")
Back to Journals » International Journal of Nanomedicine » Volume 11
Transportan in nanocarriers improves skin localization and antitumor activity of paclitaxel
Authors Pepe D, Carvalho V, McCall M, de Lemos D, Lopes L
Received 7 October 2015
Accepted for publication 16 January 2016
Published 11 May 2016 Volume 2016:11 Pages 2009—2019
DOI https://doi.org/10.2147/IJN.S97331
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Webster
Dominique Pepe,1 Vanessa FM Carvalho,2 Melissa McCall,1 Débora P de Lemos,2 Luciana B Lopes1,2
1Department of Pharmaceutical Sciences, Albany College of Pharmacy and Health Sciences, Albany, NY, USA; 2Department of Pharmacology, Institute of Biomedical Sciences, University of São Paulo, São Paulo, Brazil
Abstract: In this study, the ability of nanocarriers containing protein transduction domains (PTDs) of various classes to improve cutaneous paclitaxel delivery and efficacy in skin tumor models was evaluated. Microemulsions (MEs) were prepared by mixing a surfactant blend (polyoxyethylene 10 oleoyl ether, ethanol and propylene glycol), monocaprylin, and water. The PTD transportan (ME-T), penetratin (ME-P), or TAT (ME-TAT) was added at a concentration of 1 mM to the plain ME. All MEs displayed nanometric size (32.3–40.7 nm) and slight positive zeta potential (+4.1 mV to +6.8 mV). Skin penetration of paclitaxel from the MEs was assessed for 1–12 hours using porcine skin and Franz diffusion cells. Among the PTD-containing formulations, paclitaxel skin (stratum corneum + epidermis and dermis) penetration at 12 hours was maximized with ME-T, whereas ME-TAT provided the lowest penetration (1.6-fold less). This is consistent with the stronger ability of ME-T to increase transepidermal water loss (2.4-fold compared to water) and tissue permeability. The influence of PTD addition on the ME irritation potential was assessed by measuring interleukin-1α expression and viability of bioengineered skin equivalents. A 1.5- to 1.8-fold increase in interleukin-1α expression was induced by ME-T compared to the other formulations, but this effect was less pronounced (5.8-fold) than that mediated by the moderate irritant Triton. Because ME-T maximized paclitaxel cutaneous localization while being safer than Triton, its efficacy was assessed against basal cell carcinoma cells and a bioengineered three-dimensional melanoma model. Paclitaxel-containing ME-T reduced cells and tissue viability by twofold compared to drug solutions, suggesting the potential clinical usefulness of the formulation for the treatment of cutaneous tumors.
Keywords: microemulsion, nanocarriers, protein transduction domains, paclitaxel, skin, transdermal
Introduction
It is well known that drug delivery into/across the skin is restricted by the cutaneous barrier provided mainly (but not exclusively) by the stratum corneum (SC).1 Drugs displaying molecular weight <500 g/mol and log P between 0.5 and 2.5 are considered good candidates for topical delivery.2 However, the majority of the drugs available do not meet these requirements, and penetration-enhancing strategies are needed to improve their skin penetration. One example is the antitumor agent paclitaxel; as a large and lipophilic drug (MW =853.9 g/mol; log P =3.5), its penetration is limited.3 Only formulations for systemic use are currently available, but despite their efficacy against tumors affecting the skin, severe systemic adverse effects limit their use in dermatology.4–6 Thus, nanocarriers capable of improving paclitaxel cutaneous localization while limiting its transdermal permeation could potentially allow the use of this drug for the treatment of skin tumors.3,5,7,8 In this context, this study was aimed at evaluating the cutaneous delivery, safety, and efficacy of paclitaxel nanocarriers in bioengineered models of healthy and tumor-containing skin.
Different types of nanocarriers have been investigated for topical paclitaxel delivery; however, cutaneous localization of the drug is not often given priority over transdermal delivery during formulation design.3,7–10 Thus, in the first part of this study, the ability of a selected nanocarrier to improve the amount of drug delivered into skin layers was optimized. Microemulsions (MEs) were selected as nanocarriers due to their simple preparation (since there is no special requirement in terms of energy supply), thermodynamic stability, and versatility of composition, which can reduce costs.11,12 Considering that drug penetration can be enhanced when cationic lipids, protein transduction domains (PTDs), and pore-forming antimicrobial peptides are included in nanocarriers,13–16 we evaluated the effects of adding three different types of PTDs on the microemulsion safety and penetration-enhancing ability.
The selection of PTDs was based on their classification as primary, secondary, and nonamphipathic compounds.17 As a primary amphipathic PTD, transportan was selected. This type of PTD generally contains 20 or more amino acids and has sequential cationic and hydrophobic residues and affinity for neutral and anionic lipid membranes.18 Transportan seems to bind and insert into membranes, reduce surface tension at certain concentrations, and even induce phase transformation.17,19,20 Penetratin, representative of secondary amphipathic PTDs, was used due to its penetration-enhancing effect demonstrated by our group and others.13,21 Secondary amphipathic PTDs display their amphipathic properties after interacting with certain lipids and adoption of a helical structure separating charged and uncharged residues.17 TAT, one of the most studied PTDs, was selected to represent nonamphipathic peptides. These compounds generally have a large number of cationic amino acid residues and bind to negatively charged sulfated glycosaminoglycans of the surface of mammalian cells at low micromolar concentrations, followed by clustering and endocytic uptake.22,23 Macropinocytosis has been demonstrated to mediate TAT cell entrance.23
Our results demonstrate that the nature of PTD plays an important role in the penetration-enhancing properties of topical nanocarriers. Among the PTDs studied, addition of transportan optimized drug cutaneous localization without compromising formulation safety. The transportan-containing ME improved drug efficacy against cutaneous tumor cells and three-dimensional (3D)-bioengineered skin cancer models, indicating the potential clinical usefulness of the nanocarrier for the treatment of cutaneous tumors.
Material and methods
Materials
BRIJ 97 (polyoxyethylene 10 oleoyl ether) and propylene glycol were obtained from Sigma-Aldrich Co. (St Louis, MO, USA). Monocaprylin was kindly supplied by Abitec Corporation (Janesville, WI, USA), and myvacet oil (diacetylated monoglycerides from soybean oil) was obtained from Quest (Norwich, NY, USA). Acetonitrile, ethanol, and methanol were purchased from Mallinckrodt Baker (Phillipsburg, NJ, USA), and paclitaxel from Polymed Therapeutics (Houston, TX, USA). Penetratin, transportan, and TAT were purchased from Anaspec (Fremont, CA, USA).
