Back to Journals » Journal of Pain Research » Volume 12
Transcriptome profiling of dorsal root ganglia in a rat model of complex regional pain syndrome type-I reveals potential mechanisms involved in pain
Authors Yin C ![]() , Hu Q, Liu B, Tai Y, Zheng X
, Hu Q, Liu B, Tai Y, Zheng X ![]() , Li Y, Xiang X, Wang P, Liu B
, Li Y, Xiang X, Wang P, Liu B ![]()
Received 26 September 2018
Accepted for publication 27 February 2019
Published 12 April 2019 Volume 2019:12 Pages 1201—1216
DOI https://doi.org/10.2147/JPR.S188758
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Katherine Hanlon
Chengyu Yin,1,2,* Qimiao Hu,1,* Boyu Liu,1,* Yan Tai,3 Xiaoli Zheng,1 Yuanyuan Li,1 Xuaner Xiang,1 Ping Wang,4 Boyi Liu1
1Department of Neurobiology and Acupuncture Research, The Third Clinical Medical College, Zhejiang Chinese Medical University, Key Laboratory of Acupuncture and Neurology of Zhejiang Province, Hangzhou, People’s Republic of China; 2College of Life Science, Zhejiang Chinese Medical University, Hangzhou, People’s Republic of China; 3Academy of Chinese Medicine Sciences, Zhejiang Chinese Medical University, Hangzhou, People’s Republic of China; 4Department of Pathology, School of Basic Medical Science, Zhejiang Chinese Medical University, Hangzhou, People’s Republic of China
*These authors contributed equally to this work
Purpose: Complex regional pain syndrome type-I (CRPS-I) is a progressive and devastating pain condition, which remains clinically challenging. The mechanisms of CRPS-I still remain largely unknown. We aim to identify transcriptome profiles of genes relevant to pain mechanisms and major pathways involved in CRPS-I.
Methods: A rat model of chronic post-ischemia pain (CPIP) was established to mimic CRPS-I. RNA-sequencing (RNA-Seq) was used to profile transcriptome of L4-6 dorsal root ganglia (DRGs) of a rat model of CRPS-I.
Results: CPIP model rats developed persistent mechanical/thermal hyperalgesia in ipsilateral hind paw. RNA-Seq identified a total of 295 differentially expressed genes (DEGs), including 195 up- and 100 downregulated, in ipsilateral DRGs of CPIP rats compared with sham rats. The expression of several representative genes was confirmed by qPCR. Functional analysis of DEGs revealed that the most significant enriched biological processes of upregulated genes include response to lipopolysaccharide, inflammatory response and cytokine activity, which are all important mechanisms mediating pain. We further screened DEGs implicated in pain progress, genes enriched in small- to medium-sized sensory neurons and enriched in TRPV1-lineage nociceptors. By comparing our dataset with other published datasets of neuropathic or inflammatory pain models, we identified a core set of genes and pathways that extensively participate in CPIP and other neuropathic pain states.
Conclusion: Our study identified transcriptome gene changes in DRGs of an animal model of CRPS-I and could provide insights into identifying promising genes or pathways that can be potentially targeted to ameliorate CRPS-I.
Keywords: RNA-Seq, pain, CRPS-I, dorsal root ganglion, neuropathic pain
Introduction
Complex regional pain syndrome type-I (CRPS-I) is a progressive and devastating pain condition that can be initiated by surgery, ischemia or fractures to the extremities.5,15 CRPS-I is usually induced after an initial noxious stimulus and followed with edema, skin blood flow changes, thermal and mechanical hyperalgesia or allodynia in affected body parts. CRPS-I can develop into chronic pain condition that dramatically affects the patient’s life quality.49 Current available treatment options, such as nonsteroidal anti-inflammatory drugs, corticosteroids and physiotherapy, etc cannot produce satisfactory relieving effects on CRPS-I, rendering it a clinically difficult to treat pain condition.27
At present, the mechanisms underlying CRPS-I are still not fully elucidated. Ischemia/reperfusion injury is among one of the causes of CRPS-I.4,15 In order to gain more insights into the molecular mechanisms of CRPS-I, Coderre et al established a rat chronic postischemic pain (CPIP) model by applying prolonged hind paw ischemia and reperfusion to mimic CRPS-I.16 The CPIP model manifests many key CRPS-I-like symptoms, including early swelling, hyperemia, hind paw warmth, and long-lasting neuropathic pain condition.16,35,39 Therefore, the CPIP model is a frequently used animal model for mechanistic study of CRPS-I.
Recently, the advances in RNA-sequencing (RNA-Seq) technique and bioinformatics have made it possible to perform whole transcriptome analysis of gene changes. RNA-Seq technique has been used to profile transcriptome of sensory ganglia in physiological conditions as well as to explore gene changes under inflammatory or neuropathic pain conditions.2,22,24,28 The dorsal root ganglion (DRG) contains primary sensory neurons, which are responsible for receiving pain signals in response to noxious stimulation and conveying pain signals to spinal cord dorsal horn and play an important role in pain signal integration, transduction and peripheral sensitization.3 In order to investigate the molecular mechanisms of CRPS-I-induced pain and explore possible therapeutic options, we performed transcriptome analysis of the DRGs that innervate the affected hind limb of CPIP model rats. We determined the overall profiles of differentially expressed genes (DEGs) in CPIP model and screened the genes that are possibly involved in pain mechanisms and nociception. We further investigated the major pathways or functions that these DEGs are possibly involved in. Finally, we compared our findings with previously published microarray datasets of neuropathic and inflammatory pain models and we identified a core set of genes and pathways that extensively participate in CPIP and other neuropathic pain states. Therefore, our study will further advance our understanding of the peripheral neuronal mechanisms of CRPS-I-induced pain symptom and help to identify promising genes that can be targeted for CRPS-I treatment.
Material and methods
Animals
Male Sprague Dawley rats (from Laboratory of Animal Research Center, Zhejiang Chinese Medical University, Hangzhou, China), weighting 300–320 g, were used in this study. The rats were housed five per cage on a 12 hour light/dark cycle with controlled temperature. Food and water were provided ad libitum. The rats were given minimum of 1 week to adapt to the new environment before experiment. All experimental procedures were approved by the Animal Ethics Committee of Zhejiang Chinese Medical University and carried out according to the National Institutes of Health guide for the care and use of Laboratory animals (NIH Publications No. 8023, revised 1978).
CPIP rat model establishment
CPIP was established through exposure to prolonged hind paw ischemia and reperfusion as described previously.16 Anesthesia was induced in all rats with an intraperitoneal injection of 50 mg/kg of sodium phenobarbital and was maintained with an infusion of sodium phenobarbital at 20 mg/kg/hour. An O-ring with 7/32 internal diameter was tightly passed around the right hind limb just proximal to the ankle joint. The O-ring was then cut off 3 hours later for reperfusion. Sham rats received the same anesthetic procedure but the ankle was surrounded with a cut O-ring which did not block blood flow.
Behavioral test
Rats were habituated to the test environment daily for a consecutive 3 days before baseline test. Rats were individually placed in transparent plexiglas chambers on an elevated mesh floor and were habituated for 30 minutes before test. The mechanical allodynia was determined using a series of von Frey filaments (UGO Basile, Italy) applied perpendicularly to the mid plantar surface of the hind paws, with sufficient force to bend the filament slightly for 3–5 seconds according to methods previously described.9 An abrupt withdrawal of the paw and licking and vigorously shaking in response to stimulation were considered pain-like responses. The threshold was determined using the up-down testing paradigm, and the 50% paw withdrawal threshold was calculated by the nonparametric Dixon test.29,30
The Plantar Test Apparatus (Ugo Basile) was used to evaluate thermal hyperalgesia according to methods previously described.7 A radiant light beam generated by a light bulb was directed into the right hind paw in order to determine the paw withdrawal latency (the time spent to remove the paw from the stimulus). A 25 second cutoff threshold was set to avoid excessive heating to cause injury. Significant decreases in paw withdrawal latency were interpreted as thermal hyperalgesia. All the above behavior tests are conducted by an experimenter blinded to experimental conditions.
Tissue collect and RNA extraction
At day 7, rats were deeply anesthetized with sodium pentobarbital (40 mg/kg) and perfused through the ascending aorta with saline. After the perfusion, the L4-6 DRG segments were collected. Total RNA from the CPIP and sham groups was extracted using TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA) following the manufacturer’s instructions, and then treated with DNase I to degrade contaminating DNA. The purity and concentration of the samples was evaluated using Nanodrop spectrophotometer (NanoDrop Products, CA, USA). The RNA integrity number was measured using an Agilent 2100 BioAnalyzer (Agilent Technologies, Santa Clara, CA, USA).
RNA-Seq library establishment and RNA-Seq
Total mRNAs from three rats of from CPIP group and three rats from sham groups were isolated.
