Back to Journals » Drug Design, Development and Therapy » Volume 15
Translational Regulation in Hepatocellular Carcinogenesis
Authors Bracic Tomazic S, Schatz C, Haybaeck J
Received 3 May 2021
Accepted for publication 4 October 2021
Published 14 October 2021 Volume 2021:15 Pages 4359—4369
DOI https://doi.org/10.2147/DDDT.S255582
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Manfred Ogris
Suzana Bracic Tomazic,1,2,* Christoph Schatz,3,* Johannes Haybaeck3,4
1Department of Pathology, Hospital Graz II, Graz, 8020, Austria; 2Faculty of Medicine, University of Maribor, Maribor, 2000, Slovenia; 3Institute of Pathology, Neuropathology and Molecular Pathology, Medical University of Innsbruck, Innsbruck, 6020, Austria; 4Diagnostic & Research Center for Molecular BioMedicine, Institute of Pathology, Medical University Graz, Graz, 8010, Austria
*These authors contributed equally to this work
Correspondence: Johannes Haybaeck
Institute of Pathology, Neuropathology and Molecular Pathology, Medical University of Innsbruck, Müllerstraße 44, Innsbruck, 6020, Austria
Tel +43 512 9003 71300
Fax +43 512 9003 73301
Email [email protected]
Abstract: The mortality of hepatocellular carcinoma (HCC) is distributed unevenly worldwide. One of the major causes is hepatitis B or hepatitis C virus infection and the development and progression of liver cirrhosis. The carcinogenesis of HCC is among others regulated via the mTOR (mechanistic target of rapamycin) signaling pathway and represents a possible method of targeted treatment. The aim of our article was to address the most recent clinical advances and findings of basic studies on the mTOR signaling pathway and the involved factors. Risk factors play a key role in dysregulation of the signaling pathway, where both mTORCs are upregulated and protein synthesis is altered. eIFs and, to a lesser extent, eEFs play an essential role in this process. Whether the factor will be upregulated or downregulated, among others, depends on hepatitis B/C virus infection. The amount of a particular factor in a patient sample lets us know whether HCC recurrence will occur, what is the likelihood of chemoresistance, and what outcome is predicted for patients with an increased value. Our analysis shows that in addition to mTOR, eIF3, eIF4, and eIF5 play an important role, as they can serve as biomarkers for non- and virus-related HCC.
Keywords: mTOR, virus related HCC, non-virus related HCC, cancer, translation initiation, liver
Introduction
Hepatocellular carcinoma (HCC) is a major health problem, as it was the third most common cause of mortality worldwide in 2020.1 Between 1990 and 2016 it ranked as the fifth most common cause of cancer in men and the seventh in women.2
Major risk factors include chronic infection with hepatitis B (HBV) or hepatitis C virus (HCV), exposure to aflatoxin B1, non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH), alcohol-related liver disease like alcoholic steatohepatitis (ASH), diabetes and obesity.1,3,4
Development of HCC begins with exposure to one or more risk factors, which trigger inflammation of liver parenchyma with fibrotic deposition. When cirrhosis and decreased activity occur, the likelihood of developing HCC increases. Changes in the genome, as well as epigenetic modifications, play an important role in further development. Although several authors have already dealt with the molecular profile of HCC and the associated progression or recurrence of the disease,5,6 two main molecular groups were identified: proliferative and non-proliferative HCC.7 A classification based on metabolism was presented.8
HCC classification, treatment, and survival follow the Barcelona Clinic Liver Cancer staging and treatment strategy (BCLC). BCLC presents five prognostic subclasses and specific treatments for each: surgical resection, liver transplantation, ablation, chemoembolization, and systemic therapy.9 Despite the known risk factors and various options for monitoring and diagnosis, 40% of patients with an early stage (stage 0 and A) are suitable for treatment with resection, transplantation or local ablation. Stage B patients profit from chemoembolization and patients with advanced cancer (stage C) receive systemic therapy using sorafenib.10,11 BCLC staging could be upgraded with additional molecular-based information as it is associated with patient recurrence and outcome. Dysregulation of the signaling pathways participating in HCC development could represent additional information for therapy decision making.12
Dysregulation in multiple signaling pathways such as phosphoinositide 3-kinase/protein kinase B/mechanistic target of rapamycin (PI3K/AKT/mTOR), Ras mitogen-activated protein kinase (Ras/Raf/MAPK), Hedgehog (HH), Wnt/β-catenin, Janus kinase-signal transducer activator of transcription factor (JAK/STAT) signaling pathway was found in HCC carcinogenesis.13,14
PI3K/AKT/mTOR signaling pathway has been extensively studied in the development of HCC with and without viral background. Dysregulation of this pathway is key in hepatocarcinogenesis.15
The present review article addresses advances in the mTOR signaling pathway and its factors, allowing us to better understand the treatment possibilities of patients with HCC.
