue various approaches are possible. Immunohistochemistry (IHC), which can be applied to paraffin-embedded material, has been described as highly sensitive to, and specific for, the detection of V600E mutations12 (Figures 1–3). The drawback of IHC is that mutations other than V600E are negative. Therefore, it has been recommended to combine IHC with one of the molecular techniques like pyrosequencing or high resolution melting (HRM) analysis, starting with IHC and – if negative or uninterpretable – adding the molecular procedure.13,14 It has also been reported that if, in a single patient, several samples were tested for the presence of BRAF mutation (Sanger DNA sequencing), discordant results have been obtained in 13.5% of those patients.15 In case of discordance, both constellations were observed: wild-type in the primary tumor and V600E in metastases and, vice versa, V600E in primary tumor and wild-type in metastases. The presence of a BRAF mutation cannot be ruled out with certainty by a single test.15
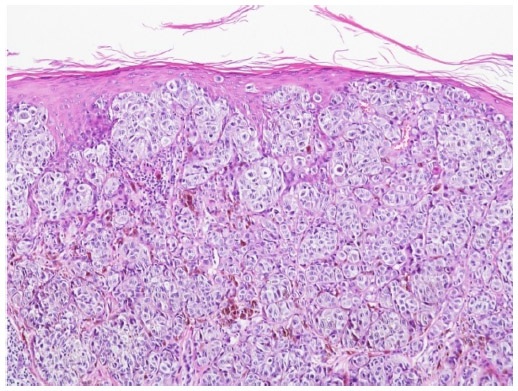 | Figure 1 Melanoma of superficial spreading type. |
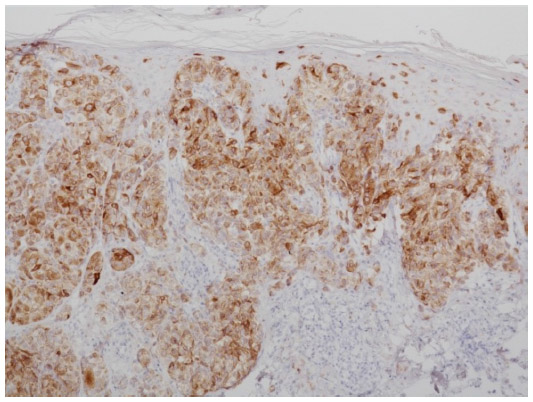 | Figure 2 Immunohistochemistry using antibodies against melanocytic antigen (melan-A). |
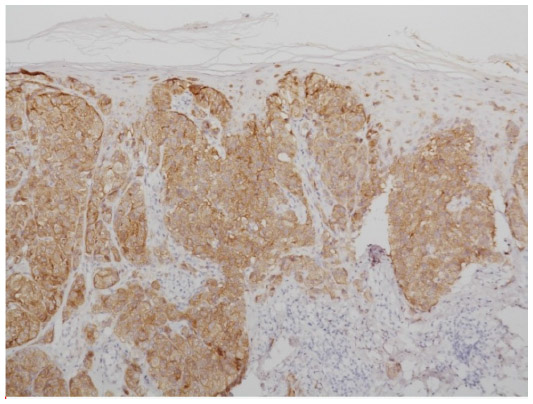 | Figure 3 Immunohistochemistry using antibodies against mutated BRAF. |
Sorafenib as a drug, which acts as a pan-inhibitor of BRAF, has largely failed in the treatment of melanoma.16 The breakthrough of targeted therapy in melanoma has been achieved with the introduction of mutation-specific inhibitors of BRAF, namely vemurafenib and later dabrafenib, both of them now approved in various countries for the treatment of BRAF-mutated metastatic melanoma.
