Back to Journals » Journal of Inflammation Research » Volume 19
Trained Immunity–Like Memory in Vascular Structural Cells: Metabolic–Epigenetic Reprogramming as a Driving Mechanism of Atherosclerosis and Residual Cardiovascular Risk
Authors Dai J, Zhou X, Yuan K, Huang K, Zhang Y
Received 10 February 2026
Accepted for publication 14 March 2026
Published 21 March 2026 Volume 2026:19 599156
DOI https://doi.org/10.2147/JIR.S599156
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Chengming Fan
Jingxuan Dai,1,* Xuancheng Zhou,1,* Kun Yuan,1,* Keming Huang,2,* Ying Zhang3
1Department of Clinical Medical College, Southwest Medical University, Luzhou, Sichuan, 646000, People’s Republic of China; 2Department of Endocrinology and Metabolism, The Affiliated Hospital of Southwest Medical University, Luzhou, Sichuan, 646000, People’s Republic of China; 3Department of Basic Medical Sciences, Southwest Medical University, Luzhou, Sichuan, 646000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Ying Zhang, Email [email protected]
Abstract: Despite major advances in lipid-lowering therapies and widespread achievement of optimal LDL-C targets, a substantial burden of residual cardiovascular risk (RCVR) persists, underscoring fundamental gaps in current preventive strategies. Existing mechanistic frameworks have largely centered on professional immune cells as the primary drivers of chronic vascular inflammation, insufficiently accounting for the durability and tissue specificity of vascular pathology after risk factor correction. Emerging evidence supports a paradigm in which vascular structural cells (VSCs), notably endothelial cells (ECs) and vascular smooth muscle cells (VSMCs), acquire a trained immunity–like state that enables them to act as a long-term reservoir of pathogenic molecular memory. Repeated metabolic, inflammatory, or mechanical priming induces persistent phenotypic switching in VSCs through a tightly coupled metabolic–epigenetic axis. This process is characterized by PFKFB3-driven glycolytic reprogramming, rewired mitochondrial metabolism with accumulation of tricarboxylic acid cycle intermediates such as succinate and fumarate, and the establishment of stable epigenetic scars, including H3K4me1, H3K27ac, and histone lactylation. These epigenetic imprints lower activation thresholds and sustain exaggerated inflammatory and proliferative responses, providing a mechanistic basis for chronic vascular remodeling in clinical entities such as in-stent restenosis and cardiac allograft vasculopathy. Targeting vascular molecular memory by erasing maladaptive epigenetic programs, using bromodomain and extraterminal domain inhibitors or metabolic modulators such as metformin, represents a promising therapeutic avenue to mitigate RCVR beyond conventional lipid-centric approaches.
Keywords: trained immunity–like response, vascular structural cells, metabolic–epigenetic axis, epigenetic scars, residual cardiovascular risk
Introduction
Atherosclerosis (AS) is a chronic inflammatory disease characterized by endothelial dysfunction, oxidative stress, and dysregulated lipid metabolism, and it constitutes the major pathological basis of cardiovascular diseases such as coronary heart disease, myocardial infarction, and stroke.1–3 Over recent decades, lipid-lowering therapies such as statins and PCSK9 inhibitors have substantially reshaped the prevention and management of ASCVD by markedly reducing low-density lipoprotein cholesterol (LDL-C) levels.4–7 However, extensive evidence from clinical trials indicates that even after achieving recommended LDL-C targets with statins, non-statin agents, or combination therapies, patients continue to experience recurrent cardiovascular events, a phenomenon known as “residual risk”.8–10 Accumulating evidence indicates that chronic low-grade inflammation is a key driver of residual risk and that anti-inflammatory interventions can partially reduce cardiovascular events.11,12 The CANTOS trial provided the first proof that targeting interleukin-1β (IL-1β) lowers cardiovascular events independently of lipid reduction.13 Nevertheless, broad immunosuppression increases infection risk, underscoring the need to identify upstream mechanisms sustaining vascular inflammation.14
Traditionally, immunological memory—defined as the ability to mount faster and stronger responses upon re-encounter with previously encountered pathogens—has been regarded as an exclusive feature of the adaptive immune system, namely T and B lymphocytes.15,16 The concept of “trained immunity” (also referred to as innate immune memory), first proposed by Netea et al, challenged this dogma by demonstrating that innate immune cells also possess memory-like properties.17 In contrast to adaptive immunity, which relies on antigen-specific gene rearrangements, trained immunity is mediated by metabolic reprogramming (such as enhanced glycolysis) and epigenetic remodeling (including modifications of H3K4me1 and H3K27ac), enabling innate immune cells—such as monocytes, macrophages, and natural killer cells—to mount amplified nonspecific inflammatory responses to subsequent stimuli following transient exposure to exogenous (e g., Candida albicans, BCG vaccination) or endogenous (e g., oxidized LDL, hyperglycemia) triggers.18–20
Current research on trained immunity in atherosclerosis has primarily focused on circulating monocytes and bone marrow progenitor cells.21,22 However, this immune cell–centric perspective may be insufficient to fully explain the long-term persistence of vascular inflammation and lesions. Vascular structural cells (VSCs), particularly endothelial cells (ECs) and vascular smooth muscle cells (VSMCs), are not inert scaffolds but active participants in the maintenance of vascular homeostasis and pathological remodeling.23,24 Their longevity renders them plausible candidates for sustaining long-term pathogenic imprinting. Observations of “metabolic memory” in diabetic vascular and renal complications suggest durable epigenetic imprinting within long-lived structural cells.25–28 Consistently, single-cell transcriptomic and epigenomic analyses indicate that ECs and VSMCs undergo stable chromatin remodeling after disturbed flow or pro-atherogenic lipid exposure, adopting persistent pro-inflammatory or dedifferentiated states reminiscent of trained immunity.29,30
At this point, it is essential to clarify conceptual boundaries. Classical trained immunity refers to enhanced responsiveness in short-lived innate immune cells, sustained in part through progenitor reprogramming in the bone marrow. By contrast, the memory-like phenotypes observed in vascular structural cells arise in long-lived, tissue-resident cells that are not primarily specialized for pathogen defense. Rather than generating acute inflammatory amplification, these cells tend to develop persistent pro-inflammatory and pro-remodeling states under chronic metabolic or mechanical stressors.31,32 Although such states similarly involve epigenetic stabilization and metabolic rewiring, their functional consequences are predominantly linked to structural remodeling and chronic vascular pathology. Accordingly, although mechanistically analogous to classical trained immunity, these vascular phenotypes do not fully conform to its canonical immunological definition and are therefore more appropriately designated as “trained immunity-like” responses. A conceptual comparison between classical trained immunity in professional immune cells and TI-like states in vascular structural cells is summarized in Table 1.
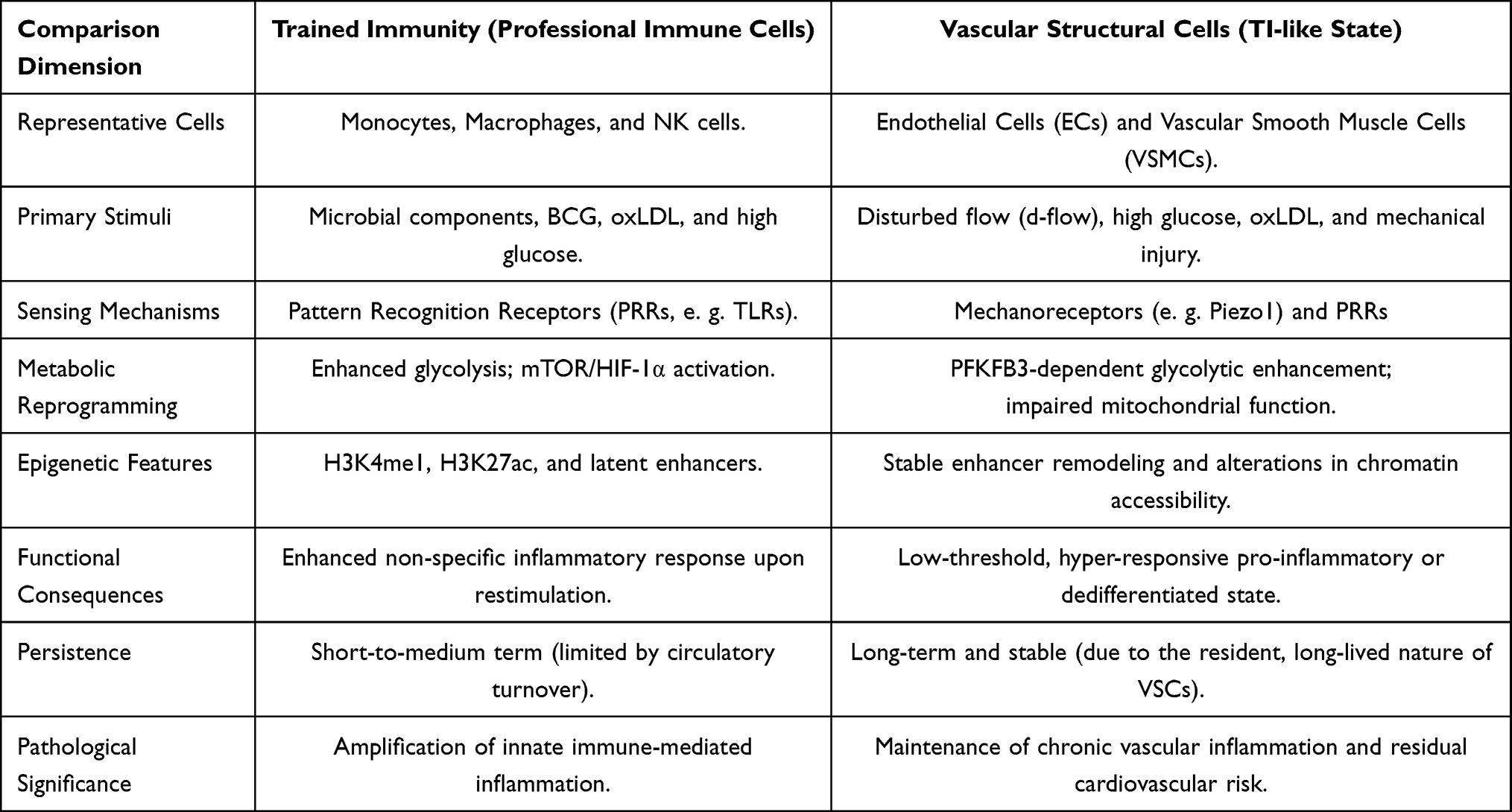 |
Table 1 Trained Immunity in Immune and Vascular Cells This Table Illustrates a Comparative Overview of Trained Immunity in Professional Immune Cells and Trained Immunity–Like States in Vascular Structural Cells. In Both Cell Types, Transient Inflammatory or Metabolic Stimuli Induce Metabolic Rewiring and Epigenetic Remodeling That Lower Activation Thresholds Upon Restimulation. While Immune Cells Exhibit Enhanced Glycolysis and Transient Epigenetic Changes Limited by Cellular Turnover, Endothelial Cells and Vascular Smooth Muscle Cells Develop More Stable Chromatin Reprogramming Due to Their Long-Lived, Tissue-Resident Nature. These Persistent Molecular Imprints Sustain Exaggerated Inflammatory and Phenotypic Responses, Providing a Mechanistic Basis for Chronic Vascular Inflammation and Residual Cardiovascular Risk Beyond Traditional Lipid-Centered Paradigms |
Importantly, analogous memory-like reprogramming has been described in other long-lived structural cells beyond the vasculature. Cardiac fibroblasts exposed to transient stress can develop sustained pro-fibrotic programs, while renal mesangial cells demonstrate persistent epigenetic alterations following hyperglycemic exposure.33,34 Comparable findings in epithelial and stromal compartments suggest that durable chromatin imprinting after environmental stress may represent a broader feature of structural cell biology rather than a vascular-restricted phenomenon. From a comparative biological perspective, such imprinting may reflect a potentially conserved stress-adaptation program in long-lived tissue-resident cells. Under conditions of chronic metabolic or inflammatory burden, however, this adaptive imprinting may become maladaptive, promoting sustained inflammation and pathological remodeling.
