Back to Journals » International Journal of Nanomedicine » Volume 15
Transferrin Conjugated pH- and Redox-Responsive Poly(Amidoamine) Dendrimer Conjugate as an Efficient Drug Delivery Carrier for Cancer Therapy
Authors Hu Q, Wang Y, Xu L, Chen D, Cheng L
Received 14 November 2019
Accepted for publication 21 March 2020
Published 22 April 2020 Volume 2020:15 Pages 2751—2764
DOI https://doi.org/10.2147/IJN.S238536
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Qing Hu,1,2,* Yifei Wang,1,* Lu Xu,1,* Dawei Chen,1,3 Lifang Cheng1
1Department of Pharmaceutics, College of Pharmaceutical Sciences, Soochow University, Suzhou 215123, People’s Republic of China; 2Department of Pharmaceutics, College of Pharmaceutical Sciences, Fujian Medical University, Fuzhou 350122, People’s Republic of China; 3School of Pharmacy, Shenyang Pharmaceutical University, Shenyang 110016, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Lifang Cheng Email [email protected]
Introduction: A multifunctional redox- and pH-responsive polymeric drug delivery system is designed and investigated for targeted anticancer drug delivery to liver cancer.
Methods: The nanocarrier (His-PAMAM-ss-PEG-Tf, HP-ss-PEG-Tf) is constructed based on generation 4 polyamidoamine dendrimer (G4 PAMAM). Optimized amount of histidine (His) residues is grafted on the surface of PAMAM to obtain enhanced pH-sensitivity and proton-buffering capacity. Disulfide bonds (ss) are introduced between PAMAM and PEG to reach accelerated intracellular drug release. Transferrin (Tf) was applied to achieve active tumor targeting. Doxorubicin (DOX) is loaded in the hydrophobic cavity of the nanocarrier to exert its anti-tumor effect.
Results: The results obtained from in vitro and in vivo evaluation indicate that HP-ss-PEG-Tf/DOX complex has pH and redox dual-sensitive properties, and exhibit higher cellular uptake and cytotoxicity than the other control groups. Flow cytometry and confocal microscopy display internalization of HP-ss-PEG-Tf/DOX via clathrin mediated endocytosis and effective endosomal escape in HepG2 cancer cells. Additionally, cyanine 7 labeled HP-ss-PEG-Tf conjugate could quickly accumulate in the HepG2 tumor. Remarkably, HP-ss-PEG-Tf/DOX present superior anticancer activity, enhanced apoptotic activity and lower heart and kidney toxicity in vivo.
Discussion: Thus, HP-ss-PEG-Tf is proved to be a promising candidate for effective targeting delivery of DOX into the tumor.
Keywords: poly(amidoamine) dendrimers, histidine, transferrin, doxorubicin, pH and redox sensitivity
Introduction
Polyamidoamine (PAMAM) dendrimers have been widely investigated as carriers for drug delivery due to their unique dendrite structure and globular architecture features. PAMAM dendrimers are highly branched macromolecules possessing a well-defined core, an interior hydrophobic region, and a large number of end amino groups.1–4 The interior region could be employed to encapsulate hydrophobic drugs, and the surface amine of PAMAM could be attached to many substances including drugs, polypeptide, antibody and functional groups.5 The PAMAM itself has cytotoxicity and hemolysis toxicity, so it is often modified with polyethylene glycol (PEG). PEGylation could reduce its own toxicity and prolong the circulation time in blood via enhanced permeability and retention (EPR) effect.6–8 Unfortunately, excessive PEGylation can prevent the extent of interactions of PAMAM with target cells, resulting in decreased cellular uptake. To overcome this problem, various ligands such as transferrin,9,10 folic acid,5,11 peptides12–14 and antibodies15,16 are introduced at the terminal ends of PEG moieties to produce active tumor targeting and increased binding. Anticancer drugs that are encapsulated within ligand modified PEG-PAMAM has been proven to have higher cytotoxicity and even improved therapeutic efficacy, compared with non-targeted PEG-PAMAM conjugate.9,10,15-17 However, the introduction of PEG or ligand-PEG may limit the drug release from PEG-PAMAM conjugates. Inadequate or slow intracellular drug release is still a key challenge in the development of drug delivery system.18,19 Therefore, it is highly desired to develop nanocarriers which are stable in circulation but rapidly respond to intracellular environment for quick drug release.
Glutathione (GSH) is the most abundant thiol species (2–10 mM) in the cytoplasm and subcellular compartment, while GSH in plasma is low (2–10 μΜ) because of rapid enzymatic degradation. Furthermore, GSH in the tumor tissues is at least four times higher than the normal tissues.20,21 The significant differences in GSH concentration provide evidence for disulfide-linked conjugate as potential carrier for intracellular drug delivery.22,23 For example, Zhong et al11 reported a biodegradable micelle based on disulfide-linked poly(ethylene glycol)-b-poly(epsilon-caprolactone) (PEG-SS-PCL) diblock copolymer. The micelle was proved to be stable in water while fast disaggregation due to the cleavage of the disulfide bonds under reducing conditions (10 mM dithiothreitol). The PEG-SS-PCL showed higher cumulative release of DOX and anti-tumor effect than those of PEG-PCL. It is known that tumor tissue has, in the extracellular media, a reduced average pH of about 6.5 to 7.0, and the pH on the surface of cancer cells may even fall to 5, whereas the pH in normal tissue and in blood is about 7.2 to 7.5.24,25 Additionally, nanoparticles may undergo different extents of pH decrease when internalized into cells via different endocytic pathways, such as the pH of the early lysosomes is between 5.0 and 6.5 and the late lysosomes is between 4.5 and 5.0.26–28 Although PAMAM dendrimer is regarded as “proton sponges” polymer and showed the characteristic of pH-sensitive, it is still not efficient enough to escape from endosomes/lysosomes to release drug in cytoplasm. Modification of PAMAM with histidine residues is one of the solutions to assist PAMAM to overcome the barriers indicated above.29,30 Histidine is an essential amino acid which has an imidazole functional group. The imidazole group is a weak base with a pKa around 6, which has the ability to absorb protons when the pH of the environment drops below 6. Thus, histidine displays positive charges in the acidic environment of endocytosis vesicles or endosomes.29–32 They can produce electrostatic interactions with endocytic vesicles or endosomal membrane, thereby inducing water and ions to enter the endosome, and leading to the swelling of lysosome. Finally, lysosomes broken up, drug carriers escape from endosome to avoid drug degradation in lysosomes. The introduction of histidine promoted the “proton sponges” effect and accelerate the drug release.33 Based on the high GSH and low pH microenvironment characteristics of tumors, an excellent pH and redox-sensitive drug delivery system can be constructed by introduction of disulfide bonds and histidine to PAMAM, aiming in enhanced acid sensitivity and faster drug release compared to PAMAM alone.
In this study, we report a highly effective drug delivery system base on redox-sensitive nanocarrier (PAMAM-ss-PEG) that developed previously in our laboratory.34 Here, we sought to further functionalize this nanocarrier for high efficiency targeted drug delivery. Histidine (His) was first conjugated with the PAMAM to improve the acidic sensitivity of nanocarrier. Then, disulfide bonds were introduced between His-PAMAM (HP) and PEG to achieve pH and redox dual-responsive HP-ss-PEG conjugate. Transferrin (Tf) was grafted on the end of PEG (HP-ss-PEG-Tf) to obtain active tumor targeting. Doxorubicin (DOX, desalt), a widely used anticancer drug, was loaded into the interior hydrophobic region of PAMAM to exert its antitumor activity. Various measurements, including 1H NMR, DLS and UV-vis, were performed to characterize the structure, size and drug loading capacity. In vitro drug release behavior, cytotoxicity, cellular uptake, subcellular localization and uptake mechanism, in vivo imaging and antitumor activity were studied in detail.