DNase I was used to degrade double-stranded and single-stranded DNA contaminant in RNA samples. mRNA molecules were purified from total RNA using oligo(dT)-attached magnetic beads. mRNA were fragmented into small pieces using fragmentation reagent. First-strand cDNA was generated using random hexamer-primed reverse transcription, followed by a second-strand cDNA synthesis. The synthesized cDNA was subjected to end-repair and then was 3ʹ adenylated. Adaptors were ligated to the ends of these 3ʹ adenylated cDNA fragments. This process is to amplify the cDNA fragments with adaptors from previous step. PCR products are purified with the SPRI beads, and dissolved in EB solution. The double stranded PCR products were heat denatured and circularized by the splint oligo sequence. The single-strand circle DNA (ssCir DNA) was formatted as the final library. Library was validating on the Agilent Technologies 2100 bioanalyzer. The library was amplified with phi29 to make DNA nanoball (DNB) which have more than 300 copies of one molecular. The DNBs were loaded into the patterned nanoarray and single end 50 base reads were generated in the way of sequenced by synthesis. Finally, the fragments were enriched by PCR amplification to construct a library ready for sequencing using BGISEQ-500 by BGI Group (Shenzhen, China). The raw data of gene expression changes is provided as
Bioinformatics analysis
Primary sequencing data produced by RNA-Seq (raw reads) were subjected to quality control (QC). Raw reads were filtered into clean reads by internal software SOAPnuke (version 1.5.2), followed by: Remove reads in which unknown bases (N) are more than 10%; Remove reads with adaptors; Remove low quality reads (we define the low quality read as the percentage of base which quality is less than 15 and greater than 50% in a read). QC of alignment was performed to determine if re-sequencing was needed. If the alignment result passed QC, downstream analysis including gene expression, differentially expressed genes, cluster analysis, gene ontology (GO) enrichment analysis, KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway enrichment analysis, etc proceeded.
Cluster analysis and screening of differentially expressed genes
Distances of expressed genes were calculated using the Euclidean method.12 The sum of the squared deviations algorithm was used to calculate distance. The cluster analysis and heat map visualization of gene expression patterns was performed using “pheatmap” package in the R software of Bioconductor. Differentially expressed mRNAs with statistical significance were identified through Volcano Plot filtering.
The threshold required for the results to be considered significant was as follows: Q-value ≤0.01 and absolute value of |log2 (Fold Change)| ≥1.0.
Functional enrichment analysis of DEGs
Functional enrichment analysis was performed by functional annotation package “clusterProfiler” in R studio software (RStudio, Boston, MA, USA). GO and KEGG enrichment analysis was also conducted. For each enriched function term, P-value of enriched functions and the P-value by multiple testing corrections were calculated by “clusterProfiler” package in R studio software.
Real‑time quantitative PCR (qPCR)
The extracted total RNA from the DRGs was reverse-transcribed into cDNA using random hexamers primers (TaKaRa Bio Inc., Shiga, Japan) according to the manufacturer’s instruction. The sequences of all primers used were shown in Table 1. qPCR was performed using DRG tissues from rats other than the ones used for RNA-Seq. Gapdh was used as an internal reference gene. qPCR was performed using the Fast Start Universal SYBR Green Master kit (TaKaRa Bio Inc.) with 25 µL reaction system according to the manufacturer’s protocol by CFX96 Real-Time System (Bio-Rad Laboratories Inc., Hercules, CA, USA). Each reaction was performed in triplicates and normalized to Gapdh gene expression. The CT value of each well was determined using the CFX96 Real-Time System software and the average of the triplicates was calculated. The relative quantification was determined by the ΔΔCT method.8,38
 | Table 1 Sequences of the primers used for qPCR validation of RNA-Seq data |
Source of microarray data
Pain genes (PG): The pain gene list was retrieved from a previous study using the same list to predict potential genes involved in mediating masseter inflammation-induced pain.13 This pain gene list contains 685 genes, which mainly includes human genes related to transmission of pain signals, central and peripheral inflammatory responses and pain mood and affect (see
Sensory neuronal genes (SN): The sensory neuronal gene list was retrieved from a previous study using magnetic cell sorting (MACS) to separate neuronal cells from nonneuronal cells from mouse DRG.48 In this study, Thakur et al demonstrated that MACS successfully segregated neuronal cells from nonneuronal cells from mouse DRGs. This procedure can enrich neuronal population up to 95% compared with 10% before sorting.
Transcriptome analysis further confirmed that many genes were enriched in the neuronal population collected through MACS. We selected genes that showed increase in expression by >40% according to criterion described by Chung et al13 (see
TRPV1-lineage neuronal genes (VL): TRPV1-lineage neuronal genes list was derived from a recent study which identified a group of genes enriched in TRPV1 expressing nociceptive sensory neurons.24 By using mouse lines selectively labeling or ablating TRPV1 lineage neurons, Goswami et al identified a group of genes enriched in VL sensory neurons. We screened genes that are enriched >3-fold in VL neurons compared to non-VL neurons according to a study by Chung et al13 (see
SNL and SNI neuropathic microarray: Data was downloaded from GEO (Gene Expression Omnbius) dataset, at the website of
Protein-protein interaction (PPI) network analysis
The search tool for the retrieval of interacting genes (STRING) is used to provide information regarding predicted and experimental interactions of proteins and the prediction method of this database is from neighborhood, gene fusion, co‑occurrence, co‑expression experiments, databases, and text mining. By setting the Combination score >0.4 as the reliability threshold value, the web based STRING database (
Statistical analysis
qPCR and behavioral results were analyzed by unpaired Student’s t-test or two-way ANOVA followed by Tukey’s post hoc test. Data in graphs are expressed as means ± SEM. Comparison is considered significantly different if the P-value is less than 0.05.
Results
The establishment of the CPIP rat model
The rat CPIP model is a frequently used animal model in laboratory to mimic human CRPS-I. We followed the methods described by Coderre et al to establish the rat CPIP model. An O-ring tourniquet was applied to the right ankle joint for 3 hours to reduce the blood flow to the hind paw.16 During the treatment, the hind paw showed cyanosis, indicating tissue hypoxia (Figure 1A, left panel). Ten minutes after the reperfusion, the ipsilateral hind paw showed obvious edema compared with contralateral hind paw (Figure 1A, middle panel). Seven days later, the treated hind paw showed signs of excoriation and dryness (Figure 1A, right panel). The ipsilateral hind paw showed obvious edema after the treatment but gradually returned to normal 72 hours later (Figure 1B). We measured the pain responses of CPIP model rats for a consecutive 14 days. As shown in Figure 1C and D, CPIP model rats exhibited rapid and persistent thermal and mechanical hyperalgesia compared with Sham group rats. Thermal and mechanical hyperalgesia both appeared 1 day after model establishment and lasted until 14 days of our observation time frame. We sacrificed the rats on day 7, a time point when rats exhibited stable pain responses and then harvested ipsilateral L4–6 dorsal root ganglia (DRGs) for RNA-Seq.