Vemurafenib has been found to show high response rates in a Phase I study, where the dosage of 960 mg twice daily was established.17 In the following Phase III study (BRIM3)18 vemurafenib was compared to the former most commonly applied chemotherapeutical agent dacarbazine, 1,000 mg dacarbazine given intravenously every three weeks. The response rates were 48% for vemurafenib compared to 5% in dacarbazine. In this study, which was published rapidly, the overall survival rate was calculated as early as 6 months after initiation, showing an increased overall survival rate for the vemurafenib group. An update of the results of the BRIM3 study presented some months later showed a median overall survival rate of 13.9 months for vemurafenib compared to 9.6 months in the dacarbazine group. Other studies underlined the impressive response rates in vemurafenib-treated patients.19 After decades of a frustrating search for new drugs for metastatic melanoma, the published results were sensational.
The side effects of the BRAF inhibitor therapy completely differ from those observed in conventional chemotherapy. The most important side effects concerning frequency and intensity during treatment with vemurafenib were the development of a rash, in some associated with a severe photosensitivity (UVA), arthralgia, and the development of keratinocytic neoplasia (namely keratoacanthoma; squamous cell carcinoma; wart-like proliferation of the skin).18,19 The development of epithelial tumors is explained by a paradoxical activation of the MAPK-pathway in BRAF wild-type cells.10 These tumors are controlled by excision or, if larger areas are affected, by photodynamic therapy. The occurrence of epithelial tumors during the treatment with vemurafenib does not require a discontinuation of the therapy. There are also a few reports that secondary melanomas may develop under therapy. These melanomas have been found to be mainly of BRAF wild-type.20,21 However, it has been questioned and discussed whether all of the melanocytic lesions developed under BRAF inhibitor therapy biologically described as melanoma really represent melanoma or if at least some of those lesions might be melanocytic nevi which have been activated by the drug, as seen in other situations like UV exposure of nevi.22,23
The second substance directed against mutated BRAF, and already approved in many countries, is dabrafenib. Like vemurafenib this substance showed comparably high response rates and a progressive-free survival advantage compared to standard therapy.24 In contrast to the very similar efficacy of both BRAF inhibitors, the toxicity and the side effects differ considerably. Dabrafenib-treated patients far less frequently develop epithelial tumors and photosensitivity, while under therapy with dabrafenib fever is more frequently found. However, compared to chemotherapy the side effects are usually mild and manageable.25
A newer inhibitor, LGX818, has been found, in a Phase I trial, to also show a high response rate (58%) and even a response rate of 11% in patients pretreated with a BRAF inhibitor.26 The major objective in developing new BRAF inhibitors is to overcome the paradoxical activation of the Erk1/2 pathway in wild-type cells with the subsequent development of epithelial and melanocytic neoplasms.
Some patients with BRAF-mutated melanoma do not respond to therapy with these specific inhibitors. This may be due to either a primary or secondary resistance to the drug, both of great importance in targeted therapy. BRAF-mutated melanoma contain a lot of additional mutations which have been shown to overcome the effects of BRAF (and MEK) inhibitors and lead to a primary resistance to the drug (reviewed by Shtivelman et al).7 Secondary resistance develops after a period of successful medication. This secondary resistance may be due to an alteration of the initial mutation in the BRAF gene or the activation of alternative pathways.7 Cell growth and proliferation are controlled by a network of pathways consisting of various key proteins which might be mutated and then influence each other, either by activation or inhibition. Consequently, the interaction at one single checkpoint will not be successful in the long-term. There is some evidence that intermittent dosing of vemurafenib after the maximum response to therapy has been reached may contribute to prolonging the response duration.27 Nevertheless, most patients developed a resistance after a certain period of treatment and the relapse of the disease is characterized by a very rapid regrowth of the metastatic disease.
Therefore, in patients with a proven BRAF mutation and rapid progress of the disease, these inhibitors should preferably be applied to achieve a fast reduction of the tumor mass.