This review integrates recent advances to elucidate how ECs and VSMCs establish and maintain pathological memory through metabolic-epigenetic coupling and explores the implications of these “epigenetic scars” in in-stent restenosis, transplant vasculopathy, and progressive atherosclerosis.
Vascular Structural Cells: Experimental Evidence and Phenotypic Heterogeneity of Non-Professional Immune Memory
The vascular wall is not merely a passive victim of atherogenic stress but rather an active, highly plastic responsive unit. Emerging evidence indicates that vascular endothelial cells (ECs) and vascular smooth muscle cells (VSMCs), following transient pathological insults, can convert short-lived stimuli into durable functional alterations through molecular-level “imprints,” a process that closely mirrors trained immunity in innate immune cells in both functional characteristics and molecular underpinnings.
Endothelial Cells: Metabolic Memory and the Hemodynamic “Legacy Effect”
Endothelial cells (ECs), serving as the physical interface between circulating blood and underlying tissues, are the primary sensors of systemic metabolic disturbances. In diabetic vascular complications, this early stress sensing is frequently translated into persistent functional dysfunction, a phenomenon termed “metabolic memory” or the “legacy effect”.35,36 The landmark UK Prospective Diabetes Study (UKPDS) demonstrated that early intensive glycemic control confers cardiovascular protection lasting for decades, with benefits persisting even after subsequent normalization of blood glucose levels, suggesting the existence of a pathogenic “record” within the vascular wall that is independent of contemporaneous glycemic status.37 In a seminal study, El-Osta et al uncovered an epigenetic mechanism underlying hyperglycemia-induced metabolic memory: following transient high-glucose exposure, human aortic endothelial cells maintain sustained transcriptional activation of NF-κB p65 even after restoration of normoglycemia, resulting in prolonged upregulation of downstream inflammatory genes such as MCP-1 and VCAM-1.38 Overall, the core mechanism involves a hyperglycemia-induced burst of reactive oxygen species (ROS), which activates the methyltransferase Set7 to promote epigenetic modification of NF-κB p65 and induces stable enrichment of H3K4me1 at the promoters of inflammation-related genes (e g., VCAM1 and MCP1), thereby sustaining chronic activation of inflammatory transcriptional programs.38–40 These epigenetic modifications function like “bookmarks” imprinted on the genome, enabling endothelial cells to retain a heightened pro-inflammatory phenotype even after metabolic stress is relieved, thereby continuously promoting monocyte adhesion and early plaque formation.
Beyond metabolic stress, endothelial cells also exhibit a pronounced feature of “mechanical priming,” whereby endothelial cells exposed to disturbed flow (d-flow) retain pro-inflammatory properties long after flow patterns are normalized, resulting in sustained leukocyte adhesion and increased susceptibility to atherosclerosis.41,42 Evidence indicates that disturbed flow activates the downstream transcription factors ETS1 and c-JUN via the mechanosensitive ion channel Piezo1, leading to specific upregulation of the histone demethylase KDM5B, which remodels the endothelial epigenetic landscape by reducing H3K4me3 levels and ultimately drives endothelial inflammation and accelerates atherosclerotic plaque formation.43 In addition, at arterial branches and curvatures, disturbed flow can trigger Piezo1-mediated calcium influx, subsequently activating the Ca2⁺/calmodulin (CaM)/CaMKII signaling pathway, thereby promoting endothelial pro-inflammatory responses and dysfunction.44 At the metabolic level, disturbed flow induces metabolic reprogramming in endothelial cells characterized by enhanced glycolysis and impaired mitochondrial oxidative capacity, a process dependent on the stabilization and activity of hypoxia-inducible factor-1α (HIF-1α).45 At the epigenetic level, disturbed flow alters chromatin accessibility and regulatory element utilization, accompanied by remodeling of transcription factor regulatory networks, including reduced activity of homeostasis-associated transcription factors such as KLF4 and KLF2 and suppression of their protective gene programs.46,47 This endothelial “susceptible phenotype” shaped by local hemodynamic features explains why, under identical lipid conditions, endothelial cells located at flow-disturbed regions such as arterial branches and curvatures exhibit exaggerated pro-inflammatory responses to mild inflammatory stimuli, thereby contributing—under the combined influence of multiple risk factors—to the characteristic spatial distribution of atherosclerotic lesions at these anatomical sites.48,49 (Figure 1).
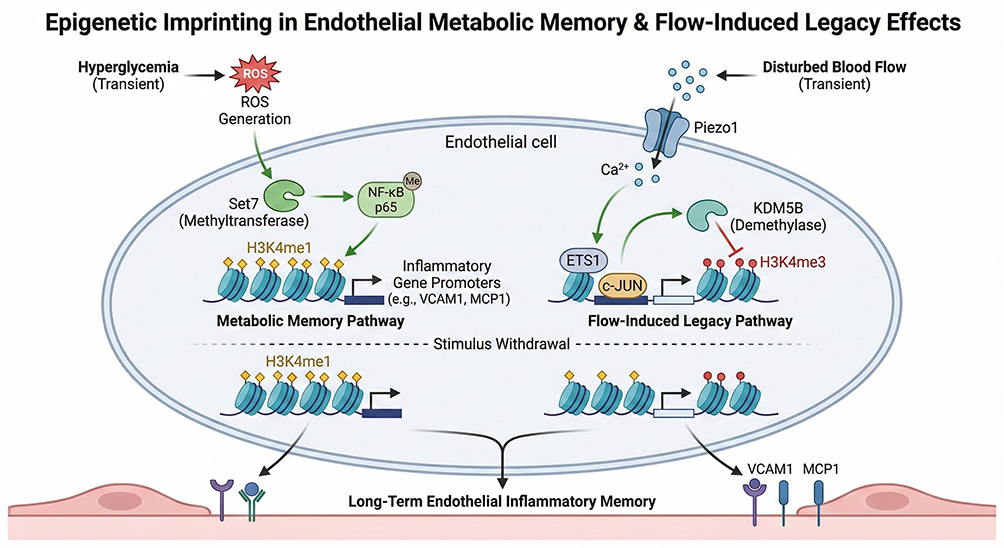 |
Figure 1 Endothelial Metabolic Memory and Flow Legacy This figure illustrates how endothelial cells convert transient metabolic and hemodynamic stress into persistent dysfunction through trained immunity–like mechanisms. Short-term hyperglycemia and disturbed flow are sensed via reactive oxygen species–dependent pathways and mechanosensitive channels, leading to glycolytic reprogramming and mitochondrial impairment. These changes drive stable epigenetic remodeling at promoters and enhancers of inflammatory genes, including sustained enrichment of H3K4me1 and H3K27ac. Notably, these epigenetic imprints persist after normalization of metabolic or flow conditions, maintaining a pro-inflammatory endothelial phenotype that promotes leukocyte adhesion and regional susceptibility to atherosclerosis. |
Vascular Smooth Muscle Cells: Epigenetic Scars During Phenotypic Switching
In vascular smooth muscle cells (VSMCs), phenomena resembling the functional features of “trained immunity” are primarily manifested by their pronounced and persistent phenotypic plasticity. Under physiological conditions, VSMCs maintain a highly differentiated contractile phenotype, characterized by expression of contractile genes such as MYH11 and ACTA2, thereby preserving vascular tone and structural integrity. However, under pathological conditions such as inflammatory stimulation, oxidized lipid accumulation, or vascular injury, VSMCs can undergo dedifferentiation and transition toward synthetic or macrophage-like phenotypes, marked by downregulation of contractile genes and upregulation of genes associated with inflammation, phagocytosis, and migration, a phenomenon particularly prominent in atherosclerotic lesions.50,51 In recent years, lineage tracing and single-cell transcriptomic studies have demonstrated that, in multiple experimental models and in human atherosclerotic lesions, a substantial proportion of foam cells do not originate from the classical monocyte–macrophage lineage but instead arise from VSMCs through phenotypic switching.52,53 These VSMC-derived foam cells play critical roles in plaque formation, stability regulation, and disease progression.