Materials and Methods
Materials
Generation 4 PAMAM (G4.0 PAMAM) dendrimer was purchased from Dendritech, Inc. Michigan (Midland, MI, USA). Transferrin (Tf) was obtained from Qcbio Technology Co., Ltd. (Shanghai, China). Fmoc-His(Trt)-OH was purchased from GL Biochem Ltd. (Shanghai, China). Methoxy-PEG-Succinimidyl carboxymethyl ester (mPEG2000-SCM), Methoxy-PEG-Thiol (mPEG2000-SH) and Thiol-PEG-Carboxymethyl (HS-PEG2000-COOH) were purchased from JenKem Technology Co., Ltd. (Beijing, China). N-hydroxybenzotriazole (HOBt), 2-(1H-benzotriazole-1-yl)-1, 1, 3, 3-tetramethyluronium hexafluorophosphate (HBTU) and Triisopropylsilane were purchased from Aladdin Chemistry Co. Ltd. N, N-diisopropylethylamine (DIPEA), Glutathione (GSH) and Trifluoroacetic acid were purchased from Sinopharm Chemical Reagent Co. Ltd (Shanghai, China). Doxorubicin hydrochloride (DOX·HCl) was purchased from Beijing Hua Feng United Technology Co., Ltd (Beijing, China). The HepG2 Cell line was purchased from Cell Bank of Chinese Academy of Sciences.
Synthesis of Histidine Activated PAMAM (His-PAMAM, HP)
Histidine activated PAMAM dendrimer (His-PAMAM, HP) was prepared using a previously reported method with minor modification.29,35 Briefly, HP was synthesized by the reaction between primary amine group of PAMAM and carboxyl group of Fmoc-His(Trt)-OH. PAMAM, Fmoc-His(Trt)-OH, HOBt, HBTU, and DIPEA were dissolved in anhydrous DMF, with a molar ratio of 1:32:32:32:64. The mixture was reacted for 24 h with stirring at room temperature, followed by twice precipitation in an excess amount of cold diethyl ether. The obtained product was re-dissolved in a piperidine/DMF (v/v, 30/70) solution, and the reaction was continued for 6 h to remove the Fmoc-protecting group. The cold diethyl ether precipitated the product again and the precipitated intermediate was re-dissolved with a THF/triisopropylsilane/H2O (v/v/v, 95/2.5/2.5) solution and reacted for 1 h to deprotect the trt group on the histidine unit. The final product was dialyzed against purified water for 48 h (MWCO = 3500) and lyophilized to obtain HP. The HP was characterized by 1H NMR in D2O.
Synthesis of His-PAMAM-Ss-PEG (HP-Ss-PEG), His-PAMAM-PEG (HP-PEG) and PAMAM-PEG Conjugates
His-PAMAM-ss-PEG (HP-ss-PEG) conjugate was synthesized using the previously reported method in our laboratory.34 Briefly, HP and SPDP (mol/mol, 1/16) were both dissolved in methanol (with 10 μL TEA) and the reaction mixture was stirred for 5 h in the dark at room temperature. Then, mPEG-HS (HP/mPEG-HS, mol/mol, 1/16) was added and the reaction carried on for another 24 h. The product was dialyzed (MWCO = 8000–14,000) against purified water for 48 h and lyophilized to obtain off-white solid powder. HP-ss-PEG was characterized by 1H NMR in D2O.
His-PAMAM-PEG (HP-PEG) without disulfide bond was prepared in one step. The mPEG-NHS was added to a solution of HP (mPEG-NHS/HP, mol/mol, 16/1) in 4 mL phosphate buffer (0.1 M, pH8.2) and allowed to react for 24 h with gentle stirring at room temperature. The resulting solution was dialyzed (MWCO = 8000–14,000) against purified water for 48 h to remove the unreacted PEG. The final product was retrieved by freeze drying and characterized by 1H NMR in D2O. The synthesized method of PAMAM-PEG (P-PEG) without histidine residues was same as HP-PEG and it was also characterized by 1H NMR in D2O.
Synthesis of His-PAMAM-Ss-PEG-Tf (HP-Ss-PEG-Tf) Conjugate
Briefly, HP was crosslinked with HS-PEG-COOH via a disulfide bond to obtain the HP-ss-PEG-COOH. The synthesized method was same to HP-ss-PEG. Next, HP-ss-PEG-COOH (0.1 μmol), EDC and NHS were dissolved in phosphate buffer (0.1 M, pH 8.2) and stirred for 1 h in the dark at room temperature. Then, Tf (0.3 μmol) was added and the mixture was stirred for another 12 h. The resulting crude product was placed in an ultrafiltration tube (MWCO = 100 KDa) and centrifuged at 6000 rpm for 10 min (7–8 times) to remove free Tf. The supernatant was collected and lyophilized to obtain HP-ss-PEG-Tf. HP-ss-PEG-Tf was characterized by 1H NMR in D2O. The conjugated number of Tf on per PAMAM was determined by BCA protein assay.
Preparation and Characterization of DOX-Loaded Complexes
The P-PEG/DOX, HP-PEG/DOX, HP-ss-PEG/DOX and HP-ss-PEG-Tf/DOX were prepared as previously described. Briefly, DOX·HCl was dissolved in methanol and appropriate TEA was added to obtain DOX. Then, each conjugate was mixed with DOX solution at a ratio of 1:30 (mol/mol) and the reaction mixture was stirred at room temperature for 24 hrs. Afterwards, the methanol was removed completely by rotary evaporation under reduced pressure and a small amount of purified water was added to dissolve the complex. The solution was placed in an ultrafiltration tube (MWCO = 100 KDa) and centrifuged at 10,000 rpm for 30 min to remove free DOX. Finally, the supernatant was lyophilized for 48 h to obtain the DOX-loaded complexes.
The particle size and Zeta potential of all the complexes were measured by dynamic light scattering (DLS) using NicompTM 380 ZLS (PSS Nicomp, Santa Barbara, USA). Samples were dissolved in PBS (0.01 M pH 7.4) and filtered through 0.45 μm cellulose acetate membrane before measurement, respectively. All measurements were performed at 37°C and carried out for three times.
The entrapment efficiency (EE) and drug loading (DL) of DOX were determined by UV-vis spectrophotometer (UV-2600, Shimadzu, Japan) at the wavelength of 497 nm. The calibration curve was generated using known concentration of DOX. The EE and DL were calculated by the following equations:
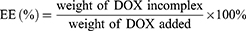
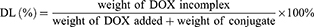
In vitro Redox- and pH-Triggered Release of DOX-Loaded Complexes
The redox and pH sensitivity of DOX-loaded complexes were investigated in vitro using a dialysis method. The release conditions were ① acetate buffer (0.1 M, pH 5.0), ② PBS buffer (0.1 M, pH 7.4) and ③ PBS buffer with 10 mM GSH (0.1 M, pH 7.4 + 10 mM GSH). The P-PEG/DOX, HP-PEG/DOX, HP-ss-PEG/DOX and HP-ss-PEG-Tf/DOX (0.5 mg DOX, n=3) were dissolved in 2 mL of release medium and placed in a dialysis bag (MWCO = 3500), respectively. The dialysis bag was then immersed into 20 mL of release medium and incubated in a shaking incubator with a stirring speed of 100 rpm at 37°C. 4.0 mL of release solution was taken out at predetermined times (0.5, 1, 2, 4, 6, 8, 12 and 24 h) and the same volume of fresh buffer was added. The amount of released DOX was analyzed using a UV-vis spectrophotometer at a wavelength of 497 nm.
In vitro Cytotoxicity Assay
The cytotoxicity of nanocarrier and DOX-loaded complexes against HepG2 cells was evaluated by the MTT assay. The tested concentration ranges were 25–800 µg/mL for PAMAM, P-PEG, HP-PEG, HP-ss-PEG and HP-ss-PEG-Tf, and 0.01–15 µg/mL DOX-conc. for free DOX, P-PEG/DOX, HP-PEG/DOX, HP-ss-PEG/DOX and HP-ss-PEG-Tf/DOX. Briefly, HepG2 cells were seeded in a 96-well plate at a density of 1×104 cells/well and incubated for 24 hrs. Then, 100 μL of the fresh medium containing various amounts of nanocarrier or DOX-loaded complexes were added. After 48 h incubation, 100 µL of MTT solution (0.5 mg/mL) was added and incubated for another 4 h. The solution was removed carefully and 100 µL DMSO was added into each well to dissolve the received blue formazan crystals. The optical density (OD) was measured at 490 nm using a microplate reader (MultiskanMK3, Thermo, USA).