PI3K/AKT/mTOR Pathway
The PI3K/AKT/mTOR pathway is an intracellular signaling pathway that is activated by insulin and various growth factors (Figure 1). PI3K activation phosphorylates and activates AKT, which, among others, activates mTOR, affecting the transcription of p70 or 4EBP1. mTOR has two complexes, mTORC1 and mTORC2. Activation of the signal pathway results in fatty acid synthesis, glycogen synthesis, cell survival, proliferation, autophagy, glycolysis and protein synthesis.16
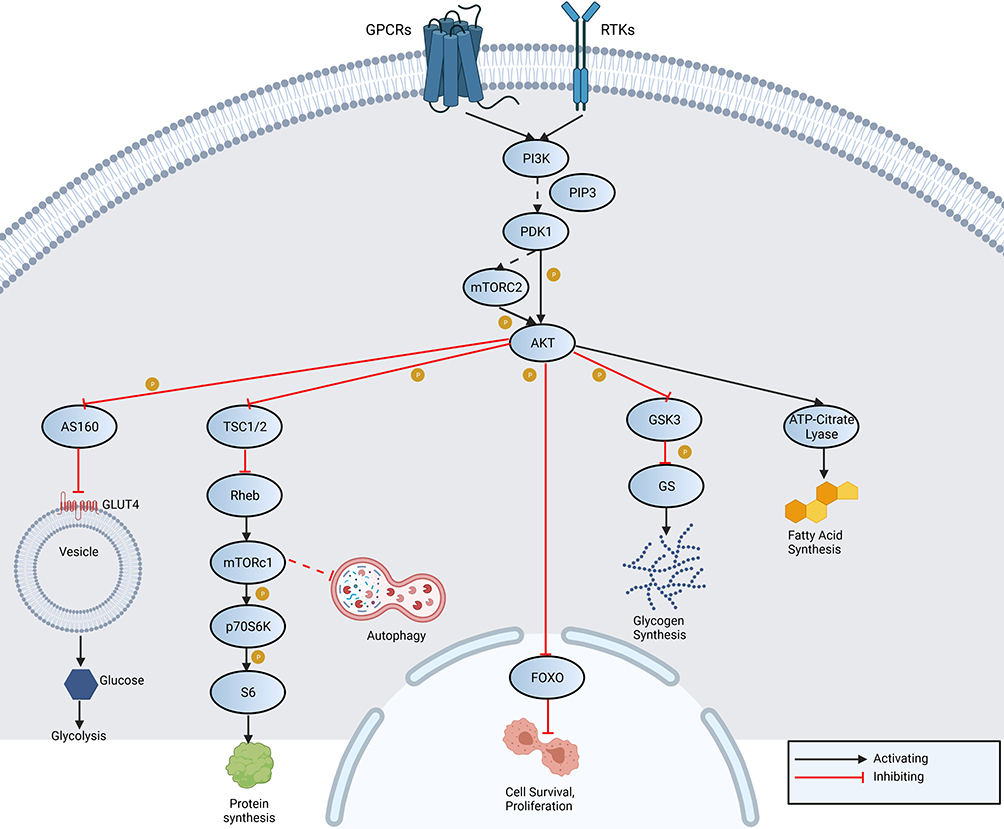 |
Figure 1 An overview of PI3K/AKT/mTOR signaling pathway shows activation of PI3K through receptor tyrosine kinases (RTK) and G-protein coupled receptors (GPCR). It acts directly on AKT or indirectly via mTORC2. Activation of inhibition of adenosine triphosphate (ATP)-citrate lyase, glycogen synthase kinase 3 (GSK3), forkhead box protein O (FOXO), Rab GTPase-activating protein (AS160) and tuberous sclerosis 1/2. As a result fatty acid synthesis, glycogen synthesis, cell survival, proliferation, autophagy, glycolysis and protein synthesis occur. Abbreviations: PIP3, phosphatidylinositol (3,4,5)-trisphosphate; GS, glycogen synthase; GLUT4, glucose transporter type 4; Rheb, Ras homolog enriched in brain; p70S6K, ribosomal protein S6 kinase beta-1; S6, ribosomal protein S6. |
The PI3K/AKT/mTOR signaling pathway is essential for protein synthesis, which is divided into four steps: initiation, elongation, termination, and ribosome recycling. Eukaryotic translation initiation factors (eIFs) thus enter the initiation phase followed by elongation, which is accompanied by eukaryotic translation elongation factors (eEFs). mTOR phosphorylation leads via additional phosphorylation steps to the phosphorylation of 4E-BP-1. 4E-BP1 dissociates and allows the eIF4F complex formation. This results in a cap-dependent activation of translation initiation. The formation of a 43S pre-initiation complex consisting of a 40S small ribosomal unit, eIF3, eIF1, eIF1A, eIF2 and eIF5 starts. Later, the eIF4F complex recognizes the 5’(prime) end of mRNA and binds to the 43S pre-initiation complex. The scanning of the start codon begins. After recognition of the start codon, the recruitment of the 60S ribosome and finally the formation of the 80S ribosome follows. Subsequently, all eIFs are released.17,18
mRNA chain then enters the elongation phase, where amino acids are added into a growing polypeptide chain. There are binding sites for tRNA on the ribosome, namely P (peptidyl) and A (aminoacyl). Upon completion of initiation, Met-tRNAiMet is bound to the P site of the ribosome. The Met-tRNAiMet anticodon is paired with the initial messenger RNA (mRNA) codon, and the second open reading frame (ORF) codon is in place. Elongation begins with aminoacyl-tRNA (RNA transfer) to the A site of the ribosome. Following guanosine triphosphate (GTP) binding, the eukaryotic eEf1A translation elongation factor is activated, and a ternary complex is formed. The eEF1A/GTP/aminoacyl-tRNA complex binds to site A. A peptide bond is formed between amino acids during peptidyl transfer with the amino acid cleaving from the tRNA located at the site P. The mRNA shifts from the 5’ to 3’ end. tRNA that was previously at site A is moved to site P; from site P to exit point (site E), where it leaves the ribosome. This task is performed with an elongation factor 2, which hydrolyzes the GTP.19–21
PI3K/AKT/mTOR in HCC
HCC PI3K/AKT/mTOR signaling (Figure 2) is dysregulated, and both mTOR complexes are involved: mTORC1 regulates protein synthesis, autophagy, glucose and lipid metabolism, and mTORC2 stimulates liver tumorigenesis. Both mTOR complexes were reported to be upregulated in 40–50% of HCC patients.22
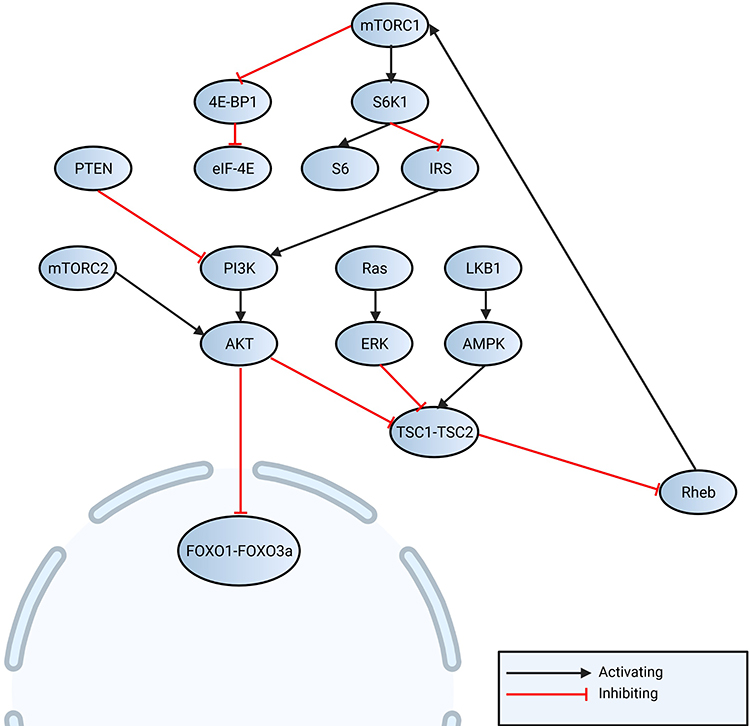 |
Figure 2 Non-virus associated regulation of mTOR is regulated by PI3K-AKT, which regulates FOXO (1 and 3a) as also protein translation by S6 and eIF4E. Liver kinase B1 (LKB1) - adenosine monophosphate-activated protein kinase (AMPK) pathway can activate TSC1-TSC2 complex, which regulates Ras (small GTPase) homolog enriched in brain (Rheb) and activates mTORC1. Activation of mTORC1 can be regulated by mitogen-activated protein kinases/extracellular signal-regulated kinases (MAPK/ERK) pathway. |