MEK
In the MAPK pathway MEK is a downstream target and the only known substrate for BRAF.28 In BRAF-mutated melanoma the orally available MEK inhibitor trametinib (GSK1120212) was compared to chemotherapy and clearly showed improvement in the progression-free survival rate (4.8 months versus 1.5 months).29 The overall toxicity is mild, including rash, cardiac dysfunction, ocular side effects, diarrhea, peripheral edema, and hypertension. Other MEK inhibitors successfully tested in melanoma are selumetinib and MEK162.30,31 Patients who did not respond to vemurafenib usually did not show any improvement after MEK inhibitors either.
Combination of BRAF and MEK inhibitor
Around 50% of the patients treated with BRAF inhibitors show a progression of disease after several months.19,24 The resistance to BRAF inhibitors is associated with a rapid recovery of the MAPK pathway corresponding to a rapid clinical progression. A complete inhibition of the MAPK pathway is obtained by the combination of BRAF and MEK inhibitors which may delay or prevent MAPK-dependent resistance.25 Another advantage of this combination is that the paradoxical activation resulting in BRAF wild-type tumors may be reduced. The combination of dabrafenib with trametinib showed a significantly higher response rate (76% versus 54%) and significantly longer progression-free survival (9.4 months versus 5.8 months) than after therapy with dabrafenib alone.32 The incidence of epithelial tumors (squamous cell carcinoma/keratoacanthoma) was impressively reduced to 2% in the group receiving 2×150 mg dabrafenib and 1 mg trametinib and to 7% in the group receiving 2×150 mg dabrafenib and 2 mg trametinib, compared to 19% after monotherapy with 2×150 mg dabrafenib alone.32 Also, the frequency of rashes was lower in patients receiving the combination. In summary, these results are very promising, indicating a clear advantage of the combination of targeted therapies which seems to be more effective, equally or less toxic, but surely more cost intensive.
NRAS
NRAS mutations are observed in about 20% of melanoma. Compared to NRAS/BRAF wild-type patients, those with NRAS mutations seem to have a worse prognosis.33 So far, there is no single targeted substance to treat NRAS-mutated melanoma, but targeting the downstream effectors of NRAS may be a treatment option.7 Monotherapy with MEK inhibitor MEK162 revealed a response rate of 21% in NRAS-mutated melanoma.31 In the near future, more data concerning the combination of MEK inhibitors with targeted substances to other pathways (PI3K; mTOR) are expected for the treatment of NRAS melanomas.
KIT
Mutations in the receptor tyrosine kinase KIT are especially found in melanomas from special sites, namely acral melanoma, mucosal melanoma, and melanoma located in sun-damaged skin (lentigo maligna melanoma). KIT mutations in melanoma are quite rare. Most of them are observed either in exon 11 or exon 13.34 Comparing the immunohistochemical analysis of KIT expression and the mutational status in melanomas in KIT-typical sites, 81% of tumors showed at least a partial expression in IHC, but in only 15% of the investigated cases a KIT mutation could be proven by molecular techniques.35 Because immunohistochemical-negative tumors did not carry a mutation, a statistically significant association of immunohistochemistry status and mutational status was found in this study. However, for the therapeutic strategy, differentiation between KIT amplification and KIT mutation is of great importance. Selective inhibitors of KIT (imatinib; sunitinib; nilotinib; dasatinib) which were developed for and used for the treatment of other types of cancer (such as gastrointestinal stroma tumor [GIST] or chronic myeloid leukemia) became of interest concerning melanoma treatment. Sunitinib has shown a clinical response in three out of four KIT-mutated melanomas, but only in one out of six melanomas with KIT amplification only.36 In a Phase II trial using imatinib in selected melanomas (acral; mucosal; sun-damaged skin), again the majority of response has been observed in cases with KIT mutation (77%) compared to KIT amplification (18%).37 As with other targeted substances, trials combining various substances are under way, so even better response rates may be expected for this type of melanoma in the future.