Methods
Preparation of MEs
The ME used in this study was previously developed by our group based on the water titration method.13 A surfactant mixture was composed of BRIJ:ethanol:propylene glycol at 2:1:1 (w/w/w) and was mixed with monocaprylin (oil phase) at 1.3:1 ratio, followed by addition of the aqueous phase at 43%. This concentration of water has been demonstrated to be sufficient to create an oil-in-water system.13 The pH of the formulations was kept between 5 and 5.5, because the skin surface pH ranges between 5 and 5.9, and formulations displaying pH closer to this range are generally considered safe.24
Paclitaxel was incorporated in the oil phase to obtain a final concentration of 0.5% (w/w), whereas penetratin, transportan, or TAT was added to the aqueous phase to obtain a final concentration of 1 mM. Other peptides were able to increase drug penetration when not covalently attached at this concentration, which justifies its selection.25 It is important to note that, in this study, PTDs were mixed within the ME; it was envisioned that PTDs would function as penetration enhancers and diffuse out of the formulation and into the skin to modulate the barrier function. This approach differs from using noncovalent interactions to modify the surface of nanocarriers with PTDs, such as using metal chelating lipids that take advantage of interactions between chelated divalent metal ions and a short sequence of histidine residues added to the N- or C-terminus of proteins.26 Interaction of PTDs with the oil–water interface of ME droplets might be possible depending on peptide concentration and amphiphilic properties. Penetratin, for example, seemed to intercalate between monoolein hydroxyl headgroups and promote shrinkage in the reversed hexagonal phase lattice parameter.21
The MEs containing paclitaxel and either TAT (ME-TAT), penetratin (ME-P), or transportan (ME-T) were characterized for their isotropicity using polarized light microscopy (Axiotop; Zeiss, Thornwood, NY, USA). Internal phase diameter and zeta potential were determined using a Zetasizer nanoseries instrument (Malvern Instruments, Malvern, UK) at room temperature.
In vitro skin penetration
Skin penetration of paclitaxel from the selected MEs was studied using Franz diffusion cells (diffusion area of 1 cm2; Laboratory Glass Apparatus, Inc, Berkeley, CA, USA), with porcine ear skin as model tissue and a receptor phase (3 mL) that consisted of 100 mM phosphate buffer (pH 7.4) containing 20% of ethanol (v/v) maintained at 37°C under constant stirring. Ethanol was added to aid drug solubility in aqueous medium as previously described.8
MEs (100 mg) were placed in the donor compartment of diffusion cells for 1–12 hours. At the end of the experiment, skin samples were rinsed with water to remove excess formulation, and the SC was separated from the epidermis (E) and dermis (D) by tape stripping. Fifteen pieces of tape were used; the first was discharged and the others were placed in conical tubes containing 4 mL of methanol for drug extraction.8 The remaining skin (viable epidermis + dermis, ED) was cut in small pieces, placed into conical tubes containing 2 mL of methanol, homogenized using a tissue homogenizer (Biospec Products, Bartlesville, OK, USA), and bath sonicated for 20 minutes. Using this method, drug recovery from skin layers was within 85%–93%.7 Paclitaxel delivered into SC, ED, and receptor phase was assayed by high-performance liquid chromatography (HPLC) after sample filtration (0.45 μm pore PTFE membranes). The assay was performed using a Shimadzu Prominence HPLC system equipped with a pump model LC-20AB, an autosampler model SIL-20A, a photodiodoarray detector model SPD-M20A set at 228 nm, and a Phenomenex C18 column (maintained at 25°C). The mobile phase was composed of 6:4 (v/v) acetonitrile:water at a flow rate of 1.0 mL/min.
To investigate whether delivery of paclitaxel occurred throughout the surface of the skin or was limited to certain structures the distribution of a fluorescent derivative of paclitaxel (Flutax, an Oregon Green 488 conjugate; Thermo Fisher Scientific, Waltham, MA, USA) in the skin was studied using fluorescence microscopy. The fluorescent derivative was incorporated into ME-T (since it delivered the largest amount of paclitaxel, as detailed in the “Results” section). After a 12-hour application of 100 mg of the fluorescent drug-loaded formulation, the diffusion area of the skin was carefully cleaned, frozen, embedded in Tissue-Tek OCT compound (Pelco International, Redding, CA, USA) and sectioned at 14 μm. The sections were analyzed under a 20× objective in a fluorescence microscope equipped with a filter for fluorescein isothiocyanate (Olympus Corporation, Tokyo, Japan). Time of exposure was fixed to minimize interference of autofluorescence, which was assessed with untreated skin sections.
ME effect on the skin barrier
Transepidermal water loss was assessed as an index of the ability of MEs to induce changes in the cutaneous barrier.27,28 Transepidermal water loss was measured before and after treatment with unloaded (not containing drug) plain ME, ME-P, ME-T, ME-TAT, or water using a closed chamber evaporimeter (Vapometer; Delfin Technologies Ltd., Kuopio, Finland) equipped with an adaptor to fit the diffusion cell (diffusion area of 1 cm2) opening.28 Skin sections mounted in diffusion cells were treated with 150 mg of the formulations for 5 minutes or 8 hours. The 8-hour period was selected as it was the earliest time point at which differences in the barrier-disrupting effect of solutions and MEs could be observed.29 After each period, skin samples were carefully wiped with tissue paper to remove the formulations, replaced back on the diffusion cells, and left undisturbed for 15 minutes before measuring the transepidermal water loss (10 seconds).30 The skin was not rinsed since previous results demonstrated the influence of this procedure on transepidermal water loss.31,32 The measurements at 5 minutes were performed to account for any effect of tissue wiping on the integrity of the skin barrier, and results were expressed as Δ transepidermal water loss, calculated as the difference in transepidermal water loss values after treatment for 8 hours and 5 minutes.
Influence of PTD on the irritation potential of formulations
In an attempt to differentiate the potential irritation induced by ME-T, ME-P, and ME-TAT, reconstructed human epidermis (MatTek Corporation, Ashland, MA, USA) was subjected to treatment with these formulations in comparison to PBS (negative control) or Triton (positive control) (25 mg). The protocols using human cells were considered as exempt for requiring approval by the Institutional Review Board of Albany College of Pharmacy and Health Scineces since sources are publicly available. Tissues were incubated at 37°C and 5% CO2 for 2 hours, 5 hours, or 12 hours. After each treatment period, tissues were rinsed with PBS and incubated with 300 μL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution (1 mg/mL) for 3 hours at 37°C and 5% CO2 to account for tissue viability. MTT was then extracted by immersing the tissues in 2 mL of extracting solution (provided by the MatTek Corporation with the tissue kit) overnight. The optical density of the extracted samples was determined at 570 nm (background reading at 650 nm was subtracted from the readings). Tissue viability (%) was plotted as a function of time.