Under physiological conditions, the liver is an organ highly involved in lipid metabolism. The liver transforms glucose into fatty acids, which is regulated by the PI3K/AKT/mTOR pathway with insulin and enzymes. It was shown that upregulation of lipid synthesis causes hepatosteatosis and consequently NASH and HCC.23 mTORC2 stimulates the uptake of fatty acids directly from the blood or is involved in de novo synthesis of fatty acids.24
NAFLD is caused by free fatty acids and their accumulation in hepatocytes. 30–40% of patients who later develop NASH25 Center for pathogenesis of NASH are free fatty acids produced by lipolysis in adipose tissue. In the case of over elevated levels of free fatty acids, lipotoxic elements can be formed, which leads to endoplasmic reticulum stress, oxidant stress and activation of inflammation cells.26 Elevated levels of fatty acids can induce NAFLD, which might progress to NASH and lead to HCC development.25,27 It was also shown that increased resistance to insulin was connected to decreased mTORC2-activity.25
Chronic alcohol consumption can lead to liver cirrhosis due to oxidative stress and the formation of steatosis by promoting hepatic lipogenesis. Increased phosphorylation of mTOR, activation of mTORC1 and downstream kinase S6K1 in hepatocytes has to take place.28 Occasional alcohol consumption promotes autophagy as a protective mechanism against hepatocyte damage from alcohol.28,29 Chronic alcohol consumption is frequently associated with oxidative stress, inhibition of hepatocellular autophagy and apoptosis through inhibition of AMPK and activation of mTOR.30
Dysregulated bile acids are associated with liver diseases as increased cell proliferation and autophagy are mediated by AMPK/mTOR pathway. Due to a protective mechanism enhanced cancer cell viability, and consequently, metastases could be formed.31
One of the risk factors for HCC is a chronic infection with HBV or HCV. It is typical for viruses that require their own translation of the host’s proteins, for which they use several different mechanisms, PI3K/AKT/mTOR, for example. As mentioned, this signal pathway pertains to the cap-dependent translation, where 4E-BP is needed for the regulation of the creation of the eIF4F complex. The interaction between eIF4E and 4E-BP is regulated, on the one hand, by phosphorylation and, on the other hand, by viruses, both in negative and positive ways.32 The activation of this pathway, caused by viruses, inhibits apoptosis and promotes the survival of infected cells for further viral replication.33
HCV uses the PI3K/AKT signal pathway to enter a cell. It has been evidenced that the HCV NS5A protein binds itself to PI3K and activates the PI3K/AKT signaling pathway (Figure 3).34,35
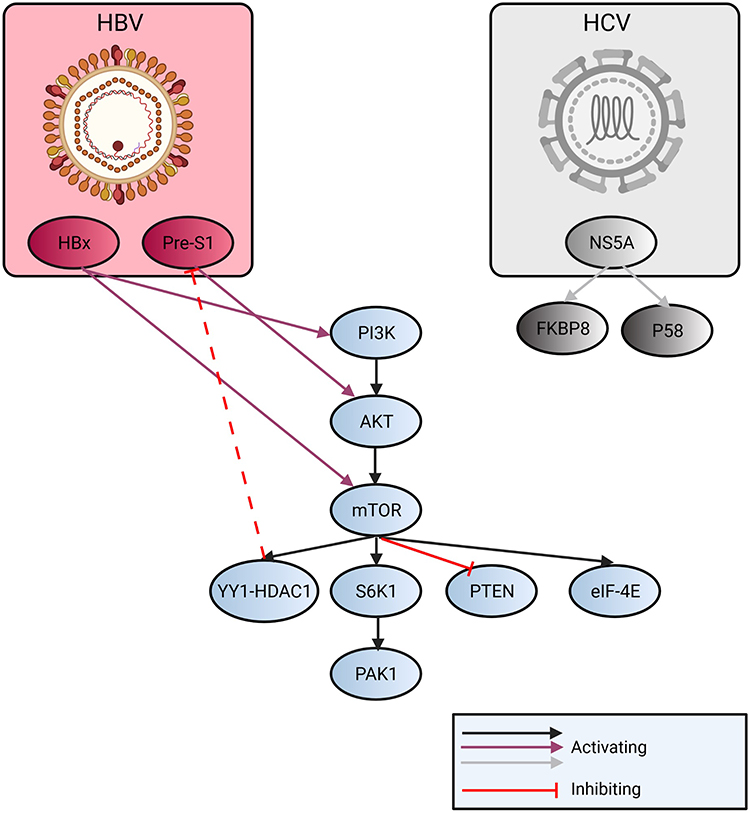 |
Figure 3 Overview of the regulation of mTOR related to HBV and HCV infection. HCV infection activates NS5A (nonstructural protein 5A). Its P85 form modulates with endoplasmic reticulum membrane or dysregulates association of FKBP5 (FK506 binding protein 5) with mTOR and PTEN is downregulated. HBV upregulates VEGFR-2 (vascular endothelial growth factor) by pre-S1 promoter and the AKT/mTOR pathway is activated. As a negative feedback, YY1-HDAC1 (mammalian zinc-finger transcription factor - Histone deacetylase 1) can inhibit pre-S1 transcription. HBx can activate PI3K and downstream pathway. |
Even though the use of the PI3K/AKT/mTOR is useful to the viruses, it at the same time limits the procreation of HBV,36 because the lessened RNA-transcription and lessened the HBV X (HBx) protein plays an essential role in pathogenesis since it encourages cell cycle progression, deactivates negative growth factors and has an effect on the tumor-suppressing genes.37 Upon HBx-transfection, autophagy is heightened by activating the PI3K/AKT/mTOR signal pathway.38 In cases of HCV-coinfection, the HBV-replication is suppressed.39
Translation Control Upon Viral Infection and as Response to Non-Virally Triggered Stress
eIFs
eIFs are part of the translation process and together with other PI3K/AKT/mTOR pathway members are dysregulated in malignancies,40 but the studies involving eIFs and eEFs in HCC are scarce.