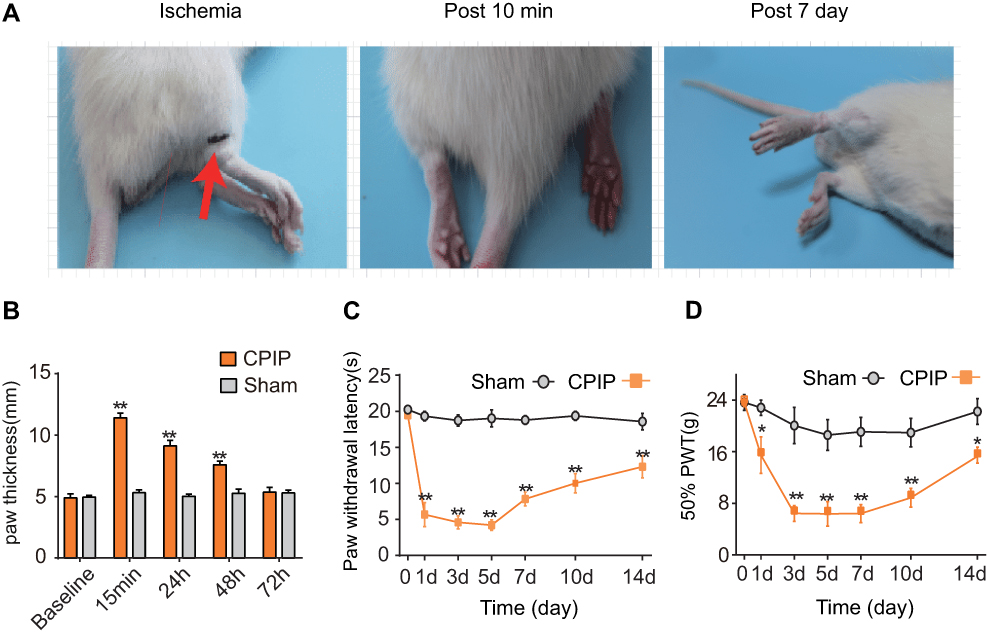 | Figure 1 The CPIP rat model exhibits persistent mechanical and thermal hyperalgesia. (A) Representative photographs of rat hind paw taken during CPIP model establishment, 10 minutes and 7 days after model establishment. The red arrow indicates the O ring clamped right above the ankle. (B) Thickness evaluation of ipsilateral hind paws of CPIP and Sham group rats. (C) Paw withdraw latency of ipsilateral hind paws of Sham and CPIP group rats. (D) 50% paw withdraw threshold of ipsilateral hind paw of Sham and CPIP group rats. N=6 rats/group. *P<0.05 and **P<0.01 vs Sham group. Abbreviation: CPIP, chronic post-ischemic pasin. |
Overview of gene expression profiles of DRGs after CPIP by RNA-Seq
We then used RNA-Seq to explore gene expression profiles of ipsilateral L4–6 DRGs 7 days after CPIP. The sequencing produced approximate 24.1 million raw reads per sample and the clean reads ratio was nearly 99.0% (Table 2). More than 89% of bases had a quality score ≥Q30 and over 95% of the clean reads data were mapped to the rat genome (Table 2). Eventually, 15,250 genes were successfully mapped and identified from RNA-Seq. Next, we utilized the hierarchical clustering analyses to get an overview of gene expression profiles of CPIP model rats compared with Sham rats. Hierarchical clustering analysis of all genes showed that three3 CPIP model samples and three3 Sham group samples were clustered in separate groups (Figure 2A). The clear segregation and clustering of the sequencing data indicated a distinct RNA expression profile existed between CPIP and Sham groups but not within groups. We then filtered out the DEGs with criteria of fold change ≥2 and q-value ≤0.01. The volcano plot indicated the number of DEGs in the CPIP group compared with the Sham group (Figure 2B). A total of 295 DEGs (195 up- and 100 downregulated) were identified (Figure 2B). The identified DEGs were further summarized and illustrated in a heat map with cluster analysis, which indicated a high level of consistency in either CPIP or Sham group samples (Figure 2C). The above data demonstrated that the rat CPIP model was accompanied with significant gene changes in ipsilateral DRGs.
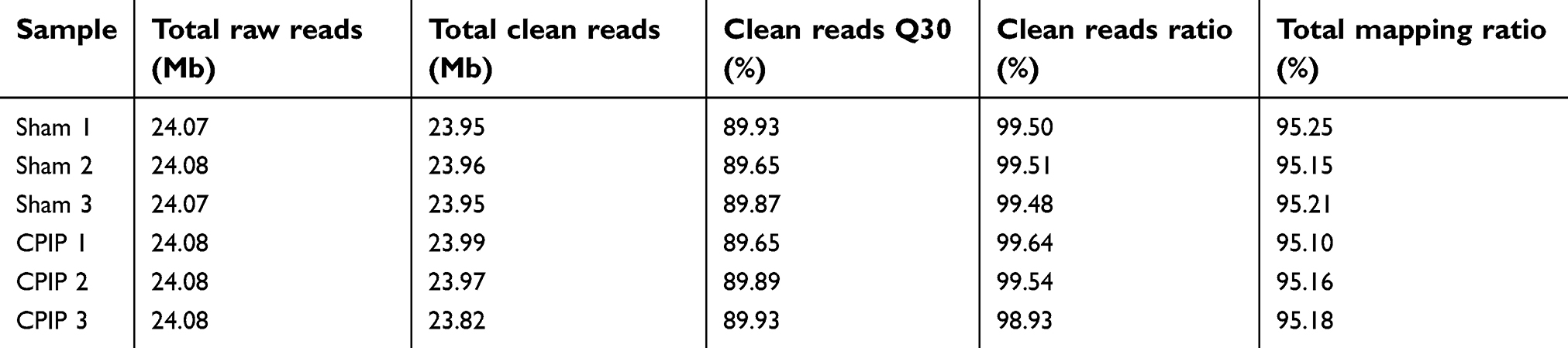 | Table 2 The information of total reads and mapping ratio for Sham and CPIP groups in RNA-Seq |
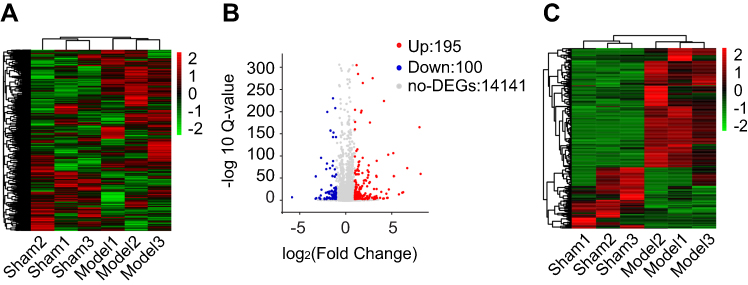 | Figure 2 RNA-Seq reveals transcriptome profile of gene expression changes in DRGs induced by CPIP. (A) Heat map illustration of hierarchical clustering of overall gene expression profile of DRGs from Sham and CPIP group rats. (B) Volcano plot showing gene expression profiles in DRGs of CPIP group compared with Sham group. Red and blue spots indicate up- and downregulated DEGs, respectively, whereas grey spots indicate non-DEGs. (C) Heat map illustration of hierarchical clustering of DEGs from CPIP and Sham groups. Abbreviations: RNA-Seq, RNA-sequencing; DRGs, dorsal root ganglions; CPIP, chronic post-ischemic pain; DEGs, differentially expressed genes. |
Analysis of DEGs in DRGs of CPIP model
Among the DEGS we have obtained, some of the genes are well established to be involved in inflammation and pain processes. These genes include: Vip (vasoactive intestinal polypeptide, fold change=261.4), IL6 (interleukin-6 precursor, fold change=163.1), Cxcl13 (C-X-C motif chemokine 13 precursor, fold change=5.6), Ccl2 (C-C motif chemokine 2 precursor, fold change =2.7), Nlrp3 (NLR family pyrin domain containing 3, fold change=2.6) and MMP9 (matrix metalloproteinase-9 precursor, fold change=2.4), etc. In addition, many other genes, whose roles in inflammation and pain have not yet been identified, were dramatically altered as well. We analyzed the data and found that 38 genes showed expression changes more than 10-fold, with 37 upregulated genes and one downregulated gene, such as Sprr1a (small proline-rich protein 1A, fold change=477.4), Crisp1 (cysteine-rich secretory protein 1 precursor, fold change=451.2), Lce1f (late cornified envelope 1F, fold change=372.4); 39 genes showed expression changes between 5- and 10-fold, which include 29 upregulated and 10 downregulated genes. Tables 3 and 4 illustrated the detailed information of the top 20 up- and 20 downregulated DEGs.