To further differentiate the irritation potential among the formulations, extracellular interleukin-1α (IL-1α) levels were assessed in the culture medium using enzyme-linked immunosorbent assay (ELISA) (Thermo Fisher Scientific) according to the manufacturer’s instructions. This cytokine was selected because its rise is consistent with induction of an inflammatory cascade by irritants.33
Cytotoxic effect of ME-T containing paclitaxel against basal cell carcinoma culture
Based on its superior ability to promote paclitaxel localization in the skin without compromising safety, ME-T was selected for subsequent studies aimed at assessing formulation efficacy.
In this experiment, the cytotoxicity of ME-T against basal cell carcinoma cells was evaluated as an index of efficacy. Basal cell carcinoma cells were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA) and grown at 37°C and 5% CO2 atmosphere in Dulbecco’s Modified Eagle’s Medium (ATCC) containing 10% fetal bovine serum (Thermo Fisher Scientific) and additional penicillin and streptomycin (1%). For the cellular viability assay, cells were plated in 96-well plates (6,000 cells/well) and treated with either ME-T or propylene glycol containing paclitaxel diluted in cell culture medium (10–100 μg/mL of formulation in medium; paclitaxel concentration in medium varying from 0.05–0.5 μg/mL) for 24 hours.7,34 Considering that paclitaxel is a cycle-specific drug (cells are arrested in G2/M phase), an extra step was performed before the viability assessment to ensure time for the drug to act and to maximize cytotoxicity: following treatment, cells were rinsed with PBS and incubated in fresh medium (without treatment) for 36 hours.7,35 In previous studies, cells were maintained for periods of up to 6 days to maximize cytotoxicity.35 Cell viability after treatment with unloaded propylene glycol and ME-T diluted in cell culture medium (10–100 μg/mL) was also assessed to account for the effects of unloaded formulation.
Viability was determined using a cell proliferation assay reagent (CellTiter 96 Aqueous One Solution; Promega Corporation, Fitchburg, WI, USA). Cell culture medium (100 μL) containing 20 μL of the cell proliferation assay reagent was added to each well, and after incubation for 2 hours at 37°C and 5% CO2, absorbance was recorded at 490 nm using a plate reader (SpectraMax; Molecular Devices LLC, Sunnyvale, CA, USA).
Cytotoxic effect of ME-T containing paclitaxel against reconstructed skin models of melanoma
To verify whether ME-T would be a viable strategy for treatment of skin tumors, its efficacy against more complex 3D models of the disease was assessed. The only commercially available bioengineered model is a melanoma model. It is acknowledged that the melanoma model presents distinct characteristics compared to nonmelanoma tumors (including basal cell carcinoma, squamous cell carcinoma) and other pretumor lesions that are considered treatable by topical formulations (such as low risk, superficial disease, and when the patient is a poor surgical candidate).36,37 However, use of this model was valuable as it allowed us to evaluate whether paclitaxel penetrates into the tissue at sufficient amounts to cause cytotoxic effects. Reconstructed skin models of melanoma (MLNM-FT-A375; MatTek Corporation) were incubated at 37°C and 5% CO2 and treated with ME-T and myvacet oil (control vehicle) either unloaded or loaded with paclitaxel (25 mg) at 0.5% (w/w) every 3 days for 2 hours during a total period of 12 days. In this regimen, after the 2-hour treatment period, tissues were rinsed with PBS and incubated without formulation until the next treatment. The 2-hour treatment period was chosen as unloaded ME-T induced no significant reductions in tissue viability compared to PBS (see Results, item “Irritation potential of PTD-containing formulations”). The viability of the tissues was determined using MTT as described in the “Influence of PTD on the irritation potential of formulations” section.
In an attempt to establish a relationship between tissue concentration of the drug and effect, paclitaxel was quantified in a separate set of tissues submitted to the same treatment scheme. At the end of the experimental period, tissues were rinsed with PBS, homogenized with 1.5 mL of methanol, and the drug was assayed by HPLC as described in the “In vitro skin penetration” section.
Statistical analyses
The results are reported as mean ± standard deviation (SD). Data were statistically analyzed using the analysis of variance test followed by Tukey’s post hoc test (GraphPad Prism software). Values were considered significantly different when P<0.05.
Results
Formulation characterization
PTD addition did not preclude ME formation, and all systems were fluid and isotropic as observed by polarized light microscopy. Droplet size was in the nanometer range, varying between 32.3 nm and 40.7 nm (Table 1, polidispersity index <0.27), and all formulations displayed a small positive charge, with TAT-containing formulations displaying the highest value of zeta potential (+6.8 mV). As previously reported, the plain ME displayed a similar size (39.5±2.1 nm) but slight negative zeta potential (−1.8±0.1 mV), demonstrating that PTD addition imparted a small positive charge in the system.13 All PTD-containing formulations were cationic, and thus, differences among the nature of charge (cationic versus anionic) should not interfere with formulation-mediated delivery as suggested by Baspinar and Borchert.38
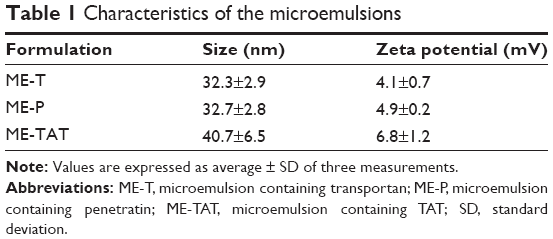 | Table 1 Characteristics of the microemulsions |
In vitro skin penetration
Independent of the presence and type of PTD, paclitaxel was detected mostly in the SC at 1 hour, suggesting a fast penetration in this layer. Skin penetration increased with time, and significantly larger amounts (P<0.01) of drug were quantified in the SC and viable layers of the skin (ED) at 12 hours compared to 1 hour for all formulations (Figure 1).
