Back to Journals » OncoTargets and Therapy » Volume 13
Transcriptomics Analysis of the Tumor-Inhibitory Pathways of 6-Thioguanine in MCF-7 Cells via Silencing DNMT1 Activity
Authors Li H, An X, Zhang D, Li Q, Zhang N, Yu H ![]() , Li Z
, Li Z ![]()
Received 29 October 2019
Accepted for publication 24 January 2020
Published 11 February 2020 Volume 2020:13 Pages 1211—1223
DOI https://doi.org/10.2147/OTT.S236543
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjeev K. Srivastava
Hao Li,1,* Xinglan An,1,* Daoyu Zhang,1 Qi Li,1 Nan Zhang,1 Hao Yu,2 Ziyi Li1
1Key Laboratory of Organ Regeneration and Transplantation of Ministry of Education, First Hospital, Jilin University, Changchun 130021, People’s Republic of China; 2College of Animal Sciences, Jilin University, Changchun 130062, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Ziyi Li
Key Laboratory of Organ Regeneration and Transplantation of Ministry of Education, First Hospital, Jilin University, Changchun 130021, People’s Republic of China
Tel +86-431-8783-6187
Email [email protected]
Hao Yu
College of Animal Sciences, Jilin University, Changchun 130062, People’s Republic of China
Tel +86-159-4306-5187
Email [email protected]
Background: 6-thioguanine (6-TG), as a conventional “ancient” drug for the treatment of acute leukemia, has been proved to have extensive anti-tumor roles. This study was created to investigate the hidden function of 6-TG on the MCF-7 breast cancer cell line (ER+, PR+) and its mechanisms.
Methods: MCF-7 cells were treated with 6-TG, and the IC50 value was measured by a cell counting kit-8 assay. Differentially expressed genes (DEGs) were confirmed by RNA-seq analysis. Apoptosis and cell cycle consequences were determined by flow cytometry and Western blot analyses.
Results: The results showed that colony formation decreased markedly and the percentage of cell apoptosis increased after 6-TG treatment. DNMT1 mRNA and protein expression decreased, and FAS expression increased. Moreover, 6-TG also induced MCF-7 cells to undergo G2/M phase cell cycle arrest and upregulated CDKN1A (p21).
Conclusion: Overall, our results suggest that 6-TG may induce FAS-mediated exogenous apoptosis and p21-dependent G2/M arrest by inhibiting the activity of DNMT1 in MCF-7 breast cancer cells.
Keywords: 6-TG, MCF-7 breast cancer cells, apoptosis, cell cycle, DNMT1
Introduction
Breast cancer is one of the most common cancers among women.1 According to the report, breast cancer accounts for 30% of all cancers in women. Approximately 75% of breast cancer is of the luminal subtype, in which estrogen receptor is positive, and estrogen receptor-positive breast cancer in young women has increased gradually.2 The treatment for breast cancer is still expensive in some developing countries such as trastuzumab which is a main component of therapy for human positive breast cancer. A real-world study from China has shown that more than 40% patients never received trastuzumab therapy in regions with limited access to those expensive targeted drugs.3 Especially among backward countries, breast cancer still accounts for the highest mortality rate of female tumor patients. Therefore, it is an alternative strategy to turn a safe and cheap drug for other diseases to breast cancer treatment.
In recent years, multiple tumor-suppressor genes that exhibit regional aberrant DNA hypermethylation have been identified as central issues for carcinogenesis.4 The majority of transcriptional genes are regulated by DNA methyltransferase 1 (DNMT1) through methylation-dependent or -independent mechanisms. In mammalian animal cells, DNMT1 mainly participates in the transcriptional silencing and activation of genes.5 DNMT1 is a member of the DNA methyltransferase family that plays a crucial role in epigenetic gene regulation.5 Increasing evidence suggests that the DNMT1-dependent DNA methylation-mediated silencing of tumor-suppressor genes is essential for tumor development and progression.6 In addition to maintaining DNA methylation, DNMT1 can also bind to histone deacetylates to establish a repressive transcription complex and directly target genes.7 TP53 is one of the target genes that DNMT1 regulates at the transcriptional level by forming a transcription complex, and DNMT1 can bind to p53 to regulate the expression of downstream target genes.8 A clinicopathologic study has shown that the expression of DNMT1 is higher than fibroadenoma.9 DNMT1 is also essential for the maintenance and tumorigenesis of breast cancer and cancer stem cells.10 This is visible evidence that it is feasible to use DNMT1 inhibitor for breast cancer.
A clinical study showed that the majority of females with breast cancer were diagnosed with chronic myeloid leukemia (CML) and the risk of CML specifically increased within the first 5 years.11 6-thioguanine (6-TG) not only inhibits the activity of DNMT1, but also has remarkable success in the early treatment of acute and chronic myeloid leukemia.12 However, inhibition of the proliferation of breast cancer cells by 6-TG inhibition of DNMT1 activity has not been reported. Even more encouraging is the recent breakthrough in the molecular mechanisms associated with carcinogenesis and therapeutic pathways with the development of high-throughput sequencing technologies. Therefore, it is promising that high-throughput sequencing technology is an effective tool to deepen the understanding of tumor treatment mechanisms and to predict hub biomarkers and pathways for cancer diagnosis, treatment and prognosis.
Hence, we selected 6-TG as a candidate drug that inhibits the activity of DNMT1 to investigate the effect of 6-TG on MCF-7 cells. Then, we utilized RNA-seq and bioinformatics technology to systematically identify the signal pathway of exerting an antitumor effect via the inhibition of DNMT1. The results provided a feasible strategy for rediscovery and further development of old drugs in the treatment of breast cancer.
Materials and Methods
Reagents
6-TG belongs to the thiopurine family of drugs which are examples of antimetabolites, and it is a purine analog of the nucleobase guanine.13 6-TG was purchased from Selleck (USA), dissolved in DMSO at a concentration of 50 mM, stored at −20°C and prepared the application concentration with the medium. CCK-8 solution was purchased from US EVERBRIGHT®, INC. (USA). TRIzol reagent was purchased from Invitrogen (USA).