In vitro Cellular Uptake Study
In vitro cellular uptake, study was evaluated by both fluorescence microscope and flow cytometry. HepG2 cells were seeded in a 6-well culture plate at a density of 5×104 cells/well and incubated for 24 hrs. Serum-free culture medium with P-PEG/DOX, HP-PEG/DOX, HP-ss-PEG/DOX or HP-ss-PEG-Tf/DOX (5.0 μg/mL DOX-conc.) was added and then incubated for 2 hrs. After the incubation, DOX-containing medium was removed and the cells were washed three times with cold PBS. Fluorescence microscope: the treated cells were visualized under fluorescence microscope (IX51, Olympus, Japan); Flow cytometry: the treated cells were harvested and resuspended in 0.5 mL of PBS, and a total of 10,000 was analyzed using flow cytometer (BD, USA).
Subcellular Localization and Cellular Uptake Mechanism
To trace the intracellular trafficking of the synthetic nanocarrier HP-ss-PEG-Tf at the cellular level, we used rhodamine B isothiocyanate (RB) as a fluorescent probe to prepare the RB-HP-ss-PEG-Tf conjugate. HepG2 cells were grown on 22-mm glass coverslip in 6-well plates at a density of 5 × 104 cells/well and incubated for 24 hrs at 37 °C. Afterwards, cells were incubated with RB-HP-ss-PEG-Tf conjugate for 2 hrs and 6 hrs. After incubation, hoechst 33,342 (5 μg/mL, 15 mins) and lysotracker Green DND-26 (80 nM, 30 mins) were used to visualize the nuclei and lysosome, respectively. The cells were washed three times with cold PBS and observed by a confocal laser scanning microscope (CLSM, LSM710, Zeiss, Germany).
To study the uptake pathways of HP-ss-PEG-Tf/DOX complex, cells were individually pretreated with the following inhibitors:36,37 chlorpromazine (10 μg/mL), sucrose (150 mg/mL), filipin (5 μg/mL), genistein (200 μM) and colchicine (40 μg/mL). After incubation of 1 h, HP-ss-PEG-Tf/DOX (5 μg/mL DOX) was added and incubated for another 1 h. Subsequently, the cells were harvested, and a total of 10,000 was analyzed using flow cytometer (BD, USA). The cell without inhibitors treatment was used as a control and the value was set as 100%.
In vivo Imaging
All experiments of animal study were performed in compliance with the relevant laws and institutional guidelines of Soochow University, and the institutional committee had approved the experiments. Female BALB/c nude mice (18–22 g) were purchased from SLAC (Shanghai, China), and all of the animals were kept in standard housing conditions with free access to food and water. To examine the in vivo biodistribution of Cy7-labeled HP-ss-PEG and HP-ss-PEG-Tf conjugates, the model of HepG2 tumor-bearing mice was established. HepG2 cells (1× 107) were injected subcutaneously in the abdomen of female BALB/c nude mice. When the tumor reached approximately 150–200 mm3, 0.1 mL of Cy7-HP-ss-PEG and Cy7-HP-ss-PEG-Tf were intravenously injected through a tail vein, respectively. The biodistribution of conjugates in HepG2 tumor-bearing nude mice was imaged by near-infrared fluorescence imaging system (IVIS Lumina, Caliper, USA, excitation filter, 745 nm; emission filter, 805 nm.) at the time points of 1, 6, 12, 24 and 48 hrs post-injection. After 48 hrs, mice were sacrificed and tumor, heart, liver, spleen, lung and kidney were collected for fluorescent intensity measurement. The fluorescence intensity of tumor at each time point was calculated using the region of interest (ROI) function of Living image® version 4.3.1 software.
In vivo Antitumor Efficacy of DOX-Loaded Conjugates
The anti-tumor efficacy of DOX-loaded complexes was investigated using the xenograft tumor model. HepG2 cells (1× 107) were implanted subcutaneously in the abdomen of nude mice (18–22 g). When the tumor volume reached approximately 80 mm3, the mice were randomly divided into six groups (n = 6) and treated with 200 μL of saline, DOX, P-PEG/DOX, HP-PEG/DOX, HP-ss-PEG/DOX and HP-ss-PEG-Tf/DOX (10 mg/kg DOX) via tail vein injection every 2 days for five times. The body weights and tumor sizes were measured and recorded every day during the process of the treatment. After treatment, mice were sacrificed and tumor, heart, liver, spleen, lung and kidney were collected for histological analysis. The cell apoptosis of tumor tissue was detected by the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay according to the manufacture’s protocol. The cell nucleus was stained by Hoechst 33,342 and the samples were analyzed under CLSM (LSM710, Zeiss, Germany). Tumor volume was calculated using the following formula: V = (a × b2)/2, where a and b is the major and minor axes of the tumor, respectively.
Statistical Analysis
All experiments were performed in triplicate and the data were presented as mean ± standard deviation (SD). Multiple comparisons between the groups were performed using the Student-Newman-Keuls method. P-values < 0.05 were considered statistically significant.
Results and Discussions
Synthesis and Characterization of HP-Ss-PEG-Tf Conjugate
The synthesis route of HP-ss-PEG-Tf conjugate was shown in Scheme 1. HP was first synthesized and the structure of it was confirmed by 1H NMR (Figure 1). Figure 1A was the 1H NMR spectrum of G5 PAMAM. 1H NMR spectrum of HP showed signals at δ 2.45–3.30 attributable to the methylene protons of PAMAM as well as resonance at δ 7.00 and 7.60 to the protons of imidazole group of His moiety (Figure 1B). The comparison of the integrals of signals at δ 2.45 and 7.60 revealed an average of 25 His molecules per PAMAM dendrimer. Then HP was activated by SPDP and crosslinked with HS-PEG-COOH to obtain HP-ss-PEG. As shown in Figure 1C, the peaks appeared at δ ≈ 3.60–3.80 ppm belong to the protons of -CH2CH2O- repeat units of PEG, which demonstrated the successful synthesis of HP-ss-PEG-COOH. The PEGylation degree was calculated by the proton integration method, and the conjugated number of PEG molecule was estimated to be 12. Finally, Tf was conjugated to HP-ss-PEG-COOH at a theoretical molar ratio of 3 via amide reaction. BCA assays confirmed that the Tf substitution degree (Tf %) of terminal amino groups (64 terminal amino groups per PAMAM molecular) was 2.4% (molar ratio), which corresponds to 1.5 Tf molecules per PAMAM on average. The structures of P-PEG and HP-PEG were also confirmed by 1H NMR and the results suggested that they were successfully synthesized (Figure S1 and Table 1).
 |
Scheme 1 The synthesis route of HP-ss-PEG-Tf conjugate. |
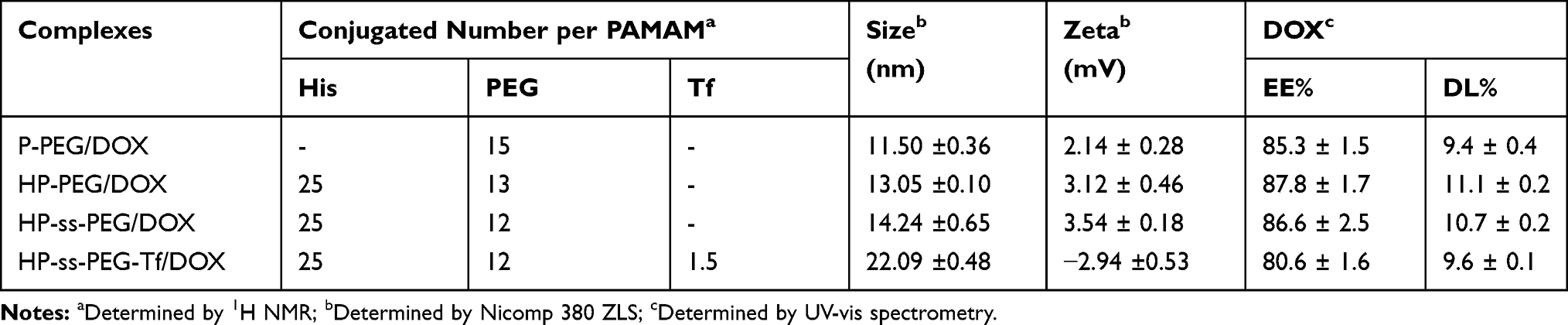 |
Table 1 Characteristics of DOX-Loaded Complexes |
HP-ss-PEG-Tf/DOX was conveniently prepared through a co-incubation method. As shown in Table 1, the particle size and size distribution were measured by DLS in aqueous solution at room temperature. The average diameter of HP-ss-PEG-Tf/DOX was 22.09 ± 0.48 nm, larger than that of HP-ss-PEG/DOX (14.24 ± 0.65 nm). With regard to the nanoparticle, particle size is an important factor which may affect their in vivo performance. In our study, the DOX-loaded complexes were all smaller than 200 nm with a narrow distribution, indicating that these NPs could avoid the clearance of RES during the blood circulation and accumulate at the tumor site through EPR effect. In the measurement of Zeta potential, the HP-ss-PEG-Tf/DOX showed negative potential at around −3 mV, suggesting potential capacity for prolonging the circulation time in blood. Because positive surface charges of NPs are likely to interact with negative charge of serum protein in the blood, leading to agglomeration. In addition, both DL% and EE% are around 10 wt% and above 80%, respectively, indicating DOX was able to effectively incorporate inside the conjugates.
