Back to Journals » OncoTargets and Therapy » Volume 11
Traditional Chinese medicine Astragalus polysaccharide enhanced antitumor effects of the angiogenesis inhibitor apatinib in pancreatic cancer cells on proliferation, invasiveness, and apoptosis
Authors Wu J, Wang J, Su Q, Ding W, Li T, Yu J, Cao B ![]()
Received 15 November 2017
Accepted for publication 20 March 2018
Published 9 May 2018 Volume 2018:11 Pages 2685—2698
DOI https://doi.org/10.2147/OTT.S157129
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Ingrid Espinoza
Jun Wu,1 Jing Wang,1 Qiang Su,1 Wei Ding,2 Teng Li,1 Junxian Yu,3 Bangwei Cao1
1Cancer Center, Beijing Friendship Hospital, Capital Medical University, Beijing, China; 2Department of Pharmacy, Beijing Friendship Hospital, Capital Medical University, Beijing, China; 3Department of Biochemistry and Molecular Biology, Capital Medical University, Beijing, China
Background: Traditional chemotherapy and molecular targeted therapy have shown modest effects on the survival of patients with pancreatic cancer. The current study aimed to investigate the antitumor effects of apatinib, Astragalus polysaccharide (APS), and the combination of both the drugs in pancreatic cancer cells and further explore the molecular mechanisms in vitro.
Materials and methods: Expression of vascular endothelial growth factor receptor-2 (VEGFR-2) in human pancreatic cancer cell lines ASPC-1, PANC-1, and SW1990 was detected by Western blotting. Cell proliferation was measured by MTS, and migration and invasion were detected by wound-healing and Transwell assays, respectively. Cell apoptosis rate was determined by flow cytometry and cellular autophagy level affected by apatinib, and APS was analyzed by Western blotting.
Results: Human pancreatic cancer cell lines ASPC-1 and PANC-1 expressed VEGFR-2, but VEGFR-2 was not detected in SW1990. Either apatinib or APS inhibited cell proliferation in a dose-dependent manner in ASPC-1 and PANC-1. APS in combination with apatinib showed enhanced inhibitory effects on cell migration and invasion compared with apatinib monotherapy in ASPC-1 and PANC-1. Meanwhile, APS combined with apatinib strongly increased cell apoptosis percentage. Western blotting showed that the combination of APS and apatinib significantly enhanced the downregulation of phosphorylated protein kinase B (AKT) and extracellular signal-regulated kinase (ERK) (p-AKT and p-ERK) as well as matrix metalloproteinases-9 (MMP-9) expression. In addition, both apatinib and APS induced cellular autophagy. However, the expression of autophagy-related proteins was not further elevated in the combination group.
Conclusion: The study first demonstrated that apatinib showed potentially inhibitory effects in pancreatic cancer cells and that APS enhanced the antitumor effects of apatinib through further downregulating the expression of phosphorylation of AKT and ERK as well as MMP-9.
Keywords: AKT, ERK, digestive tumor, molecular mechanisms, molecular targeted therapy, basic medical research, anti-angiogenesis drug
Corrigendum for this paper has been published.
Introduction
Pancreatic cancer is one of the most lethal human cancers in the world. It is the fourth leading cause of cancer death in the United States with a 5-year survival rate of <6% because of its aggressive biological nature and lack of effective treatments.1 In China, it has been estimated that 91,000 cases of pancreatic cancer were diagnosed in 2015 and that 79,400 died as a result of the devastating disease, which ranked 9th and 6th, respectively, in cancer incidence and mortality.2 The majority of pancreatic cancers are pancreatic adenocarcinomas, and most of the patients are diagnosed at the late stage with metastasis due to lack of early clinical symptoms and sensitive biomarkers. Only ~15% of patients are candidates for the curative surgery, but the prognosis remains relatively poor even after tumor resection.3
Angiogenesis plays very important roles in cancer progression. Vascular endothelial growth factor (VEGF) acting with its corresponding receptors (VEGFR1, VEGFR2, and VEGFR3) plays a pivotal role in angiogenesis, cell proliferation, migration, and invasion.4 Therefore, VEGF and its receptors have been identified as potential targets for novel cancer therapies. Blocking VEGF/VEGFR pathway is a feasible strategy for pancreatic cancer therapy because of high levels of VEGF/VEGFR expression detected in pancreatic cancers in vitro and in vivo, which correlates with poor prognosis.5,6 As an orally small-molecule inhibitor of VEGFR-2, apatinib is currently being studied in multiple tumor types. The novel tyrosine kinase inhibitor not only specifically competes with the ATP-binding site of VEGFR2 but also targets Ret, c-kit, c-src, and platelet-derived growth factor receptors (PDGFR), which subsequently blocks the downstream signal transduction and angiogenesis.7 In addition, apatinib has been proved effective in improving the overall survival and progression-free survival in the clinical Phase III trial for treating advanced gastric cancers.8 Besides convincing evidence on gastric cancer treatment, apatinib was also discovered with promising therapeutic effects against diverse tumor types, including breast cancer, non-small-cell lung cancer (NSCLC), hepatocellular carcinoma, melanoma, intrahepatic cholangiocarcinoma, and extrahepatic bile duct cancer.9–13 However, whether apatinib has an antitumor effect on human pancreatic cancer remains unknown. Chinese traditional herbal medicines have been widely used to treat diseases including cancers. Astragalus polysaccharide (APS), the active component extracted from Astragalus membranaceus, has been found to exert a marked inhibitory effect on human solid tumors by potentiating immune system, inducing apoptosis and inhibiting cancer cell growth.14,15 It is also reported that APS increased the sensitivity of chemotherapeutics and counteracted the side effects of chemotherapeutic drugs.16,17
In the current study, we investigated the effects of apatinib and APS in pancreatic cancer cells and further explored the underlying mechanisms of the synergetic effect of APS on apatinib.