As already published by our group, eIF4E and eIF3I are involved in non-virus related HCC formation. Additionally, eIF4E and eIF3I were downregulated for HBV- and HCV-related HCC, as well as for chronic hepatitis B and C. eIF5 was downregulated in non-virus related HCC, HCV-related HCC and chronic hepatitis C, but was overexpressed in HBV-related HCC. The eIF subunits p-2a, 2a, 3B, 3C, 3D, 3J, p-4B, 4G and 6 were upregulated in HCV-associated HCC. With the help of eIF3 subunits B, C, I, eIF4E, eIF4G and eIF5, a reduction in protein expression in HCV was observed. In HBV-associated HCC and chronic hepatitis B, overexpression of p-eIF2a, p-eIF4B and eIF4G was seen. Downregulation of eIF2a, eIF3D, eIF3H, eIF3I, eIF3J, eIF4E and eIF6 in HBV-associated HCC and chronic hepatitis B was observed.41
Another component of the translation, eIF3, is a protein complex that can act as a tumor suppressor or tumor promoting factor.42 In HCC, overexpression of eIF3h and eIF3i was observed. Interaction of overexpressed eIF3i with AKT1 prevents PP2A-mediated dephosphorylation and consequently activation of AKT1 signaling. If eIF3i specific inhibitors are applied, tumorigenesis is suppressed.43
Overexpression of eIF4E is associated with a higher risk of HCC recurrence, but the factor is overexpressed in other liver cancers as well.40 Another factor, namely eIF4AIII, was found to be overexpressed in HCC and promoted tumor cell proliferation and migration.44 eIF4AII was identified as a host factor supporting HCV replication in different replication systems, in contrast to eIF4AI where an opposite function was observed.45 A study on HepG2 cells showed that eIF4E phosphorylation with Mnk inhibitor blockage could be a new therapeutic option against chemoresistant HCC.46 It has also been suggested that patients with HCC who have elevated eIF4E factor in tumor samples have an increased likelihood of tumor recurrence.40 In a case where sorafenib, a kinase inhibitor, was used as a line of therapy, drug resistance can occur. It was shown that the eIF4E-eIF4G complex reduces the effectiveness of therapy.47
The eIF5 factor was shown to be increased in HCC.48,49 In patients with elevated expression level of eIF5A1, multiple tumor nodules were associated, and in eIF5A2, increased venous infiltration was associated. Inhibition of both isoforms was manifested by reduced HCC proliferation and invasiveness.48 In patients receiving chemotherapy, HCC cells go through epithelial–mesenchymal transition (EMT) and, as a result, chemotherapy is less effective, but a link to eIF5A2 has been found in chemoresistant samples. If eIF5A2 is inhibited, EMT occurs to a lesser extent, and chemotherapy is more effective.50 As suggested by Tang et al, eIF5A2 in the case of chemoresistance suppresses HCC autophagic cell death.51 It was also shown that eIF5A2 could promote cell proliferation and trigger metabolic pathways for glucose metabolism and fatty acid biosynthesis in HCC. The factor could thus not only be a new target for therapy but could also serve as a new prognostic and predictive marker.52 eIF5A2 is considered to be a factor that promotes metastasis. Inhibition of the eIF5A2 results in a minor invasion and metastasis, for which the reactive oxygen species pathway is thought to be responsible.53,54 For another factor from eIF5 family, namely eIF5B, it was shown that its expression was increased in HCC tissue samples. The factor is associated with a poorer outcome. The proliferation and invasion of cancer cells is accelerated by ASAP1 expression.55 In general, eIF5 is believed to serve as a biomarker for non-virus-related and virus-related HCC.41
To our knowledge, no study can be found to directly indicate the expression profile of eIF6 in HCC. However, it was shown that through competitively binding with miR-588 eIF6 expression is promoted and PI3K/AKT/mTOR signaling pathway activated by p38-inhibited cutaneous squamous cell carcinoma-associated lincRNA (PICSAR) in HCC.56
eEFs
Recently, more attention has been paid to eEF1 and eEF2 and their subunits and complexes, as they are involved in the elongation step.
eEF1A1 regulates cell cycle and promotes malignant cells proliferation. In the case of HCC, downregulation of eEF1A1 inhibited tumor cell proliferation.57 Based on mRNA study, it was shown that eEF1A was increased in HCC compared to non-tumor liver. In cell lines, eEF1A1 and eEF1A2 are correlated with cell growth and differentiation stage.58
It was shown that eEF2 promotes angiogenesis in tumor progression of HCC59 and is a potential drug target.60 Additionally, it might serve as a prognostic marker.60
Ongoing Clinical Trials
Regarding the extent and nature of HCC atezolizumab, bevacizumab (first line) or sorafenib, lenvatinib (first line), regorafenib (second line), pembrolizumab or nivolumab, hormonal compounds (tamoxifen, antiandrogens, and seocalcitiol) and systemic chemotherapy are standard therapy to treat HCC61 (Figure 4).
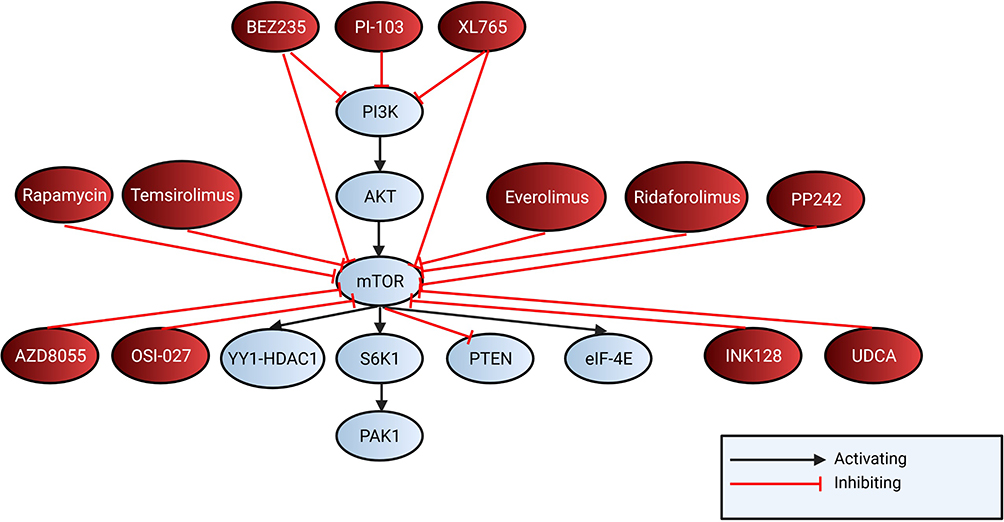 |
Figure 4 Shows mTOR and PI3K inhibitors targeting the mTOR pathway. BEZ235 and XL765 both target mTOR and PI3K and PI-103 targets PI3K. Rapamycin, temsirolimus, everolimus, ridaforolimus, PP242, AZD8055, OSI-027, INK128 and UDCA inhibit mTOR. |
Current ongoing clinical trials (*clinicaltrials.gov) investigating the role of different mTOR inhibitors are summarized in Tables 1 and 2.