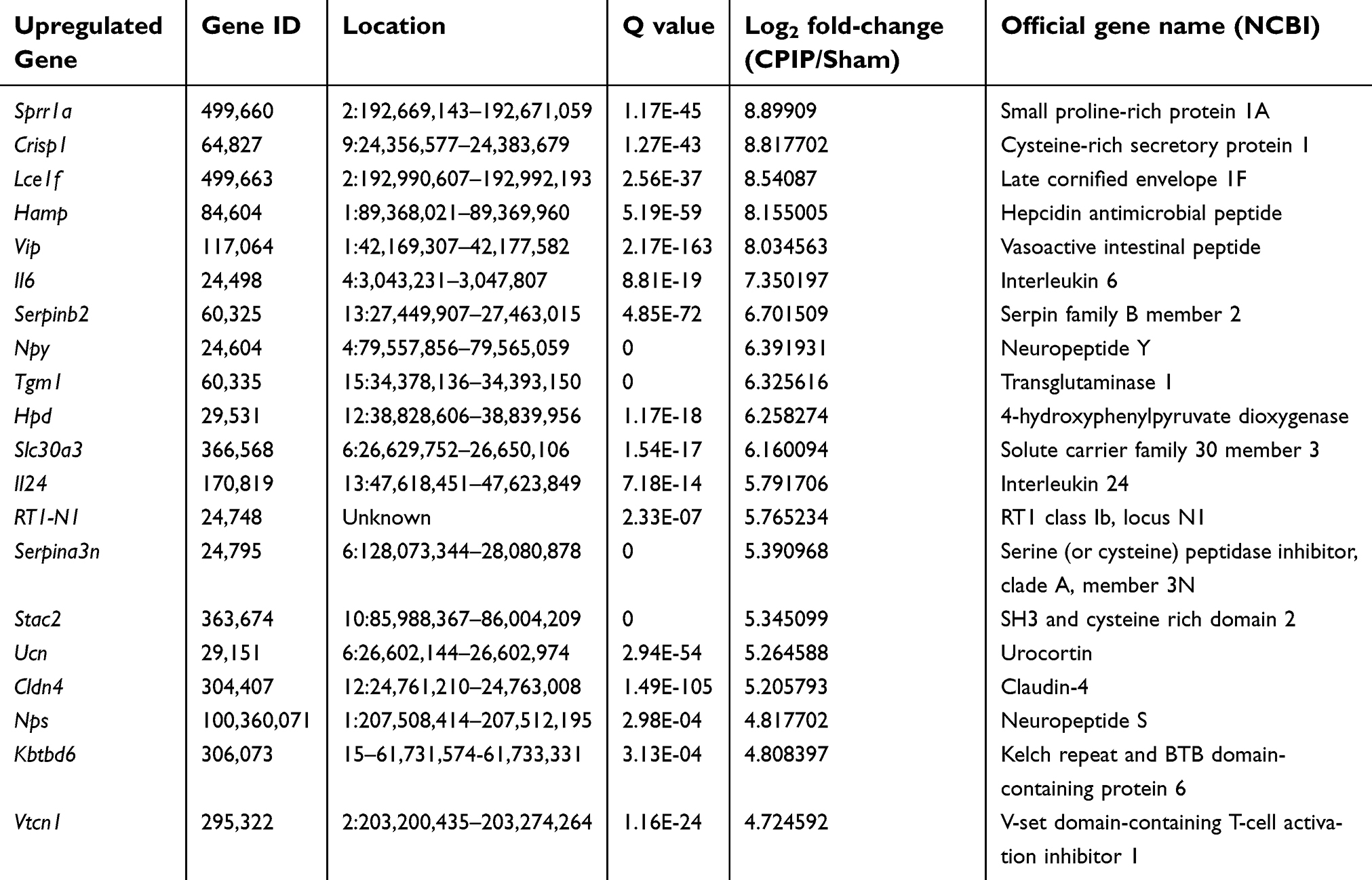 | Table 3 The detailed information of the top 20 upregulated DEGs |
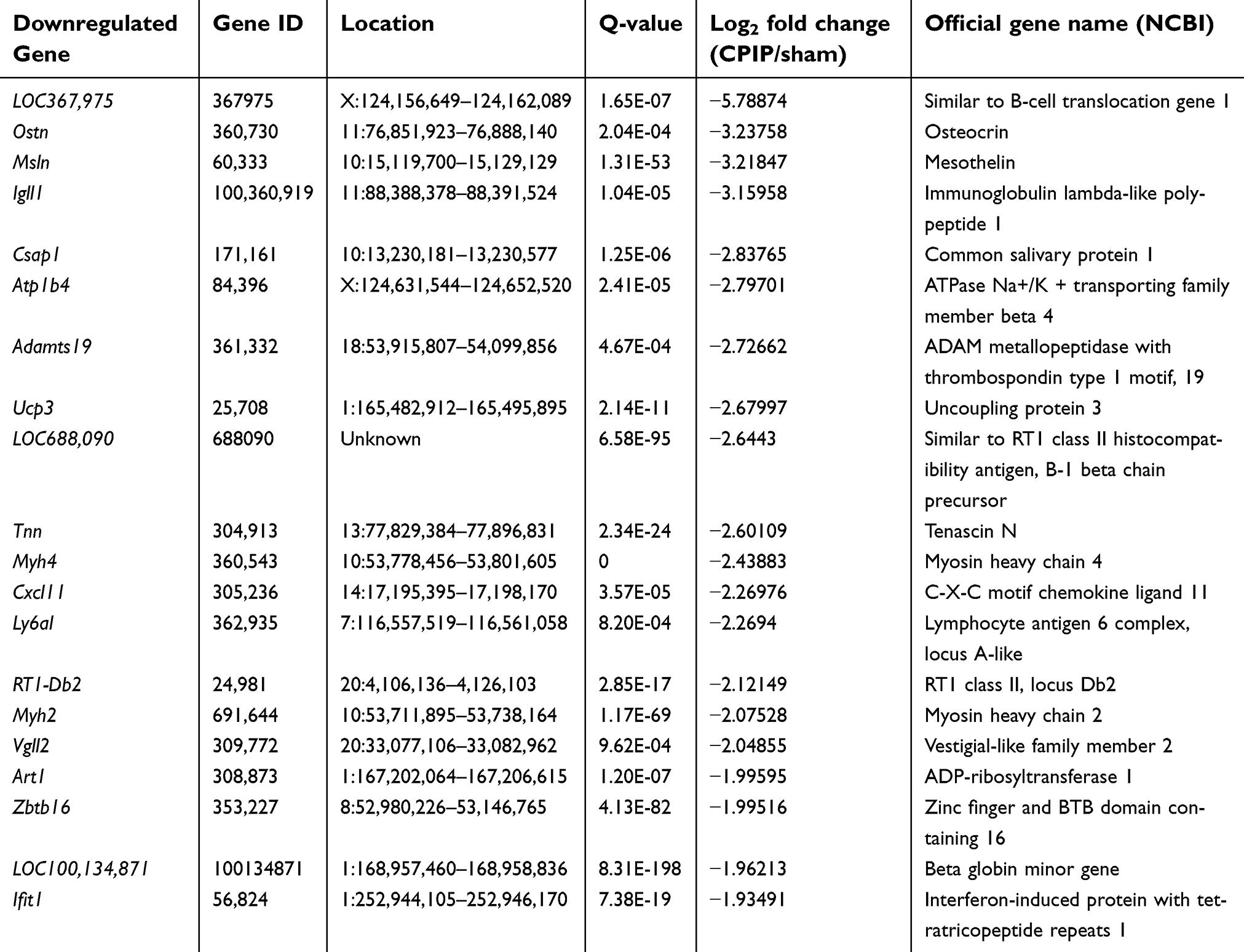 | Table 4 The detailed information of the top 20 downregulated DEGs |
Validation of RNA-Seq data by real-time quantitative PCR (qPCR)
We proceeded to verify the RNA-Seq data by qPCR. Two upregulated genes (Hpd and Vip) and two downregulated genes (Zbtb16 and Ostn) from DEGS were randomly selected and analyzed by qPCR. qPCR results indicated that the expression of Hpd and Vip were significantly increased, whereas Zbtb16 and Ostn were significantly decreased in DRGs of the CPIP model group compared with the Sham group (Figure 3A and B). These qPCR results were consistent with RNA-Seq data. We further evaluated some representative genes whose roles in inflammation or pain had been documented, such as Il24, Il6, Nps and Cxcl13 by qPCR. As a control, we also included Sprr1a, the gene which showed the highest increase fold in RNA-Seq, in our test. qPCR results showed that the expression of Il24, Il6, Nps, Cxcl13 as well as Sprr1a were all significantly increased in DRGs of the CPIP model group compared with the Sham group (Figure 3C), which was consistent with RNA-Seq data. Therefore, the qPCR assay confirmed the reliability of the RNA-Seq data.
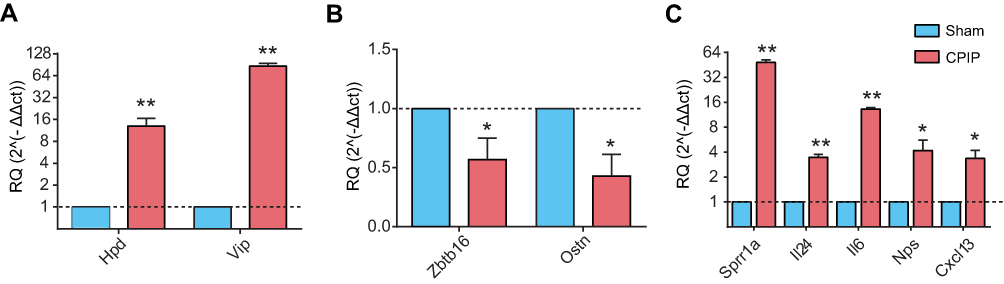 | Figure 3 The validation of RNA-Seq results using qPCR. (A) The expression of two randomly selected upregulated DEGs from RNA-seq detected by qPCR. (B) The expression of two randomly selected downregulated DEGs from RNA-seq detected by qPCR. (C) The expression of representative genes in inflammation and pain detected by qPCR. N=6 rats/group. *P<0.05, **P<0.01 vs Sham group. Abbreviations: RNA-Seq, RNA-sequencing; DEGs, differentially espressed genes. |
Pathway analysis of DEGs of CPIP model
To further investigate the molecular mechanisms underlying CRPS-I, we carried out GO analysis of the DEGs in DRGs of the CPIP model and the Sham groups. Results obtained from the GO analysis indicated that the most significantly enriched biological process of upregulated DEGs in the CPIP model group was response to lipopolysaccharide (LPS), inflammatory response, defense response to fungus, neuropeptide signaling pathway and positive regulation of ERK1 and ERK2 cascade, etc (Figure 4A). The most significantly enriched molecular function of upregulated DEGs in the CPIP model group was G-protein coupled receptor binding, cytokine activity, and calcium ion binding, etc (Figure 4B). The most significantly enriched cellular function of upregulated DEGs in the CPIP model group was extracellular space and extracellular region, etc (Figure 4C). In addition, the most significantly enriched biological process of downregulated DEGs in CPIP model group was muscle contraction, response to mechanical stimulus, ossification, striated muscle contraction, etc (Figure 4D). The most significantly enriched molecular function of downregulated DEGs in the CPIP model group was actin binding, microfilament motor activity, and motor activity, etc (Figure 4E). The most significantly enriched cellular function of downregulated DEGs in the CPIP model group was myofibril, myosin complex, and extracellular region, etc (Figure 4F).