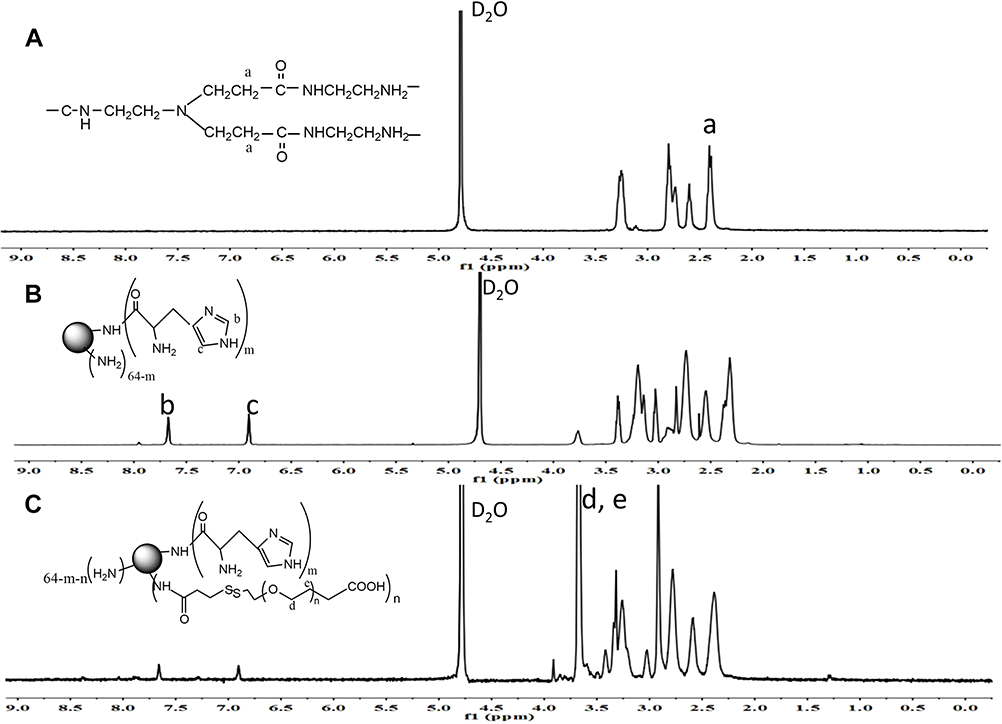 |
Figure 1 1H NMR spectrum of PAMAM (A), His-PAMAM (B) and HP-ss-PEG-COOH (C). |
In vitro Redox- and pH-Triggered Release of DOX
In vitro release behaviors of DOX from complexes were carried out at pH5.0, pH7.4 and pH7.4 + 10 mM GSH. To confirm the pH response, in vitro drug release behaviors of the P-PEG/DOX, HP-PEG/DOX, HP-ss-PEG/DOX and HP-ss-PEG-Tf/DOX were studied at both physiological pH of 7.4 and acidic pH of 5.0, respectively. As shown in Figure 2A, the drug release rate was influenced by the pH value. The lower the pH value was, the faster the drugs were released. The accumulative release amount of P-PEG/DOX at pH 5.0 (47%) was higher than that at pH 7.4 (35%) after 24 h. The pH-responsive release behavior was partly due to the protonation and deprotonation of terminal amines of PAMAM and the conformation change of PAMAM at different pH conditions,38 which promoted rapid release of DOX from the complexes. Notably, the value of accumulated DOX released from the P-PEG/DOX was 47% at pH of 5.0, which increased significantly to 64% in HP-PEG/DOX. The observed pH-triggered acceleration of DOX might be attributed to the protonation and deprotonation of terminal imidazole of Histidine. These findings revealed that the pH-responsive property of complex was enhanced by the modification of Histidine. In addition, the introduction of both disulfide bond and Tf neither reduce nor increase DOX release.
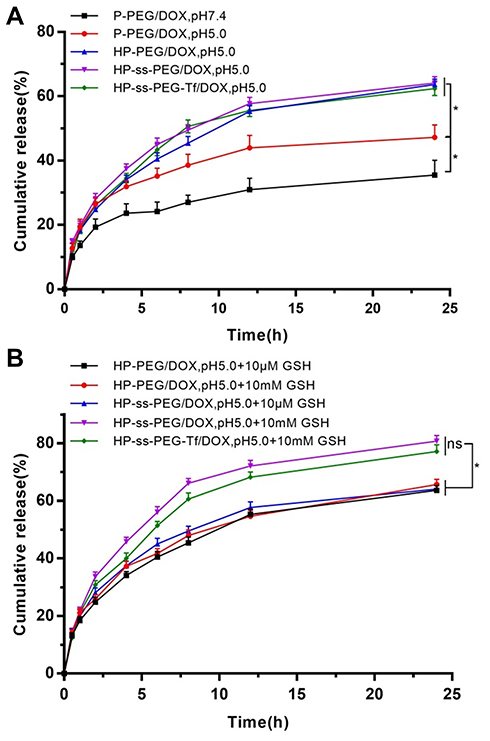 |
Figure 2 In vitro release behaviors of DOX-loaded complexes at pH5.0 and pH 7.4 (A) and pH7.4 with10 mM GSH and with GSH (B). Data are presented as mean ± SD (n = 3, *P <0.05, N.S.: no significance). |
Redox responsive of the HP-PEG/DOX, HP-ss-PEG/DOX and HP-ss-PEG-Tf/DOX complexes was also confirmed by in vitro drug release with or without 10 mM GSH. GSH is a strong reducing agent which can break the disulfide bond between PEG and PAMAM. The results were summarized in Figure 2B. The GSH did not affect the drug release behavior of HP-PEG/DOX, but HP-ss-PEG/DOX showed redox dependence. The accumulative release of DOX from the HP-ss-PEG/DOX reached approximately 38% after 24 h at pH 7.4, and up to 50% at pH 7.4 with 10 mM GSH. This is attributed to the breakage of disulfide bond under redox environment, resulting in detachment of outer PEG corona from PAMAM, and showed enhanced release of DOX. Similarly, the conjugated Tf had no effect on the DOX release. These results indicated that the carrier of HP-ss-PEG-Tf had sustained drug release behavior and pH and redox sensitivity. This carrier is highly desirable for treatment of cancer, since tumor tissues are known to be acidic, and the concentration of GSH in cytoplasm of tumor cells is higher than the level in the bloodstream and healthy cells, which can cleave disulfide bonds.
In vitro Cytotoxicity Assay
The cytotoxicity of nanocarrier and DOX-loaded complexes against HepG2 cells was evaluated using MTT assay, and the results were presented in Figure 3. PAMAM dendrimers showed significant cytotoxicity against HepG2 cells. Introduction of PEG, His and Tf reduced the cytotoxicity of PAMAM, and more than 90% of the cells were still alive even at the highest concentration (800 μg/mL, Figure 3A). Figure 3B showed the cytotoxicity of free DOX and DOX-loaded complexes after incubation with HepG2 cells for 48 h. The free DOX and DOX-loaded complexes significantly inhibited cancer cell proliferation in a concentration-dependent manner. The cytotoxicity of HP-ss-PEG/DOX complex was higher than that of HP-PEG/DOX and P-PEG/DOX. The half-maximal inhibitory concentration (IC50) values for P-PEG/DOX, HP-PEG/DOX, and HP-ss-PEG/DOX were calculated to be 1.568, 0.743 and 0.449 μg/mL, respectively. The higher cytotoxicity observed for HP-ss-PEG/DOX could be attributed to rapid intracellular DOX release in the cytoplasm with high concentration of GSH. As expected, the HP-ss-PEG-Tf/DOX had a lower IC50 (0.243 μg/mL) than the HP-ss-PEG/DOX. The conjugated Tf did enhance the cytotoxicity of the complex, most likely via their targeting effects for HepG2 cells. It was worth noting that the IC50 value of free DOX was 0.082, which was lower than the other DOX-loaded complexes. This may be due to the sensitivity of HepG2 and the diffusion mechanism of DOX through cell membrane. Time-, redox- and pH-dependent drug release characteristics of HP-ss-PEG-Tf/DOX caused a delay effect, resulting in lower cytotoxicity than free DOX.