Immunologic therapy
Melanoma is a highly immunogenic tumor which may undergo regression. Immunological strategies in therapy of localized and metastatic melanoma have a long tradition. So far, therapies for disseminated disease modulating the immunologic response like interferons, interleukins and others have been quite disappointing. Also, vaccine therapy directed against melanoma targets has not shown significant clinical responses as yet.38
CTLA-4
As a control mechanism, around 48 hours after the initial T-cell activation by interaction of B7 on antigen presenting cells (APC) with the CD28 T-cell receptor, the cytotoxic T-lymphocyte antigen 4 (CTLA-4) binds with a higher affinity to B7 interrupting the activation. Applying an antibody against CTLA-4 means “to remove the foot from the brake” and to further activate the T-cell.39 Ipilimumab (Ipi) is a fully humanized IgG 1 recombinant monoclonal antibody, which in a Phase III trial showed, compared to gp100 peptide vaccine, a significantly improved overall survival in unresectable stage III and stage IV melanoma. In this study, three groups randomized in a 3:1:1 ratio (Ipi plus gp100; Ipi alone; gp100 alone) were compared. An improvement in the overall survival of 3.6 months in patients receiving Ipi was observed.40 The Ipi dosage given was 3 mg/kg bodyweight four times every 3 weeks. However, in this study the immune-related adverse effect rate (grade 3 and 4) was 10%–15% compared to 3% in the group given gp100 alone. The total number of immune-related toxicities during therapy with Ipi must be expected to be around 60%. The most frequently observed events were gastrointestinal immune-related events (diarrhea; colitis) and other autoimmune inflammations (hypophysitis; thyroiditis; hepatitis); in such cases an early intervention with corticosteroids is mandatory.40 With increasing experience with this therapy such severe immune responses can be detected at an earlier stage, resulting in a lower grade 3 and grade 4 toxicity. If there is a response to Ipi treatment at all, some of them are long-term responses. Ipi was also found to be effective in patients with brain metastases, especially in the subgroup of non-symptomatic (small) metastases.41 The presence of antibodies to NY-ESO-1, an antigen expressed in advanced melanoma, and a rise in absolute lymphocyte count in the blood, increase the likelihood of a response to Ipi.42
Comparing the treatment of previously untreated melanoma patients either with Ipi plus dacarbazine or dacarbazine alone revealed a higher overall survival rate with the combination Ipi with dacarbazine (11.2 months versus 9.1 months, respectively). The survival rates after 3 years were 20.8% in the Ipi/dacarbazine group compared to 12.2% in the dacarbazine group.43 The combination also showed a clearly higher rate of adverse events (grade 3 or 4) with 56.3% compared to 27.5%. In this study, Ipi was given at a dose of 10 mg/kg bodyweight.43 The response to Ipi as an immunologic reaction appears slowly but may be long lasting. On the other hand, the response of BRAF inhibitors in BRAF-mutated melanoma appears early, but may be soon overcome by resistance. Consequently, Ipi and BRAF inhibitors were given in combination in patients with BRAF-mutated melanoma.44 Out of the 28 patients who were treated first with vemurafenib and subsequently with Ipi, 12 had a rapid disease progression resulting in death with a median survival of only 5.7 months. However, those patients who were able to complete treatment with Ipi showed an overall survival rate of 18.6 months, indicating that the sequential use of these substances may improve the patients’ outcome.44
PD-1/PD-L1
Another interesting key point in immunologic tumor response is the PD-1 receptor located on T-cells and its ligand PD-L1 located on cells of solid tumors like melanoma. The interaction between the PD-1 and its ligand results in a downregulation/termination of the immune response against the tumor. The inhibition of the interaction is associated with increased antitumor activity.45,46 The human antibody nivolumab is directed against PD-1, which in a Phase I trial produced a response in 28% of the melanoma patients. More than half of these responders were long-term and lasted 1 year or longer. The best response was observed with a dosage of 3 mg/kg bodyweight. The overall toxicity rate (296 patients) was 41%, but grade 3 or 4 toxicity was observed in only 6% of these cases.46 Most often, diarrhea, rush, and pruritus were observed. The most severe side effect was the development of pneumonitis, which caused three drug-related deaths.46 Interestingly, the response to PD-1 antibodies may, at least partially, be predicted by the immunohistochemical detection of the PD-L1 on the tumor cells.46,47
Lambrolizumab is a humanized IgG4 antibody directed against PD-1. In a Phase I trial with 135 melanoma patients it showed a response rate of 38% across all dose cohorts.48 The highest response rate was 52% and was observed using the dose of 10 mg/kg bodyweight given every 2 weeks. Most of the responses became evident after 12 weeks and were long lasting (In 81%, lasting more than 11 months). The response was not dependent on whether the patient had received Ipi before or not. Adverse effects were reported in 79% of patients, most frequently fever, chills, fatigue, and myalgia, but mild forms thereof.