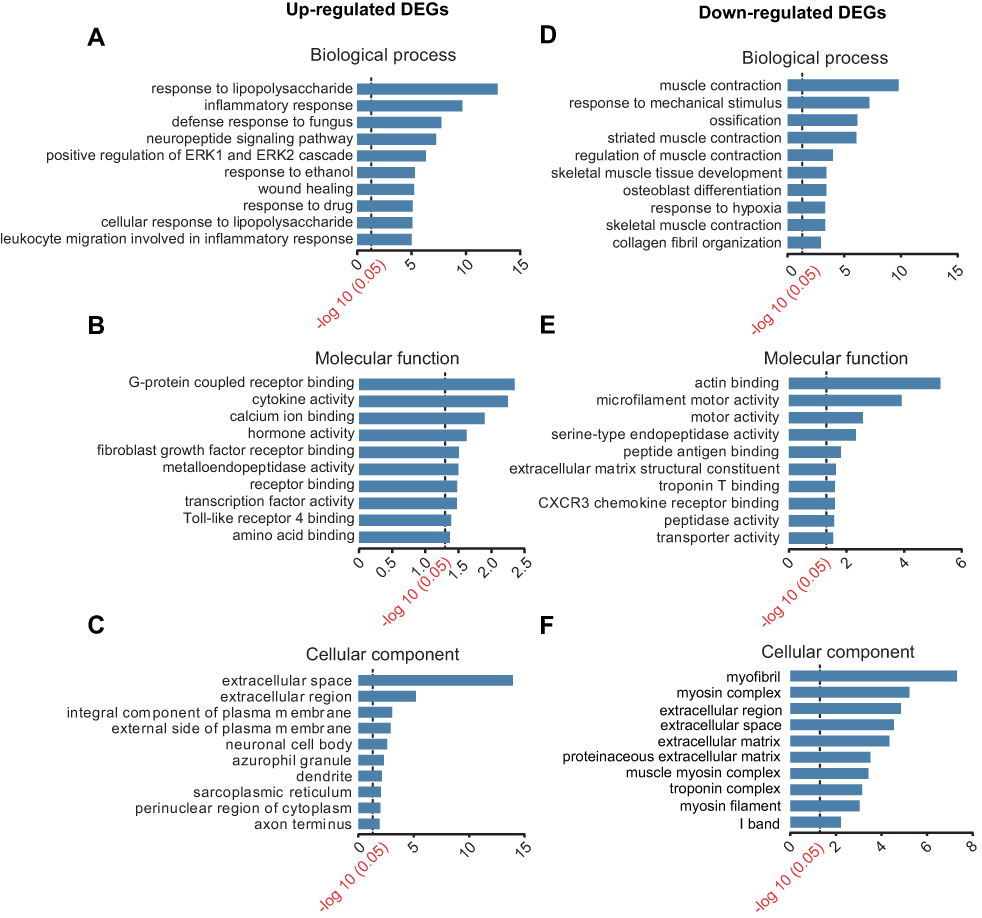 | Figure 4 GO pathway analysis of DEGs. (A–C) The top 10 significant biological processes, molecular functions and cellular components of upregulated DEGs. (D–F) The top 10 significant biological processes, molecular functions and cellular components of downregulated DEGs. The dotted line indicated P-value of 0.05. Abbreviations: GO, gene ontology; DEGs, differentially expressed genes. |
We further performed gene enrichment analysis using KEGG pathway analysis. As shown in Figure 5A, the upregulated DEGs in DRGs of the CPIP model were mainly involved in phagosome, cell adhesion molecules, herpes simplex infection, rheumatoid arthritis, and TNF signaling pathway, etc. The downregulated DEGs in DRGs of the CPIP model mainly involved human papillomavirus infection, focal adhesion, and protein digestion and absorption, etc (Figure 5B).
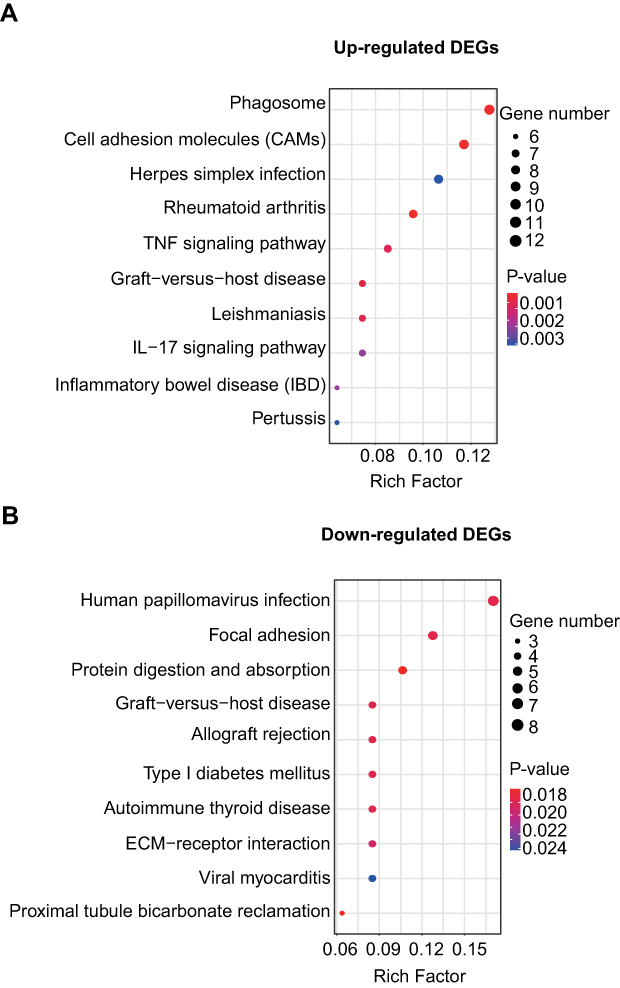 | Figure 5 KEGG pathway analysis of DEGs. (A) Bubble plots showing the top 10 significant pathways for upregulated DEGs. (B) Bubble plots showing the top 10 significant pathways for downregulated DEGs. Larger bubbles indicate higher number of genes. The color of each bubble reflects significance (P-value). Abbreviations: KEGG, Kyoto Encyclopedia of Genes and Genomes; DEGs, differentially expressed genes. |
To focus our analysis on the genes more related with pain and nociception, we searched previously published microarray datasets about genes involved in pain processing (PG), genes enriched in small-diameter primary sensory DRG neurons (SN) and genes enriched in TRPV1-lineage nociceptors (VL) from previously published datasets.11,19,37 We found that 37 DEGs are related to pain processing and Vip, Il6 and Il1a are among the pain-related genes that are most highly upregulated (Figure 6A,
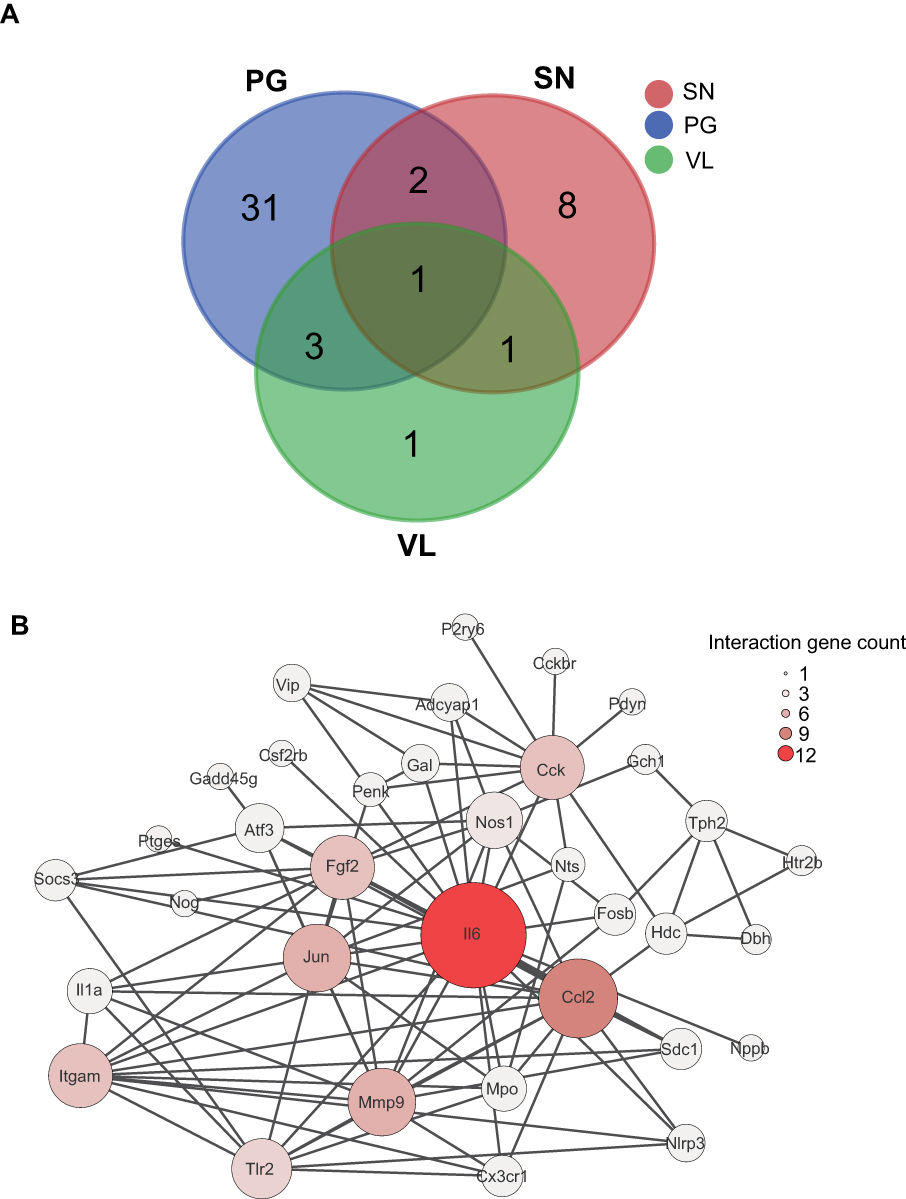 | Figure 6 Overlapping of DEGs with genes involved in pain processing and nociception. (A) Venn diagram showing the overlapping of DEGs with genes involved in pain processing (PG, blue color), genes enriched in small-diameter DRG neurons (SN, pink color) and genes enriched in TRPV1-lineage nociceptors (VL, green color). (B) PPI network analysis of the DEGs overlapped with PG, SN and VL gene datasets. Larger circles and deeper colors reflect more interactions. Abbreviations: DEGs, differentially expressed genes; DRG, dorsal root ganglion; PPI, protein-protein interaction. |
Comparison of our RNA-Seq dataset with other published datasets of neuropathic or inflammatory pain models
The CPIP rat model is an animal model that mimics human CRPS-I, a neuropathic pain condition. However, it is still not known whether the CPIP model shows any typical neuropathic pain-related gene expression profiles. Therefore, we compared our CPIP RNA-Seq data with the microarray data obtained from two well-known neuropathic pain models, namely, the rat models of SNI (spared nerve injury) and SNL (spinal nerve ligation). The same criteria (fold change ≥2, q ≤0.01) were imposed upon both SNI and SNL microarray datasets for identifying DEGs. We observed that the CPIP model group had an overlap of 34 and 41 genes with SNI and SNL models, respectively, which accounts for 11.5% and 13.9% of all DEGs of the CPIP model. In addition, the three groups had a core set of 19 genes that were overlapped (Figure 7A,
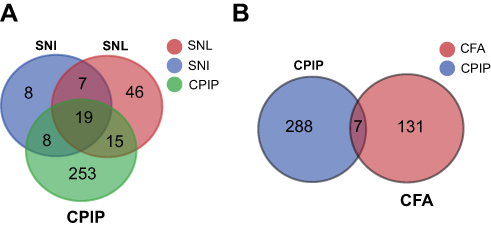 | Figure 7 Comparison of our present CPIP RNA-Seq datasets with other published microarray datasets of neuropathic and inflammatory pain models. (A) Venn diagram showing the overlapping of DEGs in DRGs from CPIP rat model with rat model of SNI and SNL neuropathic pain. (B) Venn diagram showing the overlapping of DEGs in DRGs from CPIP rat model with rat model of CFA inflammatory pain. Abbreviations: CPIP, chonic post-ischemic pain; RNS-Seq, RNA-sequencing; DEGs, differentially expressed genes; DRGs, dorsal root ganlions. |
Discussion
In the present study, we performed transcriptome profiling of the DRGs which innervate the affected hind limb of the CPIP model rats by means of RNA-Seq. We analyzed the genes that are differentially regulated and confirmed their expression by qPCR. We further sorted out the genes that are possibly involved in pain mechanisms or enriched in nociceptors. Lastly, by comparing with published microarray datasets of rat neuropathic and inflammatory pain models, we identified key pathways shared by the rat CRPS-I model with two other neuropathic pain models. Distinct patterns of gene expression in different levels of the nociceptive system are critically involved in the development and maintenance of neuropathic pain.29,36 Therefore, we presented by far, the first study to profile the transcriptome changes and key pathways in the primary sensory ganglia of a rat model of CRPS-I using RNA-Seq.