Cell Lines and Cell Culture
The breast cancer cell line MCF-7 and the human mammary epithelial cell line MCF-10A were purchased from the Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences (Beijing, China). MCF-7 breast cancer cells were cultured in high-glucose DMEM (Gibco, USA) supplemented with 10% fetal bovine serum (BI, Israel), 1% MEM non-essential amino acids (Gibco, USA), 1% GluMAX and 1% penicillin-streptomycin solution (Gibco, USA). MCF-10A cells were cultured in DMEM/F12 medium (Procell, China). All cells were maintained at 37°C in a cell incubator with 5% CO2.
Cell Proliferation and Viability Assay
The effects of 6-TG on cell viability in vitro were tested by the CCK-8 (Cell-Counting Kit-8) assay. Cells were assayed in 96-well plates (6500 cells/well) in a volume of 100 μL/well of media. After overnight incubation at 37°C and 5% CO2, cells were treated with various concentrations of 6-TG (0 μM, 1 μM, 5 μM, or 10 μM). After treatment with 6-TG, 10 μL of CCK-8 solution was added to each well. Cells in 96-well plates were incubated for 1–4 h, and the optical density was measured at 450 nm using an ELISA (BIO-RAD, USA).
To determine the half-maximal inhibitory concentration (IC50) of 6-TG for MCF-10A and MCF-7 cells, at least three replicates of cells were added to 96-well plates (6500 cells/well). After overnight incubation at 37°C and 5% CO2, serial dilutions of 6-TG at the appropriate range were added to 96-well plates. After 48 h of treatment, 10 μL of CCK-8 solution was added to the media, and the optical density was measured at 450 nm.
Cell Apoptosis Assay
Cell apoptosis was detected by a TransDetectTM Annexin V-EGFP/PI Cell Apoptosis Detection Kit (TRAN, China). Briefly, 2×105 MCF-7 breast cancer cells were added to the 6-well plates overnight, and MCF-7 cells were harvested after 6-TG treatment for 48 h. Subsequently, the cells were carefully washed with prechilled PBS 2 times and centrifuged at 500 g at 4°C for 5 min. Then, the cells were resuspended in 100 μL of prechilled 1× annexin V-binding buffer, and 5 μL of annexin V-EGFP and 5 μL of PI were added and reacted in the dark at room temperature for 15 min. Finally, 200 μL of 1× annexin V-binding buffer was added. The samples were detected by flow cytometry (BD, FACS Fortessa) and the cell apoptosis was analyzed using FlowJo software.
Cell Cycle Distribution Analysis
Then, 2×105 MCF-7 breast cancer cells were added to the 6-well plates overnight, and MCF-7 cells were harvested after 6-TG treatment for 48 h. Cells were carefully washed 2 times with prechilled PBS and centrifuged at 500 g at 4°C for 5 min, and the supernatant was discarded. Subsequently, the cells were fixed with 5 mL of prechilled 70% ethanol for at least 18 h at 4°C and centrifuged at 500 g at 4°C for 5 min. Next, the cells were washed twice with PBS and treated with 300 μL of PI/RNase staining buffer solution in the dark at room temperature for 15 min. Eventually, the samples were detected by a flow cytometer (BD, FACS Fortessa). The data were analyzed by Modfit software.
Colony Formation Assay
MCF-7 breast cancer cells (200) were cultured in 6-well plates at 37°C and 5% CO2 for 24 h. Next, the cells were washed in PBS and treated with 6-TG in a 37°C cell incubator. After 8 days, the cells were stained with 1× Giemsa stain (Sigma G4507) after being washed 3 times with PBS and fixed with methanol for 5 min. Finally, the colony formation numbers were counted under an inverted microscope.
RNA Isolation and qPCR and PCR Array Analysis
Total RNA was extracted from MCF-7 breast cancer cells using TRIzol reagent according to the manufacturer’s instructions. The RNA was reverse transcribed into cDNA with TransScript All-in-One First-Strand cDNA Synthesis SuperMix for qPCR (TRAN, China). The quantitative amplification of cDNA was performed in 96-well optical reaction plates using SYBR® Premix Ex TaqTM reagents (TaKaRa, Japan) and a Light Cycle® 96 Real-Time PCR System (Roche, Switzerland). The qPCR volume was 20 μL and included 10 μL of SYBR Green premix, 7 μL of dH2O, 1 μL of cDNA and 2 μL of primers (forward and reverse primers). The qPCR conditions were as follows: 30 s denaturation at 95°C, 45 cycles of PCR for the quantitative analysis (95°C for 5 s and 60°C for 30 s), one cycle for the melting curve analysis (95°C for 5 s, 60°C for 1 min, and 95°C for 1 s) and cooling at 4°C. The relative expression level of each gene was calculated by using the 2−∆∆CT method. Apoptosis was evaluated by PCR array analysis after 6-TG treatment. The transcription of genes associated with apoptosis was detected by Human Apoptosis PCR Array (QIAGEN, China).
Western Blot
Total protein from MCF-7 breast cancer cells was extracted after 6-TG treatment. The protein samples were fractionated on a BiofurawTM Precast Gel (Tanon, China) and electrically transferred onto a polyvinylidene difluoride (PVDF) membrane. Next, the membranes were blocked with 5% non-fat milk and incubated overnight at 4°C with a primary antibody (DNMT1 antibody from GeneTex, USA; p53 antibody from Affinity, USA; FAS/CD95anyibody from Proteintech, China). The membranes were washed 3 times with TBST and incubated for 1–2 h at room temperature (RT) with a secondary antibody (horseradish peroxidase-conjugated goat anti-rabbit IgG and goat anti-mouse IgG from Proteintech, China). Finally, the membranes were washed 3 times with TBST and visualized by using a Tanon 5200 automatic fluorescence/chemiluminescence imaging analysis system (Tanon, China).