A Phase I trial using the human IgG4 antibody against PD-L1 (BMS-936559) in 55 patients with melanoma showed a response of 17% across all dose cohorts with the best response (29%) after 3 mg/kg bodyweight.49 Toxicity was generally mild and manageable, but 9% of patients had grade 3 toxicity. A second antibody against PD-L1 which has been under investigation in a Phase I trial is MPDL3280A showing an overall response rate of 29% in 44 patients treated for melanoma.50
Taken together, these second-generation immune checkpoint inhibitors are characterized by an enhanced efficacy and less toxicity compared to ipilimumab.25
Intralesional immunotherapy
If an immunotherapy is applied topically/intralesionally, a regression of the treated metastasis should be achieved, but also an immunologic response at distant sites is desired.51 As early as around 50 years ago, the first studies using Bacillus Calmette-Guérin were performed in oncology. In 1974, a study with 151 patients who were treated with Bacillus Calmette-Guérin was published, reporting a local response rate of 90%, a response at distant sites in 17%, and some long-lasting remissions.52 Despite some promising results, the therapy has been abandoned because of some more severe side effects and some other studies which could not confirm the initial results. Other substances used for a topical immunomodulatory therapy were GM-CSF and the interferons alpha and beta.53 Again, at least in some cases, systemic responses were found in addition to local tumor control.54
Local tumor response was also observed using intralesional interleukin-2, which showed a complete response in 78% of treated lesions.55
One of the more recently-developed substances for local treatment is allovectin (velimogene aliplasmid), which consists of a plasmid encoding for HLA-B7 and beta2-microglobulin. Transfected tumor cells are able to induce an immune response by induction of cytotoxic T-cells and macrophages, which is locally effective in non-treated tumor tissue.56 Another development is the treatment with genetically manipulated oncolytic viruses.51 Talimogene laherparepvec (T-Vec) is a herpes simplex virus type 1, in which ICP34.5 has been replaced by ICP47 (a sequence encoding for human GM-CSF). This replacement results in a tumor-selective replication and in an increase of GM-CSF.57 A Phase III randomized trial comparing T-Vec with GM-CSF vaccination revealed a clear increase only in the long-term response rate (survival more than 6 months; 2.1% versus 26.4%) in the T-Vec arm.58 CAVATAK™ consists of the genetically unmodified coxsackie virus A21 which has an enhanced affinity to melanoma cells compared to normal cells. This is due to the fact that the decay-accelerating factor, which interacts with the virus, is upregulated on melanoma cells. Studies treating patients with advanced melanoma with CAVATAK™ are under way.51
Future perspectives
During the past few years the management of patients with metastatic melanoma has undergone an impressive shift.59 Individualized targeted therapy shows response rates that have never been described in melanoma before. However, early development of resistance is the major drawback of this therapy. This might be overcome by the combination of various targeted substances. Combined therapy leads to increased response rates, but not necessarily to increased toxicity as observed after the combination of chemotherapeutic substances. Responses to immunomodulatory treatment need more time to become evident but, if the tumor responds, the time of duration might be longer. The best therapy, therefore, is not a question of tumor stage, but is dependent on the individual mutational status and how rapidly the tumor progresses. Currently, knowledge of the various factors which may be involved in tumor escape is limited. Besides, a genetic heterogeneity of melanoma autocrine properties and a modulation of the tumor environment by tumor cells must be considered.60 Sequencing various therapies, finding of new immunologic key points, and developing further targeting substances, in addition to an advanced understanding of resistance mechanisms, will certainly further improve the treatment of melanoma disease in the near future. Beyond the well-established therapy with interferon-alpha, new approaches are also expected for the adjuvant treatment of melanoma.61
Disclosure
The authors report no conflict of interest in this work.