Notably, phenotypic switching of VSMCs is not a simple or readily reversible functional adjustment but is accompanied by relatively stable epigenetic regulatory alterations, including remodeling of transcription factor regulatory networks and systematic changes in chromatin accessibility and histone modification states. These epigenetic alterations provide a molecular basis for the establishment and maintenance of specific gene expression programs.54–56 For example, atherosclerosis-related stimuli such as oxidized low-density lipoprotein (oxLDL) induce upregulation of the transcription factor KLF4, which represses contractile phenotype genes including MYH11 and ACTA2 while activating gene programs associated with dedifferentiation and inflammation, thereby driving VSMCs toward synthetic or macrophage-like states.57,58 Meanwhile, alterations in the expression of epigenetic regulators such as TET2 are closely associated with VSMC phenotypic states, with TET2 downregulation correlating with changes in DNA hydroxymethylation and reduced expression of contractile genes, suggesting that TET2 may participate in the long-term stabilization of these gene expression programs.59 Even when subsequent pathological stimuli are attenuated or removed, these epigenetically mediated changes may keep VSMCs locked in a persistent pro-inflammatory and synthetic state, characterized by elevated cytokine expression and enhanced migratory and proliferative capacities.60,61 On this basis, VSMC phenotypic switching and its potential “memory-like” features provide a novel conceptual framework for understanding the sustained aberrant reactivity of VSMCs in in-stent restenosis following percutaneous coronary intervention (PCI). (Figure 2).
 |
Figure 2 Epigenetic Scars in VSMC Phenotypic Switching This figure illustrates the establishment of long-lasting epigenetic scars during phenotypic switching of vascular smooth muscle cells. Transient exposure to inflammatory cytokines, oxidized lipids, hypoxia, or vascular injury induces metabolic rewiring and activation of transcriptional regulators such as KLF4, resulting in repression of contractile gene programs and induction of synthetic or macrophage-like phenotypes. These transitions are accompanied by stable changes in chromatin accessibility and DNA hydroxymethylation, often associated with TET2 downregulation. Even after stimulus withdrawal, these epigenetic alterations persist, locking smooth muscle cells in a hyper-responsive state that contributes to plaque progression and restenosis. |
Molecular Cascades of Trained Immunity in the Vascular Wall: From Stress Sensing to Epigenetic Imprinting
The “trained immunity–like responses” observed in vascular structural cells (VSCs) are not driven by a single signaling event but instead represent a multilevel regulatory process operating across temporal and spatial scales.19 Accumulating evidence suggests that this process can be conceptually abstracted as a regulatory axis comprising stress sensing, metabolic reprogramming, and epigenetic remodeling, whereby external physical or chemical stresses are first sensed and transduced into intracellular signals, subsequently induce rewiring of metabolic pathways, and ultimately establish relatively stable transcriptional regulatory states at the chromatin level through epigenetic mechanisms, thereby shaping responses to subsequent stimuli.62,63 Thus, trained immunity is not simply a consequence of sustained signaling pathway activation but more likely reflects long-term remodeling of chromatin regulatory states. A summary of the major stimuli, sensing pathways, metabolic rewiring, epigenetic remodeling, and functional consequences associated with trained immunity–like responses in vascular structural cells is presented in Table 2.
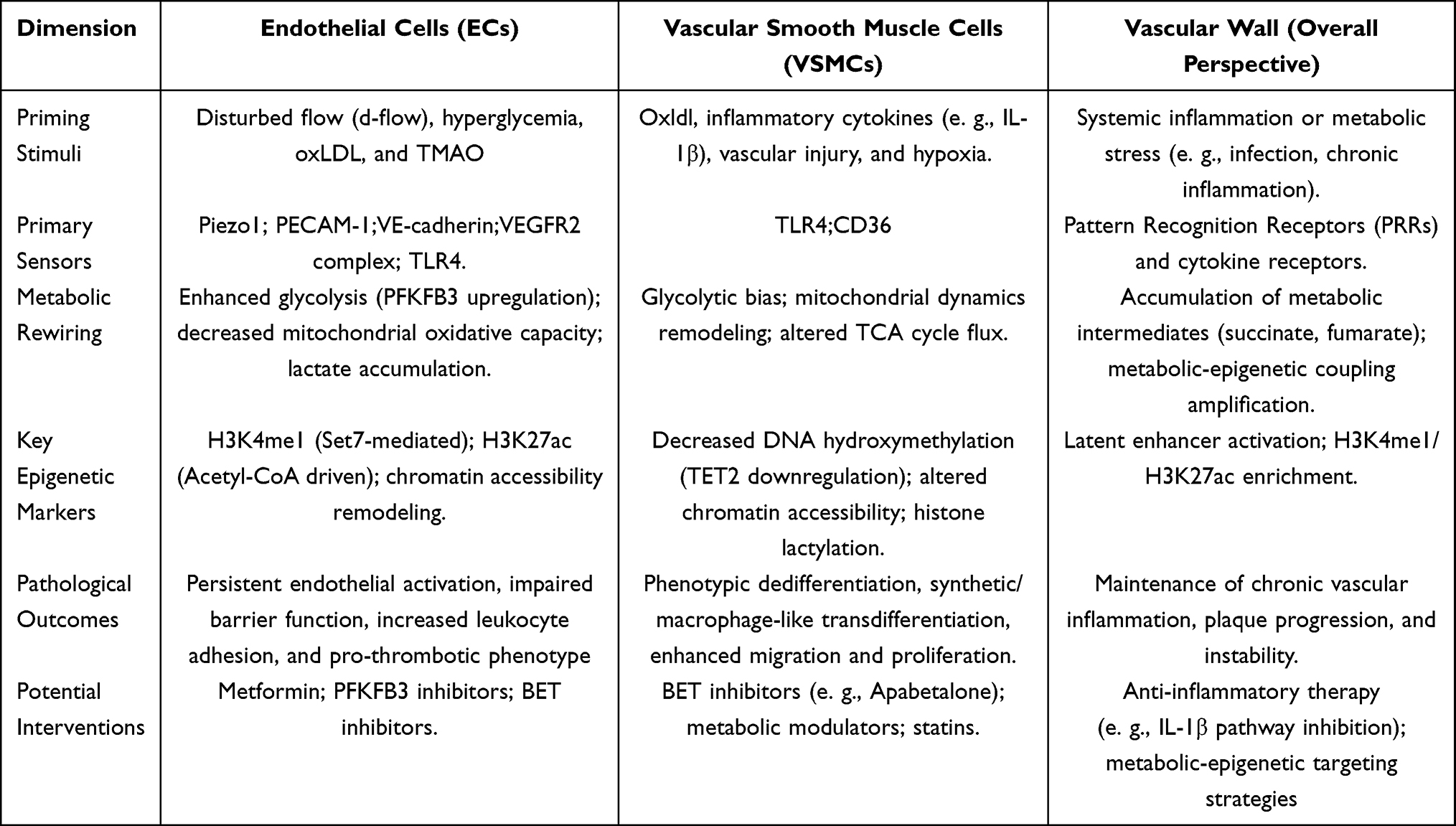 |
Table 2 Molecular Features of Vascular Trained Immunity This Table Summarizes the Defining Molecular Features and Pathological Consequences of Trained Immunity–Like States in Vascular Structural Cells. In Endothelial Cells, Mechanical and Metabolic Stress Induces PFKFB3-Dependent Glycolytic Enhancement, Mitochondrial Dysfunction, and Enrichment of H3K4me1 and H3K27ac, Sustaining Endothelial Activation. In Vascular Smooth Muscle Cells, Inflammatory and Lipid Stimuli Promote Metabolic Bias and Stable Epigenetic Remodeling Associated with Altered Chromatin Accessibility and TET2 Downregulation. Together, These Persistent Cellular Programs Maintain Chronic Vascular Inflammation and Contribute to Residual Cardiovascular Risk |
Stress Sensing and Signal Initiation: Integration of Mechanical and Metabolic Stimuli
Vascular wall cells are chronically exposed to complex mechanical and chemical stressors, and endothelial cells (ECs) and vascular smooth muscle cells (VSMCs) transduce these external cues into intracellular signals through multiple sensing mechanisms. Disturbed flow (d-flow) represents one of the most characteristic mechanical stimuli in vascular pathological conditions. Studies have shown that the mechanosensitive ion channel Piezo1 senses fluid shear stress and mediates calcium (Ca2⁺) influx into endothelial cells.64 Under disturbed flow or mechanical stress, activation of this channel induces intracellular Ca2⁺ accumulation, which subsequently activates calcium/calmodulin-dependent kinase signaling (including CaMKII) and other downstream pathways, thereby contributing to endothelial inflammatory responses and metabolic adaptation processes.44,65 In addition, at endothelial cell–cell junctions, a mechanosensory complex composed of PECAM-1, VE-cadherin, and VEGFR2 is considered a central structure for shear stress signal transduction.66 Upon shear stress stimulation, this complex cooperatively transmits mechanical signals and activates multiple downstream pathways, including NF-κB and Akt/eNOS, thereby influencing endothelial homeostasis and inflammation-related gene expression.67,68
In addition to mechanical stimuli, vascular wall cells also sense metabolic and inflammatory signals through pattern recognition receptors. Oxidized low-density lipoprotein (oxLDL) activates NF-κB–dependent inflammatory responses via the TLR4–MyD88 pathway,69,70 while the gut microbiota–derived metabolite trimethylamine N-oxide (TMAO) has also been shown to amplify endothelial inflammatory signaling and promote atherosclerosis progression.71,72 Together, these physical and chemical stimuli constitute a “first-hit” signal in the vascular wall, providing the initial conditions for subsequent metabolic state alterations and epigenetic remodeling. Available evidence suggests that these changes preferentially accumulate at key regulatory elements such as enhancers and promoters and are accompanied by alterations in chromatin accessibility, rendering subsequent transcriptional responses more readily reactivated and potentially giving rise to self-reinforcing regulatory architectures. (Figure 3).