Illumina Deep Sequence and Data Processing for the Analysis of DEGs
The RNA samples from the 6-TG and control groups were sequenced on an Illumina HiSeqTM 2500 system. Demultiplexing, adapter trimming, and initial quality filtering were performed with default parameters within the Illumina NextSeq base calling software. Quality filtered RNA-seq reads were mapped against the human genome using Kallisto.14 Differentially expressed genes were identified using DESeq2 after adjustment for false-discovery rate (FDR q values <0.05) and an absolute value of log2 fold change (FC) ≥ 2.15 The volcano plot was constructed using the ggplot2 package in R.16 We conducted additional hierarchical clustering and principal component analysis (PCA) before differential expression analysis for an overall check of gene-expression profiling of all the samples by factoMineR package in R.17 KEGG pathway analysis of the DEGs was performed using package “clusterProfiler” of R 3.4.3 software. All the mRNA transcripts of the RNA-seq were used to perform the GSEA enrichment analysis.18 We identified the enrichment function of these mRNAs according to their position in different pathways and calculated the false discovery rate (FDR). Gene sets with FDR <0.05 were regarded as significant. Raw sequencing reads generated from this study are deposited in the Gene Expression Omnibus (NCBI) under accession number GSE130161.
PPI Network Construction and Molecular Analysis
The Search Tool for the Retrieval of Interacting Genes (STRING) database provides information regarding the predicted and experimental interactions of proteins.19 In our study, the DEGs with a confidence score ≥0.04 were mapped into PPIs with the STRING database. The PPI network was constructed by using Cytoscape software.
Survival Analysis of Hub Genes
To estimate the time of death and survival probability, data on the effects of genes on the survival of cancer patient samples were downloaded from the human protein atlas (https://www.Proteinatlas.org). Survival analysis, including the P score and 5-year survival rate, is shown in the graph.
Primer and Probe Design and Validation
The primer pairs and probes were designed using Allele ID 7 software. All sequences were tested for potential secondary structure and dimerization formation using the OligoAnalyzer 3.1 program. The specificity of the primers was identified using the Primer-blast program. Validated sequences are shown in Supplementary table S1.
Statistical Analysis
All experimental data are expressed as the mean value ± standard deviation (SD). The qPCR data were analyzed with Prism 7 (GraphPad, LA Jolla, CA) by Student’s t-test and one-way ANOVA. P < 0.05 was considered statistically significant. For the gene integration analysis, the false discovery rate (FDR)-adjusted p-value ≤0.05 and an absolute value of log2 fold change (FC) ≥2 were used as the thresholds to judge the significance of differences in gene expression.
Results
6-TG Inhibited the mRNA and Protein Expression of DNMT1 in the MCF-7 Breast Cancer Cells
The chemical structure of 6-TG is shown in Figure 1A. To explore the effect of 6-TG on DNMT1 expression in breast cancer cells, qPCR and Western blotting assays were used. The results showed that the mRNA and protein levels of DNMT1 in MCF-7 breast cancer cells were significantly decreased after treatment with 6-TG for 48 h (Figure 1B and C). The results suggested that the 6-TG could effectively reduce the activity of DNMT1 in MCF-7 cells.
 |
Figure 1 The inhibition of DNMT1 expression by 6-TG in MCF-7 cells. (A) Chemical structure of 6-thioguanine. (B) The mRNA expression level of DNMT1. GAPDH was used as a control. The data are presented as the mean ± SD of three replicates. ***p<0.001 compared with the control column. (C) The protein level of DNMT1 relative to that of GAPDH. |
Evaluation of the Inhibition Effect of 6-TG on Cell Viability and Proliferation in MCF-7 Breast Cancer Cells
A CCK-8 assay was used to evaluate the effect of 6-TG on the viability of the MCF-7 breast cancer cell line and the MCF-10A epithelial breast cell line. We obtained cell proliferation curves at different concentrations. MCF-7 breast cancer cells were treated with 6-TG for 48 h, and 5 μM 6-TG treatment to MCF-7 cells significantly reduced cell viability compared to other concentrations (Figure 2A). The inhibition effect was dependent on time. After treatment with 6-TG at a concentration gradient with a difference of 10 times for 48 h, the results showed that the treatment of MCF-7 cells caused significant inhibition of the cell growth effect, but the treatment of MCF-10A cells did not cause any remarkable inhibitory effect. As shown in Figure 2B and C, the MCF-7 cell line treated with 6-TG showed a reduction in cell viability, with IC50 values of 5.481 μM (the IC50 value of MCF-10A cells was 54.16 μΜ). Given the cell proliferation curves and the IC50 values, we selected a concentration of 6 μM for 48 h for subsequent experiments. Based on the data showing different IC50 values for MCF-7 and MCF-10A cells, our results suggested that 6-TG can reduce the viability of MCF-7 breast cancer cells and that MCF-10A epithelial breast cells are not sensitive to 6-TG.