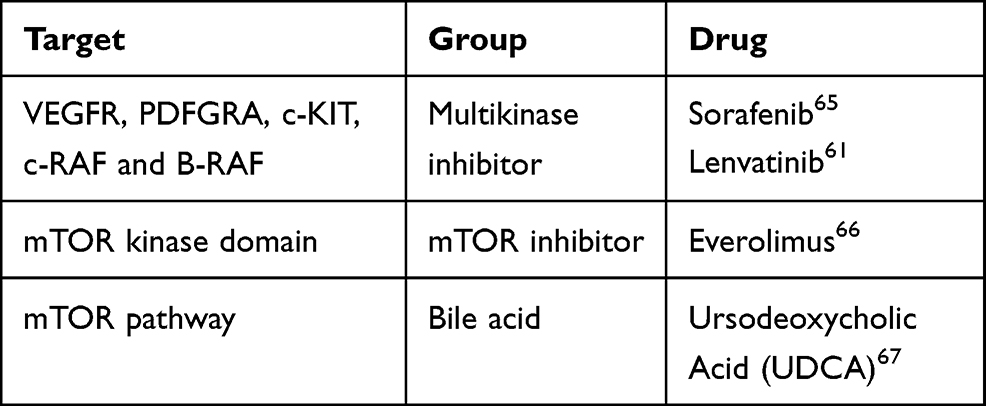 |
Table 1 Drugs and Its Targets |
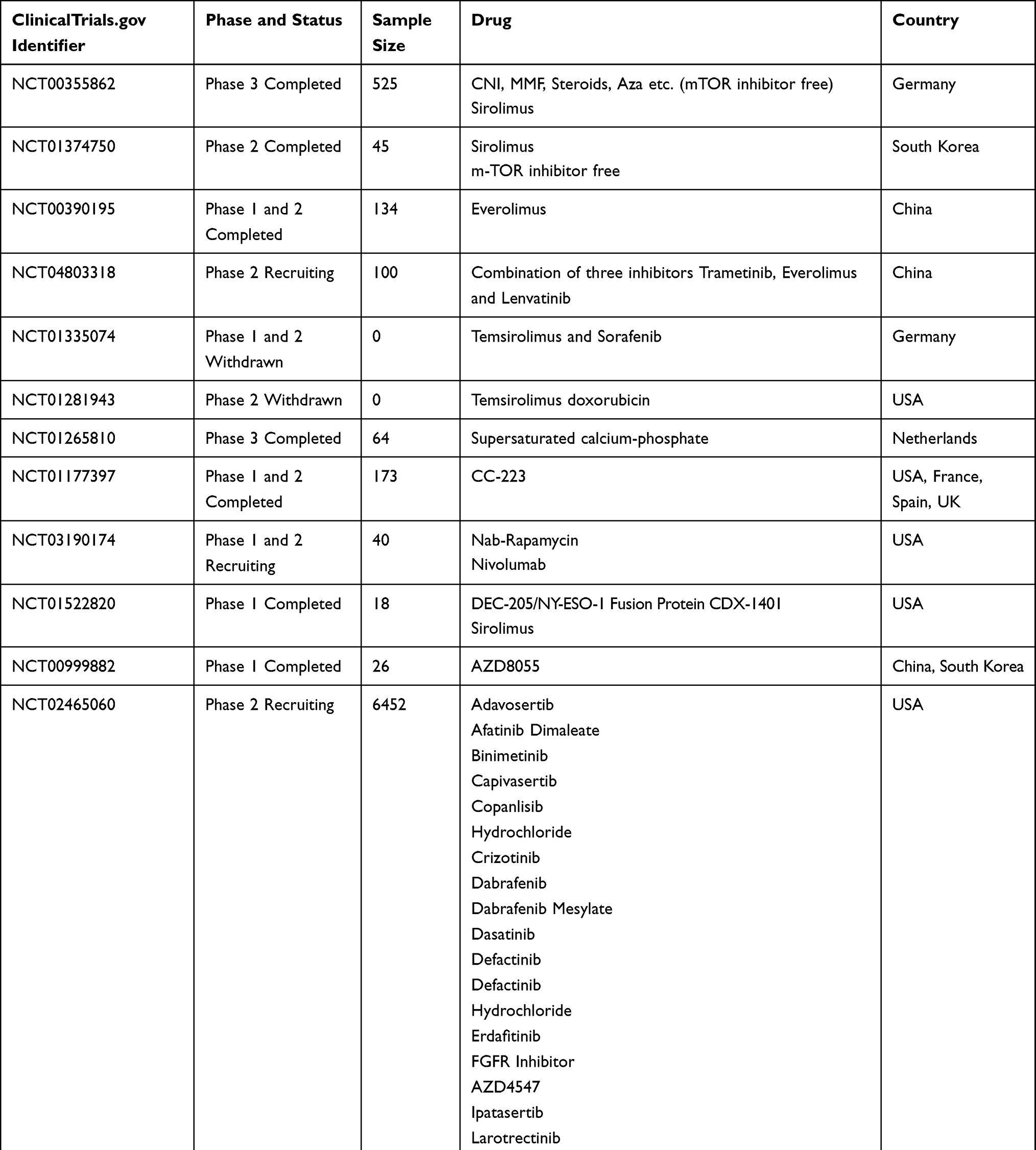 |
Table 2 Clinical Trials for HCC and mTOR |
In a prospective randomized trial, the effect of sirolimus (rapamycin, an mTOR inhibitor) on the HCC recurrence after liver transplantation was studied (Clinicaltrials.gov: NCT00355862). Results showed that treatment with mTOR-inhibitor for ≥3 months improved the outcome. A subgroup of patients with elevated alpha-fetoprotein where better outcome was observed, indicating the usefulness of mTOR-inhibitors in this group of patients.62
Another prospective-randomized open-label international trial (ClinicalTrials.gov NCT00355862) investigated the effect of sirolimus in liver transplanted patients. It was shown that patients could benefit from sirolimus as recurrence-free survival and overall survival were better in the first 3 to 5 years.63
An open-label, parallel-group, noncomparative, randomised study investigated the maximum tolerated dose of the mTOR-inhibitor everolimus for patients with advanced HCC. It was recommended that 7.5 mg of everolimus should be taken daily with acceptable tolerability and evidence of efficiency (ClinicalTrials.gov NCT00390195).64
Future Consideration
Although the risk factors for hepatocellular carcinogenesis are known, treatment is successful to a low extent as HCC-based mortality is still high. As described, different signaling pathways play an important role in HCC development, also the PI3K/AKT/mTOR pathway with and without viral interplay.