In this study, a rat model of CPIP was used to mimic CRPS-I. Ischemia/reperfusion is among one of the possible factors leading to CRPS-I.4,15 It is reported that around 45% and 70% of CRPS-I patients develop signs of thermal and mechanical hypersensitivity, respectively. Thermal and mechanical hypersensitivities are among the major symptoms affecting CRPS-I patients.25,42,43,47 A recent study demonstrated CPIP model rats developed obvious signs of thermal hyperalgesia, which correlates with our present findings.46 Therefore, the CPIP rat model could be used as a preclinical animal model for studying mechanisms underlying both thermal and mechanical hypersensitivity of CRPS-I patients.
According to GO analyses of the DEGs in DRGs, we discovered that the significantly enriched biological processes and molecular functions of upregulated genes in CPIP vs Sham groups were mostly involved in response to LPS, inflammation, G-protein coupled receptor binding, cytokine activity, and neuropeptide signaling. These findings suggest that neuroinflammation is likely to be a predominant process involved in the pathophysiology of the CPIP model. This result also coincided with previous studies showing that neuroinflammation, in terms of glial activation, immune cell infiltration, and inflammatory mediator release in the sensory system, is critically involved in neuropathic pain mechanisms. In addition, positive regulation of ERK1 and ERK2 cascade was also among the most significantly enriched pathways of upregulated genes by GO analysis. In fact, ERK1 and ERK2 cascade has been suggested to play a crucial role in mediating neuropathic pain as well as many other pain conditions.7,18,51 The above findings suggest that targeting neuroinflammation and ERK1/2 cascade may represent effective approaches to treat CRPS-I.
GO functional analysis also showed that downregulated DEGs might associate with muscle contraction, response to mechanical stimulus, ossification, striated muscle contraction, etc. At this time, we have no idea how these processes might be related with neuropathic pain or DRG function. But we also noticed that similar biological process, like skeletal muscle contraction, also showed up in the downregulated gene list of GO analysis in a study investigating DEGs in spinal cord dorsal horn upon SNL neuropathic pain.31 But how the above biological processes are related with CPIP-induced pain or DRG functions needs to be further investigated.
KEGG analysis shows that most of the upregulated genes are involved in phagosome. We noticed that phagosome was also observed in another study investigating DEGs in spinal dorsal horn involved in CCI-induced neuropathic pain model.21 One of the genes involved in phagosome is Itgam, which is a marker for macrophage. Neuropathic pain is usually accompanied with infiltration of macrophages in peripheral sensory ganglia.19,52 The infiltrated macrophages produce oxidative stress and release ROS products, including H2O2 and lipid peroxidation products, which in turn activate TRPA1 and cause pain.1,37 Therefore, our data suggest that macrophages may be involved in the pain mechanisms of CPIP model. In addition, KEGG analysis also shows that most of the downregulated genes are involved in human papillomavirus infection. However, at this moment we do not know how these genes are related to CPIP-induced pain or DRG functions and further studies are needed to verify this finding.
The pathophysiological mechanisms of CRPS still remain poorly understood. Studies have been undertaken to monitor the inflammatory mediators in local blister fluid of CRPS patients.6 It is found that levels of IL-6 and TNF-α are highly elevated in blister fluid of CRPS patients.6 In our study, we found that IL-6 is among one of the most upregulated pain-related genes in DRGs of the CPIP model rats. Our subsequent qPCR results confirmed a more than 10-fold upregulation of IL6 in DRGs of the CPIP model rats. Furthermore, by performing PPI network analysis, we found that IL6 was the hub gene that showed the most interactions with other pain or nociception-related genes. Recently, the expression levels of IL-6 and its receptor have been found to be upregulated in DRGs and spinal cord in pathological pain and nerve injury conditions.26,34,45,54 Moreover, application of IL-6 neutralizing antibody ameliorated these pain-related behaviors.26,34,41 Therefore, our findings, together with others, suggest that the upregulation of IL-6 in both primary sensory ganglia and local tissue may play an important role in mediating CRPS-I.