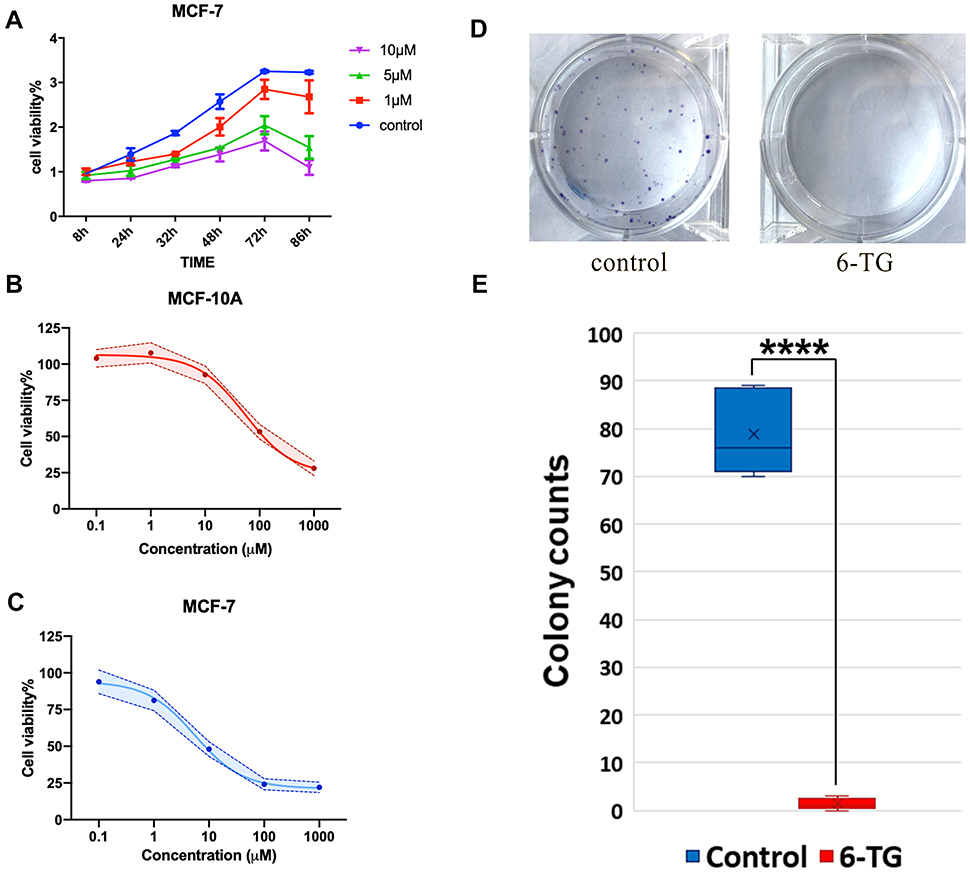 |
Figure 2 6-TG inhibits proliferation and colony formation in the MCF-7 cells. (A) The proliferation curve of MCF-7 cells. Cells were incubated with 0 μM, 1 μM, 5 μM, or 10 μM 6-TG. Cell viability was reduced at 5 μM. (B) The IC50 values of the MCF-10A epithelial cell line following treatment with 6-TG. Each data point represents an average of 3 replicates. (C) The IC50 values of the MCF-7 breast cancer cell line following treatment with 6-TG. Each data point represents an average of 3 replicates. (D, E) Effect of 6-TG on the colony formation ability of MCF-7 cells. MCF-7 cells were exposed to 6 μM 6-TG for 8 days. Representative images and summarised data indicate the inhibition of colony formation. The data are presented as the mean ± SD of three replicates. ****p<0.0001 compared with the control column. |
Next, to determine the inhibitory effect of 6-TG on proliferation in the MCF-7 breast cancer cell line, a colony formation assay was conducted. After treatment with 6-TG for 8 days, colony formation was nearly invisible in the 6-TG group, while in the control group, colony formation was clearly observed (Figure 2D and E). Our results indicated that 6-TG treatment can strongly inhibit colony formation in MCF-7 breast cancer cells.
Identification and Functional Enrichment Analysis of DEGs in MCF-7 Breast Cancer Cell Line
There was a significant difference in the MCF-7 and the MCF-7-6-TG groups. The comparison of MCF-7 with MCF-7 6-TG groups revealed a high number of significant differences in the volcano plot. 1382 upregulated DEGs and 597 downregulated DEGs were identified (Figure 3A), which suggests that these genes could play important roles during the process of 6-TG treatment.
 |
Figure 3 Identification and enrichment analysis of the DEGs. (A) Volcano plot of DEGs in the MCF-7 and MCF-7 6-TG groups. Blue represented low expression levels, and red represented high expression levels. (B) Significant pathway enrichment of the upregulated DEGs was ranked by Q-value. A Q-value <0.05 was regarded as significant. (C) Significant pathway enrichment of the downregulated DEGs was ranked by Q-value. A Q-value <0.05 was regarded as significant. (D) GSEA enrichment plot of the p53 signaling pathway. (E) GSEA enrichment plot of apoptosis. (F) GSEA enrichment plot of the cell cycle. |
According to the KEGG enrichment analysis, the results indicated that the DEGs tended to be involved in diverse biological pathways. Among pathways in cancer, the DEGs were mainly involved in the apoptosis and cell cycle pathways mediated by the p53 signaling pathway (Supplementary Fig S1). Indeed, based on the Q-value and differential expression gene number, the upregulated DEGs mainly participated in the apoptosis and p53 signaling pathway, and the downregulated DEGs were significantly related to the cell cycle (Figure 3B and C). GSEA plots further showed the overall gene enrichment on each pathway (Figure 3D–F). Taken together, the above results suggested that apoptosis, p53 signaling pathway and cell cycle are potential candidate pathways responsible for the inhibition of cell viability and proliferation.
PPI Network Construction of Candidate Pathway
6-TG had an inhibited effect of DNMT1, and apoptosis, the p53 signaling pathway and the cell cycle were selected as the main biological pathways according to the functional enrichment analysis. Therefore, to determine the relationship between DNMT1 and the DEGs related to apoptosis, the effect of DNMT1 on the apoptosis pathway after treatment with 6-TG was determined by constructing a PPI network. The PPI network comprised DNMT1 and 34 genes connected with apoptosis in MCF-7 breast cancer cells and was constructed by using Cytoscape. According to the PPI network, 3 genes directly interacted with DNMT1, and hub genes, including TP53 (degree=16) and FAS (degree=9), were identified. DNMT1 was shown to interact with FAS and p53, and p53 also targeted FAS (Figure 4A). These results indicated that FAS, the key gene that triggers extrinsic apoptosis, might be activated by DNMT1 directly or by DNMT1-mediated TP53.