Recently, new techniques have opened up additional possibilities for studying the carcinogenesis of HCC. One of them is liquid biopsy, which is one of the least studied in HCC.68,69 Another promising area is molecular profiling of the tumor, which would allow us an individual approach to treatment. However, there is a problem with inter- and intraheterogeneity of the tumor, as well as the heterogeneity of the tumor in its development. Studies of molecular mechanisms as well as clinical studies suggest that much remains unclear in the field of hepatocellular carcinogenesis as well as targeted therapy and offers us opportunities to research and develop new target drugs in the future.70,71
Disclosure
The authors declare no conflicts of interest.
References
1. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;1–41. doi:10.3322/caac.21660.
2. Fitzmaurice C, Akinyemiju TF, Al Lami FH, et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 29 cancer groups, 1990 to 2016: a systematic analysis for the global burden of disease study global burden of disease study. JAMA Oncol. 2018;4(11):1553–1568. doi:10.1001/jamaoncol.2018.2706
3. Liberal R, Grant CR. Cirrhosis and autoimmune liver disease: current understanding. World J Hepatol. 2016;8(28):1157–1168. doi:10.4254/wjh.v8.i28.1157
4. Schlesinger S, Aleksandrova K, Pischon T, et al. Diabetes mellitus, insulin treatment, diabetes duration, and risk of biliary tract cancer and hepatocellular carcinoma in a European Cohort. Ann Oncol. 2013;24(9):2449–2455. doi:10.1093/annonc/mdt204
5. Huang Y-K, Qiu F. Current perspectives of recurrence and progression in hepatocellular carcinoma. J Xiangya Med. 2017;2(9):68. doi:10.21037/jxym.2017.09.01
6. Hoyos S, Escobar J, Cardona D, et al. Factors associated with recurrence and survival in liver transplant patients with HCC - A single center retrospective study. Ann Hepatol. 2015;14(1):58–63. doi:10.1016/s1665-2681(19)30801-4
7. Chiang DY, Villanueva A, Hoshida Y, et al. Focal gains of vascular endothelial growth factor A and molecular classification of hepatocellular carcinoma. Cancer Res. 2008;68(16):6779–6788. doi:10.1158/0008-5472.CAN-08-0742.Focal
8. Yang C, Huang X, Liu Z, Qin W, Wang C. Metabolism-associated molecular classification of hepatocellular carcinoma. Mol Oncol. 2020;14(4):896–913. doi:10.1002/1878-0261.12639
9. Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet. 2018;391(10127):1301–1314. doi:10.1016/S0140-6736(18)30010-2
10. Llovet JM, Real MI, Montaña X, et al. Arterial embolisation or chemoembolisation versus symptomatic treatment in patients with unresectable hepatocellular carcinoma: a randomised controlled trial. Lancet. 2002;359(9319):1734–1739. doi:10.1016/S0140-6736(02)08649-X
11. Bruix J, Sherman M. Management of hepatocellular carcinoma: an update. Hepatology. 2011;53(3):1020–1022. doi:10.1002/hep.24199
12. Quetglas IM, Moeini A, Pinyol R, Llovet JM. Integration of genomic information in the clinical management of HCC. Best Pract Res Clin Gastroenterol. 2014;28(5):831–842. doi:10.1016/j.bpg.2014.08.004
13. Psyrri A, Arkadopoulos N, Vassilakopoulou M, Smyrniotis V, Dimitriadis G. Pathways and targets in hepatocellular carcinoma. Expert Rev Anticancer Ther. 2012;12(10):1347–1357. doi:10.1586/era.12.113
14. Dimri M, Satyanarayana A. Molecular signaling pathways and therapeutic targets in hepatocellular carcinoma. Cancers (Basel). 2020;12(2):491. doi:10.3390/cancers12020491
15. Matter MS, Decaens T, Andersen JB, Thorgeirsson SS. Targeting the mTOR pathway in hepatocellular carcinoma: current state and future trends. J Hepatol. 2014;60(4):855–865. doi:10.1016/j.jhep.2013.11.031.Targeting
16. Xie J, Wang X, Proud CG. mTOR inhibitors in cancer therapy. F1000Research. 2016;5:2078. doi:10.12688/f1000research.9207.1
17. Ali MU, Ur Rahman MS, Jia Z, Jiang C. Eukaryotic translation initiation factors and cancer. Tumor Biol. 2017;39(6):101042831770980. doi:10.1177/1010428317709805
18. Spilka R, Ernst C, Mehta AK, Haybaeck J. Eukaryotic translation initiation factors in cancer development and progression. Cancer Lett. 2013;340(1):9–21. doi:10.1016/j.canlet.2013.06.019
19. Voorhees RM, Ramakrishnan V. Structural basis of the translational elongation cycle. Annu Rev Biochem. 2013;82:203–236. doi:10.1146/annurev-biochem-113009-092313
20. Kapp LD, Lorsch JR. The molecular mechanics of eukaryotic translation. Ann Rev Biochem. 2004;73:657–704. doi:10.1146/annurev.biochem.73.030403.080419
21. Bhat M, Robichaud N, Hulea L, Sonenberg N, Pelletier J, Topisirovic I. Targeting the translation machinery in cancer. Nat Rev Drug Discov. 2015;14(4):261–278. doi:10.1038/nrd4505
22. Sahin F, Kannangai R, Adegbola O, Wang J, Su G, Torbenson M. mTOR and P70 S6 kinase expression in primary liver neoplasms. Clin Cancer Res. 2004;10(24):8421–8425. doi:10.1158/1078-0432.CCR-04-0941
23. Lu X, Paliogiannis P, Calvisi DF, Chen X. Role of the mammalian target of rapamycin pathway in liver cancer: from molecular genetics to targeted therapies. Hepatology. 2021;73(S1):49–61. doi:10.1002/hep.31310