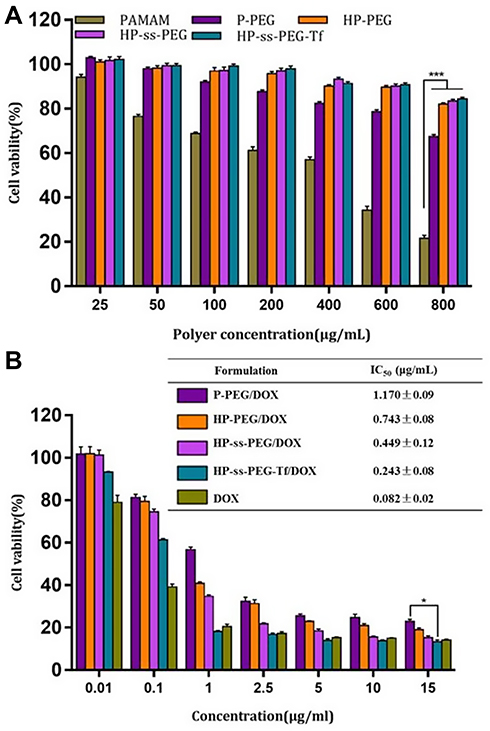 |
Figure 3 In vitro cytotoxicity of blank conjugates (A), and free DOX and DOX-loaded complexes (B) against HepG2 cells after treatment for 48 h. Data are presented as mean ± SD (n = 3, *P <0.05, ***P<0.001). |
In vitro Cellular Uptake Study
The cellular uptake efficiency of DOX-loaded complexes was investigated by flow cytometry and fluorescence microscope. Figure 4A showed the mean fluorescence intensity (MFI) values of HepG2 cells incubated with P-PEG/DOX, HP-PEG/DOX, HP-ss-PEG/DOX and HP-ss-PEG-Tf/DOX at 37°C for 2 h, respectively. On the basis of fluorescent intensity, the uptake rate of the complexes follows the order: HP-ss-PEG-Tf/DOX > HP-ss-PEG/DOX ≈ HP-PEG/DOX > P-PEG/DOX. The MFI value for the cells treated with HP-ss-PEG/DOX (51.25) or HP-PEG/DOX (48.93) was statistically higher than those treated with P-PEG/DOX (32.45) (P<0.01), indicating His-modified conjugates could enhance membrane fusion with cells, promote endocytosis, and increase cell uptake. The cellular uptake of HP-ss-PEG-Tf/DOX (97.31) was 1.9-fold higher than HP-ss-PEG/DOX (P<0.001). The conjugated Tf on the PEG terminal could bind preferentially to the highly expressed Tf receptor (TfR) on the surface of HepG2 cells, and thus efficiently internalized into the cells. Similar results were observed on the images of fluorescence microscope (Figure 4B). Strong red fluorescences were presented in the group of HP-PEG/DOX, HP-ss-PEG/DOX and especially in HP-ss-PEG-Tf/DOX, but weak in P-PEG/DOX group. These results further confirmed efficient and specific cellular uptake of HP-ss-PEG-Tf/DOX.
In our study, the His-PAMAM conjugates with different molar ratios of His to PAMAM (8:1, 16:1 and 32:1) were prepared at first. Then, His-PAMAM/DOX complexes were optimized by the experiments of drug release, cytotoxicity, and cellular uptake. The obtained results showed that the pH-sensitivity, cytotoxicity against HepG2 cells and cellular uptake of complexes increased significantly with the degree of His modification (Figure S2). Therefore, we chose His-PAMAM (32:1, molar ratio) as a basis to construct other conjugates including HP-PEG, HP-ss-PEG and HP-ss-PEG-Tf.
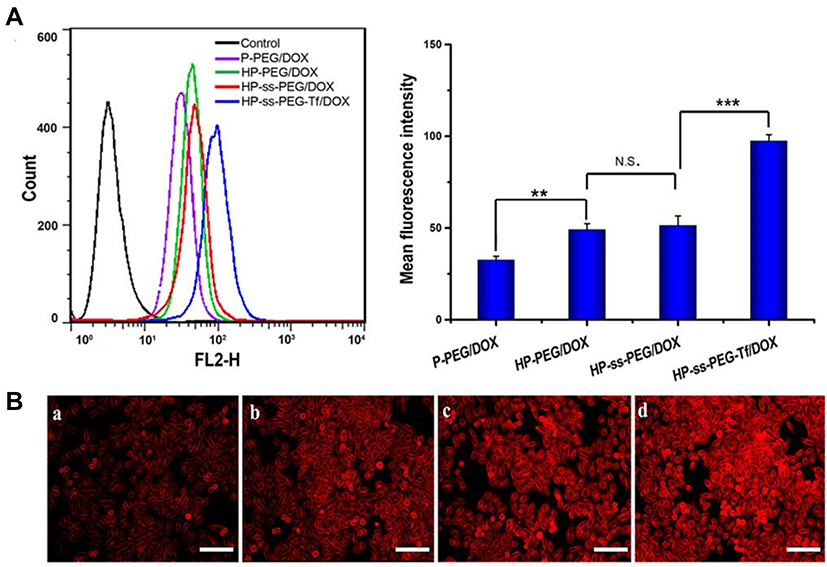 |
Figure 4 (A) Flow cytometry of HepG2 cells after 2 h incubation with the DOX-loaded complexes. Data are presented as mean ± SD. (n = 3, **P <0.01, ***P <0.001, N.S.: no significance). (B) Fluorescence microscope images of HepG2 cells after 2 h incubation with the DOX-loaded complexes (×100): (a) P-PEG/DOX, (b) HP-PEG/DOX, (c) HP-ss-PEG/DOX and (d) HP-ss-PEG-Tf/DOX (bar: 100 μm). |
Subcellular Localization and Uptake Mechanism
As shown in Figure 5A, CLSM observations showed that RB-HP-ss-PEG-Tf (red) mostly colocalized with late endosomes/lysosomes (green) of HepG2 cells after 2 hrs incubation. At 6 hrs, strong red fluorescence owing to RB-HP-ss-PEG-Tf was observed in the merged image, indicating that most part of RB-HP-ss-PEG-Tf has escaped from endosomes. These results indicate that HP-ss-PEG-Tf can effectively escape from endo/lysosomes. The high endosome disruption activity of HP-ss-PEG-Tf is likely due to the fact that protonation of HP-ss-PEG-Tf (PAMAM and His) in acidic endosomes that promote fusion with endosomal membranes.
The cytotoxicity of specific concentrations of inhibitors was first detected by MTT assay. Figure S3 showed that more than 95% of the cells were alive at these concentrations of inhibitors. Figure 5B presented the results of cellular uptake of HP-ss-PEG-Tf/DOX complexes in the presence of various inhibitors. Cells were exposed with hypertonic sucrose or chlorpromazine which is known to disrupt the formation of clathrin-coated pits.36,39,40 The cell uptake was reduced by 51% and 54% when treated with sucrose and chlorpromazine, respectively, indicating that HP-ss-PEG-Tf/DOX was taken up by clathrin mediated endocytosis. The effects of caveolin mediated endocytosis and macropinocytosis on the internalization of the complex was evaluated using filipin, genistein and colchicine,39 and no significant uptake difference was observed compared with the control. This suggested that caveolin mediated endocytosis and macropinocytosis might not be involved in the endocytosis process of HP-ss-PEG-Tf/DOX. Furthermore, uptake of HP-ss-PEG-Tf/DOX was competitively inhibited by free Tf, in which the mean fluorescence intensity was decreased to 48% of control value. The results revealed that HP-ss-PEG-Tf/DOX was possibly internalized by clathrin and TfR mediated endocytosis and subsequently delivered to the lysosomal compartments.