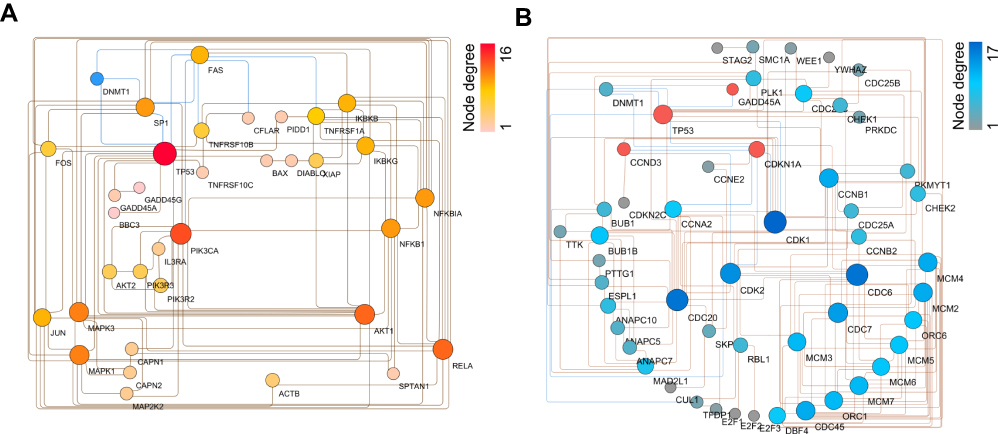 |
Figure 4 PPI network of DEGs in the MCF-7 cancer cell line compared with the 6-TG treatment group. (A) PPI network of DEG interactions based on DNMT1 and apoptosis in MCF-7 cells. (B) PPI network of DEG interactions based on DNMT1 and the cell cycle. |
Furthermore, a PPI network including DNMT1, the cell cycle and the p53-signaling pathway was constructed to determine the relationship between DNMT1 and DEGs related to the cell cycle, which exhibited a complicated interaction relationship with the p53 signaling pathway. In addition, through KEGG pathway enrichment analysis of the DEGs involved in the cell cycle and the p53 signaling pathway, these upregulated genes, such as TP53, CDKN1A and CCND3, in the p53-signaling pathway were identified in the cell cycle process (Supplementary Fig S2). As shown in the PPI network, DNMT1 interacts with these four upregulated genes related to the p53 signaling pathway and then regulated cell cycle progression. Among these DEGs involved in the cell cycle, CDK1 and CDK2 were considered hub genes, with degrees of 17 and 14, respectively. DNMT1 and p53 and CDKN1A had a direct interaction (Figure 4B). These results indicated that treatment of MCF-7 breast cancer cells with 6-TG affected cell cycle progression, and the process might be induced by DNMT1 directly or by DNMT1-mediated CDKN1A to regulate hub gene expression.
Detection of 6-TG-Induced Apoptosis and Cell Cycle in MCF-7 Breast Cancer Cell Line
To determine whether 6-TG treatment can induce apoptosis in the MCF-7 breast cancer cell line, the percentage of apoptotic MCF-7 cells was estimated by flow cytometry after 48 h of exposure to 6-TG. The flow cytometry data showed that the percentage of apoptotic cells in the 6-TG treatment group (18.55%) was higher compared to that of the control group (10.66%). Moreover, after treatment with 6-TG, the early apoptosis rate of MCF-7 cells significantly increased from 1.39% to 9.43%. **p value <0.01 compared with the control group (Figure 5A and B). In addition, the apoptosis results of reference concentration (3 μM and 12 μM) were shown in the Supplementary Fig S3. These results revealed that the apoptosis of MCF-7 cells was induced by 6-TG.
 |
Figure 5 6-TG induced cell apoptosis and cell cycle arrest in MCF-7 breast cancer cells. (A) MCF-7 cell apoptosis was assessed by flow cytometric analysis. In the control group, the apoptosis rate was 10.66%, while the apoptosis rate in the 6-TG treatment group was 18.55%. Each data point represents an average of three replicates. (B) Quantitation of the percentage of MCF-7 cells in the two groups. The data are presented as the mean ± SD of three replicates. **p value <0.01 compared with the control column. (C) The MCF-7 cell cycle progression was assessed by flow cytometric analysis. (D) Quantitation of the percentage of MCF-7 cells in different cell cycle stages. The data are presented as the mean ± SD of three replicates. MCF-7 cells caused G2/M cell cycle arrest after 6-TG treatment. *p value <0.05 compared with the control column. |
Next, to investigate whether 6-TG treatment can induce cell cycle arrest in the MCF-7 breast cancer cell line, flow cytometry was used to detect the effect of 6-TG on the cell cycle in MCF-7 cells. Compared with the control group, 6-TG treatment resulted in an inhibitory effect on cell cycle progression. There was an increase in the number of cells in the G2/M phase, which was followed by a decrease in the number of cells in the G0/G1 and S phase. *p-value <0.05 compared with the control group (Figure 5C and D). The cell cycle results of reference concentration (3 μM and 12 μM) were shown in the Supplementary Fig S4. These results indicated that treatment with 6-TG could induce cell cycle arrest at the G2/M phase in MCF-7 breast cancer cells. From the results, the effect of 6-TG on the apoptosis of MCF-7 cells was more obvious than the effect of cell cycle progression in MCF-7 cells.
Validation of 6-TG-Induced Apoptosis in the MCF-7 Breast Cancer Cells
To further determine the apoptotic response of MCF-7 cells induced by 6-TG, Human Apoptosis PCR Array (PAHS-012Z) was also employed. Our data revealed that the treatment of MCF-7 cells with 6-TG significantly stimulated the transcriptional levels of genes related to apoptosis compared with those of the control group. After treatment with 6-TG, the heatmap showed that the expression of most apoptosis genes, such as TP53, FAS, Caspase 3 and Caspase 9, was increased. Moreover, the expression of the anti-apoptosis gene Bcl 2 was decreased (Figure 6A). The results of PCR array were applied to the apoptosis pathway by KEGG enrichment analysis (Figure 6B). Since the PPI network shows that DNMT1 and FAS have a direct targeted relationship, we suspected that 6-TG might activate the extrinsic apoptosis pathway by activating FAS mediated by DNMT1.
 |
Figure 6 Apoptosis in MCF-7 breast cancer cells induced by 6-TG. (A) Heatmap of gene expression changes in the apoptosis array in MCF-7 and MCF-7-6-TG cells. Blue represents low expression, and red represents high expression. (B) The 6-TG-induced apoptosis pathway graph. Red molecules represent high expression levels. Green molecules represent low expression levels. (C) The mRNA expression level of FAS and TP53 was measured by qPCR. GAPDH was used as a control. The data are presented as the mean ± SD of three replicates. ****p value <0.0001 compared with the control column. (D) The protein level of FAS and p53 relative to that of GAPDH. (E) The data of Western blotting are presented as the mean ± SD of three replicates. ***p value <0.001 compared with the control column. |
To verify the effect of 6-TG treatment on the mRNA and protein expression levels of apoptotic hub genes, qPCR and Western blotting assays were used. The data indicated that 6-TG treatment for 48 h increased the mRNA and protein levels of TP53 and FAS (Figure 6C–E). The results of reference concentration (3 μM and 12 μM) were shown in the Supplementary Fig S5A and B. Although the increase of p53 protein level was not significant, the mRNA and protein levels of FAS increased significantly. Therefore, these results illustrated that 6-TG significantly affected the mRNA level of TP53 and mainly regulated FAS expression to induce apoptosis in the MCF-7 cells.