 |
Figure 3 Integration of Mechanical and Metabolic Stress Sensing This figure illustrates how vascular structural cells integrate mechanical and metabolic stimuli to initiate trained immunity–like responses. Endothelial cells and vascular smooth muscle cells sense disturbed flow, shear stress, and vascular injury through mechanosensitive structures, including Piezo1 and junctional complexes, leading to Ca2⁺-dependent inflammatory signaling. In parallel, metabolic and inflammatory cues such as oxidized LDL and hyperglycemia activate pattern recognition receptors, including TLR4. The convergence of these signals constitutes a critical priming phase that establishes permissive conditions for downstream metabolic reprogramming and epigenetic imprinting. |
Metabolic Reprogramming: An “Amplifier” Linking Transient Stimuli to Persistent Memory
Sustained stress signals do not need to persist over long periods to induce profound alterations in vascular wall cells; rather, the metabolic state itself serves as a central amplification and transduction hub that links transient stimuli to long-term phenotypic memory. Under pathological conditions such as inflammation, hyperglycemia, or disturbed flow, both ECs and VSMCs commonly exhibit a glycolysis-biased metabolic profile, with 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3) recognized as a key rate-limiting node driving glycolytic flux reprogramming.73,74 Enhanced glycolysis not only fulfills cellular energy demands but also markedly reshapes the intracellular landscape of metabolic intermediates. The resulting increase in acetyl–coenzyme A provides a direct substrate for histone acetyltransferases (HATs), preferentially targeting promoters and enhancers of inflammation-related genes, thereby amplifying and stabilizing the transcriptional programs induced by the initial stimulus at the chromatin level.75–77
Meanwhile, mitochondrial metabolism and dynamics are also profoundly remodeled. Under chronic stress or pathological stimuli—such as oxidative stress, inflammation, or metabolic reprogramming—mitochondria tend to shift from a fused toward a fragmented state, accompanied by alterations in tricarboxylic acid (TCA) cycle flux and accumulation of intermediates such as succinate and fumarate.78,79 Extensive experimental evidence indicates that these TCA intermediates act as competitive inhibitors of α-ketoglutarate (α-KG), thereby suppressing the activity of α-KG–dependent dioxygenases—including histone and DNA demethylases—disrupting demethylation processes and favoring the maintenance of a pro-inflammatory chromatin state.80,81 In addition, lactate produced under hypoxic or hyperglycemic conditions can induce histone lactylation, providing a novel mechanism by which metabolic states directly reshape chromatin architecture, and has been implicated in the stabilization of synthetic or fibrotic phenotypes in VSMCs.82–84 (Figure 4).
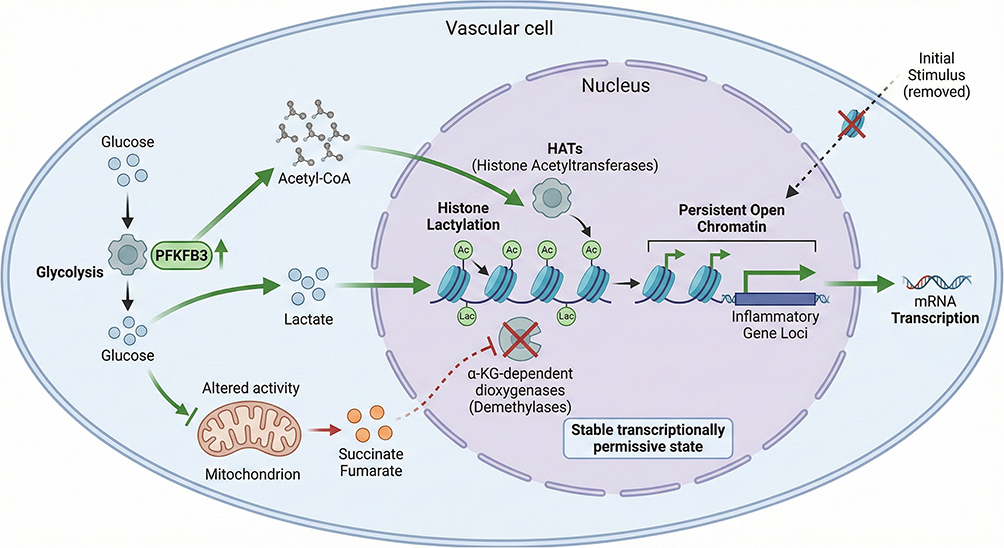 |
Figure 4 Metabolic Reprogramming as a Memory Amplifier This figure illustrates metabolic reprogramming as a central amplifier linking transient stimuli to persistent cellular memory in the vascular wall. Pathological stressors induce a glycolysis-biased metabolic shift driven by PFKFB3 upregulation, accompanied by impaired mitochondrial oxidative metabolism. Increased production of metabolites such as acetyl–coenzyme A, lactate, succinate, and fumarate directly influences chromatin-modifying enzymes, promoting histone acetylation, lactylation, and impaired demethylation. Through these mechanisms, metabolic rewiring stabilizes pro-inflammatory epigenetic states and sustains trained immunity–like phenotypes in vascular structural cells. |
Epigenetic Engraving: Locking Mechanisms of Trained-Like Memory
Metabolism-driven chromatin modifications are ultimately “engraved” at enhancer and promoter regions as relatively stable epigenetic states, thereby endowing vascular wall cells with a transcriptional potential characterized by a lowered activation threshold and heightened responsiveness. Classic studies have demonstrated that under hyperglycemic or lipotoxic conditions, the histone methyltransferase Set7/9 mediates the deposition of H3K4me at promoters of inflammatory genes such as CCL2. Notably, this modification can persist after stimulus withdrawal, giving rise to the so-called “metabolic memory” or “legacy effect”.38,85 More recently, chromatin accessibility profiling and single-cell omics studies have revealed the existence of so-called “latent enhancers” in models of trained immunity and chronic stimulation.86 These regulatory elements acquire enhancer-associated marks following an initial stimulus and remain partially accessible during the resting state, enabling cells to rapidly and robustly activate associated gene expression upon subsequent mild stimulation.87 Although this mechanism was originally described in innate immune cells, the fundamental principle of “enhancer preprogramming and reactivation” provides an important conceptual framework for understanding how vascular structural cells acquire trained-like memory under chronic or repetitive stimuli. In VSMCs, the transcription factor KLF4 is considered a key molecular link between phenotypic dedifferentiation and epigenetic remodeling. By repressing contractile gene expression and reshaping epigenetic regulatory networks, KLF4 drives the transition toward synthetic or pro-inflammatory states. Concurrently, alterations in DNA demethylation–related regulators, such as TET2, may contribute to the long-term maintenance of these states by modulating the global DNA methylation landscape.56,88
Taken together, these studies indicate that trained immunity–like responses in the vascular wall do not rely on sustained signaling inputs, but are more likely maintained by transcriptional programs “locked in” through metabolism-driven epigenetic remodeling.
Clinical Implications and Therapeutic Strategies: Erasing Epigenetic Scars in the Vascular Wall
The recognition that vascular structural cells (VSCs) can acquire trained immunity–like properties provides a unified and mechanistically informative pathophysiological framework to explain multiple long-standing “failure modes” in cardiovascular interventions, particularly adverse outcomes that persist despite adequate lipid control. Among these, in-stent restenosis (ISR) and neointimal hyperplasia represent some of the most clinically relevant manifestations. Although drug-eluting stents (DES) have markedly reduced the incidence of early luminal loss, the mechanical overexpansion imposed on the vascular wall during stent deployment constitutes a potent “first hit,” which may induce durable epigenetic reprogramming in residual vascular smooth muscle cells (VSMCs).89,90 Studies have shown that the epigenetic regulator TET2, which is critical for maintaining the contractile phenotype of VSMCs, is downregulated under multiple pathological conditions and is strongly associated with the transition of VSMCs from a contractile phenotype toward synthetic or dedifferentiated states—a phenotypic switch considered a key epigenetic hallmark of pro-inflammatory and proliferative VSMC activation.91–93 Consequently, even when intensive postoperative statin therapy reduces low-density lipoprotein cholesterol (LDL-C) to very low levels, these epigenetically “primed” smooth muscle cells may still exhibit exaggerated proliferative and migratory responses upon subsequent exposure to mild systemic inflammatory stimuli (a “second hit”), thereby providing a biologically plausible explanation for restenosis beyond lipid burden alone.94,95
Similar pathological memory–like phenomena are also observed in cardiac allograft vasculopathy (CAV), a leading cause of long-term graft failure after heart transplantation. During donor organ procurement and implantation, the vasculature inevitably undergoes ischemia–reperfusion injury (IRI), a process that induces mitochondrial reactive oxygen species (ROS) production and leads to sustained endothelial activation.96,97 Injured endothelial cells can release extracellular vesicles enriched in miR-155, paracrinally promoting monocyte polarization toward a pro-inflammatory M1 phenotype, thereby propagating inflammatory signaling within the vascular microenvironment and accelerating atherosclerotic progression.98,99 Importantly, this non-coding RNA–mediated epigenetic remodeling may establish a trained immunity–like sensitized state in endothelial cells, predisposing graft vessels to amplified inflammatory responses upon subsequent immune stress from the host and thereby markedly accelerating the development of occlusive vasculopathy.