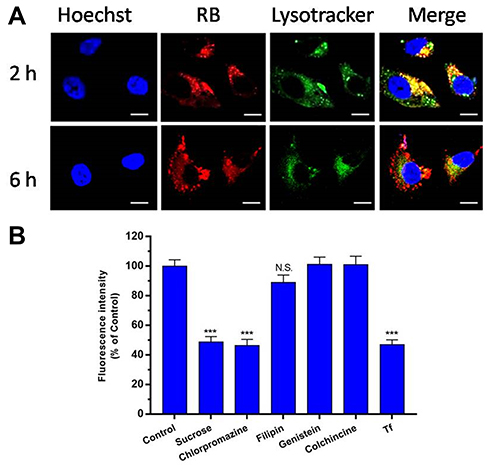 |
Figure 5 (A) Endosomal escape of Cy7 labeled HP-ss-PEG-Tf in HepG2 cells following 2 or 6 h incubation observed by confocal microscopy (bar: 10 μm). (B) Influence of endocytic inhibitors on cellular uptake after pre-treating the HepG2 cells with specific endocytic inhibitors for 1 h followed by co-treatment with HP-ss-PEG-Tf/DOX for 1 h. The data are reported as the mean ± SD (n = 3), ***p < 0.001, N.S.: no significance. |
In vivo Imaging
A non-invasive near-infrared optical imaging technique (NIR) was used to visualize the biodistribution of Cy7-labeled HP-ss-PEG (Cy7-HP-ss-PEG) and HP-ss-PEG-Tf (Cy7-HP-ss-PEG-Tf) in the HepG2 tumor-xenograft nude mice. As shown in Figure 6A, both Cy7-HP-ss-PEG and Cy7-HP-ss-PEG-Tf displayed a remarkable fluorescence signal at the tumor site after 1 h post-injection. The fluorescence intensity continued to increase and reached the maximum at 12 h after injection, indicating superior accumulation in the tumor. Furthermore, the fluorescence signal of Cy7-HP-ss-PEG-Tf was stronger than that of Cy7-HP-ss-PEG over 48 h, signifying the importance of ligand-receptor interaction for HP-ss-PEG-Tf in achieving high target ability and accumulation in the HepG2 tumor in vivo.
HepG2 tumor bearing mice were sacrificed at 48 h post-injection, tumor and main organs (heart, liver, spleen, kidney, and lung) were excised and tissue fluorescence was measured. Cy7-HP-ss-PEG-Tf exhibited higher tumor uptake than Cy7-HP-ss-PEG as shown in Figure 6B. The result supported that HP-ss-PEG-Tf could efficiently target to HepG2 tumor. Notably, the fluorescence in the kidney of both groups was stronger than the other tissues, indicating intravenously administered HP-ss-PEG-Tf and HP-ss-PEG could be trapped by the renal glomeruli to achieve long circulation. Overall, both HP-ss-PEG-Tf and HP-ss-PEG can efficiently accumulate in the tumor through the EPR effect, and the tumor accumulation was markedly increased by the interaction of Tf-TfR.
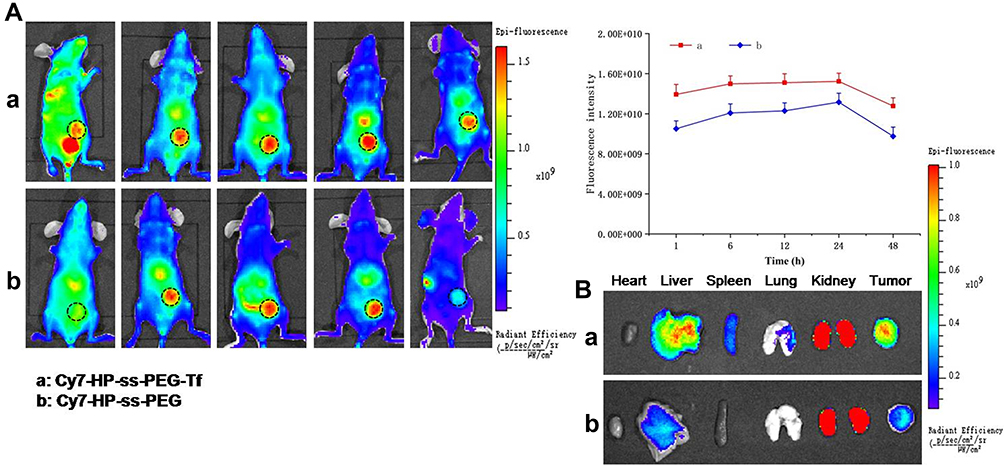 |
Figure 6 In vivo biodistribution of Cy7 labeled HP-ss-PEG-Tf and HP-ss-PEG conjugates. (A) In vivo fluorescence imaging of mice bearing HepG2 tumor after intravenous injection with Cy7-labeled HP-ss-PEG-Tf and HP-ss-PEG conjugates, respectively. The black dotted circle indicates the tumor region. (B) After 48 h, HP-ss-PEG-Tf and HP-ss-PEG conjugates treated mice were sacrificed, the major organs (heart, liver, spleen, kidney and lung) and tumors extracted, and measured by NIR. |
In vivo Antitumor Efficacy of DOX-Loaded Complex
The therapeutic effect of DOX-loaded complexes was studied in HepG2 hepatic carcinoma bearing nude mice at a dosage of 10 mg/kg DOX. The drug was given every 2 days for five injections in total. As expected, saline-treated group showed fast tumor growth (Figure 7A). The tumor volume of the mice treated with different DOX formulation followed the order: Control (saline) > P-PEG/DOX > HP-PEG/DOX > HP-ss-PEG/DOX > HP-ss-PEG-Tf/DOX ≈ free DOX. Both HP-ss-PEG-Tf/DOX and free DOX group almost completely suppressed tumor growth, but severe body weight loss, mild diarrhea and humpback were observed in free DOX group (Figure 7B), indicating the severe systemic toxicity of free DOX solution. The other treated groups had barely any body weight loss, indicating that the treatments are well tolerated. The tumor growth inhibition rate was determined by comparing the weights of tumors at the end of treatment to that of the control receiving saline only. The tumor inhibition rates of P-PEG/DOX, HP-PEG/DOX, HP-ss-PEG/DOX, HP-ss-PEG-Tf/DOX and free DOX were 41.8%, 55.7%, 70.9%, 81.9% and 80.8%, respectively (Figure 7C and D). The result indicated more efficient intracellular drug release and better tumor targeting ability could improve the antitumor effect in vivo.