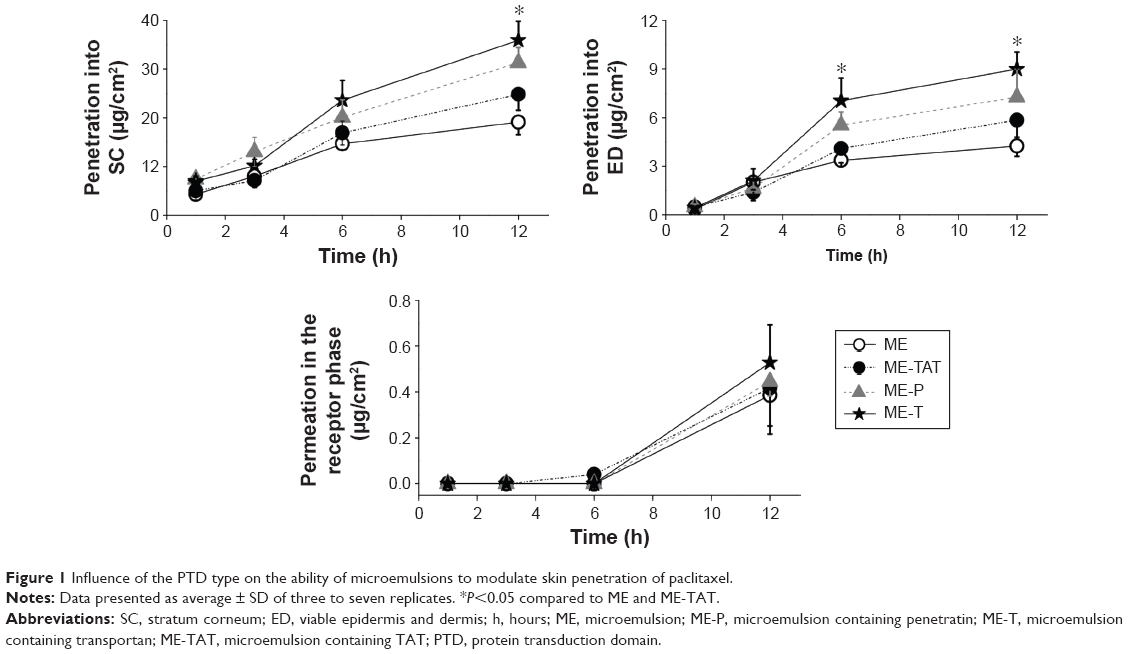 | Figure 1 Influence of the PTD type on the ability of microemulsions to modulate skin penetration of paclitaxel. |
Comparing the penetration mediated by PTD-containing MEs with the plain formulation, only ME-T increased paclitaxel delivery into ED at both 6 hours and 12 hours in a significant manner (P<0.05). At 12 hours, ME-P also promoted a significant (P<0.05) increase in paclitaxel delivery to ED, while ME-TAT failed to do so. Among the PTD-containing formulations, paclitaxel skin (SC + ED) penetration at the longest time point studied (12 hours) was maximized with ME-T, whereas ME-TAT provided the lowest penetration (1.6-fold less). ME-T-mediated paclitaxel penetration into ED was significantly (P<0.05) higher at both 6 hours and 12 hours (1.6- to 1.8-fold) compared to ME-TAT. ME-P-mediated delivery was intermediate, and no significant difference (P>0.05) was observed compared to the either ME-T or ME-TAT at any of the time points studied.
Taken together, these results support previous observations that, although the presence of positive charge in nanocarriers has been associated with improved penetration,38 it may not be the only factor influencing the penetration-enhancing effect of the studied MEs. The nature of the PTD seems to play a role. When it comes to transdermal delivery, there was no significant difference among the formulations (delivery of paclitaxel varied between 0.36 μg/cm2 and 0.44 μg/cm2).
To better understand the influence of the various PTDs on the kinetics of drug transport into the skin, we evaluated the rate of paclitaxel penetration into the SC and viable skin layers (Figure 2). Considering penetration into SC, all formulations displayed a maximum rate at 1 hour, suggesting fast drug penetration into this layer. ME-P and ME-T displayed similar maximum rates, while lower values were displayed by ME-TAT and the plain ME. Maximum rates of paclitaxel penetration into viable skin layers were achieved at a longer period of time (6 hours), with ME-T displaying the highest rate (1.75 μg/cm2/h). These results suggest that the presence and nature of the PTD do not influence the time necessary to achieve the maximum rate of paclitaxel penetration; however, they influence the maximum rate value obtained, especially into viable layers. Transportan-containing MEs displayed the most pronounced effect.
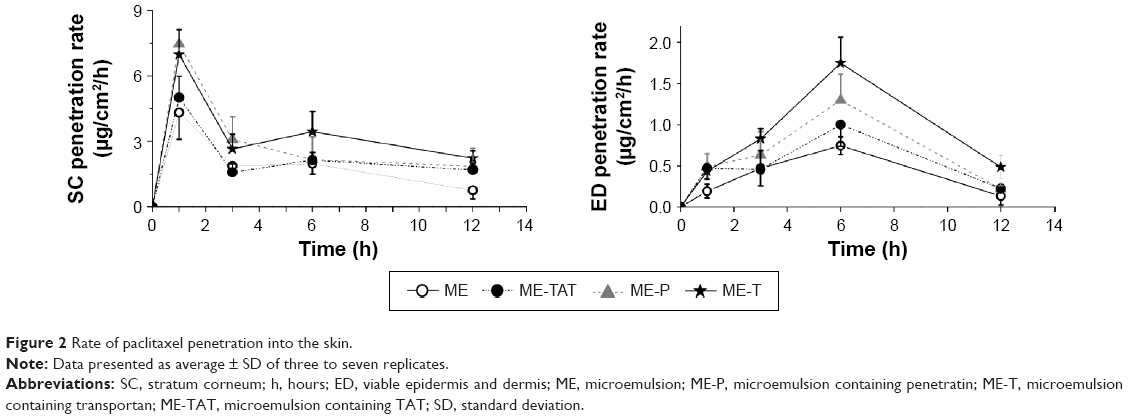 | Figure 2 Rate of paclitaxel penetration into the skin. |
Considering that ME-T delivered the largest amounts of paclitaxel, drug distribution in the skin promoted by this formulation was studied using fluorescence microscopy. After administration of ME-T containing the fluorescent derivative of paclitaxel for 12 hours, the fluorescence seemed fairly homogenously dispersed on the tissue surface and viable skin layers, suggesting that ME-mediated delivery is not limited to certain skin structures/shunts but may occur throughout the SC (Figure 3).