Based on these findings, targeting and “erasing” or remodeling epigenetic memory in the vascular wall has emerged as a novel direction in cardiovascular pharmacology. Bromodomain and extraterminal domain (BET) inhibitors, such as apabetalone (RVX-208), suppress inflammatory transcriptional programs driven by epigenetically primed vascular structural cells (VSCs) by blocking the recognition of acetylated histones by reader proteins including BRD4100–102 (Figure 5). In the Phase III BETonMACE trial, although apabetalone did not significantly reduce major adverse cardiovascular events (MACE) in the overall population, a clear benefit signal was observed in the subgroup of patients with type 2 diabetes and concomitant chronic kidney disease.103 These findings suggest that pharmacological interventions targeting metabolic–epigenetic regulation may offer clinical benefits in selected high-risk populations. Beyond dedicated epigenetic therapies, conventional metabolic modulators may also indirectly influence the establishment and maintenance of trained immunity–like states. Metformin, by activating AMP-activated protein kinase (AMPK) and inhibiting mTOR signaling, limits sustained glycolytic flux and reshapes cellular metabolism,104,105 potentially modulating acetyl-CoA availability and associated epigenetic modifications, thereby providing a molecular basis for its anti-inflammatory and vasculoprotective effects beyond glucose lowering.106–108 (Figure 6)
 |
Figure 5 Epigenetic Targeting of Vascular Memory This figure illustrates therapeutic targeting of trained immunity–like vascular memory through inhibition of bromodomain and extraterminal proteins. In epigenetically primed endothelial cells and vascular smooth muscle cells, sustained histone acetylation supports BRD4-dependent transcription of pathogenic gene programs. BET inhibitors, such as apabetalone, disrupt the interaction between acetylated histones and BET proteins, thereby suppressing enhancer-driven transcription. By modulating epigenetically locked transcriptional states rather than acute inflammatory signaling, BET inhibition attenuates chronic vascular inflammation and cellular hyper-responsiveness. |
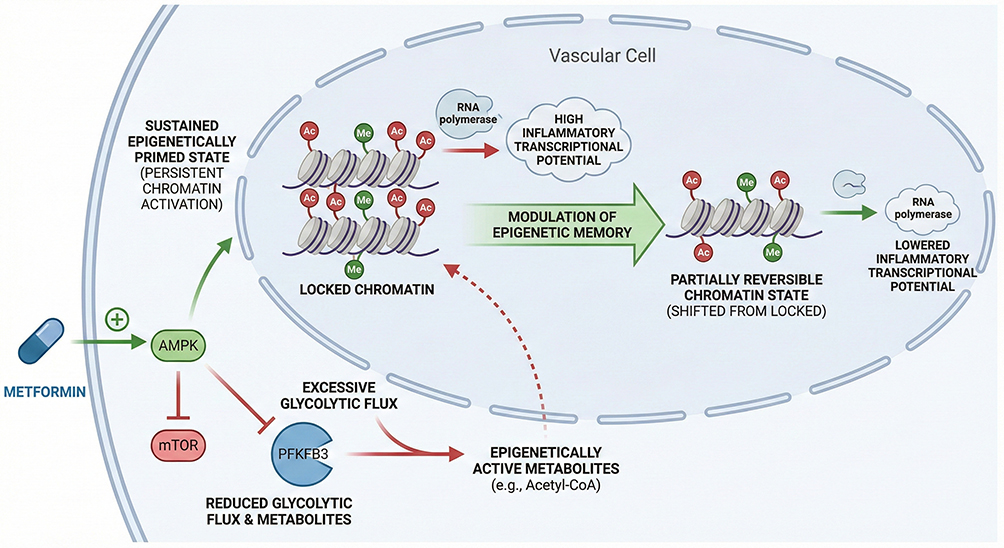 |
Figure 6 Metabolic Modulation of Vascular Memory This figure illustrates how metabolic intervention reshapes trained immunity–like responses in vascular structural cells. Metformin activates AMP-activated protein kinase and inhibits mTOR signaling, suppressing excessive glycolytic flux and PFKFB3-driven metabolic reprogramming. These effects limit the availability of key epigenetic substrates, including acetyl–coenzyme A, thereby attenuating histone acetylation and pro-inflammatory transcriptional programs. Through modulation of metabolism–epigenetic coupling, metabolic therapy reduces vascular cell hyper-responsiveness beyond glucose and lipid lowering. |
At the therapeutic level, local delivery strategies offer new opportunities to selectively remodel vascular memory while minimizing systemic adverse effects. Functionalized nanoparticle platforms targeting endothelial adhesion molecules, such as vascular cell adhesion molecule-1 (VCAM-1), have been developed to deliver pharmacological agents or epigenetic modulators directly to atherosclerotic lesions. Multiple studies in experimental models of atherosclerosis have demonstrated that VCAM-1–targeted nanotherapeutic approaches achieve enhanced lesion accumulation and exhibit anti-inflammatory potential, thereby providing experimental support for precision interventions based on epigenetic regulation109–111 (Figure 7). In parallel, endogenous anti-inflammatory metabolites and their derivatives, including 4-octyl itaconate (4-OI), have been shown to activate the Nrf2 antioxidant pathway via KEAP1 alkylation, while suppressing NLRP3 inflammasome–driven inflammatory responses and cellular pro-inflammatory capacity.112–115 (Figure 8)
 |
Figure 7 Targeted Nanotherapy for Memory Remodeling This figure illustrates nanoparticle-based strategies for selectively targeting trained immunity–like states within the vascular wall. Nanoparticles functionalized with endothelial adhesion molecules, such as VCAM-1, preferentially accumulate at sites of endothelial activation and atherosclerotic lesions. Localized delivery of metabolic or epigenetic modulators attenuates inflammatory signaling and epigenetically primed transcriptional programs in vascular structural cells while minimizing systemic exposure. This precision approach offers a strategy to remodel pathological vascular memory. |
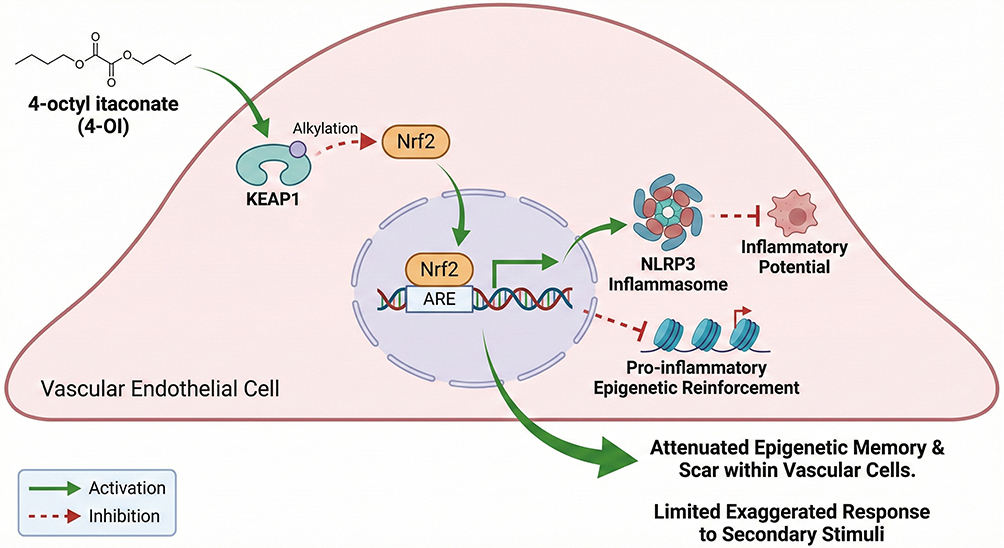 |
Figure 8 Endogenous Metabolites Limiting Inflammatory Memory This figure illustrates the role of endogenous immunometabolic regulators in suppressing trained immunity–like vascular inflammation. Itaconate and its derivative 4-octyl itaconate activate the Nrf2 antioxidant pathway via KEAP1 alkylation while inhibiting NLRP3 inflammasome activation and downstream cytokine production. By integrating redox regulation with inflammatory control, these metabolites reduce inflammatory responsiveness of vascular structural cells, highlighting a physiological mechanism to attenuate vascular inflammatory memory. |
Looking ahead, cardiovascular risk assessment is expected to evolve from reliance on single circulating biochemical markers toward multidimensional evaluation of vascular epigenomic features. The integration of single-cell assay for transposase-accessible chromatin sequencing (scATAC-seq) with spatial transcriptomic technologies enables the identification of key cellular subpopulations within atherosclerotic plaques that reside in a “primed” or hyper-responsive state. Enrichment patterns of enhancer-associated epigenetic marks, such as H3K4me1 and H3K27ac, may aid in predicting plaque stability and therapeutic responsiveness, thereby advancing a new paradigm of precision cardiovascular medicine that targets molecular memory rather than terminal clinical events.
Knowledge Gaps and Conceptual Challenges
Despite growing mechanistic support for trained immunity-like phenotypes in vascular structural cells, important conceptual and translational uncertainties remain. A key question is whether these vascular adaptations fulfill the strict definition of memory. Classical immunological memory implies a return to baseline followed by an augmented response upon re-exposure. By contrast, vascular structural cells often display sustained activation without a clearly defined resting phase, suggesting that these states may represent persistent maladaptive reprogramming rather than inducible memory in the traditional sense.
In addition, longitudinal human evidence is limited. Much of the current support derives from in vitro systems, animal models, or cross-sectional observations. Direct proof of durable epigenetic imprinting within human vascular tissues over time-particularly in response to defined metabolic or mechanical stressors-remains scarce, leaving the temporal stability and clinical relevance of these phenotypes incompletely established.
The issue of reversibility further complicates this framework. Although epigenetic modifications are theoretically dynamic, it remains unclear whether established vascular reprogramming can be therapeutically reversed or whether prolonged disease exposure results in progressively fixed chromatin states. Clarifying this point is crucial for determining whether vascular memory constitutes a modifiable driver of pathology or merely a biomarker of irreversible damage.
Moreover, causal relationships are not yet fully resolved. While metabolic-epigenetic remodeling strongly associates with endothelial dysfunction and smooth muscle phenotypic switching, definitive evidence demonstrating that such reprogramming is necessary and sufficient to drive atherosclerotic progression remains limited. Addressing this gap will require temporally controlled interventions and more precise mechanistic models.
Finally, distinguishing adaptive from maladaptive forms of vascular memory is essential. Transient priming responses may serve protective functions, whereas chronic epigenetic stabilization may promote sustained inflammation and structural remodeling. Resolving these conceptual challenges will be critical for refining the trained immunity-like framework and for translating it into effective strategies to reduce residual cardiovascular risk.
Conclusion
Despite substantial advances in lipid-lowering and blood pressure–controlling therapies, a considerable residual cardiovascular risk persists even when traditional risk factors are optimally managed. This clinical reality underscores the need to move beyond trigger-centered strategies and to address the intrinsic cellular programs that sustain vascular inflammation and remodeling. Vascular structural cells, endowed with remarkable metabolic and epigenetic plasticity, emerge as central orchestrators of this persistent pathological state.