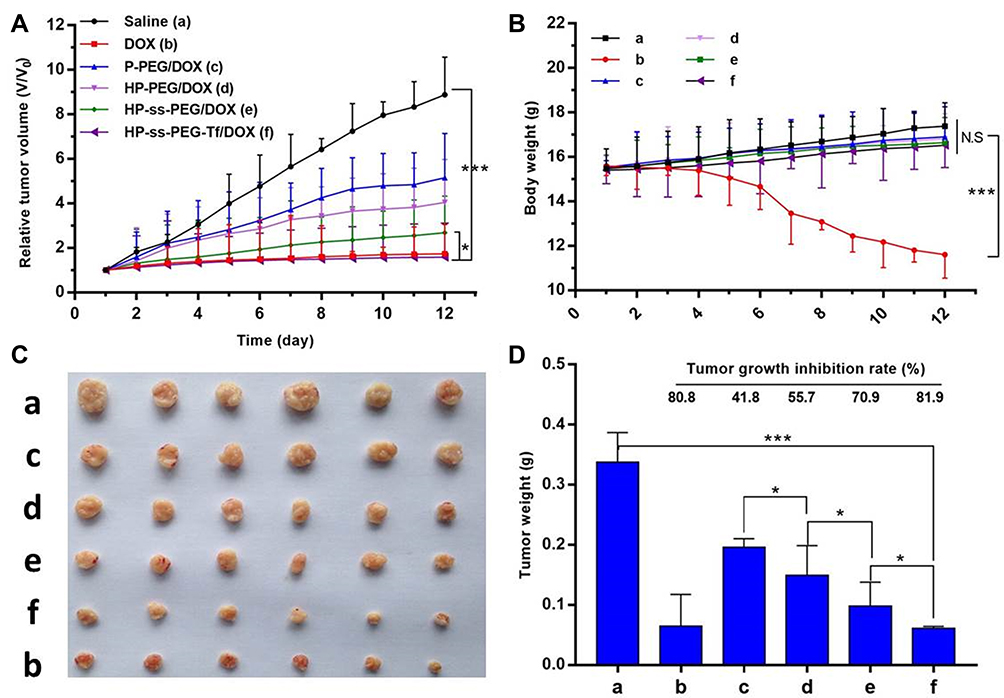 |
Figure 7 In vivo antitumor efficacy of saline, P-PEG/DOX, HP-PEG/DOX, HP-ss-PEG/DOX, HP-ss-PEG-Tf/DOX and free DOX in HepG2 tumor bearing nude mice. (A) Relative tumor volume change; (B) Mice body weight change; (C) Tumor tissue images extracted from the mice at the end of treatment by various formulations; (D) Tumor growth inhibition of the different treatment groups. During the in vivo experiment, no mice died. The survival rate of tumorigenic mice models was 100%. Data were presented as mean ± SD (n = 6, *p< 0.05, ***p< 0.001, N.S.: no significance). |
The systemic toxicity of DOX-loaded complexes and free DOX was further investigated by histopathology assay. As shown in Figure 8A, in the saline group, the tumor cells were spindle and round, with rich cytoplasm and more nuclear division. The groups treated with DOX-loaded complexes and free DOX showed spotty necrosis and nuclear fission in the tumor section, especially obvious in HP-ss-PEG-Tf/DOX and free DOX. The H&E staining of major organs recovered from mice treated with normal saline and DOX-loaded complexes revealed no significant damage or pathological features. However, the mice group treated with free DOX showed more acute inflammatory cell infiltration with obvious organ damage of necrosis in heart and kidney tissues than in other mice. Furthermore, the apoptosis in tumor cells was detected by TUNEL staining assay, in which the apoptotic cells stained green (Figure 8B). The TUNEL staining revealed significantly more apoptotic cells in both HP-ss-PEG-Tf/DOX and free DOX group. These findings demonstrated that HP-ss-PEG-Tf/DOX significantly improves the safety, targetability and treatment effects of DOX to TfR positive HepG2 tumor xenografts.
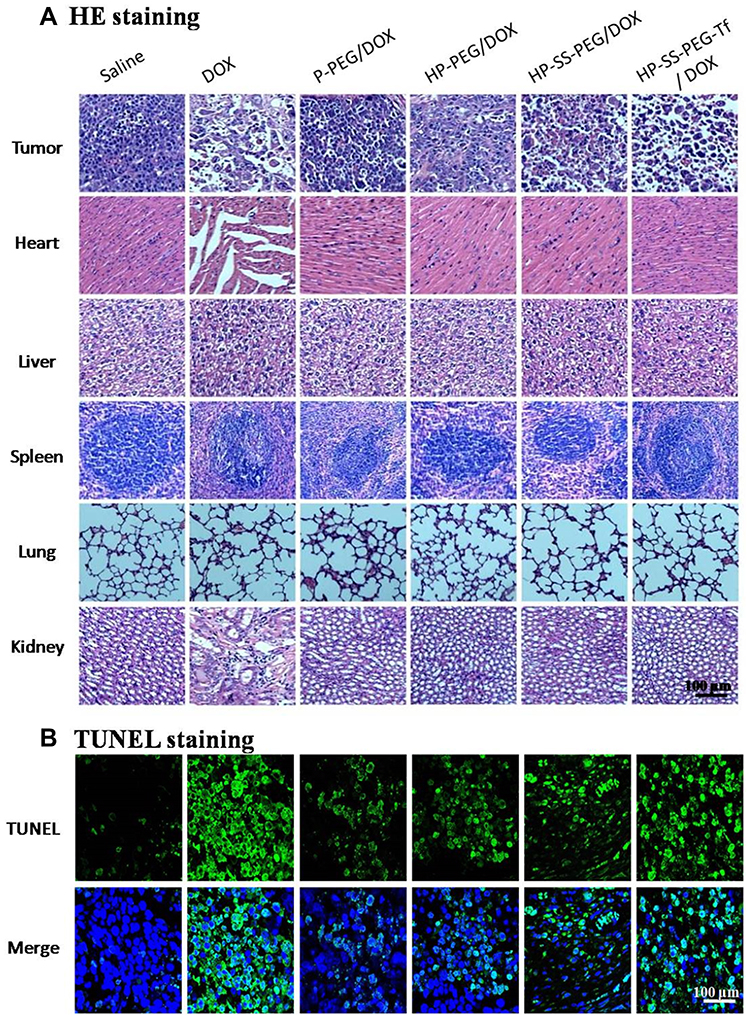 |
Figure 8 (A) H&E staining images of the major organs and tumor of mice treated with different formulations (×100). (B) TUNEL staining images of the tumor of mice treated with different formulations (×100, apoptotic cells: green, nuclei: blue). |
Conclusion
In this study, a pH and redox dual-responsive Tf modified conjugate was successfully synthesized and used as functional nanocarrier to fabricate HP-ss-PEG-Tf/DOX complex. The multifunctional complex was systematically evaluated in vitro and in vivo. The HP-ss-PEG-Tf/DOX with high drug content and excellent tumor cell uptake, and demonstrated that DOX could be responsively released in pH- and redox-dependent manner. After intravenous injection, the HP-ss-PEG-Tf/DOX could accumulate at the tumor site through the EPR effect, and significantly internalized into HepG2 cells through specific interaction between Tf and TfR, escape from the lysosomes via a proton sponge effect, and de-shield the PEG shells triggered by high concentration of intracellular GSH and finally release the DOX to the cytoplasm to exert its anti-tumor activity. These results suggest the HP-ss-PEG-Tf conjugate may be an appropriate carrier for anticancer drug delivery in tumors.
Acknowledgment
This work was supported by Suzhou Science and Technology Development Project (SYS201713), National Natural Science Foundation of China (81302719) and Natural Science Foundation of Jiangsu Province (BK20151224). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Svenson S. Dendrimers as versatile platform in drug delivery applications. Eur J Pharmaceutics Biopharm. 2009;71(3):445. doi:10.1016/j.ejpb.2008.09.023
2. Svenson S, Tomalia DA. Dendrimers in biomedical applications—reflections on the field. Adv Drug Deliv Rev. 2005;57(15):2106. doi:10.1016/j.addr.2005.09.018
3. Hu Q, Ding B, Yan X, et al. Polyethylene glycol modified PAMAM dendrimer delivery of kartogenin to induce chondrogenic differentiation of mesenchymal stem cells. Nanomed Nanotechnol Biol Med. 2017;13(7):2189. doi:10.1016/j.nano.2017.05.011
4. Nigam S, Chandra S, Newgreen DF, Bahadur D, Chen Q. Poly (ethylene glycol)-modified PAMAM-Fe3O4-doxorubicin triads with the potential for improved therapeutic efficacy: generation-dependent increased drug loading and retention at neutral pH and increased release at acidic pH. Langmuir. 2014;30:1004. doi:10.1021/la404246h
5. Cheng L, Hu Q, Cheng L, et al. Construction and evaluation of PAMAM–DOX conjugates with superior tumor recognition and intracellular acid-triggered drug release properties. Colloids Surf B. 2015;136:37. doi:10.1016/j.colsurfb.2015.04.003
6. Karthikeyan R, Kumar PM, Kumar PV. PEGylated dendritic nanoarchitechture improves mean survival time of BDF1 mice bearing myelogenous k −562 leukemia. J Acute Dis. 2013;2(4):327. doi:10.1016/S2221-6189(13)60153-5
7. Fang J, Nakamura H, Maeda H. The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63(3):136. doi:10.1016/j.addr.2010.04.009
8. Zhu S, Qian L, Hong M, Zhang L, Pei Y, Jiang Y. RGD-modified PEG-PAMAM-DOX conjugate: in vitro and in vivo targeting to both tumor neovascular endothelial cells and tumor cells. Adv Mater. 2011;23(12):H84. doi:10.1002/adma.201003944
9. Li Y, He H, Jia X, Lu WL, Lou J, Wei Y. A dual-targeting nanocarrier based on poly(amidoamine) dendrimers conjugated with transferrin and tamoxifen for treating brain gliomas. Biomaterials. 2012;33:3899. doi:10.1016/j.biomaterials.2012.02.004
10. Xu Q, Liu Y, Su S, Li W, Chen C, Wu Y. Anti-tumor activity of paclitaxel through dual-targeting carrier of cyclic RGD and transferrin conjugated hyperbranched copolymer nanoparticles. Biomaterials. 2012;33(5):1627. doi:10.1016/j.biomaterials.2011.11.012
11. Wang M, Hu H, Sun Y, et al. A pH-sensitive gene delivery system based on folic acid-PEG-chitosan – PAMAM-plasmid DNA complexes for cancer cell targeting. Biomaterials. 2013;34(38):10120. doi:10.1016/j.biomaterials.2013.09.006
12. Wang Y, Luo Y, Zhao Q, Wang Z, Xu Z, Jia X. An enzyme-responsive nanogel carrier based on PAMAM dendrimers for drug delivery. ACS Appl Mater Interfaces. 2016;8(31):19899. doi:10.1021/acsami.6b05567