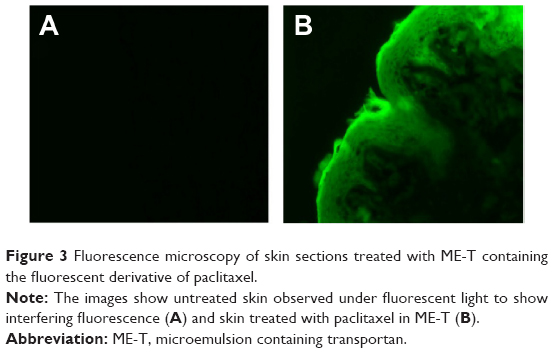 | Figure 3 Fluorescence microscopy of skin sections treated with ME-T containing the fluorescent derivative of paclitaxel. |
ME effect on the skin barrier
To investigate whether the superiority of transportan-containing ME to improve paclitaxel penetration could be attributed to a stronger disruption in the skin barrier, the transepidermal water loss was evaluated and compared with water and the other formulations (Figure 4). Compared to water, all MEs increased transepidermal water loss, but the effect of ME-T was the most pronounced (P<0.001). These results suggest that transportan addition increased the disruptive effect of the ME more strongly. Additionally, the results also suggest that formulation-induced alterations in the barrier function could be detected at an earlier time point compared to a previous study in which the effect of PTD-containing nanocarriers was studied for 12 hours.13
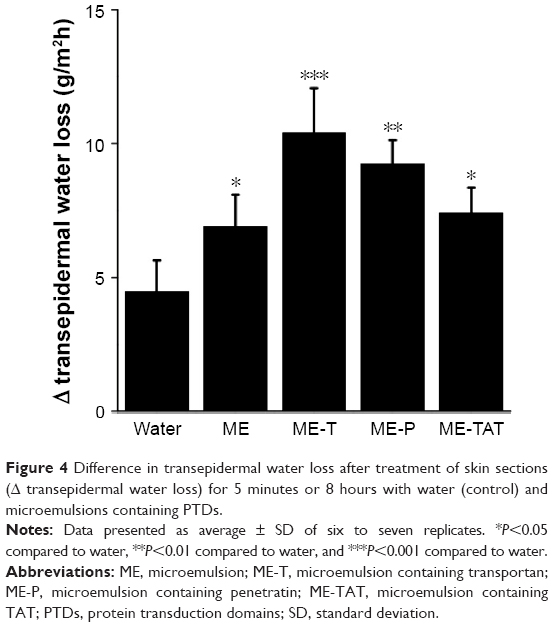 | Figure 4 Difference in transepidermal water loss after treatment of skin sections (Δ transepidermal water loss) for 5 minutes or 8 hours with water (control) and microemulsions containing PTDs. |
Irritation potential of PTD-containing formulations
We next evaluated whether the type of PTD influenced the irritation potential of the MEs. PBS is considered safe and, as previously observed,8 did not reduce tissue viability during the time period studied (Figure 5A). Compared to PBS, the viability of the tissues treated with Triton was significantly (P<0.05) reduced to 73.9%±4.5% after 2 hours, while a longer period of time (5 hours) was required for the PTD-containing MEs to display similar effects. At 5 hours, Triton-treated tissues displayed significantly lower (P<0.05) viability than ME-treated tissues, suggesting the lower irritation potential of the formulations. Comparing the MEs among each other, ME-T-treated tissues displayed lower, but not significantly different, viability at 12 hours compared to ME-P or ME-TAT.
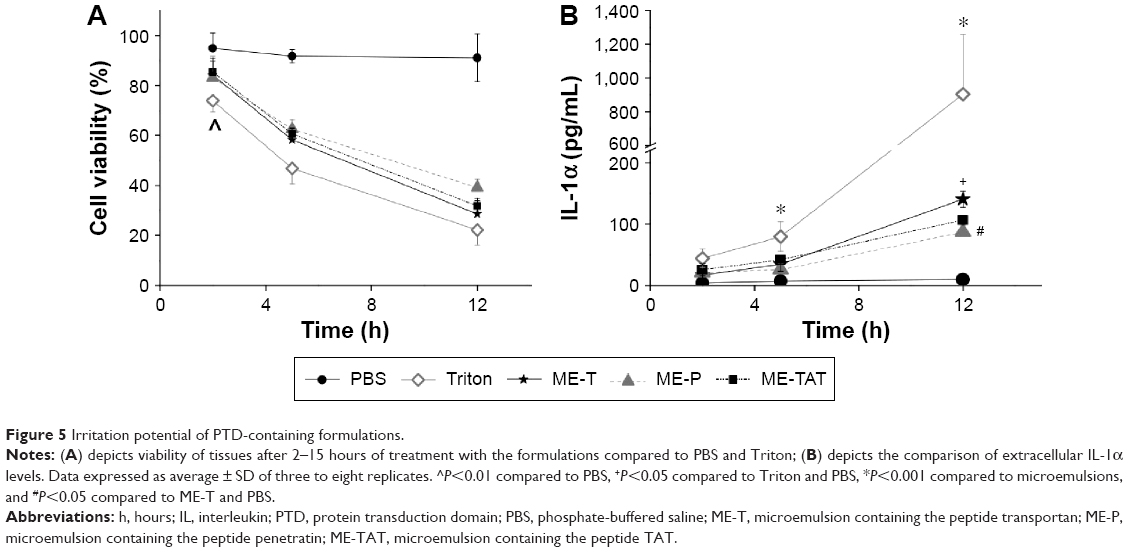 | Figure 5 Irritation potential of PTD-containing formulations. |
IL-1α is believed to be one of the cytokines involved in the initiation of the inflammatory cascade in response to skin exposure to irritants. Measurements of IL-1α released from keratinocytes of epidermal skin equivalents have been used to predict and rank the irritation potential of formulations.39 We assessed the extracellular levels of this cytokine in an attempt to further differentiate the influence of the various PTDs on the formulation irritation potential (Figure 5B). At the longest time point studied (12 hours), ME-T promoted the most pronounced increase in IL-1α release among the MEs (1.8- and 1.5-fold compared to ME-P and ME-TAT, P<0.05), but these cytokine levels were still 5.8-fold lower than those induced by Triton. These results suggest that the transportan-containing ME displays the most pronounced irritation potential among the MEs, which is not unexpected since it also presented a stronger ability to increase transepidermal water loss. However, given the fact that its effects on IL-1α release were less than those of Triton, it is reasonable to suggest that ME-T should be better tolerated by the skin, and thus, addition of transportan does not compromise formulation safety.
Cytotoxicity of ME-T containing paclitaxel against basal cell carcinoma culture
Since ME-T maximized the cutaneous delivery of paclitaxel and was safer than the moderate irritant Triton, the efficacy of this formulation against basal cell carcinoma cells was studied and compared to a drug solution in propylene glycol. Cell viability after treatment with the unloaded formulations at any of the concentrations used was at least 80% (Figure 6). Paclitaxel-loaded solution and ME-T led to significant reductions in cell viability (P<0.05) compared to the unloaded formulations, suggesting that the stronger cytotoxicity of the loaded formulations derive from the presence of the drug and that incorporation in the ME did not hinder drug activity.