Accumulating evidence suggests that endothelial cells (ECs) and vascular smooth muscle cells (VSMCs) can acquire stable metabolic–epigenetic reprogramming in response to transient mechanical, metabolic, or inflammatory stressors, thereby adopting trained immunity–like phenotypes.116,117 These adaptations may provide a mechanistic explanation for sustained inflammatory activation, plaque progression, in-stent restenosis, and transplant vasculopathy despite apparent risk factor control. Targeting the metabolic and epigenetic circuits underlying these memory-like states therefore represents a promising therapeutic direction.
Looking forward, several key areas warrant focused investigation. High-resolution single-cell multi-omics approaches integrating transcriptomic, epigenomic, and metabolic profiling will be essential to delineate cell-type–specific memory landscapes within the vascular wall. Lineage-tracing strategies are needed to determine the durability and clonal propagation of reprogrammed vascular cells over time, thereby clarifying whether these phenotypes represent transient activation or stable heritable states. In parallel, emerging epigenome-editing technologies offer the possibility of directly testing causality and selectively reversing pathogenic chromatin configurations.
From a translational perspective, the identification of reliable clinical biomarkers reflecting vascular trained immunity–like activity will be critical for patient stratification and therapeutic monitoring. Furthermore, the development of cell-specific targeting strategies—including nanoparticle-based delivery systems and precision epigenetic modulators—may enable selective modulation of pathological memory without compromising systemic immune function.
In summary, the vascular wall should be viewed not merely as a passive structural barrier but as a dynamic tissue capable of molecular memory imprinting. Elucidating, monitoring, and selectively reshaping pathological trained immunity–like programs may represent a transformative frontier in cardiovascular prevention, with the potential to meaningfully reduce residual cardiovascular risk. (Figure 9).
 |
Figure 9 Vascular Trained Immunity and Residual Risk This figure summarizes the central concept of the review, positioning trained immunity–like memory in vascular structural cells as a unifying mechanism underlying residual cardiovascular risk. Transient metabolic, inflammatory, or mechanical stimuli induce durable metabolic and epigenetic reprogramming in endothelial cells and vascular smooth muscle cells, maintaining a hyper-responsive phenotype despite risk factor control. Targeting these maladaptive memory programs represents a conceptual shift toward restoring vascular cellular homeostasis. |
Author Contributions
Jingxuan Dai: Conceptualization, Data curation, Validation, Visualization, Writing – original draft, Writing – review & editing. Xuancheng Zhou: Methodology, Formal analysis, Writing – original draft, Writing – review & editing. Kun Yuan: Investigation, Data curation, Writing – original draft, Writing – review & editing. Keming Huang: Conceptualization, Funding acquisition, Supervision, Writing – original draft, Writing – review & editing. Ying Zhang: Conceptualization, Data curation, Validation, Visualization, Writing – original draft, Writing – review & editing.All authors took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the College Student Innovation and Entrepreneurship Training Program (Grant No. 202510632004).
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Akhtar S, Sharma A. Endothelial dysfunction sustains immune response in atherosclerosis: potential cause for ineffectiveness of prevailing drugs. Int Rev Immunol. 2022;41(2):123–18. doi:10.1080/08830185.2020.1866568
2. Gao S, Wang X, Meng L-B, et al. Recent progress of chronic stress in the development of atherosclerosis. Oxid Med Cell Longev. 2022;2022:4121173. doi:10.1155/2022/4121173
3. Hartley A, Haskard D, Khamis R. Oxidized LDL and anti-oxidized LDL antibodies in atherosclerosis - Novel insights and future directions in diagnosis and therapy. Trends Cardiovasc Med. 2019;29(1):22–26. doi:10.1016/j.tcm.2018.05.010
4. Kim BK, Hong S-J, Lee Y-J, et al. Long-term efficacy and safety of moderate-intensity statin with ezetimibe combination therapy versus high-intensity statin monotherapy in patients with atherosclerotic cardiovascular disease (RACING): a randomised, open-label, non-inferiority trial. Lancet. 2022;400(10349):380–390. doi:10.1016/S0140-6736(22)00916-3
5. Grundy SM, Stone NJ, Bailey AL, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: a Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;139(25):e1082–e1143. doi:10.1161/CIR.0000000000000625
6. Hummelgaard S, Vilstrup JP, Gustafsen C, et al. Targeting PCSK9 to tackle cardiovascular disease. Pharmacol Ther. 2023;249:108480. doi:10.1016/j.pharmthera.2023.108480
7. Garwood CL, Cabral KP, Brown R, et al. Current and emerging PCSK9-directed therapies to reduce LDL-C and ASCVD risk: a state-of-the-art review. Pharmacotherapy. 2025;45(1):54–65. doi:10.1002/phar.4635
8. Sabatine MS, Giugliano RP, Keech AC, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376(18):1713–1722. doi:10.1056/NEJMoa1615664
9. Hoogeveen RC, Ballantyne CM. Residual Cardiovascular Risk at Low LDL: remnants, Lipoprotein(a), and Inflammation. Clin Chem. 2021;67(1):143–153. doi:10.1093/clinchem/hvaa252
10. Mach F, Visseren FLJ, Cater NB, et al. Addressing residual risk beyond statin therapy: new targets in the management of dyslipidaemias-A report from the European Society of Cardiology Cardiovascular Round Table. J Clin Lipidol. 2024;18(5):e685–e700. doi:10.1016/j.jacl.2024.07.001
11. Punnanithinont N, Kambalapalli S, Iskander B, et al. “Anti-inflammatory Therapies in Atherosclerosis - Where are we going?”. Curr Atheroscler Rep. 2024;27(1):19. doi:10.1007/s11883-024-01267-7
12. Ajala ON, Everett BM. Targeting inflammation to reduce residual cardiovascular risk. Curr Atheroscler Rep. 2020;22(11):66. doi:10.1007/s11883-020-00883-3
13. Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377(12):1119–1131. doi:10.1056/NEJMoa1707914
14. Naganuma M. Solving the questions regarding 5-aminosalitylate formulation in the treatment of ulcerative colitis. J Gastroenterol. 2020;55(11):1013–1022. doi:10.1007/s00535-020-01713-8
15. Bonilla FA, Oettgen HC. Adaptive immunity. J Allergy Clin Immunol. 2010;125(2 Suppl 2):S33–40. doi:10.1016/j.jaci.2009.09.017
16. Chi H, Pepper M, Thomas PG. Principles and therapeutic applications of adaptive immunity. Cell. 2024;187(9):2052–2078. doi:10.1016/j.cell.2024.03.037
17. Shu G, Tang Y, Zhou Y, et al. Zac1 is a histone acetylation-regulated NF-kappaB suppressor that mediates histone deacetylase inhibitor-induced apoptosis. Cell Death Differ. 2011;18(12):1825–1835. doi:10.1038/cdd.2011.51
18. Geller AE, Shrestha R, Woeste MR, et al. The induction of peripheral trained immunity in the pancreas incites anti-tumor activity to control pancreatic cancer progression. Nat Commun. 2022;13(1):759. doi:10.1038/s41467-022-28407-4
19. Netea MG, Joosten LAB, Latz E, et al. Trained immunity: a program of innate immune memory in health and disease. Science. 2016;352(6284). doi:10.1126/science.aaf1098.
20. Netea MG, Domínguez-Andrés J, Barreiro LB, et al. Defining trained immunity and its role in health and disease. Nat Rev Immunol. 2020;20(6):375–388. doi:10.1038/s41577-020-0285-6
21. Zhong C, Yang X, Feng Y, et al. Trained immunity: an underlying driver of inflammatory atherosclerosis. Front Immunol. 2020;11:284. doi:10.3389/fimmu.2020.00284
22. Georgescu A, Simionescu M. Extracellular vesicles: versatile nanomediators, potential biomarkers and therapeutic agents in atherosclerosis and COVID-19-related thrombosis. Int J Mol Sci. 2021;22(11):5967. doi:10.3390/ijms22115967
23. He Z, Wang G, Wu J, et al. The molecular mechanism of LRP1 in physiological vascular homeostasis and signal transduction pathways. Biomed Pharmacother. 2021;139:111667. doi:10.1016/j.biopha.2021.111667
24. Bialkowska K, Szpak D, Verbovetskiy D, et al. Kindlin-2 DELETION IN MURAL CELLS LEADS TO VASCULAR INSTABILITy. FASEB J. 2025;39(14):e70823. doi:10.1096/fj.202402977R
25. Li B, Zhao X, Xie W, et al. Causal association of circulating metabolites with diabetic retinopathy: a bidirectional Mendelian randomization analysis. Front Endocrinol. 2024;15:1359502.