Materials and methods
Cell culture and reagents
Human umbilical vein endothelial cells (HUVECs) and human pancreatic cancer cell lines ASPC-1, PANC-1, and SW1990 were obtained from the Department of Experimental Research Center in Beijing Friendship Hospital (Beijing, China). The Department of Experimental Research Center in Beijing Friendship Hospital purchased the above cells from American Type Culture Collection (ATCC, Manassas, VA, USA). HUVEC and SW1990 were routinely cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Corning Incorporated, Corning, NY, USA), and ASPC-1 and PANC-1 cells were cultured in Roswell Park Memorial Institute (RPMI)-1640 (Corning Incorporated) supplemented with 10% fetal bovine serum (FBS; Biological Industries, Tel Aviv, Israel) and 1% penicillin/streptomycin (KeyGen, Nanjing, China) at 37°C in a humidified atmosphere of 5% CO2. Apatinib was obtained from Jiangsu Hengrui Medicine Co, Ltd (Jiangsu, China). APS was purchased from Nanjing ZeLang Biotech Ltd (Nanjing, China). Recombinant human vascular endothelial growth factor 165 (rhVEGF-165) was purchased from R&D Systems, Inc. (Minneapolis, MN, USA).
Cell proliferation assay
A CellTiter-96 Aqueous-One Solution Cell Proliferation (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt, [MTS]) Assay Kit (Promega Corporation, Fitchburg, WI, USA) was used. Cells were seeded in 96-well plates at 5×103 cells per well and then treated for conditions according to the experimental design for 24 hours. For ASPC-1 and PANC-1 cells, rhVEGF-165 at 20 ng/mL was added. The MTS reagent was added into the culture medium and maintained for 1 hour at 37°C. The plates were subjected to measurements of the absorption at 490 nm with the ELx808™ Absorbance Microplate Reader (BioTek, Winooski, VT, USA). The A490 value is directly proportional to the viability of cell proliferation. The concentration to inhibit cell proliferation by 50% (IC50) was determined by using a linear regression equation. Triplicate independent experiments were performed in a parallel manner for each concentration point.
Migration assay
Wound-healing assay was used to determine cell migration. Cells were seeded into 12-well plates. When the cell confluency reached about 90%, the wound was created by drawing a straight line across the cells with a 200 μL pipette. After washing with phosphate-buffered saline (PBS) twice to remove cell debris, the cells were treated with different agents suspended in RPMI-1640 without FBS, and then the wound healing was observed at 0 and 24 hours at the scratch lines. Representative fields were photographed by using an inverted phase contrast microscope equipped with a digital camera (Olympus Corporation, Tokyo, Japan). Cell motility was evaluated according to the following formula: cell motility = (distance 24 hours − distance 0 hours)/distance 0 hours. Each experiment was performed in triplicate. ImageJ Plus (National Institutes of Health, Bethesda, MD, USA) was used to quantify the motility images of the wound-healing analysis. Quantification of cell migration was performed by measuring the area migrated at indicated time points compared to the scratch area at 0 hours.
Invasion assay
The invasion assay was performed by using Transwell co-culture chambers (24 wells, 8 μm pore size, Costar; Corning Incorporated). First, pre-chilled upper inserts were coated with 60 μL thawed Matrigel (Corning Incorporated) per well (1:10 and 1:20 in dilution for ASPC-1 and PANC-1, respectively) and then set for 6 hours at 37°C in a 5% CO2 humidified incubator. The dilution agent was pre-chilled in RPMI-1640 medium without FBS. Before adding the cells, upper inserts were hydrated by RPMI-1640 without FBS for 1 hour in room temperature, and then 200 μL serum-free medium with ASPC-1 (2×104/well) and PANC-1 (3×104/well) suspension containing different treatment agents were added into the upper insert. About 600 μL of RPMI-1640 per well with 10% FBS as a chemoattractant was added in the lower chamber. After 24 hours of incubation at 37°C, cells in the upper chamber were removed gently by scraping with a cotton swab and washed with PBS. The invaded cells left at the bottom of chambers were fixed with 4% paraformaldehyde for 0.5 hours in room temperature and then stained with 0.5% crystal violet (Macklin, Shanghai, China) for 10 minutes. Following air-drying, crystal violet-positive invaded cells were counted. The invaded cells were counted in six random visual fields for each chamber using an inverted phase contrast microscope equipped with a digital camera (Olympus), and the average invaded cell numbers per view were calculated.
Apoptosis assay
ASPC-1 and PANC-1 were seeded into 6-well plates. The cells were incubated overnight and then treated with different agents when cell confluence reached about 60%–70%. After 24 hours, to detect the percentage of cell apoptosis, Annexin V-fluorescein isothiocyanate (FITC) and propidium iodide (PI) staining assay were performed by using an apoptosis detecting kit (KeyGen) according to the manufacturer’s instructions, and then cell apoptosis rate was detected by flow cytometry. The apoptosis rate in every group was described by Q2 (late apoptosis) + Q4 (early apoptosis).