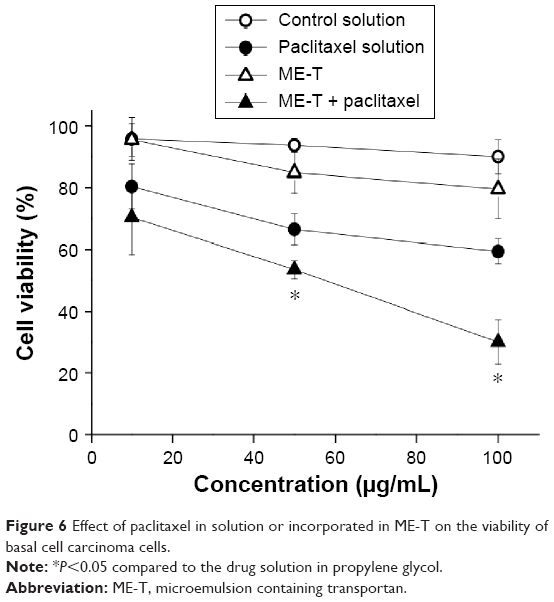 | Figure 6 Effect of paclitaxel in solution or incorporated in ME-T on the viability of basal cell carcinoma cells. |
Drug cytotoxicity was dose dependent. When the concentration of paclitaxel solution in the treatment medium was 10 μg/mL (and drug concentration was 0.05 μg/mL), cell viability was 80.4%, which represents a reduction of 16% compared to the unloaded vehicle. Increasing the concentration of drug solution to 100 μg/mL (and drug concentration to 0.5 μg/mL) further reduced cell viability to 59.4% (P<0.05). Compared to the drug solution, treatment with paclitaxel-loaded ME-T promoted a curve shifting, ie, treatment with the same concentration of the drug-loaded ME resulted in lower viability. At the largest concentration (100 μg/mL), ME-T promoted a twofold reduction in cell viability (P<0.05). Considering that there was no significant difference in cell viability when comparing the unloaded formulations at the concentrations used, the more pronounced reduction in cell viability provided by the loaded ME-T may potentially result from a more efficient drug delivery and higher uptake of the drug.
Cytotoxicity of ME-T containing paclitaxel against reconstructed skin models of melanoma
Having demonstrated the cytotoxic effects of loaded ME-T against tumor cells, we next evaluated the formulation efficacy against a reconstructed skin model of melanoma (MLNM-FT-A375; MatTek Corporation). Similar to what we observed in cells, tissue viability after treatment with paclitaxel-containing ME-T was significantly lower (2.2-fold, P<0.005) than that with the unloaded ME (Figure 7), demonstrating that the presence of paclitaxel promoted a more pronounced decrease in tissue viability.
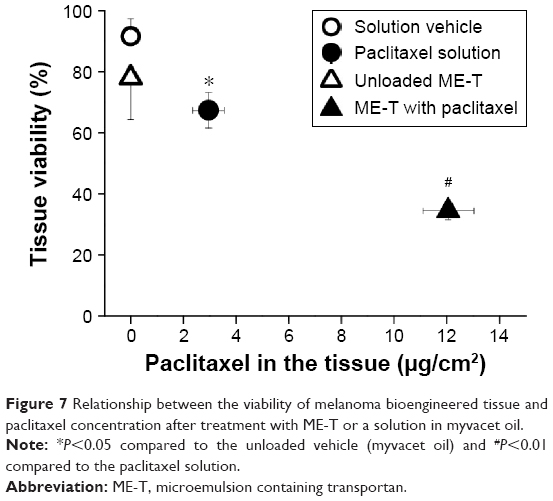 | Figure 7 Relationship between the viability of melanoma bioengineered tissue and paclitaxel concentration after treatment with ME-T or a solution in myvacet oil. |
To verify whether drug incorporation into ME-T increased cytotoxicity compared to simple solutions, tissue viability after treatment with paclitaxel-loaded myvacet oil solution at the same concentration was assessed. As can be observed in Figure 7, cell viability was approximately twofold lower after treatment with ME-T (67.4%±5.8% and 34.5%±2.9% for myvacet solution and ME-T, respectively), demonstrating the benefit of the nanocarrier.
To establish a relationship between reduction in cell viability and drug delivery to the skin, paclitaxel concentration in the tissue was assessed. Paclitaxel delivery into the skin after treatment with ME-T was 4.5-fold higher (P<0.005) compared to the solution. These results demonstrated that the more pronounced reduction in tissue viability mediated by ME-T resulted from a larger amount of drug delivered into the skin.
Discussion
The interest in topical formulations aimed at increasing localization of paclitaxel in the skin has grown over the past years. In a previous study, we assessed the skin penetration of paclitaxel from cationic MEs containing either the PTD penetration or the lipid phytosphingosine.13 In that study, only one PTD (penetratin) and one time point (12 hours) were evaluated. The superiority of the penetratin-containing ME encouraged us to compare the influence of PTDs from various classes on the skin permeability, penetration kinetics, and formulation irritation as described in this study. Furthermore, in this study, we advanced in formulation evaluation and assessed the potential clinical usefulness of a selected system by studying its efficacy against cutaneous tumor cells and a bioengineered 3D melanoma model.
Compared to other formulations studied, MEs have the advantage of easy preparation, thermodynamic stability, and versatility of composition, which can reduce costs.11,12 However, due to the high concentration of surfactants and addition of penetration enhancers (including the PTDs), MEs may display increased irritation potential.40 Thus, one of the goals of this study was to verify whether MEs containing PTDs would be tolerated by the skin. The toxicity of PTDs has been well studied in various cell lines,41,42 but to the best of our knowledge, this is the first report comparing the effects of PTDs from different classes on formulation-induced irritation in bioengineered skin. Among the formulations, the most pronounced effect on IL-1α release was displayed by ME-T. Since this effect was less pronounced than that induced by the moderate irritant Triton, it is reasonable to suggest that the formulation should be better tolerated by the skin, and thus, transportan addition does not compromise formulation safety.
The increase in the IL-1α release mediated by transportan compared to the other PTDs was counterbalanced by its ability to maximize paclitaxel cutaneous delivery. These effects may be related to the more pronounced ability of the formulation to disrupt biological barriers, as demonstrated by the increased transepidermal water loss among all studied MEs. The TAT-containing ME displayed the least effect. The following two conclusions can be drawn from these results: first, as previously observed, the presence of positive charge in nanocarriers does not seem to be the only relevant factor affecting paclitaxel delivery as all PTD-containing MEs displayed similar slight positive charges. Second, not all PTDs might penetrate the skin and function equally like penetration enhancers in a given condition.43,44 Considered a primary amphipathic PTD, transportan is long enough to theoretically span the hydrophobic core of bilayers.17 Occurrence of a connection between the two monolayers through the peptide might lead to bilayer thinning and formation of pores.45 These effects provide reasonable justification for the enhanced ability of ME-T to decrease skin barrier function, while increasing IL-1α release and paclitaxel penetration. Previous studies have suggested a correlation between amphiphilic properties and toxicity of PTDs.42,46
ME-T improved paclitaxel cytotoxicity against cultured basal carcinoma cells compared to a control drug solution, most likely due to a more efficient delivery. This result is consistent with previous reports demonstrating the efficacy of nanocarriers to improve drug cytotoxicity against cutaneous tumor cells.5,47 The stronger cytotoxic effect of paclitaxel-containing ME-T was also observed in melanoma tissues, which displayed viability <40%. This low viability suggests that paclitaxel affects not only cancer cells but also normal cells in the tissue, which is in agreement with other studies demonstrating the cytotoxic and proliferation-inhibiting effects of paclitaxel against several cell types, including normal cells.48,49 This wide-range cytotoxicity reinforces the importance of localizing paclitaxel in the lesions to prevent adverse effects in other tissues/organs. This effect was associated with a higher drug concentration in the tissue, demonstrating the relationship between increased cytotoxicity and enhanced drug delivery by ME-T. It is worth pointing out that formulation-mediated penetration would be more pronounced in the melanoma tissue than in porcine ear skin (the sum of drug in SC and viable epidermis) if we take into consideration the time of application,13 which is consistent with previous studies demonstrating a higher permeability of bioengineered tissues compared to the porcine and human skin. Higher permeation flux values of 500–800 times than those for human skin have been reported for skin equivalent models compared to human skin, and this difference seems to be more pronounced for lipophilic compounds.50,51
PTDs have been most frequently studied for systemic delivery, but the high in vitro uptake rates, the relatively low specificity, and the fast clearance rates observed in several studies suggest that their topical application presents a higher potential.52,53 Our results support the potential of selected PTDs to improve drug topical delivery, leading to a more pronounced efficacy against a bioengineered model of melanoma. Given the fact that the PTDs were simply mixed into ME, their amphiphilic properties may play an important role as demonstrated by the stronger effect of ME-T compared to ME-TAT, at least within the experimental conditions used here. Although we emphasized the effect of the PTD on the penetration-enhancing ability of the ME, it is necessary to consider the ME role on the PTD penetration. As demonstrated by Kim et al,54 the pore-forming peptide magainin alone was not able to affect skin permeability, likely due to its relatively large size and insufficient penetration into the tissue. Skin permeability to fluorescein was increased by 47-fold when magainin was combined with N-lauroyl sarcosine in a 50% ethanol solution,54 which indicates that the use of formulations capable of promoting peptide penetration aids its effect on skin permeability.