A genome-wide expression profiling in the blood from CRPS patients using microarray has been performed and identified MMP9 to be significantly upregulated in the blood samples of CRPS-I patients.32 In our study, MMP9 is also among the DEGs we identified that are significantly upregulated in the DRGs. MMP-9 belongs to a family of extracellular proteases named matrix metalloproteinases that play crucial roles in neuroinflammation and neuropathic pain in the nerve system.20 MMP9 expression is dramatically increased in DRGs of rats undergone sciatic nerve crush and MMP-9 gene deletion can protect nerve fibers from demyelination and reduce neuropathic pain after injury.11 MMP9 gene-deficient mice showed reduced neuropathic pain response.33 Therefore, our study suggested that upregulation of MMP9 gene may underlie the pain mechanisms of CRPS-I.
We also observed obvious upregulation of CXCL13 gene in the DRGs of CPIP model rats by RNA-Seq. CXCL13 is a B lymphocyte chemoattractant and activates CXCR5 receptor in the immune system.53 CXCL13 was recently found to be the most upregulated gene among all chemokines in the spinal cord tissue after SNL-induced neuropathic pain.30 Genetic knockout Cxcr5 can alleviate neuropathic pain and reduce spinal glial activation.30 More recently, CXCL13/CXCR5 signaling was found to be upregulated in DRG neurons and participate in inflammatory pain via enhancing Nav1.8 current density in DRG neurons through acting on p38 MAP kinase.50 These findings demonstrated a pivotal role of CXCL13/CXCR5 in mediating neuropathic and inflammatory pain.53 Our qPCR result further confirmed that Cxcl13 mRNA expression was significantly increased in DRGs of CPIP model rats. Therefore, our study suggests that CXCL13 may be another promising pharmacological target for CRPS-I treatment.
CRPS-I patients usually developed obvious heat and cold pain hypersensitivity and increased responses to capsaicin stimulation.25,47 TRPV1-expressing DRG neurons are mainly nociceptive sensory neurons and play crucial roles in transuding signals arising from exposure to painful heat, noxious cold, inflammatory and chemical stimuli.14,24,40 Therefore, it is likely that TRPV1-lineage DRG neurons may play a role in mediating CRPS-I pain symptom. Therefore, we mapped our identified DEGs with the recently published datasets of TRPV1-lineage genes, as well as pain-related genes, and genes enriched in sensory neurons.13,24,48 This approach can allow us to better deduce the possible contributions of these genes to the pain mechanisms of CRPS-I. After mapping, we found a number of these genes overlapped between groups. These genes we identified are very likely to be expressed in nociceptive sensory neurons and have been previously found to be related with human pain conditions. The rat CPIP model is widely used to mimic human CRPS-I and considered to be a neuropathic pain model.16,39 However, it is still not known whether the CPIP model displays any signatures of gene changes similar with other neuropathic models. In order to address this issue, we continued to compare the DEGs of the CPIP model with the rat SNI and SNL models, two well-established rat neuropathic pain models, as well as with CFA-induced inflammatory pain model. We found that the CPIP rat model shares many similar altered genes with two neuropathic pain models but little with the inflammatory pain model, suggesting CPIP model reflects a neuropathic pain-like pathology in primary sensory ganglia.
Conclusion
Our study presented by far the first study to perform transcriptome analysis of gene changes and key pathways using RNA-Seq in primary sensory ganglia of an animal model of CRPS-I. We identified a number of DEGs and pathways that may be potentially involved in pain mechanisms of CRPS-I. These findings may allow us to gain more mechanistic insights into the pain mechanisms of CRPS-I, which in turn may help to develop more effective approaches for treating pain conditions associated with CRPS-I.
Acknowledgments
This project is supported by funding from Zhejiang Provincial Natural Science Funds for Distinguished Young Scholars (LR17H270001), the National Natural Science Foundation of China (81873365 and 81603676), Qianjiang Talent Program (QJD1702020), 5151 Talent Program of Zhejiang Chinese Medical University, New Star Talent Program of Zhongshan Hospital of Zhejiang Province (2016xxrc02) to Boyi Liu, and Scientific Research Funding of Zhejiang Chinese Medical University to Boyu Liu (2018ZY19). Contents are solely the responsibility of the authors and do not necessarily represent the views of the funders.
Author contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Andersson DA, Gentry C, Moss S, Bevan S. Transient receptor potential A1 is a sensory receptor for multiple products of oxidative stress. J Neurosci. 2008;28(10):2485–2494. doi:10.1523/JNEUROSCI.5369-07.2008
2. Athie MCP, Vieira AS, Teixeira JM, et al. Transcriptome analysis of dorsal root ganglia’s diabetic neuropathy reveals mechanisms involved in pain and regeneration. Life Sci. 2018;205:54–62. doi:10.1016/j.lfs.2018.05.016
3. Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. 2009;139(2):267–284. doi:10.1016/j.cell.2009.09.028
4. Berthelot JM. Current management of reflex sympathetic dystrophy syndrome (complex regional pain syndrome type I). Joint Bone Spine. 2006;73(5):495–499. doi:10.1016/j.jbspin.2005.11.022
5. Birklein F, Ajit SK, Goebel A, Perez R, Sommer C. Complex regional pain syndrome - phenotypic characteristics and potential biomarkers. Nat Rev Neurol. 2018;14(5):272–284. doi:10.1038/nrneurol.2018.20
6. Birklein F, Drummond PD, Li W, et al. Activation of cutaneous immune responses in complex regional pain syndrome. J Pain. 2014;15(5):485–495. doi:10.1016/j.jpain.2014.01.490
7. Borges G, Berrocoso E, Mico JA, Neto F. ERK1/2: function, signaling and implication in pain and pain-related anxio-depressive disorders. Prog Neuropsychopharmacol Biol Psychiatry. 2015;60:77–92. doi:10.1016/j.pnpbp.2015.02.010
8. Caceres AI, Liu B, Jabba SV, Achanta S, Morris JB, Jordt SE. Transient receptor potential cation channel subfamily M member 8 channels mediate the anti-inflammatory effects of eucalyptol. Br J Pharmacol. 2017;174:867–879. doi:10.1111/bph.13760
9. Chai W, Tai Y, Shao X, et al. Electroacupuncture alleviates pain responses and inflammation in a rat model of acute gout arthritis. Evid Based Complement Alternat Med. 2018;2018:2598975. doi:10.1155/2018/9567061
10. Chang M, Smith S, Thorpe A, Barratt MJ, Karim F. Evaluation of phenoxybenzamine in the CFA model of pain following gene expression studies and connectivity mapping. Mol Pain. 2010;6:56. doi:10.1186/1744-8069-6-56
11. Chattopadhyay S, Myers RR, Janes J, Shubayev V. Cytokine regulation of MMP-9 in peripheral glia: implications for pathological processes and pain in injured nerve. Brain Behav Immun. 2007;21(5):561–568. doi:10.1016/j.bbi.2006.10.015
12. Chen W, Ding H, Zhou X, Lin H, Chou KC. iRNA(m6A)-PseDNC: identifying N6-methyladenosine sites using pseudo dinucleotide composition. Anal Biochem. 2018;561–562:59–65. doi:10.1016/j.ab.2018.09.002
13. Chung MK, Park J, Asgar J, Ro JY. Transcriptome analysis of trigeminal ganglia following masseter muscle inflammation in rats. Mol Pain. 2016;12. doi:10.1177/1744806916668526
14. Cobos EJ, Nickerson CA, Gao F, et al. Mechanistic differences in neuropathic pain modalities revealed by correlating behavior with global expression profiling. Cell Rep. 2018;22(5):1301–1312. doi:10.1016/j.celrep.2018.01.006
15. Coderre TJ, Bennett GJ. A hypothesis for the cause of complex regional pain syndrome-type I (reflex sympathetic dystrophy): pain due to deep-tissue microvascular pathology. Pain Med. 2010;11(8):1224–1238. doi:10.1111/j.1526-4637.2010.00911.x
16. Coderre TJ, Xanthos DN, Francis L, Bennett GJ. Chronic post-ischemia pain (CPIP): a novel animal model of complex regional pain syndrome-type I (CRPS-I; reflex sympathetic dystrophy) produced by prolonged hindpaw ischemia and reperfusion in the rat. Pain. 2004;112(1–2):94–105. doi:10.1016/j.pain.2004.08.001
17. Costigan M, Belfer I, Griffin RS, et al. Multiple chronic pain states are associated with a common amino acid-changing allele in KCNS1. Brain. 2010;133(9):2519–2527. doi:10.1093/brain/awq195
18. Cruz CD, Neto FL, Castro-Lopes J, McMahon SB, Cruz F. Inhibition of ERK phosphorylation decreases nociceptive behaviour in monoarthritic rats. Pain. 2005;116(3):411–419. doi:10.1016/j.pain.2005.05.031
19. De Logu F, Nassini R, Materazzi S, et al. Schwann cell TRPA1 mediates neuroinflammation that sustains macrophage-dependent neuropathic pain in mice. Nat Commun. 2017;8(1):1887. doi:10.1038/s41467-017-01739-2
20. Dev R, Srivastava PK, Iyer JP, Dastidar SG, Ray A. Therapeutic potential of matrix metalloprotease inhibitors in neuropathic pain. Expert Opin Investig Drugs. 2010;19(4):455–468. doi:10.1517/13543781003643486
21. Du H, Shi J, Wang M, An S, Guo X, Wang Z. Analyses of gene expression profiles in the rat dorsal horn of the spinal cord using RNA sequencing in chronic constriction injury rats. J Neuroinflammation. 2018;15(1):280. doi:10.1186/s12974-018-1220-7
22. Flegel C, Schobel N, Altmuller J, et al. RNA-Seq analysis of human trigeminal and dorsal root ganglia with a focus on chemoreceptors. PLoS One. 2015;10(6):e0128951. doi:10.1371/journal.pone.0128951
23. Franceschini A, Szklarczyk D, Frankild S, et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41(Databaseissue):D808–D815. doi:10.1093/nar/gks1094
24. Goswami SC, Mishra SK, Maric D, et al. Molecular signatures of mouse TRPV1-lineage neurons revealed by RNA-Seq transcriptome analysis. J Pain. 2014;15(12):1338–1359. doi:10.1016/j.jpain.2014.09.010
25. Grothusen JR, Alexander G, Erwin K, Schwartzman R. Thermal pain in complex regional pain syndrome type I. Pain Physician. 2014;17(1):71–79.