References
Erickson C, Driscoll MS. Melanoma epidemic: facts and controversies. Clin Dermatol. 2010;28(3):281–286. | |
Tronnier M, Semkova K, Wollina U, Tchernev G. Malignant melanoma: epidemiologic aspects, diagnostic and therapeutic approach. Wien Med Wochenschr. 2013;163(15–16):354–358. | |
Linos E, Swetter SM, Cockburn MG, Colditz GA, Clarke CA. Increasing burden of melanoma in the United States. J Invest Dermatol. 2009;129(7):1666–1674. | |
Garbe C, Leiter U. Melanoma epidemiology and trends. Clin Dermatol. 2009;27(1):3–9. | |
Volkovova K, Bilanicova D, Bartonova A, Letašiová S, Dusinska M. Associations between environmental factors and incidence of cutaneous melanoma. Environ Health. 2012;11:Suppl 1:S12. | |
Garbe C, Peris K, Hauschild A, et al; European Dermatology Forum; European Association of Dermato-Oncology; European Organization of Research and Treatment of Cancer. Diagnosis and treatment of melanoma: European consensus-based interdisciplinary guideline – Update 2012. Eur J Cancer. 2012;48(15):2375–2390. | |
Shtivelman E, Davies MQ, Hwu P, et al. Pathways and therapeutic targets in melanoma. Oncotarget. 2014;5(7):1701–1752. | |
Ackerman AB. Malignant melanoma. A unifying concept. Am J Dermatopathol. 1980;2(4):309–313. | |
Mar VJ, Wong SQ, Li J, et al. BRAF/NRAS wild-type melanomas have a high mutation load correlating with histologic and molecular signatures of UV damage. Clin Cancer Res. 2013;19(17):4589–4598. | |
Poulikakos PI, Zhang C, Bollaq G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464(7287):427–430. | |
Lang GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29(10):1239–1246. | |
Long GV, Wilmott JS, Capper D, et al. Immunohistochemistry is highly sensitive and specific for the detection of V600E BRAF mutation in melanoma. Am J Surg Pathol. 2013;37(1):61–65. | |
Colomba E, Hélias-Rodzewicz Z, von Deimling A, et al. Detection of BRAF p.V600E mutations in melanomas: comparison of four methods argues for sequential use of immunohistochemistry and pyrosequencing. J Mol Diagn. 2013;15(1):94–100. | |
Ihle MA, Fassunke J, König K, et al. Comparison of high resolution melting analysis, pyrosequenzing, next generation sequencing and immunohistochemistry to conventional Sanger sequencing for the detection of p.V600E and non-p.V600E mutations. BMC Cancer. 2014;14:13. | |
Saint-Jean M, Quéreux G, Nguyen JM, et al. Is a single BRAF wild-type test sufficient to exclude melanoma patients from vemurafenib therapy? J Invest Dermatol. 2014;134(5):1468–1470. | |
Eisen T, Ahmad T, Flaherty KT, et al. Sorafenib in advanced melanoma: a phase II randomised discontinuation trial analysis. Br J Cancer. 2006;95(5):581–586. | |
Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363(9):809–819. | |
Chapman PB, Hauschild A, Robert C, et al; BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–2516. | |
Sosman JA, Kim KB, Schuchter L, et al. Survival in BRAF V600-mutated advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366(8):707–714. | |
Göppner D, Müller J, Krüger S, Franke I, Gollnick H, Quist SR. High Incidence of Naevi-associated BRAF Wild-type Melanoma and Dysplastic Naevi under Treatment with the Class I BRAF Inhibitor Vemurafenib. Acta Derm Venereol. Epub February 14, 2014. | |