24. Guri Y, Colombi M, Dazert E, et al. mTORC2 promotes tumorigenesis via lipid synthesis. Cancer Cell. 2017;32(6):807–823.e12. doi:10.1016/j.ccell.2017.11.011
25. Byrne CD, Targher G. NAFLD: a multisystem disease. J Hepatol. 2015;62(1):S47–S64. doi:10.1016/j.jhep.2014.12.012
26. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. 2018;24. doi:10.1038/s41591-018-0104-9
27. Lally JSV, Ghoshal S, Deperalta DK, et al. Inhibition of acetyl-CoA carboxylase (ACC) by phosphorylation or by the liver-specific inhibitor, ND-654, suppresses lipogenesis and hepatocellular carcinoma. Cell Metabol. 2019;29(1):174–182. DOI:10.1016/j.cmet.2018.08.020.Inhibition
28. Chen H, Shen F, Sherban A, et al. DEPTOR suppresses lipogenesis and ameliorates hepatic steatosis and acute-on-chronic liver injury in alcoholic liver disease. Hepatology. 2018;68(2):496–514. doi:10.1002/hep.29849
29. Ceni E, Mello T, Galli A. Pathogenesis of alcoholic liver disease: role of oxidative metabolism. World J Gastroenterol. 2014;20(47):17756–17772. doi:10.3748/wjg.v20.i47.17756
30. Sid B, Verrax J, Calderon PB. Role of AMPK activation in oxidative cell damage: implications for alcohol-induced liver disease. Biochem Pharmacol. 2013;86(2):200–209. doi:10.1016/j.bcp.2013.05.007
31. Xie G, Wang X, Huang F, et al. Dysregulated hepatic bile acids collaboratively promote liver carcinogenesis. Int J Cancer. 2016;139(8):1764–1775. doi:10.1002/ijc.30219
32. Roberts LO, Jopling CL, Jackson RJ, Willis AE. Viral strategies to subvert the mammalian translation machinery. Prog Mol Biol Transl Sci. 2009;90(C):313–367. doi:10.1016/S1877-1173(09)90009-6
33. Cooray S. The pivotal role of phosphatidylinositol 3-kinase-Akt signal transduction in virus survival. J Gen Virol. 2004;85(5):1065–1076. doi:10.1099/vir.0.19771-0
34. Street A, Macdonald A, McCormick C, Harris M. Hepatitis C virus NS5A-mediated activation of phosphoinositide 3-kinase results in stabilization of cellular β-catenin and stimulation of β-catenin-responsive transcription. J Virol. 2005;79(8):5006–5016. doi:10.1128/jvi.79.8.5006-5016.2005
35. Street A, Macdonald A, Crowder K, Harris M. The hepatitis C virus NS5A protein activates a phosphoinositide 3-kinase-dependent survival signaling cascade. J Biol Chem. 2004;279(13):12232–12241. doi:10.1074/jbc.M312245200
36. Xiang K, Wang B. Role of the PI3K-AKT-mTOR pathway in hepatitis B virus infection and replication. Mol Med Rep. 2018;17(3):4713–4719. doi:10.3892/mmr.2018.8395
37. Neuveut C, Wei Y, Buendia MA. Mechanisms of HBV-related hepatocarcinogenesis. J Hepatol. 2010;52(4):594–604. doi:10.1016/j.jhep.2009.10.033
38. Wang P, Guo QS, Wang ZW, Qian HX. HBx induces HepG-2 cells autophagy through PI3K/Akt-mTOR pathway. Mol Cell Biochem. 2013;372(1–2):161–168. doi:10.1007/s11010-012-1457-x
39. Guo H, Zhou T, Jiang D, et al. Regulation of hepatitis B virus replication by the phosphatidylinositol 3-kinase-Akt signal transduction pathway. J Virol. 2007;81(18):10072–10080. doi:10.1128/jvi.00541-07
40. Wang XL, Cai HP, Ge JH, Su XF. Detection of eukaryotic translation initiation factor 4E and its clinical significance in hepatocellular carcinoma. World J Gastroenterol. 2012;18(20):2540–2544. doi:10.3748/wjg.v18.i20.2540
41. Golob-Schwarzl N, Krassnig S, Toeglhofer AM, et al. New liver cancer biomarkers: PI3K/AKT/mTOR pathway members and eukaryotic translation initiation factors. Eur J Cancer. 2017;83:56–70. doi:10.1016/j.ejca.2017.06.003
42. Silvera D, Formenti SC, Schneider RJ. Translational control in cancer. Nat Rev Cancer. 2010;10(4):254–266. doi:10.1038/nrc2824
43. Wang YW, Lin KT, Chen SC, et al. Overexpressed-eIF3I interacted and activated oncogenic Akt1 is a theranostic target in human hepatocellular carcinoma. Hepatology. 2013;58(1):239–250. doi:10.1002/hep.26352
44. Zhang L, Chen Y, Bao C, Zhang X, Li H. Eukaryotic initiation Factor 4AIII facilitates hepatocellular carcinoma cell proliferation, migration, and epithelial-mesenchymal transition process via antagonistically binding to WD repeat domain 66 with miRNA-2113. J Cell Physiol. 2020;235(11):8199–8209. doi:10.1002/jcp.29475
45. Ahmed CS, Winlow PL, Parsons AL, Jopling CL. Eukaryotic translation initiation factor 4AII contributes to microRNA-122 regulation of hepatitis C virus replication. Nucleic Acids Res. 2018;46(12):6330–6343. doi:10.1093/nar/gky262
46. Liu Y, Sun L, Su X, Guo S. Inhibition of eukaryotic initiation factor 4E phosphorylation by cercosporamide selectively suppresses angiogenesis, growth and survival of human hepatocellular carcinoma. Biomed Pharmacother. 2016;84:237–243. doi:10.1016/j.biopha.2016.09.038
47. Fang C, Xie H, Zhao J, et al. eIF4E-eIF4G complex inhibition synergistically enhances the effect of sorafenib in hepatocellular carcinoma. Anti Cancer Drugs. 2021;1–7. doi:10.1097/CAD.0000000000001074
48. Lee NP, Tsang FH, Shek FH, et al. Prognostic significance and therapeutic potential of eukaryotic translation initiation factor 5A (eIF5A) in hepatocellular carcinoma. Int J Cancer. 2010;127(4):968–976. doi:10.1002/ijc.25100