qPCR Validation and Survival Analysis of Hub Genes Related to Cell Cycle
According to the above results, although the change of cell cycle was not as obvious as apoptosis, the change of hub gene in the pathway was likely to affect the survival time of breast cancer patients, so we explored the effect of hub genes related to cell cycle that were selected by PPI network construction. To validate the differential expression among several selected hub genes, qPCR assay was used to confirm the transcript levels. The data indicated that CDKN1A and CCND3 were upregulated, and that CDK1 and CDK2 were downregulated (Figure 7A). The qPCR results of reference concentration (3 μM and 12 μM) were shown in the Supplementary Fig S5C. Data from the RNA-seq and qPCR two approaches showed agreement with each other. Combined with the PPI network, these results indicated that DNMT1 and CDKN1A had a direct interaction, which induced the upregulation of CDKN1A and ultimately downregulated CDK1 and CDK2 expression.
 |
Figure 7 qPCR data and survival curves of four hub genes related to cell cycle: (A) The mRNA expression levels of hub genes. Each data point represents the mean ± SD of three replicates. **p value <0.01, ***p value <0.001 and ****p value <0.0001 compared with the control column. (B) survival curve of CDK1. (C) survival curve of CDK2. (D) survival curve of CCND3. (E) survival curve of CDKN1A. |
The prognostic information of 4 hub genes selected from the cell cycle PPI network was available in the human protein atlas database (https://www.proteinatlas.org). It was found that the downregulation of CDK1 and CDK2 was correlated with a better survival probability for breast cancer patients, with a 5-year survival rate as high as 77% and 73%, respectively, and as low as 86% and 84%, respectively (Figure 7B and C). Moreover, high CCND3 expression was associated with a better survival probability for breast cancer patients, with a 5-year survival rate as high as 86%, and as low as 80% (Figure 7D). Although CDKN1A gene expression increased most in the 6-TG group, it had no significant effect on the survival of breast cancer patients (Figure 7E). Taken together, these results indicated that CDK1, CDK2, CCND3 might have direct effects on 6-TG-induced cell cycle arrest and CDKN1A only played a regulatory role.
Discussion
Although 6-TG has been available as an anticancer and immunosuppressive agent in medical practice for over the semicentury, it is only widely used in several lymphoid tumors.20 However, the previous report also suggested that some breast cell lines, such as MDA-MB-231 and HCC1937 (triple-negative breast cancer cell lines), had a sensitive effect on 6-TG. The role and mechanism of 6-TG in the luminal subtype breast cancer were not clear.21 Therefore, 6-TG, as a conservative drug, might be a promising anticancer therapy for breast cancer. Here, we conducted an RNA-seq analysis and used bioinformatics technology to systematically explore the effect of 6-TG on MCF-7 breast cancer cells for the first time.
Previous reports have suggested that 6-TG exerts significant activity against human leukemias via cell toxicity induced by incorporation into DNA.22 In addition, 6-TG, as an immunosuppressive drug, was used in autoimmune and chronic inflammatory diseases, such as Crohn’s disease, by regulating GTPase activity, and in pancreatic ductal adenocarcinoma (PDAC) cells, 6-TG inhibited the BRAF-MEK-ERK pathway to induce cell apoptosis.23,24 More notably, 6-TG induced an inhibitory effect on the growth of BRCA-defective tumors, similar to a PARP inhibitor, which was induced because of the inability to repair DNA damage.25,26 Nevertheless, the effect of 6-TG on the most common breast cancer MCF-7 cell line has not been investigated clearly. Consequently, through KEGG enrichment analysis, our data showed that 6-TG treatment mainly upregulated DEGs involved in two biological pathways, apoptosis and the p53 signaling pathway, in MCF-7 cells. At the same time, the downregulated DEGs mainly play important functions in the cell cycle. These three pathways play important roles during cell growth. Therefore, our results indicated that 6-TG affected apoptosis, p53 signaling pathway and the cell cycle in these three main pathways and inhibited MCF-7 cell growth.
With the activation of FAS, FAS ligand (FasL) was also up-regulated, thus triggering the exogenous apoptosis pathway, as described in the report about the effect of scutellarein on Hep3B cells.27 In addition, previous research has shown that in NIH 3T3 cells, DNMT1 regulates the CpG methylation of the FAS promotor to regulate the mRNA expression level of FAS.28 Fas and FasL, which induced cell apoptosis, were activated in a report related to the effect of lycorine hydrochloride on MCF-7 cells.29 In differentiated cells, DNMT1 activated p53 to regulate programmed cell death.30 It has been confirmed that p53, as a dominant regulator of the apoptotic framework, can regulate extrinsic and intrinsic apoptosis pathway signaling cascades.31,32 Additionally, 6-TG decreased DNMT1 expression levels in leukemic cells by inducing the proteasome-mediated degradation of DNMT1.33 6-TG is known to cause DNA damage and accumulating DNA strand breaks, ultimately leading to programmed cell death in HeLa cells.34–36 Interestingly, in our study, we found that 6-TG decreased DNMT1 expression during the process of cell apoptosis and increased FAS and p53 expression levels in MCF-7 cells. Our results also showed that DNMT1 and FAS interacted directly in the PPI network and that the down expression of DNMT1 activated FAS expression. In addition, although the expression level of p53 was only slightly increased, it enhanced the activation of FAS. Taken together, our results inferred that 6-TG caused FAS activation by DNMT1, induced the extrinsic apoptosis pathway and accelerated apoptosis in the MCF-7 breast cancer cell line.