26. Guptarak J, Wanchoo S, Durham-Lee J, et al. Inhibition of IL-6 signaling: a novel therapeutic approach to treating spinal cord injury pain. Pain. 2013;154(7):1115–1128. doi:10.1016/j.pain.2013.03.026
27. Hord ED, Oaklander AL. Complex regional pain syndrome: a review of evidence-supported treatment options. Curr Pain Headache Rep. 2003;7(3):188–196.
28. Hu G, Huang K, Hu Y, et al. Single-cell RNA-seq reveals distinct injury responses in different types of DRG sensory neurons. Sci Rep. 2016;6:31851. doi:10.1038/srep31851
29. Imai S, Ikegami D, Yamashita A, et al. Epigenetic transcriptional activation of monocyte chemotactic protein 3 contributes to long-lasting neuropathic pain. Brain. 2013;136(Pt 3):828–843. doi:10.1093/brain/aws330
30. Jiang BC, Cao DL, Zhang X, et al. CXCL13 drives spinal astrocyte activation and neuropathic pain via CXCR5. J Clin Invest. 2016;126(2):745–761. doi:10.1172/JCI81950
31. Jiang BC, Sun WX, He LN, Cao DL, Zhang ZJ, Gao YJ. Identification of lncRNA expression profile in the spinal cord of mice following spinal nerve ligation-induced neuropathic pain. Mol Pain. 2015;11:43. doi:10.1186/s12990-015-0047-9
32. Jin EH, Zhang E, Ko Y, et al. Genome-wide expression profiling of complex regional pain syndrome. PLoS One. 2013;8(11):e79435. doi:10.1371/journal.pone.0079435
33. Kawasaki Y, Xu ZZ, Wang X, et al. Distinct roles of matrix metalloproteases in the early- and late-phase development of neuropathic pain. Nat Med. 2008;14(3):331–336. doi:10.1038/nm1723
34. Kiguchi N, Maeda T, Kobayashi Y, Kondo T, Ozaki M, Kishioka S. The critical role of invading peripheral macrophage-derived interleukin-6 in vincristine-induced mechanical allodynia in mice. Eur J Pharmacol. 2008;592(1–3):87–92. doi:10.1016/j.ejphar.2008.07.008
35. Klafke JZ, da Silva MA, Rossato MF, et al. Acute and chronic nociceptive phases observed in a rat hind paw ischemia/reperfusion model depend on different mechanisms. Pflugers Arch. 2016;468(2):229–241. doi:10.1007/s00424-015-1746-9
36. LaCroix-Fralish ML, Austin JS, Zheng FY, Levitin DJ, Mogil JS. Patterns of pain: meta-analysis of microarray studies of pain. Pain. 2011;152(8):1888–1898. doi:10.1016/j.pain.2011.04.014
37. Liu B, Tai Y, Caceres AI, et al. Oxidized phospholipid OxPAPC activates TRPA1 and contributes to chronic inflammatory pain in mice. PLoS One. 2016;11(11):e0165200. doi:10.1371/journal.pone.0165200
38. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25(4):402–408. doi:10.1006/meth.2001.1262
39. Luo X, Tai WL, Sun L, et al. Crosstalk between astrocytic CXCL12 and microglial CXCR4 contributes to the development of neuropathic pain. Mol Pain. 2016;12.
40. Mishra SK, Tisel SM, Orestes P, Bhangoo SK, Hoon MA. TRPV1-lineage neurons are required for thermal sensation. EMBO J. 2011;30(3):582–593. doi:10.1038/emboj.2010.325
41. Murakami T, Kanchiku T, Suzuki H, et al. Anti-interleukin-6 receptor antibody reduces neuropathic pain following spinal cord injury in mice. Exp Ther Med. 2013;6(5):1194–1198. doi:10.3892/etm.2013.1296
42. Rasmussen VF, Karlsson P, Drummond PD, et al. Bilaterally reduced intraepidermal nerve fiber density in unilateral CRPS-I. Pain Med. 2018;19(10):2021–2030. doi:10.1093/pm/pnx240
43. Reimer M, Rempe T, Diedrichs C, Baron R, Gierthmuhlen J. Sensitization of the nociceptive system in complex regional pain syndrome. PLoS One. 2016;11(5):e0154553. doi:10.1371/journal.pone.0154553
44. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi:10.1101/gr.1239303
45. Su TF, Zhao YQ, Zhang LH, et al. Electroacupuncture reduces the expression of proinflammatory cytokines in inflamed skin tissues through activation of cannabinoid CB2 receptors. Eur J Pain. 2012;16(5):624–635. doi:10.1002/j.1532-2149.2011.00055.x
46. Tang Y, Liu L, Xu D, et al. Interaction between astrocytic colony stimulating factor and its receptor on microglia mediates central sensitization and behavioral hypersensitivity in chronic post ischemic pain model. Brain Behav Immun. 2018;68:248–260. doi:10.1016/j.bbi.2017.10.023
47. Terkelsen AJ, Gierthmuhlen J, Finnerup NB, Hojlund AP, Jensen TS. Bilateral hypersensitivity to capsaicin, thermal, and mechanical stimuli in unilateral complex regional pain syndrome. Anesthesiology. 2014;120(5):1225–1236. doi:10.1097/ALN.0000000000000220
48. Thakur M, Crow M, Richards N, et al. Defining the nociceptor transcriptome. Front Mol Neurosci. 2014;7:87. doi:10.3389/fnmol.2014.00087
49. Urits I, Shen AH, Jones MR, Viswanath O, Kaye AD. Complex regional pain syndrome, current concepts and treatment options. Curr Pain Headache Rep. 2018;22(2):10. doi:10.1007/s11916-018-0667-7
50. Wu XB, Cao DL, Zhang X, et al. CXCL13/CXCR5 enhances sodium channel Nav1.8 current density via p38 MAP kinase in primary sensory neurons following inflammatory pain. Sci Rep. 2016;6:34836. doi:10.1038/srep34836
51. Xing F, Kong C, Bai L, et al. CXCL12/CXCR4 signaling mediated ERK1/2 activation in spinal cord contributes to the pathogenesis of postsurgical pain in rats. Mol Pain. 2017;13:1744806917718753. doi:10.1177/1744806917718753
52. Zhang H, Li Y, de Carvalho-Barbosa M, et al. Dorsal root ganglion infiltration by macrophages contributes to paclitaxel chemotherapy-induced peripheral neuropathy. J Pain. 2016;17(7):775–786. doi:10.1016/j.jpain.2016.02.011
53. Zhang ZJ, Jiang BC, Gao YJ. Chemokines in neuron-glial cell interaction and pathogenesis of neuropathic pain. Cell Mol Life Sci. 2017;74(18):3275–3291. doi:10.1007/s00018-017-2513-1
54. Zhou YQ, Liu Z, Liu ZH, et al. Interleukin-6: an emerging regulator of pathological pain. J Neuroinflammation. 2016;13(1):141. doi:10.1186/s12974-016-0607-6
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.