Perier-Muzet M, Thomas L, Poulalhon N, et al. Melanoma patients under vemurafenib: prospective follow-up of melanocytic lesions by digital dermoscopy. J Invest Dermatol. 2014;134(5):1351–1358. | |
Argenziano G, Lallas A, Longo C, Moscarella E, Raucci M, Zalaudek I. Dormant melanomas or changing nevi? J Invest Dermatol. 2014;134(5):1196–1198. | |
Tronnier M, Smolle J, Wolff HH. Ultraviolet irradiation induces acute changes in melanocytic nevi. J Invest Dermatol. 1995;104(4):475–478. | |
Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358–365. | |
Menzies AM, Long GV. Recent advances in melanoma systemic therapy. BRAF inhibitors, CTLA4 antibodies and beyond. Eur J Cancer. 2013;49(15):3229–3241. | |
Dummer R, Robert C, Nyakas M, et al. Initial results from a phase I, open-label, does escalation study of the oral BRAF inhibitor LGX818 in patients with BRAF V600 mutant advanced or metatastic melanoma. J Clin Oncol. 2013;31(Suppl 15;abstract 9028). | |
Das Thakur M, Salangsang F, Landman AS, et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature. 2013;494(7436):251–255. | |
Jarkowski A 3rd, Khushalani NI. BRAF and beyond: Tailoring strategies for the individual melanoma patient. J Carcinog. 2014; 13:1. | |
Flaherty KT, Robert C, Hersey P, et al; METRIC Study Group. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367(2):107–114. | |
Adjei AA, Cohen RB, Franklin W, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J Clin Oncol. 2008;26(13):2139–2146. | |
Ascierto PA, Berking C, Agarwala, et al. Efficacy and safety of the oral MEK162 in patients with locally advanced and unresectable or metastatic cutaneous melanoma harboring BRAFV600 or NRAS mutations. J Clin Oncol. 2012;30(Suppl;abstract 8511). | |
Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhibition in melanomas with BRAF V600 mutations. N Engl J Med. 2012;367(18):1694–1703. | |
Jakob JA, Bassett RL Jr, Ng CS, et al. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer. 2012;118(16):4014–4023. | |
Woodman SE, Davies MA. Targeting KIT in melanoma: a paradigm of molecular medicine and targeted therapeutics. Biochem Pharmacol. 2010;80(5):568–574. | |
Torres-Cabala CA, Wang WL, Trent J, et al. Correlation between KIT expression and KIT mutation in melanoma: a study of 173 cases with emphasis on the acral-lentiginous/mucosal type. Mod Pathol. 2009;22(11):1446–1456. | |
Minor DR, Kashani-Sabet M, Garrido M, O’Day SJ, Hamid O, Bastian BC. Sunitinib therapy for melanoma patients with KIT mutations. Clin Cancer Res. 2012;18(5):1457–1463. | |
Hodi FS, Corless CL, Giobbie-Hurder A, et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J Clin Oncol. 2013;31(26):3182–3190. | |
Blanchard T, Srivastava PK, Duan F. Vaccines against advanced melanoma. Clin Dermatol. 2013;31(2):179–190. | |
Chambers CA, Kuhns MS, Egen JG, Allison JP. CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu Rev Immunol. 2001;19: 565–594. | |
Hodi FS, O’Day SJ, McDermott, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010; 363(8):711–723. | |
Margolin K, Ernstoff MS, Hamid O, et al. Ipilimumab in patients with melanoma and brain metastases: an open-label, phase 2 trial. Lancet Oncol. 2012;13(5):459–465. | |