26. Wang Y, Wang Y, Luo M, et al. Novel curcumin analog C66 prevents diabetic nephropathy via JNK pathway with the involvement of p300/CBP-mediated histone acetylation. Biochim Biophys Acta. 2015;1852(1):34–46. doi:10.1016/j.bbadis.2014.11.006
27. Zarzour A, Kim HW, Weintraub NL. Epigenetic regulation of vascular diseases. Arterioscler Thromb Vasc Biol. 2019;39(6):984–990. doi:10.1161/ATVBAHA.119.312193
28. Yamunadevi A, Pratibha R, Rajmohan M, et al. Basics of epigenetics and role of epigenetics in diabetic complications. J Pharm Bioallied Sci. 2021;13(Suppl 1):S336–S343. doi:10.4103/jpbs.JPBS_771_20
29. Tamargo IA, Baek KI, Kim Y, et al. Flow-induced reprogramming of endothelial cells in atherosclerosis. Nat Rev Cardiol. 2023;20(11):738–753. doi:10.1038/s41569-023-00883-1
30. Marracino L, Fortini F, Bouhamida E, et al. Adding a “notch” to cardiovascular disease therapeutics: a microrna-based approach. Front Cell Dev Biol. 2021;9:695114. doi:10.3389/fcell.2021.695114
31. Chen Z, Malek V, Natarajan R. Update: the role of epigenetics in the metabolic memory of diabetic complications. Am J Physiol Renal Physiol. 2024;327(3):F327–f339. doi:10.1152/ajprenal.00115.2024
32. Cannito S, Giardino I, D’Apolito M, et al. From metabolic to epigenetic memory: the impact of hyperglycemia-induced epigenetic signature on kidney disease progression and complications. Genes. 2025;16:1442. doi:10.3390/genes16121442
33. Le KTT, Keur N, Middelkamp H, et al. Cytokine-induced memory-like responses in endothelial cells link chronic inflammation to vascular disease risk. Mol Omics. 2025;21(6):706–722. doi:10.1039/D5MO00136F
34. Salminen A, Kaarniranta K, Kauppinen A. Tissue fibroblasts are versatile immune regulators: an evaluation of their impact on the aging process. Ageing Res Rev. 2024;97:102296. doi:10.1016/j.arr.2024.102296
35. Folz R, Laiteerapong N. The legacy effect in diabetes: are there long-term benefits? Diabetologia. 2021;64(10):2131–2137. doi:10.1007/s00125-021-05539-8
36. Laiteerapong N, Ham SA, Gao Y, et al. The legacy effect in type 2 diabetes: impact of early glycemic control on future complications (the diabetes & aging study). Diabetes Care. 2019;42(3):416–426. doi:10.2337/dc17-1144
37. Holman RR, Paul SK, Bethel MA, et al. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med. 2008;359(15):1577–1589. doi:10.1056/NEJMoa0806470
38. El-Osta A, Brasacchio D, Yao D, et al. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med. 2008;205(10):2409–2417. doi:10.1084/jem.20081188
39. Chen X, Shi C, Wang Y, et al. The mechanisms of glycolipid metabolism disorder on vascular injury in type 2 diabetes. Front Physiol. 2022;13:952445. doi:10.3389/fphys.2022.952445
40. Bai Y, Wu J, Jian W. Trained immunity in diabetes: emerging targets for cardiovascular complications. Front Endocrinol. 2025;16:1533620. doi:10.3389/fendo.2025.1533620
41. Li F, Yan K, Wu L, et al. Single-cell RNA-seq reveals cellular heterogeneity of mouse carotid artery under disturbed flow. Cell Death Discov. 2021;7(1):180. doi:10.1038/s41420-021-00567-0
42. Hu S, Zhu L. Semaphorins and their receptors: from axonal guidance to atherosclerosis. Front Physiol. 2018;9:1236. doi:10.3389/fphys.2018.01236
43. Wu L, Jiang S, Zhou X, et al. Endothelial KDM5B regulated by piezo1 contributes to disturbed flow induced atherosclerotic plaque formation. J Cell Mol Med. 2024;28(23):e70237. doi:10.1111/jcmm.70237
44. Lan Y, Lu J, Zhang S, et al. Piezo1-mediated mechanotransduction contributes to disturbed flow-induced atherosclerotic endothelial inflammation. J Am Heart Assoc. 2024;13(21):e035558. doi:10.1161/JAHA.123.035558
45. Wu D, Huang RT, Hamanaka RB, et al. HIF-1alpha is required for disturbed flow-induced metabolic reprogramming in human and porcine vascular endothelium. Elife. 2017;6:e25217
46. Andueza A, Kumar S, Kim J, et al. Endothelial reprogramming by disturbed flow revealed by single-cell rna and chromatin accessibility study. Cell Rep. 2020;33(11):108491. doi:10.1016/j.celrep.2020.108491
47. Moonen JR, Chappell J, Shi M, et al. KLF4 recruits SWI/SNF to increase chromatin accessibility and reprogram the endothelial enhancer landscape under laminar shear stress. Nat Commun. 2022;13(1):4941. doi:10.1038/s41467-022-32566-9
48. Davies PF, Civelek M, Fang Y, et al. The atherosusceptible endothelium: endothelial phenotypes in complex haemodynamic shear stress regions in vivo. Cardiovasc Res. 2013;99(2):315–327. doi:10.1093/cvr/cvt101
49. Baek KI, Ryu K. Role of flow-sensitive endothelial genes in atherosclerosis and antiatherogenic therapeutics development. J Cardiovasc Transl Res. 2024;17(3):609–623. doi:10.1007/s12265-023-10463-w
50. Rong JX, Shapiro M, Trogan E, et al. Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc Natl Acad Sci U S A. 2003;100(23):13531–13536. doi:10.1073/pnas.1735526100
51. Xiang L, Wang Y, Liu S, et al. Quercetin Attenuates KLF4-mediated phenotypic switch of vsmcs to macrophage-like cells in atherosclerosis: a critical role for the jak2/stat3 pathway. Int J Mol Sci. 2024;25(14):7755. doi:10.3390/ijms25147755
52. Grootaert MOJ, Bennett MR. Vascular smooth muscle cells in atherosclerosis: time for a re-assessment. Cardiovasc Res. 2021;117(11):2326–2339. doi:10.1093/cvr/cvab046
53. Allahverdian S, Chehroudi AC, McManus BM, et al. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation. 2014;129(15):1551–1559. doi:10.1161/CIRCULATIONAHA.113.005015
54. Zhang F, Guo X, Xia Y, et al. An update on the phenotypic switching of vascular smooth muscle cells in the pathogenesis of atherosclerosis. Cell Mol Life Sci. 2021;79(1):6. doi:10.1007/s00018-021-04079-z
55. Lambert J, Jorgensen HF. Epigenetic regulation of vascular smooth muscle cell phenotypes in atherosclerosis. Atherosclerosis. 2025;401:119085. doi:10.1016/j.atherosclerosis.2024.119085
56. Yu Y, Cai Y, Yang F, et al. Vascular smooth muscle cell phenotypic switching in atherosclerosis. Heliyon. 2024;10(18):e37727. doi:10.1016/j.heliyon.2024.e37727
57. Yap C, Mieremet A, de Vries CJM, et al. Six shades of vascular smooth muscle cells illuminated by KLF4 (Kruppel-Like Factor 4). Arterioscler Thromb Vasc Biol. 2021;41(11):2693–2707. doi:10.1161/ATVBAHA.121.316600
58. Shankman LS, Gomez D, Cherepanova OA, et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. 2015;21(6):628–637. doi:10.1038/nm.3866
59. Liu R, Jin Y, Tang WH, et al. Ten-eleven translocation-2 (TET2) is a master regulator of smooth muscle cell plasticity. Circulation. 2013;128(18):2047–2057. doi:10.1161/CIRCULATIONAHA.113.002887
60. Zhu N, Guo Z-F, Kazama K, et al. Epigenetic regulation of vascular smooth muscle cell phenotypic switch and neointimal formation by PRMT5. Cardiovasc Res. 2023;119(12):2244–2255. doi:10.1093/cvr/cvad110
61. Cao G, Xuan X, Hu J, et al. How vascular smooth muscle cell phenotype switching contributes to vascular disease. Cell Commun Signal. 2022;20(1):180. doi:10.1186/s12964-022-00993-2
62. Novakovic B, Habibi E, Wang S-Y, et al. beta-glucan reverses the epigenetic state of lps-induced immunological tolerance. Cell. 2016;167(5):1354–1368e14. doi:10.1016/j.cell.2016.09.034
63. Arts RJW, Carvalho A, La Rocca C, et al. Immunometabolic Pathways in BCG-Induced Trained Immunity. Cell Rep. 2016;17(10):2562–2571. doi:10.1016/j.celrep.2016.11.011
64. Zheng Q, Zou Y, Teng P, et al. Mechanosensitive Channel PIEZO1 Senses Shear Force to Induce KLF2/4 Expression via CaMKII/MEKK3/ERK5 Axis in Endothelial Cells. Cells. 2022;11(14):2191. doi:10.3390/cells11142191
65. Zhang X, Leng S, Liu X, et al. Ion channel Piezo1 activation aggravates the endothelial dysfunction under a high glucose environment. Cardiovasc Diabetol. 2024;23(1):150. doi:10.1186/s12933-024-02238-7
66. Tzima E, Irani-Tehrani M, Kiosses WB, et al. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature. 2005;437(7057):426–431. doi:10.1038/nature03952
67. Conway D, Schwartz MA. Lessons from the endothelial junctional mechanosensory complex. F1000 Biol Rep. 2012;4:1. doi:10.3410/B4-1
68. Power G, Ferreira-Santos L, Martinez-Lemus LA, et al. Integrating molecular and cellular components of endothelial shear stress mechanotransduction. Am J Physiol Heart Circ Physiol. 2024;327(4):H989–H1003. doi:10.1152/ajpheart.00431.2024
69. Yin YW, Liao S-Q, Zhang M-J, et al. TLR4-mediated inflammation promotes foam cell formation of vascular smooth muscle cell by upregulating ACAT1 expression. Cell Death Dis. 2014;5(12):e1574. doi:10.1038/cddis.2014.535
70. Miller YI, Viriyakosol S, Worrall DS, et al. Toll-like receptor 4-dependent and -independent cytokine secretion induced by minimally oxidized low-density lipoprotein in macrophages. Arterioscler Thromb Vasc Biol. 2005;25(6):1213–1219. doi:10.1161/01.ATV.0000159891.73193.31
71. Seldin MM, Meng Y, Qi H, et al. Trimethylamine N-oxide promotes vascular inflammation through signaling of mitogen-activated protein kinase and nuclear factor-kappaB. J Am Heart Assoc. 2016;5.
72. Liu Y, Dai M. Trimethylamine N-oxide generated by the gut microbiota is associated with vascular inflammation: new insights into atherosclerosis. Mediators Inflamm. 2020;2020:4634172. doi:10.1155/2020/4634172
73. Zhou L, Li J, Wang J, et al. Pathogenic role of PFKFB3 in endothelial inflammatory diseases. Front Mol Biosci. 2024;11:1454456. doi:10.3389/fmolb.2024.1454456
74. Zhang X, Zheng B, Zhao L, et al. KLF4-PFKFB3-driven glycolysis is essential for phenotypic switching of vascular smooth muscle cells. Commun Biol. 2022;5(1):1332. doi:10.1038/s42003-022-04302-y
75. Wellen KE, Thompson CB. A two-way street: reciprocal regulation of metabolism and signalling. Nat Rev Mol Cell Biol. 2012;13(4):270–276. doi:10.1038/nrm3305
76. Shi L, Tu BP. Acetyl-CoA induces transcription of the key G1 cyclin CLN3 to promote entry into the cell division cycle in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2013;110(18):7318–7323. doi:10.1073/pnas.1302490110
77. McDonnell E, Crown SB, Fox DB, et al. Lipids reprogram metabolism to become a major carbon source for histone acetylation. Cell Rep. 2016;17(6):1463–1472. doi:10.1016/j.celrep.2016.10.012
78. van der Bliek AM, Shen Q, Kawajiri S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb Perspect Biol. 2013;5.