Western blotting
ASPC-1 and PANC-1 cells were treated with reagents at indicated concentrations for 24 hours in 6 cm2 plates. Following the appropriate treatments, cells were lysed on ice in modified radio immunoprecipitation assay buffer (Applygen, Beijing, China) containing protease inhibitor cocktail (Amresco, Ohio, USA). The supernatants were collected by centrifugation at 12,000 × g at 4°C. Total protein concentrations were determined with a bicinchoninic acid (BCA) protein assay kit (Merck, Darmstadt, Germany). Protein samples were mixed with 5× loading buffer (Applygen) and heated in water at 95°C for 10 minutes. Equal amount of protein was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. After being transferred onto polyvinylidene fluoride membrane filters (EMD Millipore, Billerica, MA, USA) and blocked with blocking buffer (Tris-buffered saline and 0.2% Tween [TBST] containing 1.5% bovine serum albumin) for 1 hour at room temperature, the filters were blotted with primary antibodies overnight at 4°C. The following primary antibodies were used to detect the proteins: rabbit anti-human VEGFR-2 monoclonal antibody (1:1,000; Abcam, Cambridge, UK), rabbit anti-human Bcl-2 polyclonal antibody (1:500; Abclonal, Wuhan, China), rabbit anti-human Bax monoclonal antibody (1:1,000; Cell Signaling Technology [CST], Danvers, MA, USA), mouse anti-human MMP-9 monoclonal antibody (1:100; Santa Cruz Biotechnology Inc., Dallas, TX, USA), rabbit anti-human LC3 (Light Chain 3) monoclonal antibody (1:1,000; CST), rabbit anti-human monoclonal antibody phosphorylated extracellular signal-regulated kinase (ERK) (p-ERK, 1:1,000; CST), rabbit anti-human protein kinase B (AKT) monoclonal antibody (1:1,000; CST), rabbit anti-human ERK monoclonal antibody (1:1,000; CST), rabbit anti-human p-AKT polyclonal antibody (1:500; Abclonal), rabbit anti-human β-actin monoclonal antibody (1:3,000; Abcam). After being washed with TBST for three times, the membrane was incubated with the goat anti-rabbit and anti-mouse immunoglobulin G conjugated to horseradish peroxidase antibody (1:5,000; Santa Cruz Biotechnology Inc.) as the secondary antibodies at 37°C for 1 hour. The blottings were visualized for bands with an enhanced chemiluminescence (ECL; EMD Millipore) detection system.
Statistical analysis
The experimental data were presented as mean ± standard deviation from at least three independent experiments. The data were analyzed, and the statistical graphs were created by GraphPad Prism 5 (GraphPad Software, Inc., La Jolla, CA, USA). Differences between groups were analyzed by using one-way analysis of variance, and the statistical significance was determined at P-values <0.05.
Results
Expression of VEGFR-2 in pancreatic cancer cells
The cell lines were evaluated for VEGFR-2 expression by Western blotting analysis, and HUVEC cell line was used as the positive control for VEGFR-2. Results showed that VEGFR-2 was expressed in ASPC-1 and PANC-1 (Figure 1A), but not in SW1990 as shown in Figure S1A.
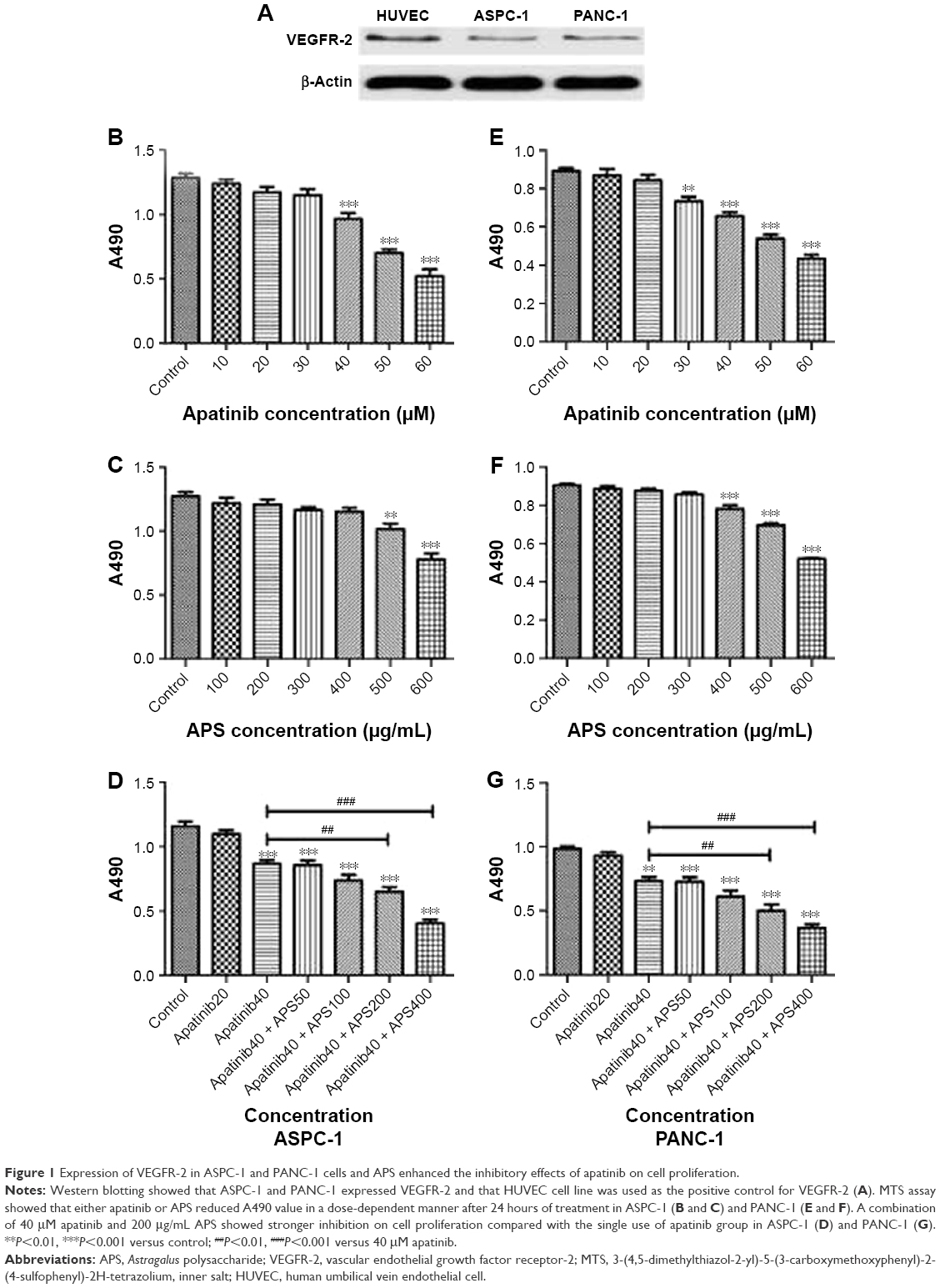 | Figure 1 Expression of VEGFR-2 in ASPC-1 and PANC-1 cells and APS enhanced the inhibitory effects of apatinib on cell proliferation. |
APS enhanced the inhibitory effects of apatinib on cell proliferation
To determine the effects of apatinib and APS on cell proliferation, ASPC-1, PANC-1, and SW1990 were incubated with various concentrations of apatinib and APS for 24 hours. We observed both apatinib and APS reduced A490 value in a dose-dependent manner in ASPC-1 and PANC-1. As shown in Figure 1, Apatinib at 40 and 30 μmol/L (μM), and APS at 500 and 400 μg/mL showed significant inhibitory effects in ASPC-1 (0.97±0.08 and 1.29±0.06, Figure 1B, P<0.001; 1.02±0.07 and 1.27±0.06, Figure 1C, P<0.01) and PANC-1 (0.73±0.04 and 0.89±0.03, Figure 1E, P<0.01; 0.78±0.03 and 0.90±0.02, Figure 1F, P<0.001) proliferation, respectively, compared with the control group. The IC50 dose of apatinib in ASPC-1 and PANC-1 was 51.3 and 47.6 μM and that of APS was 678 and 602 μg/mL, respectively. In SW1990, compared with control group, apatinib reduced A490 value at 60 μM (0.96±0.10 and 1.27±0.13, Figure S1B, P<0.05) with IC50 at 86.4 μM, which was much higher than that of the concentration