Yuan J, Adamow M, Ginsberg BA, et al. Integrated NY-ESO-1 antibody and CD8+ T-cell responses correlate with clinical benefit in advanced melanoma patients treated with ipilimumab. Proc Natl Acad Sci U S A. 2011;108(40):16723–16728. | |
Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011; 364(26):2517–2526. | |
Ascierto PA, Simeone E, Giannarelli D, Grimaldi AM, Romano A, Mozzillo N. Sequencing of BRAF inhibitors and ipilimumab in patients with metastatic melanoma: a possible algorithm for clinical use. J Translat Med. 2012;10:107. | |
Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A. 2002;99(19):11293–12297. | |
Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. | |
Grosso J, Horak CE, Inzunza D, et al; Keck School of Medicine of the University of Southern California, Los Angeles, CA; Dako North America, Inc., Carpinteria, CA. Association of tumor PD-L1 expression and immune biomarkers with clinical activity in patients (pts) with advanced solid tumors treated with nivolumab (anti-PD-1; BMS-936558; ONO4538) J Clin Oncol. 2013;31(Suppl;abstract 3016). | |
Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369(2):134–144. | |
Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–2465. | |
Hamid O, Sosman JA, Lawrence DP, et al. Clinical activity, safety, and biomarkers of MPDL3280A, an engineered PD-L1 antibody in patients with locally advanced or metastatic melanoma (mM). J Clin Oncol. 2013;31(Suppl;abstract 9010). | |
Hersey P, Gallagher S. Intralesional immunotherapy for melanoma. J Surg Oncol. 2014;109(4):320–326. | |
Morton DL, Eilber FR, Holmes EC, et al. BCG immunotherapy of malignant melanoma: summary of a seven-year experience. Ann Surg. 1974;180(4):635–643. | |
Dranoff G. GM-CSF-secreting melanoma vaccines. Oncogene. 2003;22(20):3188–3192. | |
von Wussow P, Block B, Hartmann F, Deicher H. Intralesional interferon-alpha therapy in advanced malignant melanoma. Cancer. 1988;61(6):1071–1074. | |
Temple-Oberle CF, Byers BA, Hurdle V, Fyfe A, McKinnon JG. Intra-lesional interleukin-2 therapy for in transit melanoma. J Surg Oncol. 2014;109(4):327–331. | |
Doukas J, Rolland A. Mechanisms of action underlying the immunotherapeutic activity of Allovectin in advanced melanoma. Cancer Gene Ther. 2012;19(12):811–817. | |
Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol. 2012;30(7):658–670. | |
Andtbacka RHI, Collichio FA, Amatruda T, et al; Department of Biostatistics and Epidemiology; Amgen, Inc.; Rush University Medical Center. OPTiM: A randomized phase III trial of talimogene laherparepvec (T-Vec) versus subcutaneous (SC) granulocyte-macrophage colony-stimulating factor (GM-CSF) for the treatment (tx) of unresected stage IIIB/C and IV melanoma. J Clin Oncol. 2013;31(Suppl;abstract LBA9008). | |
Srivastava N, McDermott D. Update on benefit of immunotherapy and targeted therapy in melanoma: the changing landscape. Cancer Manag Res. 2014;6:279–289. | |
Slominski AT, Carlson JA. Melanoma resistance: a bright future for academicians and a challenge for patient advocates. Mayo Clin Proc. 2014;89(4):429–433. | |
Davar D, Tarhini AA, Kirkwood JM. Adjuvant immunotherapy of melanoma and development of new approaches using neoadjuvant approach. Clin Dermatol. 2013;31(3):237–250. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.