Additionally, it has been suggested that the CDKN1A promoter contains a p53-binding site that permits p53 to transcriptionally activate CDKN1A.37 A previous report related to the role of plumbagin in MCF-7 cells showed that cells were able to arrest at the G2/M phase, mainly accompanied by the upregulation of CDKN1A if p53 is activated.38 However, our experimental results show that the downregulation of DNMT1 did not cause significant changes in p53 protein expression, and the activation of CDKN1A might come directly from the effect of DNMT1. It is known that in normal human fibroblasts, the inhibition of DNMT1 can direct induce the expression of CDKN1A to cause cell cycle arrest.39 Sophie Laget further confirmed that there was a binding site for DNMT1 at methylated CpG islands of CDKN1A.40 By interacting with the N-terminal domain or by disturbing the phosphorylation of CDK1 and CDK2, CDKN1A can inhibit CDK activity.41 In U2OS-derived cell lines, if CDK activity is strongly blocked, cell cycle progression will be inhibited. In a previous report, the upregulation of CDKN1A reduced CDK1/cyclin B complex activity and then hindered cells in the G2/M phase.41,42 CCND3 (cyclin D3) can bind to CDKs and form complex compounds, and the upregulation of Cyclin D-CDK compounds inhibits cell cycle progression in multiple myeloma.43 In the present study, we found that CDKN1A is an essential hub gene that plays a main role in the interaction between the p53 signaling pathway and the cell cycle to regulate the expression of CDK1 and CDK2. Therefore, 6-TG not only induced apoptosis but also produced G2/M phase arrest by the upregulation of CDKN1A.
In conclusion, our results indicated that 6-TG might induce FAS-mediated exogenous apoptosis and CDKN1A-dependent G2/M arrest by inhibiting the activity of DNMT1 in MCF-7 breast cancer cells. Thus, these data provide new insights for revealing the molecular mechanism of 6-TG inhibiting breast cancer development and new opportunities for breast cancer treatment.
Acknowledgments
This research was funded by National Key R&D Program of China, grant number 2017YFA0104400, and Program for Changjiang Scholars and Innovative Research Team in University, grant number IRT_16R32.
Disclosure
The authors report no conflicts of interest in this work.
References
1. DeSantis C, Ma J, Bryan L, Jemal A. Breast cancer statistics, 2013. CA Cancer J Clin. 2014;64(1):52–62. doi:10.3322/caac.21203
2. Jeong SB, Im JH, Yoon JH, et al. Essential role of polo-like kinase 1 (Plk1) oncogene in tumor growth and metastasis of tamoxifen-resistant breast cancer. Mol Cancer Ther. 2018;17(4):825–837. doi:10.1158/1535-7163.MCT-17-0545
3. Li J, Wang S, Wang Y, et al. Disparities of trastuzumab use in resource-limited or resource-abundant regions and its survival benefit on HER2 positive breast cancer: a real-world study from China. Oncologist. 2017;22(11):1333–1338. doi:10.1634/theoncologist.2017-0088
4. Baylin SB, Makos M, Wu JJ, et al. Abnormal patterns of DNA methylation in human neoplasia: potential consequences for tumor progression. Cancer Cells. 1991;3(10):383–390.
5. Lyko F. The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat Rev Genet. 2018;19(2):81–92. doi:10.1038/nrg.2017.80
6. Huang J, Stewart A, Maity B, et al. RGS6 suppresses Ras-induced cellular transformation by facilitating Tip60-mediated Dnmt1 degradation and promoting apoptosis. Oncogene. 2014;33(27):3604–3611. doi:10.1038/onc.2013.324
7. Rountree MR, Bachman KE, Baylin SB. DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat Genet. 2000;25(3):269–277. doi:10.1038/77023
8. Esteve PO, Chin HG, Pradhan S. Human maintenance DNA (cytosine-5)-methyltransferase and p53 modulate expression of p53-repressed promoters. Proc Natl Acad Sci U S A. 2005;102(4):1000–1005. doi:10.1073/pnas.0407729102
9. Yu Z, Xiao Q, Zhao L, et al. DNA methyltransferase 1/3a overexpression in sporadic breast cancer is associated with reduced expression of estrogen receptor-alpha/breast cancer susceptibility gene 1 and poor prognosis. Mol Carcinog. 2015;54(9):707–719. doi:10.1002/mc.22133
10. Pathania R, Ramachandran S, Elangovan S, et al. DNMT1 is essential for mammary and cancer stem cell maintenance and tumorigenesis. Nat Commun. 2015;6:6910. doi:10.1038/ncomms7910
11. Al-Husseini MJ, Mohamed HH, Saad AM, et al. Risk and survival of chronic myeloid leukemia after breast cancer: a population-based study. Curr Probl Cancer. 2018;43:213–221.
12. Munshi PN, Lubin M, Bertino JR. 6-thioguanine: a drug with unrealized potential for cancer therapy. Oncologist. 2014;19(7):760–765. doi:10.1634/theoncologist.2014-0178
13. Wang H, Wang Y. 6-Thioguanine perturbs cytosine methylation at the CpG dinucleotide site by DNA methyltransferases in vitro and acts as a DNA demethylating agent in vivo. Biochemistry. 2009;48(10):2290–2299. doi:10.1021/bi801467z
14. Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 2016;34(5):525–527. doi:10.1038/nbt.3519
15. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi:10.1186/s13059-014-0550-8
16. Walter W, Sanchez-Cabo F, Ricote M. GOplot: an R package for visually combining expression data with functional analysis. Bioinformatics. 2015;31(17):2912–2914. doi:10.1093/bioinformatics/btv300
17. Le S, Josse J, Husson F. FactoMineR: an R package for multivariate analysis. J Stat Softw. 2008;25(1):1–18. doi:10.18637/jss.v025.i01
18. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. doi:10.1073/pnas.0506580102
19. Ni M, Liu X, Wu J, et al. Identification of candidate biomarkers correlated with the pathogenesis and prognosis of non-small cell lung cancer via integrated bioinformatics analysis. Front Genet. 2018;9:469. doi:10.3389/fgene.2018.00469
20. Karran P, Attard N. Thiopurines in current medical practice: molecular mechanisms and contributions to therapy-related cancer. Nat Rev Cancer. 2008;8(1):24–36. doi:10.1038/nrc2292
21. Gu Y, Helenius M, Vaananen K, et al. BRCA1-deficient breast cancer cell lines are resistant to MEK inhibitors and show distinct sensitivities to 6-thioguanine. Sci Rep. 2016;6:28217. doi:10.1038/srep28217
22. Cheng HW, Armstrong RD, Sadee W. Modulation of 6-thioguanine activity by guanine in human promyelocytic leukemia HL-60 cells. Cancer Res. 1988;48(13):3648–3651.