in ASPC-1 and PANC-1, indicating apatinib-inhibited proliferation more selectively in cells with VEGFR-2 expression. APS at 600 μg/mL showed significant inhibitory effects in SW1990 compared with the control group (0.94±0.16 and 1.15±0.07, Figure S1C, P<0.05) with IC50 at 764 μg/mL. We chose apatinib at 40 μM in combination with APS at 50, 100, 200, and 400 μg/mL to clarify whether APS combined with apatinib could increase the inhibitory effects on cell proliferation. Results showed that 40 μM apatinib combined with 200 μg/mL APS showed stronger inhibition on cell proliferation compared with the single use of apatinib group in ASPC-1 (0.65±0.06 and 0.87±0.05, Figure 1D, P<0.01) and PANC-1 (0.51±0.08 and 0.73±0.05, Figure 1G, P<0.01). Moreover, the combinational group decreased the IC50 to 275.4 and 247.8 μg/mL in ASPC-1 and PANC-1, respectively. Interestingly, the single use of 200 μg/mL APS showed no obvious inhibitory effects on cell proliferation in ASPC-1 (1.21±0.07 and 1.27±0.06, Figure 1C, P>0.05) and in PANC-1 (0.88±0.02 and 0.90±0.02, Figure 1F, P>0.05). In SW1990, 40 μM apatinib combined with 400 μg/mL APS showed stronger inhibition on cell proliferation significantly (0.86±0.07 and 1.07±0.08, Figure S1D, P<0.05). The IC50 dose decreased to 463 μg/mL in the combinational group. Based on the above results, apatinib at 40 μM in combination with 200 μg/mL APS was considered as optimal conditions and was used in the subsequent experiment in ASPC-1 and PANC-1. SW1990 showed no VEGFR-2 expression; hence, this cell was not used in the following assay. Moreover, in this part, rhVEGF-165, as a stimulating factor binding with VEGFR-2, was added in ASPC-1 and PANC-1 to elicit the proliferation of pancreatic cancer cell lines. After 24 hours of incubation, at 20 and 10 μM apatinib reduced A490 value significantly in ASPC-1 (1.03±0.05 and 1.23±0.08, Figure S2A, P<0.05) and PANC-1 (0.82±0.09 and 0.98±0.04, Figure S2B, P<0.05), respectively. The IC50 dose of apatinib in ASPC-1 and PANC-1 was 42.1 and 35.4 μM, which was lower than that of the IC50 dose without rhVEGF-165. However, APS showed the same concentration compared with the groups without rhVEGF-165 (Figure S2C and D). Consistent with the former results, 20 μM apatinib combined with 200 μg/mL APS showed stronger inhibition on cell proliferation compared with the single use of apatinib group in ASPC-1 (0.84±0.06 and 1.02±0.05, Figure S2E, P<0.05) and PANC-1 (0.63±0.08 and 0.85±0.07, Figure S2F, P<0.05).
APS enhanced the inhibition of apatinib on cell migration and invasion
Migration and invasion were closely associated with cancer cell metastasis; hence, we investigated the inhibitory effects of apatinib in pancreatic cancer cells, which have a huge metastatic potential. Migration and invasion properties were assessed by wound-healing and Transwell assays, respectively. As shown in Figure 2, apatinib monotherapy effectively inhibited cell migration in ASPC-1 compared with the control group (0.92±0.02 and 0.79±0.02, Figure 2A and B, P<0.001) and both apatinib (0.25±0.03, P<0.001) and APS (0.83±0.06, P<0.05) monotherapy effectively inhibited cell migration in PANC-1 compared with the control group (0.91±0.09; Figure 2C and D). When apatinib combined with APS, the inhibitory effects were more obvious in ASPC-1 (0.33±0.03 and 0.79±0.02; Figure 2A and B, P<0.001) than in PANC-1 (0.23±0.02 and 0.25±0.03, Figure 2C and D, P>0.05). We also observed apatinib-inhibited cell invasion through Transwell assay in ASPC-1 (46±4.6 and 66±6.0, Figure 3A and B, P<0.01) and PANC-1 (36±5.0 and 80±4.5, Figure 3C and D, P<0.001) compared with the control group. Meanwhile, APS further promoted the inhibitory effects of apatinib on cell invasion in ASPC-1 (34±4.2 and 46±4.6, Figure 3A and B, P<0.05) and PANC-1 (27±5.6 and 36±5.0, Figure 3C and D, P<0.05). To confirm the above results, MMP-9 protein, which is closely related to cell migration and invasion, was detected by Western blotting. As shown in Figure 3E, apatinib was found to inhibit MMP-9 expression significantly in ASPC-1 and PANC-1 cells after 24 hours of treatment, and when apatinib was combined with APS, further inhibition in MMP-9 expression was observed (Figure 3E, P<0.05).