Acknowledgments
The authors are grateful to Dr Catherine Dreiza (Arizona State University) for language revision. This study was supported by grant number 2013/16617-7, São Paulo Research Foundation (FAPESP 2013/16617-7) and a Research Starter Grant in Pharmaceutics from PhRMA Foundation. Fellowships from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (VFM Carvalho) and São Paulo Research Foundation (DP de Lemos) are greatly appreciated.
Disclosure
The authors report no conflicts of interest in this work.
References
Menon GK. New insights into skin structure: scratching the surface. Adv Drug Deliv Rev. 2002;54(Suppl 1):S3–S17. | ||
Moser K, Kriwet K, Naik A, Kalia YN, Guy RH. Passive skin penetration enhancement and its quantification in vitro. Eur J Pharm Biopharm. 2001;52(2):103–112. | ||
Khandavilli S, Panchagnula R. Nanoemulsions as versatile formulations for paclitaxel delivery: peroral and dermal delivery studies in rats. J Invest Dermatol. 2007;127(1):154–162. | ||
Barceló R, Viteri A, Muñoz A, Gil-Negrete A, Rubio I, Lopez-Vivanco G. Paclitaxel for progressive basal cell carcinoma. J Am Acad Dermatol. 2006;54(2 Suppl):S50–S52. | ||
Paolino D, Celia C, Trapasso E, Cilurzo F, Fresta M. Paclitaxel-loaded ethosomes(R): potential treatment of squamous cell carcinoma, a malignant transformation of actinic keratoses. Eur J Pharm Biopharm. 2012;81(1):102–112. | ||
Kunstfeld R, Wickenhauser G, Michaelis U, et al. Paclitaxel encapsulated in cationic liposomes diminishes tumor angiogenesis and melanoma growth in a “humanized” SCID mouse model. J Invest Dermatol. 2003;120(3):476–482. | ||
Hosmer JM, Shin SH, Nornoo A, Zheng H, Lopes LB. Influence of internal structure and composition of liquid crystalline phases on topical delivery of paclitaxel. J Pharm Sci. 2011;100(4):1444–1455. | ||
Hosmer JM, Steiner AA, Lopes LB. Lamellar liquid crystalline phases for cutaneous delivery of Paclitaxel: impact of the monoglyceride. Pharm Res. 2013;30(3):694–706. | ||
Utreja P, Jain S, Tiwary AK. Localized delivery of paclitaxel using elastic liposomes: Formulation development and evaluation. Drug Deliv. 2011;18(5):367–376. | ||
Kilfoyle BE, Sheihet L, Zhang Z, Laohoo M, Kohn J, Michniak-Kohn BB. Development of paclitaxel-TyroSpheres for topical skin treatment. J Control Release. 2012;163(1):18–24. | ||
Lopes LB. Overcoming the cutaneous barrier with microemulsions. Pharmaceutics. 2014;6(1):52–77. | ||
McClements DJ. Nanoemulsions versus microemulsions: terminology, differences, and similarities. Soft Matter. 2012;8:1719–1729. | ||
Pepe D, McCall M, Zheng H, Lopes LB. Protein transduction domain-containing microemulsions as cutaneous delivery systems for an anticancer agent. J Pharm Sci. 2013;102(5):1476–1487. | ||
Cohen-Avrahami M, Shames AI, Ottaviani MF, Aserin A, Garti N. HIV-TAT enhances the transdermal delivery of NSAID drugs from liquid crystalline mesophases. J Phys Chem B. 2014;118(23): 6277–6287. | ||
Kim YC, Late S, Banga AK, Ludovice PJ, Prausnitz MR. Biochemical enhancement of transdermal delivery with magainin peptide: modification of electrostatic interactions by changing pH. Int J Pharm. 2008;362(1–2):20–28. | ||
Baspinar Y, Keck CM, Borchert HH. Development of a positively charged prednicarbate nanoemulsion. Int J Pharm. 2010;383(1–2): 201–208. | ||
Ziegler A. Thermodynamic studies and binding mechanisms of cell-penetrating peptides with lipids and glycosaminoglycans. Adv Drug Deliv Rev. 2008;60(4–5):580–597. | ||
Deshayes S, Plénat T, Aldrian-Herrada G, Divita G, Le Grimellec C, Heitz F. Primary amphipathic cell-penetrating peptides: structural requirements and interactions with model membranes. Biochemistry. 2004;43(24):7698–7706. | ||
Pooga M, Hällbrink M, Zorko M, Langel U. Cell penetration by transportan. FASEB J. 1998;12(1):67–77. | ||
Madani F, Lindberg S, Langel U, Futaki S, Gräslund A. Mechanisms of cellular uptake of cell-penetrating peptides. J Biophys. 2011; 2011:414729. | ||
Cohen-Avrahami M, Libster D, Aserin A, Garti N. Penetratin-induced transdermal delivery from H(II) mesophases of sodium diclofenac. J Control Release. 2012;159(3):419–428. | ||
Krämer SD, Wunderli-Allenspach H. No entry for TAT(44-57) into liposomes and intact MDCK cells: novel approach to study membrane permeation of cell-penetrating peptides. Biochim Biophys Acta. 2003;1609(2):161–169. | ||
Wadia JS, Stan RV, Dowdy SF. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat Med. 2004;10(3):310–315. | ||
Schmid-Wendtner MH, Korting HC. The pH of the skin surface and its impact on the barrier function. Skin Pharmacol Physiol. 2006;19(6):296–302. | ||
Kim YC, Ludovice PJ, Prausnitz MR. Optimization of transdermal delivery using magainin pore-forming peptide. J Phys Chem Solids. 2008;69(5–6):1560–1563. | ||
Patlolla RR, Desai PR, Belay K, Singh MS. Translocation of cell penetrating peptide engrafted nanoparticles across skin layers. Biomaterials. 2010;31(21):5598–5607. | ||