23. Tiede I, Fritz G, Strand S, et al. CD28-dependent Rac1 activation is the molecular target of azathioprine in primary human CD4+ T lymphocytes. J Clin Invest. 2003;111(8):1133–1145. doi:10.1172/JCI16432
24. Kim I, Choi YS, Song JH, et al. A drug-repositioning screen for primary pancreatic ductal adenocarcinoma cells identifies 6-thioguanine as an effective therapeutic agent for TPMT-low cancer cells. Mol Oncol. 2018;12(9):1526–1539. doi:10.1002/1878-0261.12364
25. Issaeva N, Thomas HD, Djureinovic T, et al. 6-thioguanine selectively kills BRCA2-defective tumors and overcomes PARP inhibitor resistance. Cancer Res. 2010;70(15):6268–6276. doi:10.1158/0008-5472.CAN-09-3416
26. Yalon M, Tuval-Kochen L, Castel D, et al. Overcoming resistance of cancer cells to PARP-1 inhibitors with three different drug combinations. PLoS One. 2016;11(5):e0155711. doi:10.1371/journal.pone.0155711
27. Sang Eun H, Seong Min K, Ho Jeong L, et al. Scutellarein induces fas-mediated extrinsic apoptosis and G2/M cell cycle arrest in Hep3B hepatocellular carcinoma cells. Nutrients. 2019;11(2). doi:10.3390/nu11020263.
28. Gazin C, Wajapeyee N, Gobeil S, Virbasius CM, Green MR. An elaborate pathway required for Ras-mediated epigenetic silencing. Nature. 2007;449(7165):1073–1077. doi:10.1038/nature06251
29. Ji Y, Yu M, Qi Z, et al. Study on apoptosis effect of human breast cancer cell MCF-7 induced by lycorine hydrochloride via death receptor pathway. Saudi Pharm J. 2017;25(4):633–637. doi:10.1016/j.jsps.2017.04.036
30. Loughery JE, Dunne PD, O’Neill KM, Meehan RR, McDaid JR, Walsh CP. DNMT1 deficiency triggers mismatch repair defects in human cells through depletion of repair protein levels in a process involving the DNA damage response. Hum Mol Genet. 2011;20(16):3241–3255. doi:10.1093/hmg/ddr236
31. Maheshwari A, Misro MM, Aggarwal A, Sharma RK, Nandan D. Pathways involved in testicular germ cell apoptosis induced by H2O2 in vitro. FEBS J. 2009;276(3):870–881. doi:10.1111/j.1742-4658.2008.06831.x
32. Yang G, Zhang W, Qin Q, et al. Mono(2-ethylhexyl) phthalate induces apoptosis in p53-silenced L02 cells via activation of both mitochondrial and death receptor pathways. Environ Toxicol. 2015;30(10):1178–1191. doi:10.1002/tox.v30.10
33. Yuan B, Zhang J, Wang H, et al. 6-Thioguanine reactivates epigenetically silenced genes in acute lymphoblastic leukemia cells by facilitating proteasome-mediated degradation of DNMT1. Cancer Res. 2011;71(5):1904–1911. doi:10.1158/0008-5472.CAN-10-3430
34. Hogarth LA, Redfern CP, Teodoridis JM, et al. The effect of thiopurine drugs on DNA methylation in relation to TPMT expression. Biochem Pharmacol. 2008;76(8):1024–1035. doi:10.1016/j.bcp.2008.07.026
35. Zeng X, Kinsella TJ. Mammalian target of rapamycin and S6 kinase 1 positively regulate 6-thioguanine-induced autophagy. Cancer Res. 2008;68(7):2384–2390. doi:10.1158/0008-5472.CAN-07-6163
36. Swann PF, Waters TR, Moulton DC, et al. Role of postreplicative DNA mismatch repair in the cytotoxic action of thioguanine. Science. 1996;273(5278):1109–1111. doi:10.1126/science.273.5278.1109
37. Xue WH, Fan ZR, Li LF, et al. Construction of an oesophageal cancer-specific ceRNA network based on miRNA, lncRNA, and mRNA expression data. World J Gastroenterol. 2018;24(1):23–34. doi:10.3748/wjg.v24.i1.23
38. De U, Son JY, Jeon Y, et al. Plumbagin from a tropical pitcher plant (Nepenthes alata Blanco) induces apoptotic cell death via a p53-dependent pathway in MCF-7 human breast cancer cells. Food Chem Toxicol. 2019;123:492–500. doi:10.1016/j.fct.2018.11.040
39. Young JI, Smith JR. DNA methyltransferase I (DNMT1) inhibition induces a p21 dependent cell cycle arrest. TheScientificWorldJournal. 2001;1:131. doi:10.1100/tsw.2001.232
40. Laget S, Miotto B, Chin HG, et al. MBD4 cooperates with DNMT1 to mediate methyl-DNA repression and protects mammalian cells from oxidative stress. Epigenetics. 2014;9(4):546–556. doi:10.4161/epi.27695
41. Dutto I, Tillhon M, Cazzalini O, Stivala LA, Prosperi E. Biology of the cell cycle inhibitor p21(CDKN1A): molecular mechanisms and relevance in chemical toxicology. Arch Toxicol. 2015;89(2):155–178.
42. Medema RH, Klompmaker R, Smits VA, Rijksen G. p21waf1 can block cells at two points in the cell cycle, but does not interfere with processive DNA-replication or stress-activated kinases. Oncogene. 1998;16(4):431–441. doi:10.1038/sj.onc.1201558
43. Misiewicz-Krzeminska I, Sarasquete ME, Vicente-Duenas C, et al. Post-transcriptional modifications contribute to the upregulation of cyclin D2 in multiple myeloma. Clin Cancer Res. 2016;22(1):207–217. doi:10.1158/1078-0432.CCR-14-2796
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.