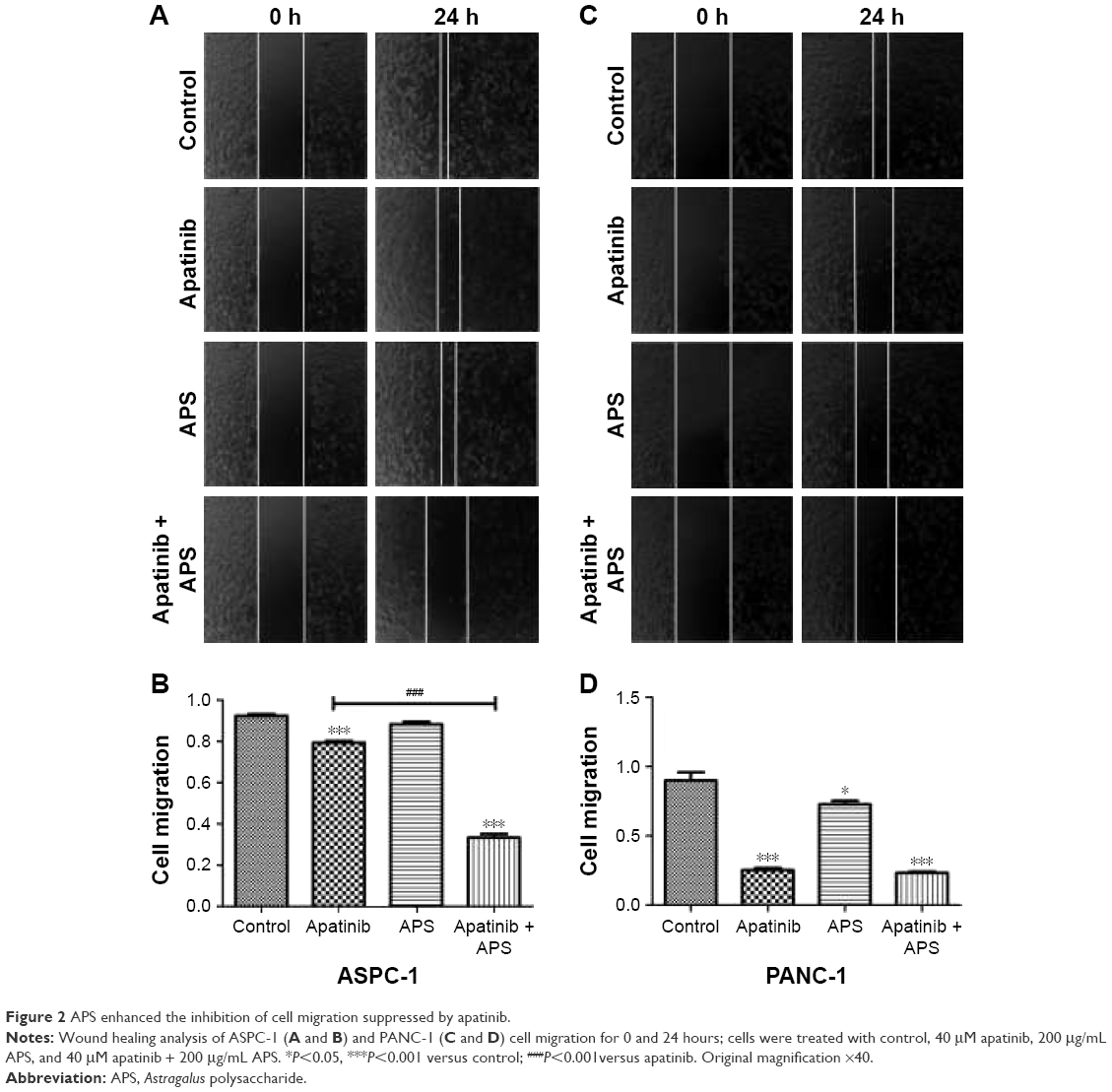 | Figure 2 APS enhanced the inhibition of cell migration suppressed by apatinib. |
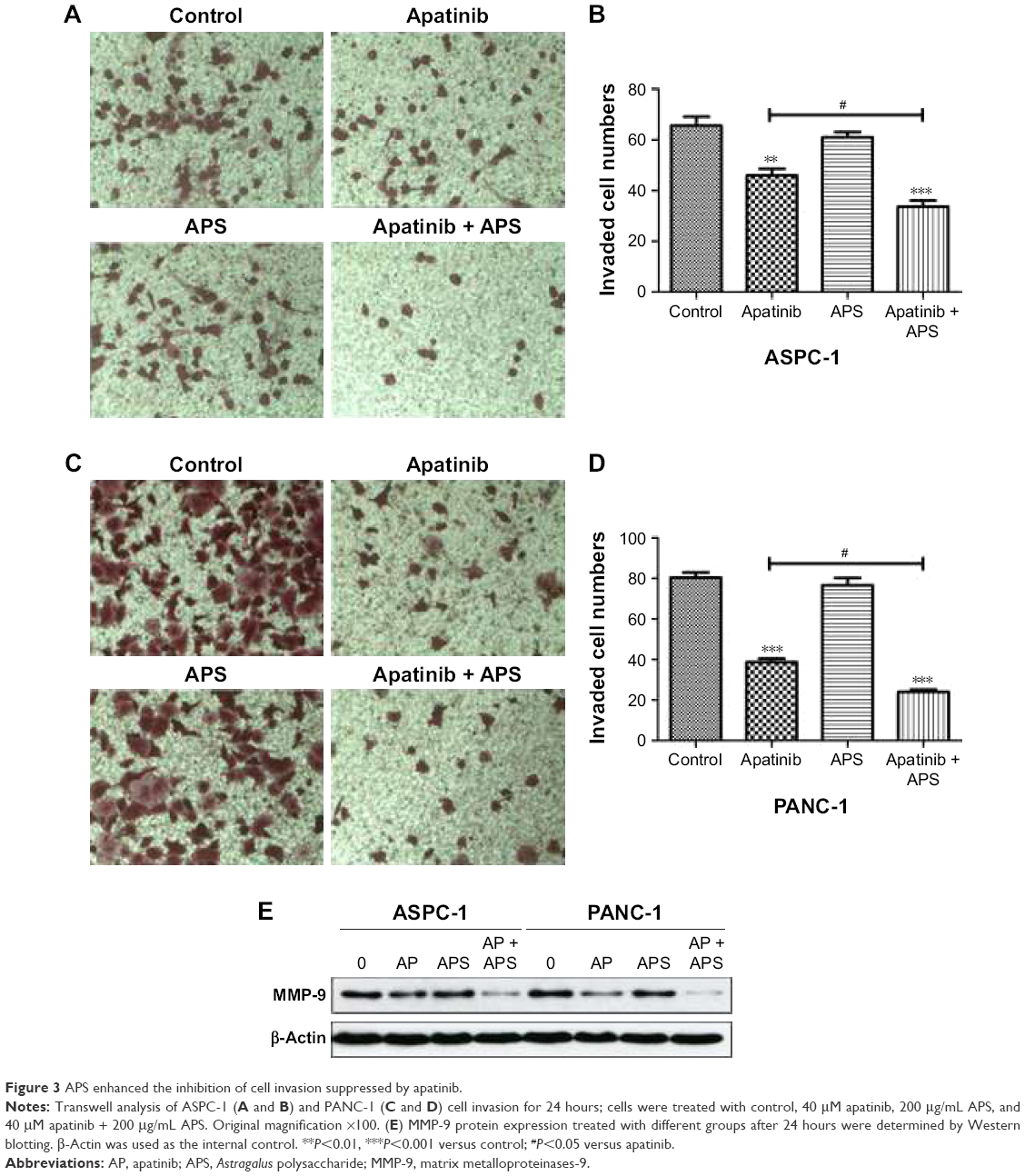 | Figure 3 APS enhanced the inhibition of cell invasion suppressed by apatinib. |
APS enhanced cell apoptosis induced by apatinib
To determine whether apatinib could induce apoptosis and also clarify that APS enhanced the apoptosis induced by apatinib in pancreatic cancer cells, Annexin V and PI staining assays were used to detect apoptosis percentage of cells undergoing various treatments after 24 hours and then results were analyzed by flow cytometry. As shown in Figure 4, apatinib increased apoptosis percentage in ASPC-1 (19.2%±1.97% [Q2: 11.5±1.67; Q4: 7.7±1.84] and 8.3%±1.60% [Q2: 5.1±2.14; Q4: 3.2±1.73], P<0.001, Figure 4A and B) and PANC-1 (17.2%±1.23% [Q2: 12.4±1.87; Q4: 4.8±1.94] and 8.7%±1.05% [Q2: 5.8±1.45; Q4: 2.9±2.12], P<0.001, Figure 4C and D) compared with the control group, and we also found that APS significantly enhanced apoptosis rate induced by apatinib in ASPC-1 (33.7%±2.61% [Q2: 22.9±2.09; Q4: 10.8±1.93] and 19.2%±1.97% [Q2: 11.5±1.67; Q4: 7.7±1.84], P<0.001, Figure 4A and B) and PANC-1 (20.7%±1.91% [Q2: 16.4±1.75; Q4: 4.3±1.97] and 17.2%±1.23% [Q2: 12.4±1.87; Q4: 4.8±1.94], P<0.05, Figure 4C and D). The single use of 200 μg/mL APS did not increase apoptosis percentage in ASPC-1 (10.8%±1.49% [Q2: 