49. Shek FH, Fatima S, Lee NP. Implications of the use of eukaryotic translation initiation factor 5A (eIF5A) for prognosis and treatment of hepatocellular carcinoma. Int J Hepatol. 2012;2012:1–6. doi:10.1155/2012/760928
50. Lou B, Fan J, Wang K, et al. N1-guanyl-1,7-diaminoheptane (GC7) enhances the therapeutic efficacy of doxorubicin by inhibiting activation of eukaryotic translation initiation factor 5A2 (eIF5A2) and preventing the epithelial-mesenchymal transition in hepatocellular carcinoma cells. Exp Cell Res. 2013;319(17):2708–2717. doi:10.1016/j.yexcr.2013.08.010
51. Tang Y, Chen K, Luan X, et al. Knockdown of eukaryotic translation initiation factor 5A2 enhances the therapeutic efficiency of doxorubicin in hepatocellular carcinoma cells by triggering lethal autophagy. Int J Oncol. 2020;57(6):1368–1380. doi:10.3892/ijo.2020.5143
52. Cao TT, Lin SH, Fu L, et al. Eukaryotic translation initiation factor 5A2 promotes metabolic reprogramming in hepatocellular carcinoma cells. Carcinogenesis. 2017;38(1):94–104. doi:10.1093/carcin/bgw119
53. Liu RR, Lv YS, Tang YX, et al. Eukaryotic translation initiation factor 5A2 regulates the migration and invasion of hepatocellular carcinoma cells via pathways involving reactive oxygen species. Oncotarget. 2016;7(17):24348–24360. doi:10.18632/oncotarget.8324
54. Tang DJ, Dong SS, Ma NF, et al. Overexpression of eukaryotic initiation factor 5A2 enhances cell motility and promotes tumor metastasis in hepatocellular carcinoma. Hepatology. 2010;51(4):1255–1263. doi:10.1002/hep.23451
55. Wang ZG, Zheng H, Gao W, et al. eIF5B increases ASAP1 expression to promote HCC proliferation and invasion. Oncotarget. 2016;7(38):62327–62339. doi:10.18632/oncotarget.11469
56. Liu Z, Mo H, Sun L, et al. Long noncoding RNA PICSAR/miR-588/EIF6 axis regulates tumorigenesis of hepatocellular carcinoma by activating PI3K/AKT/mTOR signaling pathway. Cancer Sci. 2020;111(11):4118–4128. doi:10.1111/cas.14631
57. Huang J, Zheng C, Shao J, Chen L, Liu X, Shao J. Overexpression of eEF1A1 regulates G1-phase progression to promote HCC proliferation through the STAT1-cyclin D1 pathway. Biochem Biophys Res Commun. 2017;494(3–4):542–549. doi:10.1016/j.bbrc.2017.10.116
58. Grassi G, Scaggiante B, Farra R, et al. The expression levels of the translational factors eEF1A 1/2 correlate with cell growth but not apoptosis in hepatocellular carcinoma cell lines with different differentiation grade. Biochimie. 2007;89(12):1544–1552. doi:10.1016/j.biochi.2007.07.007
59. Zhou Y, Li Y, Xu S, et al. Eukaryotic elongation factor 2 kinase promotes angiogenesis in hepatocellular carcinoma via PI3K/Akt and STAT3. Int J Cancer. 2020;146(5):1383–1395. doi:10.1002/ijc.32560
60. Pott LL, Hagemann S, Reis H, et al. Eukaryotic elongation factor 2 is a prognostic marker and its kinase a potential therapeutic target in HCC. Oncotarget. 2017;8(7):11950–11962. doi:10.18632/oncotarget.14447
61. Gordan JD, Kennedy EB, Abou-Alfa GK, et al. Systemic therapy for advanced hepatocellular carcinoma: ASCO guideline. J Clin Oncol. 2020;38(36):4317–4345. doi:10.1200/JCO.20.02672
62. Schnitzbauer AA, Filmann N, Adam R, et al. mTOR inhibition is most beneficial after liver transplantation for hepatocellular carcinoma in patients with active tumors. Ann Surg. 2020;272(5):855–862. doi:10.1097/SLA.0000000000004280
63. Geissler EK, Schnitzbauer AA, Zölke C, et al. Sirolimus use in liver transplant recipients with hepatocellular carcinoma: a randomized, multicenter, open-label phase 3 trial. Transplantation. 2016;100(1):116–125. doi:10.1097/TP.0000000000000965
64. Shiah HS, Chen CY, Dai CY, et al. Randomised clinical trial: comparison of two everolimus dosing schedules in patients with advanced hepatocellular carcinoma. Aliment Pharmacol Ther. 2013;37(1):62–73. doi:10.1111/apt.12132
65. Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–390. doi:10.1056/nejmoa0708857
66. Koeberle D, Dufour JF, Demeter G, et al. Sorafenib with or without everolimus in patients with advanced hepatocellular carcinoma (HCC): a randomized multicenter, multinational Phase II trial (SAKK 77/08 and SASL 29). Ann Oncol. 2016;27(5):856–861. doi:10.1093/annonc/mdw054
67. Huang TE, Deng YN, Hsu JL, et al. Evaluation of the anticancer activity of a bile acid-dihydroartemisinin hybrid ursodeoxycholic-dihydroartemisinin in hepatocellular carcinoma cells. Front Pharmacol. 2020;11:1–14. doi:10.3389/fphar.2020.599067
68. Pezzuto F, Buonaguro L, Buonaguro FM, Tornesello ML. The role of circulating free DNA and microRNA in non-invasive diagnosis of HBV- and HCV-related hepatocellular carcinoma. Int J Mol Sci. 2018;19(4):1007. doi:10.3390/ijms19041007
69. Maravelia P, Silva DN, Rovesti G, et al. Liquid biopsy in hepatocellular carcinoma: opportunities and challenges for immunotherapy. Cancers. 2021;1–19.
70. Kaseb AO, Sanchez NS, Sen S, et al. Molecular profiling of hepatocellular carcinoma using circulating cell-free DNA. Clin Cancer Res. 2019;25(20):6107–6118. doi:10.1158/1078-0432.CCR-18-3341
71. Demory A, Nault JC. Molecular perspectives for the treatment of hepatocellular carcinoma. Acta Gastroenterol Belg. 2020;83(2):309–312.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.