13. Feng Q, Yu MZ, Wang JC, et al. Synergistic inhibition of breast cancer by co-delivery of VEGF siRNA and paclitaxel via vapreotide-modified core–shell nanoparticles. Biomaterials. 2014;35(18):5028. doi:10.1016/j.biomaterials.2014.03.012
14. Duan Z, Chen C, Qin J, et al. Cell-penetrating peptide conjugates to enhance the antitumor effect of paclitaxel on drug-resistant lung cancer. Drug Deliv. 2017;24(1):752. doi:10.1080/10717544.2017.1321060
15. Kulhari H, Pooja D, Shrivastava S, et al. Trastuzumab-grafted PAMAM dendrimers for the selective delivery of anticancer drugs to HER2-positive breast cancer. Sci Rep. 2016;6(1):23179. doi:10.1038/srep23179
16. Lewis Phillips GD, Li G, Dugger DL, et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008;68(22):9280. doi:10.1158/0008-5472.CAN-08-1776
17. Rostami I, Zhao Z, Wang Z, et al. Peptide-conjugated PEGylated PAMAM as a highly affinitive nanocarrier towards HER2-overexpressing cancer cells. RSC Adv. 2016;6(109):107337–107343. doi:10.1039/C6RA19552K
18. Zhu S, Hong M, Zhang L, Tang G, Jiang Y, Pei Y. Erratum to: pEGylated PAMAM dendrimer-doxorubicin conjugates: in vitro evaluation and in vivo tumor accumulation. Pharm Res. 2010;27(9):2030. doi:10.1007/s11095-010-0217-4
19. Liu D, Hu H, Zhang J, Zhao X, Tang X, Chen D. Drug pH-sensitive release in vitro and targeting ability of polyamidoamine dendrimer complexes for tumor cells. Chem Pharm Bull (Tokyo). 2011;59(1):63. doi:10.1248/cpb.59.63
20. Yin M, Bao Y, Gao X, et al. Redox/pH dual-sensitive hybrid micelles for targeting delivery and overcoming multidrug resistance of cancer. J Mater Chem B. 2017;5:2964–2978.
21. Wen HY, Dong HQ, Xie WJ, et al. Rapidly disassembling nanomicelles with disulfide-linked PEG shells for glutathione-mediated intracellular drug delivery. Chem Commun. 2011;47(12):3550. doi:10.1039/c0cc04983b
22. Sun H, Guo B, Cheng R, Meng F, Liu H, Zhong Z. Biodegradable micelles with sheddable poly(ethylene glycol) shells for triggered intracellular release of doxorubicin. Biomaterials. 2009;30(31):6358. doi:10.1016/j.biomaterials.2009.07.051
23. Zhang H, Wei L, Guo X, et al. Specifically increased paclitaxel release in tumor and synergetic therapy by a hyaluronic acid–tocopherol nanomicelle. ACS Appl Mater Interfaces. 2017;9(24):20385. doi:10.1021/acsami.7b02606
24. Danhier F, Feron O, Préat V. To exploit the tumor microenvironment: passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J Controlled Release. 2010;148(2):135. doi:10.1016/j.jconrel.2010.08.027
25. Liao J, Zheng H, Fei Z, et al. Tumor-targeting and pH-responsive nanoparticles from hyaluronic acid for the enhanced delivery of doxorubicin. Int J Biol Macromol. 2018;113:737. doi:10.1016/j.ijbiomac.2018.03.004
26. Yin H, Lee ES, Kim D, Lee KH, Oh KT, Bae YH. Physicochemical characteristics of pH-sensitive poly(l-Histidine)-b-poly(ethylene glycol)/poly(l-Lactide)-b-poly(ethylene glycol) mixed micelles. J Controlled Release. 2008;126(2):130. doi:10.1016/j.jconrel.2007.11.014
27. Zhan F, Chen W, Wang Z, et al. Acid-activatable prodrug nanogels for efficient intracellular doxorubicin release. Biomacromolecules. 2011;12(10):3612. doi:10.1021/bm200876x
28. Shen H, Shi H, Xie M, et al. Biodegradable chitosan/alginate BSA-gel-capsules for pH-controlled loading and release of doxorubicin and treatment of pulmonary melanoma. J Mater Chem B. 2013;1(32):3906. doi:10.1039/c3tb20330a
29. Wen Y, Guo Z, Du Z, et al. Serum tolerance and endosomal escape capacity of histidine-modified pDNA-loaded complexes based on polyamidoamine dendrimer derivatives. Biomaterials. 2012;33(32):8111. doi:10.1016/j.biomaterials.2012.07.032
30. Tambe P, Kumar P, Karpe YA, Paknikar KM, Gajbhiye V. Triptorelin tethered multifunctional PAMAM-histidine-PEG nanoconstructs enable specific targeting and efficient gene silencing in LHRH overexpressing cancer cells. ACS Appl Mater Interfaces. 2017;9(41):35562. doi:10.1021/acsami.7b11024
31. Yu GS, Bae YM, Choi H, Kong B, Choi IS, Choi JS. Synthesis of PAMAM dendrimer derivatives with enhanced buffering capacity and remarkable gene transfection efficiency. Bioconjug Chem. 2011;22(6):1046. doi:10.1021/bc100479t
32. Qiu L, Li Z, Qiao M, et al. Self-assembled pH-responsive hyaluronic acid–g-poly(l-histidine) copolymer micelles for targeted intracellular delivery of doxorubicin. Acta Biomater. 2014;10(5):2024. doi:10.1016/j.actbio.2013.12.025
33. Wu JL, Liu CG, Wang XL, Huang ZH. Preparation and characterization of nanoparticles based on histidine–hyaluronic acid conjugates as doxorubicin carriers. J Mater Sci Mater Med. 2012;23:1921.
34. Hu W, Qiu L, Cheng L, et al. Redox and pH dual responsive poly(amidoamine) dendrimer-poly(ethylene glycol) conjugates for intracellular delivery of doxorubicin. Acta Biomater. 2017;36:241. doi:10.1016/j.actbio.2016.03.027
35. Liu Y, Bryantsev VS, Diallo MS, Iii WAG. PAMAM dendrimers undergo ph responsive conformational changes without swelling. J Am Chem Soc. 2009;131(8):2798. doi:10.1021/ja8100227
36. Wu LP, Ficker M, Mejlsøe SL, et al. Poly-(amidoamine) dendrimers with a precisely core positioned sulforhodamine B molecule for comparative biological tracing and profiling. J Controlled Release. 2017;246:88. doi:10.1016/j.jconrel.2016.12.016
37. Kitchens KM, Foraker AB, Kolhatkar RB, Swaan PW, Ghandehari H. Endocytosis and interaction of poly (amidoamine) dendrimers with Caco-2 cells. Pharm Res. 2007;24(11):2138. doi:10.1007/s11095-007-9415-0
38. Kim TI, Bai CZ, Nam K, Park JS. Comparison between arginine conjugated PAMAM dendrimers with structural diversity for gene delivery systems. J Controlled Release. 2009;136(2):132. doi:10.1016/j.jconrel.2009.01.028
39. Albertazzi L, Serresi M, Albanese A, Beltram F. Dendrimer internalization and intracellular trafficking in living cells. Mol Pharm. 2010;7(3):680. doi:10.1021/mp9002464
40. Vercauteren D, Vandenbroucke RE, Jones AT, et al. The use of inhibitors to study endocytic pathways of gene carriers: optimization and pitfalls. Mol Ther J Ame Soc Gene Ther. 2010;18(3):561. doi:10.1038/mt.2009.281
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.