7.6±1.84; Q4: 3.2±2.14] and 8.3%±1.0% [Q2: 5.1±2.14; Q4: 3.2±1.73], P>0.05, Figure 4A and B) and PANC-1 (9.1%±1.38% [Q2: 6.4±1.85; Q4: 2.7±2.23] and 8.7%±1.05% [Q2: 5.8±1.45; Q4: 2.9±2.12], P>0.05, Figure 4C and D) compared with the control group. Consistent with the trend of apoptosis rate assayed by flow cytometry, Western blotting analysis showed decreased expression of antiapoptotic protein (Bcl-2) in apatinib-treated group, and Bcl-2 expression was decreased further significantly in the apatinib + APS-treated group (Figure 4E, P<0.05). Correspondingly, the expression of proapoptotic protein (Bax) was increased in apatinib-treated group, and the elevation was more noticeable in the combination group in both ASPC-1 and PANC-1 (Figure 4E, P<0.05). To determine the molecular mechanisms of APS exerting the enhanced antitumor effects, we examined the key signaling molecules of AKT and ERK signaling pathway by Western blotting. After 24 hours of treatment, the expression of phosphorylated AKT (p-AKT) and p-ERK, which represent the activity of AKT and ERK signaling pathway, were inhibited noticeably by 40 μM apatinib (Figure 5, P<0.05), but 200 μg/mL APS-treated group singly did not influence its expression markedly (P>0.05). The combination of apatinib and APS exerted enhanced effects on p-AKT and p-ERK inhibition in ASPC-1 and PANC-1 compared with the apatinib-treated group (Figure 5, P<0.05).