Rachakonda VK, Yerramsetty KM, Madihally SV, Robinson RL Jr, Gasem KA. Screening of chemical penetration enhancers for transdermal drug delivery using electrical resistance of skin. Pharm Res. 2008; 25(11):2697–2704. | ||
Herwadkar A, Sachdeva V, Taylor LF, Silver H, Banga AK. Low frequency sonophoresis mediated transdermal and intradermal delivery of ketoprofen. Int J Pharm. 2012;423(2):289–296. | ||
Thomas S, Vieira CS, Hass MA, Lopes LB. Stability, cutaneous delivery, and antioxidant potential of a lipoic acid and alpha-tocopherol codrug incorporated in microemulsions. J Pharm Sci. 2014;103(8):2530–2538. | ||
Mohammed D, Hirata K, Hadgraft J, Lane ME. Influence of skin penetration enhancers on skin barrier function and skin protease activity. Eur J Pharm Sci. 2014;51:118–122. | ||
Voegeli D. The effect of washing and drying practices on skin barrier function. J Wound Ostomy Continence Nurs. 2008;35(1):84–90. | ||
Lehmann L, Keipert S, Gloor M. Effects of microemulsions on the stratum corneum and hydrocortisone penetration. Eur J Pharm Biopharm. 2001;52(2):129–136. | ||
Welss T, Basketter DA, Schröder KR. In vitro skin irritation: facts and future. State of the art review of mechanisms and models. Toxicol In Vitro. 2004;18(3):231–243. | ||
Fojo T, Menefee M. Mechanisms of multidrug resistance: the potential role of microtubule-stabilizing agents. Ann Oncol. 2007;18(Suppl 5):v3–v8. | ||
Desai A, Vyas T, Amiji M. Cytotoxicity and apoptosis enhancement in brain tumor cells upon coadministration of paclitaxel and ceramide in nanoemulsion formulations. J Pharm Sci. 2008;97(7):2745–2756. | ||
Rubin AI, Chen EH, Ratner D. Basal-cell carcinoma. N Engl J Med. 2005;353(21):2262–2269. | ||
Kwasniak LA, Garcia-Zuazaga J. Basal cell carcinoma: evidence-based medicine and review of treatment modalities. Int J Dermatol. 2011;50(6):645–658. | ||
Baspinar Y, Borchert HH. Penetration and release studies of positively and negatively charged nanoemulsions-Is there a benefit of the positive charge? Int J Pharm. 2012;430(1–2):247–252. | ||
Bernhofer LP, Barkovic S, Appa Y, Martin KM. IL-1alpha and IL-1ra secretion from epidermal equivalents and the prediction of the irritation potential of mild soap and surfactant-based consumer products. Toxicol In Vitro. 1999;13(2):231–239. | ||
Hosmer J, Reed R, Bentley MV, Nornoo A, Lopes LB. Microemulsions containing medium-chain glycerides as transdermal delivery systems for hydrophilic and hydrophobic drugs. AAPS PharmSciTech. 2009;10(2):589–596. | ||
Sugita T, Yoshikawa T, Mukai Y, et al. Comparative study on transduction and toxicity of protein transduction domains. Br J Pharmacol. 2008;153(6):1143–1152. | ||
El-Andaloussi S, Jarver P, Johansson HJ, Langel U. Cargo-dependent cytotoxicity and delivery efficacy of cell-penetrating peptides: a comparative study. Biochem J. 2007;407(2):285–292. | ||
Wang YH, Chen CP, Chan MH, et al. Arginine-rich intracellular delivery peptides noncovalently transport protein into living cells. Biochem Biophys Res Commun. 2006;346(3):758–767. | ||
Lopes LB, Brophy CM, Furnish E, et al. Comparative study of the skin penetration of protein transduction domains and a conjugated peptide. Pharm Res. 2005;22(5):750–757. | ||
Pourmousa M, Wong-ekkabut J, Patra M, Karttunen M. Molecular dynamic studies of transportan interacting with a DPPC lipid bilayer. J Phys Chem B. 2013;117(1):230–241. | ||
Saar K, Lindgren M, Hansen M, et al. Cell-penetrating peptides: a comparative membrane toxicity study. Anal Biochem. 2005;345(1):55–65. | ||
Fu H, Shi K, Hu G, et al. Tumor-targeted paclitaxel delivery and enhanced penetration using TAT-decorated liposomes comprising redox-responsive poly(ethylene glycol). J Pharm Sci. 2015;104(3): 1160–1173. | ||
Akhlaghi SP, Saremi S, Ostad SN, Dinarvand R, Atyabi F. Discriminated effects of thiolated chitosan-coated pMMA paclitaxel-loaded nanoparticles on different normal and cancer cell lines. Nanomedicine. 2010;6(5):9. | ||
Choritz L, Grub J, Wegner M, Pfeiffer N, Thieme H. Paclitaxel inhibits growth, migration and collagen production of human Tenon’s fibroblasts – potential use in drug-eluting glaucoma drainage devices. Graefes Arch Clin Exp Ophthalmol. 2010;248(2):197–206. | ||
Schmook FP, Meingassner JG, Billich A. Comparison of human skin or epidermis models with human and animal skin in in-vitro percutaneous absorption. Int J Pharm. 2001;215(1–2):51–56. | ||
Zghoul N, Fuchs R, Lehr CM, Schaefer UF. Reconstructed skin equivalents for assessing percutaneous drug absorption from pharmaceutical formulations. ALTEX. 2001;18(2):103–106. | ||
Sarko D, Beijer B, Garcia Boy R, et al. The pharmacokinetics of cell-penetrating peptides. Mol Pharm. 2010;7(6):2224–2231. | ||
Jarver P, Mager I, Langel U. In vivo biodistribution and efficacy of peptide mediated delivery. Trends Pharmacol Sci. 2010;31(11): 528–535. | ||
Kim YC, Ludovice PJ, Prausnitz MR. Transdermal delivery enhanced by magainin pore-forming peptide. J Control Release. 2007;122(3):375–383. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.