79. Tannahill GM, Curtis AM, Adamik J, et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature. 2013;496(7444):238–242. doi:10.1038/nature11986
80. Xiao M, Yang H, Xu W, et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012;26(12):1326–1338. doi:10.1101/gad.191056.112
81. Sarkar S, Chang C-I, Jean J, et al. TCA cycle-derived oncometabolites in cancer and the immune microenvironment. J Biomed Sci. 2025;32(1):87. doi:10.1186/s12929-025-01186-y
82. Liu L, Zhang J, Dong Z, et al. Histone lactylation-mediated metabolic remodeling in vascular smooth muscle cells aggravates aortic aneurysm and dissection by promoting lactate accumulation. Circulation. 2026;153(3):189–209. doi:10.1161/CIRCULATIONAHA.125.072576
83. Chen A, Chen Z, Huang B, et al. Hypoxia-induced histone lactylation promotes pulmonary arterial smooth muscle cells proliferation in pulmonary hypertension. Mol Cell Biochem. 2025;480(11):5685–5697. doi:10.1007/s11010-025-05342-8
84. Zhu Z, Huang C, Chen J, et al. Lactylation at the crossroads of immune metabolism and epigenetic regulation: revealing its role in rheumatic immune diseases. J Transl Med. 2025;24(1):25. doi:10.1186/s12967-025-07498-9
85. Okabe J, Orlowski C, Balcerczyk A, et al. Distinguishing hyperglycemic changes by Set7 in vascular endothelial cells. Circ Res. 2012;110(8):1067–1076. doi:10.1161/CIRCRESAHA.112.266171
86. Ostuni R, Piccolo V, Barozzi I, et al. Latent enhancers activated by stimulation in differentiated cells. Cell. 2013;152(1–2):157–171. doi:10.1016/j.cell.2012.12.018
87. van der Heijden C, Noz MP, Joosten LAB, et al. Epigenetics and Trained Immunity. Antioxid Redox Signal. 2018;29(11):1023–1040. doi:10.1089/ars.2017.7310
88. Zurek M, Aavik E, Mallick R, et al. Epigenetic regulation of vascular smooth muscle cell phenotype switching in atherosclerotic artery remodeling: a mini-review. Front Genet. 2021;12:719456. doi:10.3389/fgene.2021.719456
89. Pelliccia F, Zimarino M, Niccoli G, et al. In-stent restenosis after percutaneous coronary intervention: emerging knowledge on biological pathways. Eur Heart J Open. 2023;3(5). doi:10.1093/ehjopen/oead083.
90. Curcio A, Torella D, Indolfi C. Mechanisms of smooth muscle cell proliferation and endothelial regeneration after vascular injury and stenting: approach to therapy. Circ J. 2011;75(6):1287–1296. doi:10.1253/circj.CJ-11-0366
91. Chen R, McVey DG, Shen D, et al. Phenotypic switching of vascular smooth muscle cells in atherosclerosis. J Am Heart Assoc. 2023;12(20):e031121. doi:10.1161/JAHA.123.031121
92. Ostriker AC, Xie Y, Chakraborty R, et al. TET2 protects against vascular smooth muscle cell apoptosis and intimal thickening in transplant vasculopathy. Circulation. 2021;144(6):455–470. doi:10.1161/CIRCULATIONAHA.120.050553
93. Lee BSL, Dunn JK, Liang C, et al. Epigenetic reprogramming via TET2 prevents medial calcification and restores vascular smooth muscle cell identity. JACC Basic Transl Sci. 2025;11(1):101434. doi:10.1016/j.jacbts.2025.101434
94. Yanai H, Adachi H, Hakoshima M, et al. Molecular biological and clinical understanding of the statin residual cardiovascular disease risk and peroxisome proliferator-activated receptor alpha agonists and ezetimibe for its treatment. Int J Mol Sci. 2022;23(7):3418. doi:10.3390/ijms23073418
95. Yanai H, Adachi H, Hakoshima M, et al. Atherogenic lipoproteins for the statin residual cardiovascular disease risk. Int J Mol Sci. 2022;23(21):13499. doi:10.3390/ijms232113499
96. Xiang Q, Yi X, Zhu X-H, et al. Regulated cell death in myocardial ischemia-reperfusion injury. Trends Endocrinol Metab. 2024;35(3):219–234. doi:10.1016/j.tem.2023.10.010
97. Liu J, Man K. Mechanistic insight and clinical implications of ischemia/reperfusion injury post liver transplantation. Cell Mol Gastroenterol Hepatol. 2023;15(6):1463–1474. doi:10.1016/j.jcmgh.2023.03.003
98. He S, Wu C, Xiao J, et al. Endothelial extracellular vesicles modulate the macrophage phenotype: potential implications in atherosclerosis. Scand J Immunol. 2018;87(4):e12648. doi:10.1111/sji.12648
99. Yang WW, Li Q-X, Wang F, et al. Exosomal miR −155-5p promote the occurrence of carotid atherosclerosis. J Cell Mol Med. 2024;28(21):e70187. doi:10.1111/jcmm.70187
100. Borck PC, Guo LW, Plutzky J. BET epigenetic reader proteins in cardiovascular transcriptional programs. Circ Res. 2020;126(9):1190–1208. doi:10.1161/CIRCRESAHA.120.315929
101. Tsujikawa LM, Fu L, Das S, et al. Apabetalone (RVX-208) reduces vascular inflammation in vitro and in CVD patients by a BET-dependent epigenetic mechanism. Clin Clin Epigenet. 2019;11(1):102. doi:10.1186/s13148-019-0696-z
102. Shahid S, Pantakani M, Binder L, et al. Small molecule BRD4 inhibitors apabetalone and jq1 rescues endothelial cells dysfunction, protects monolayer integrity and reduces midkine expression. Molecules. 2022;27(21):7453. doi:10.3390/molecules27217453
103. Kalantar-Zadeh K, Schwartz GG, Nicholls SJ, et al. Effect of apabetalone on cardiovascular events in diabetes, ckd, and recent acute coronary syndrome: results from the betonmace randomized controlled trial. Clin J Am Soc Nephrol. 2021;16(5):705–716. doi:10.2215/CJN.16751020
104. Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia. 2017;60(9):1577–1585. doi:10.1007/s00125-017-4342-z
105. Bu Y, Peng M, Tang X, et al. Protective effects of metformin in various cardiovascular diseases: clinical evidence and AMPK -dependent mechanisms. J Cell Mol Med. 2022;26(19):4886–4903. doi:10.1111/jcmm.17519
106. Tang CJ, Xu J, Ye H-Y, et al. Metformin prevents PFKFB3-related aerobic glycolysis from enhancing collagen synthesis in lung fibroblasts by regulating AMPK/mTOR pathway. Exp Ther Med. 2021;21(6):581. doi:10.3892/etm.2021.10013
107. Cuyas E, Fernández-Arroyo S, Joven J, et al. Metformin targets histone acetylation in cancer-prone epithelial cells. Cell Cycle. 2016;15(24):3355–3361. doi:10.1080/15384101.2016.1249547
108. Giordo R, Posadino AM, Mangoni AA, et al. Metformin-mediated epigenetic modifications in diabetes and associated conditions: biological and clinical relevance. Biochem Pharmacol. 2023;215:115732. doi:10.1016/j.bcp.2023.115732
109. Castro R, Adair JH, Mastro AM, et al. VCAM-1-targeted nanoparticles to diagnose, monitor and treat atherosclerosis. Nanomedicine. 2024;19(8):723–735. doi:10.2217/nnm-2023-0282
110. Xu H, Li S, Liu YS. Nanoparticles in the diagnosis and treatment of vascular aging and related diseases. Signal Transduct Target Ther. 2022;7(1):231. doi:10.1038/s41392-022-01082-z
111. Distasio N, Salmon H, Dierick F, et al. VCAM‐1‐targeted gene delivery nanoparticles localize to inflamed endothelial cells and atherosclerotic plaques. Adv Ther. 2020;4(2):2000196
112. Mills EL, Ryan DG, Prag HA, et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature. 2018;556(7699):113–117. doi:10.1038/nature25986
113. Hooftman A, Angiari S, Hester S, et al. The immunomodulatory metabolite itaconate modifies nlrp3 and inhibits inflammasome activation. Cell Metab. 2020;32(3):468–478e7. doi:10.1016/j.cmet.2020.07.016
114. Zhao X, Feng R, Chen J, et al. 4-Octyl itaconate alleviates experimental autoimmune prostatitis by inhibiting the NLRP3 inflammasome-induced pyroptosis through activating Nrf2/HO-1 pathway. Prostate. 2024;84(4):329–341. doi:10.1002/pros.24652
115. Tang C, Wang X, Xie Y, et al. 4-Octyl Itaconate Activates Nrf2 Signaling to inhibit pro-inflammatory cytokine production in peripheral blood mononuclear cells of systemic lupus erythematosus patients. Cell Physiol Biochem. 2018;51(2):979–990. doi:10.1159/000495400
116. Drummer CT, Saaoud F, Shao Y, et al. Trained immunity and reactivity of macrophages and endothelial cells. Arterioscler Thromb Vasc Biol. 2021;41(3):1032–1046. doi:10.1161/ATVBAHA.120.315452
117. Yang Q, Saaoud F, Lu Y, et al. Innate immunity of vascular smooth muscle cells contributes to two-wave inflammation in atherosclerosis, twin-peak inflammation in aortic aneurysms and trans-differentiation potential into 25 cell types. Front Immunol. 2023;14:1348238. doi:10.3389/fimmu.2023.1348238
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.