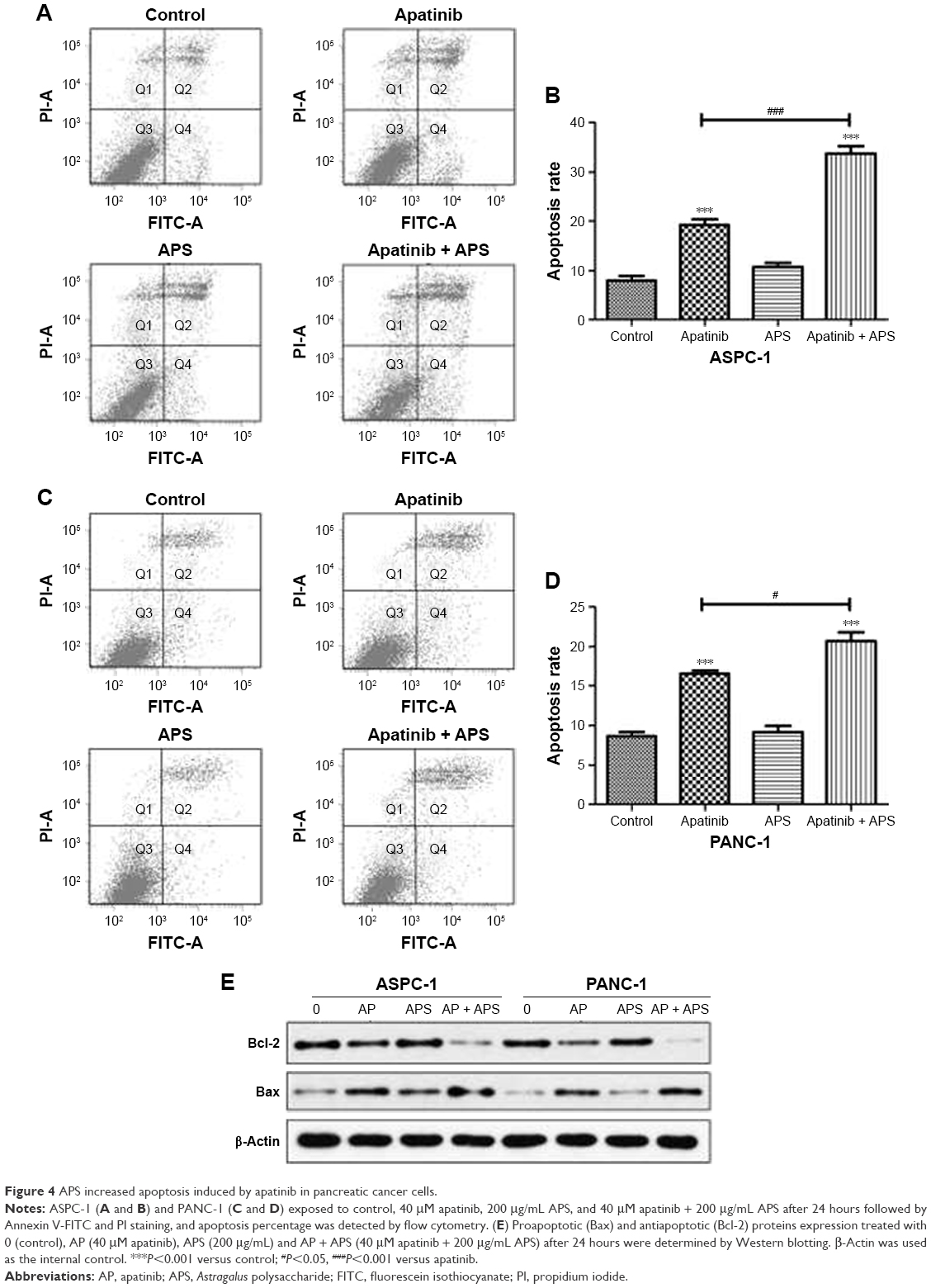 | Figure 4 APS increased apoptosis induced by apatinib in pancreatic cancer cells. |
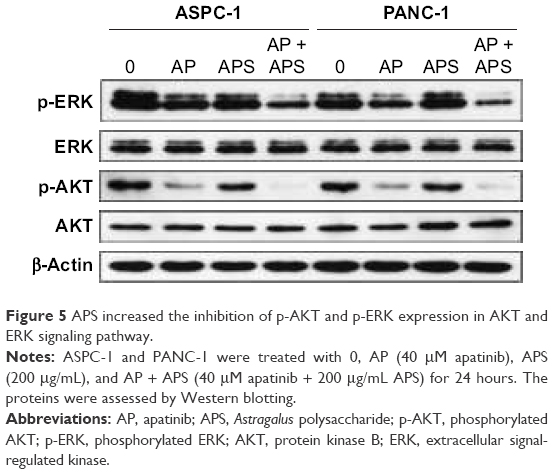 | Figure 5 APS increased the inhibition of p-AKT and p-ERK expression in AKT and ERK signaling pathway. |
Apatinib and APS induced cellular autophagy in pancreatic cancer cells
Autophagy is the process through which cells utilize lysosomal hydrolases to degrade cytoplasmic misfolded proteins and damaged organelles to maintain intracellular homeostasis. In order to learn whether apatinib and APS could change cellular autophagy level, we detected the protein expression of LC3, which is a specific marker of autophagy. As shown in Figure 6, after treatment for 24 hours, apatinib or APS singly increased LC3 expression in ASPC-1 (Figure 6A) and PANC-1 (Figure 6B) in a dose-dependent manner (P<0.05); hence, we concluded that both apatinib and APS induced cellular autophagy. However, compared with apatinib-treated group, LC3 expression was not elevated significantly when 40 μM apatinib was combined with 200 μg/mL APS (Figure 6C, P>0.05).
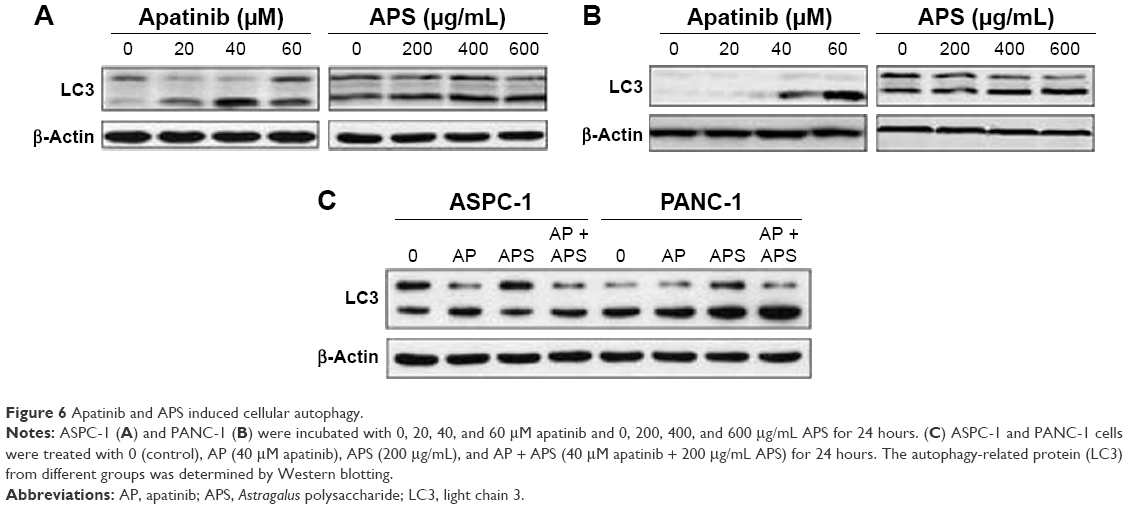 | Figure 6 Apatinib and APS induced cellular autophagy. |
Discussion
Although enormous efforts have been made in the treatment of pancreatic cancer, the overall survival has not improved substantially in the past decades due to its susceptibility to chemotherapy resistance because of the stroma-rich tumor microenvironment, tumor hypoxia, as well as the genetic complexity.18 For example, gemcitabine, the most commonly used chemotherapy drug for pancreatic cancer and the fluorouracil, oxaliplatin, irinotecan, and folinic acid regimen showed modest clinical therapeutic effects with median survival <12 months.19 The unfavorable treatment outcomes necessitate the development of novel strategies for patients with pancreatic cancer. In recent years, molecular targeted therapy showed great potentials in the systemic treatments of human cancers. Unfortunately, a series of targeted agents have failed to demonstrate effectiveness in patients with pancreatic cancer. A meta-analysis of randomized Phase III trials demonstrated that gemcitabine in combination with targeted therapy revealed no significant improvement compared to gemcitabine monotherapy in advanced pancreatic cancer.20 Moreover, VEGF-targeted therapy has not been as successful as expected in pancreatic cancer. The addition of bevacizumab (VEGF monoclonal antibody) to gemcitabine did not improve survival significantly in advanced pancreatic cancer patients.21 Therefore, there is an urgent need to develop effective therapeutic approaches targeting highly heterogeneous pancreatic cancer cells and complicated tumor microenvironments.
Yet, whether apatinib has antitumor effects in pancreatic cancers remains unclear. Now, two case reports of apatinib on human pancreatic cancer showed effectiveness, which provided an additional option for the treatment of pancreatic cancer in the future.22,23 Our study first revealed that apatinib exerted antitumor effects in human pancreatic cancer cells. It was observed that apatinib inhibited cell proliferation, migration, and invasion of ASPC-1 and PANC-1. In SW1990 cells with no VEGFR-2 expression, apatinib inhibited cell proliferation at a higher concentration compared with the concentrations in ASPC-1 and PANC-1, indicating that SW1990 was not sensitive to apatinib mainly targeting VEGFR-2. We believe that perhaps apatinib inhibits cell proliferation in SW1990 by targeting other molecules, such as Ret, c-kit, c-src, and PDGFR.7 We also found that apatinib induced cell apoptosis in ASPC-1 and PANC-1. However, drug resistance is a major obstacle to the successful treatment of cancer, and apatinib will inevitably face the same problem in the future. In recent years, extracts of Chinese herbal medicines have emerged as a promising alternative option to improve the survival time of cancer patients. APS was reported to act as an anticancer agent that might be related to immune function enhancement and apoptosis inducement.14,24 Our study showed that APS produced additive inhibitory effects on cell proliferation by MTS, and we also observed that APS in combination with apatinib showed enhanced inhibitory effects on cell migration and invasion. The relevance of apatinib in combination with APS therapy in vivo is not clear. A study showed that APS injection integrated with cisplatin improved the quality of life in advanced NSCLC patients compared with cisplatin alone.16 A meta-analysis showed that APS + FOLFOX (5-fluorouracil, leucovorin, and oxaliplatin) in patients with gastric cancer was superior in reducing adverse reactions compared with FOLFOX alone.25 Another new study in our laboratory showed that APS enhanced antitumor effects of apatinib in gastric cancer cells.26 Hence, we believe that apatinib + APS combination therapy is a promising regime for pancreatic cancer patients in the future. To elucidate why apatinib and APS have synergetic roles in ASPC-1 and PANC-1, rhVEGF-165 as a stimulating factor was added in the proliferation assay. We observed that the IC50 dose of apatinib decreased, but that of APS showed the same concentration compared with that of the treatment without rhVEGF-165. Hence, we believe that apatinib and APS may be targeting different receptors on the cancer cell lines. MMP-9 protein is closely associated with cell invasion, angiogenesis, and poor prognosis in cancer patients, playing important roles in key processes involved in metastasis as well as response to therapy.27,28 Western blotting showed that apatinib significantly inhibited the expression of MMP-9 in pancreatic cancer cells which could be further inhibited by the administration of APS. Therefore, we concluded that apatinib inhibited cell migration and invasion by inhibiting MMP-9 expression, and we inferred that APS potentiated the inhibitory effects of apatinib on cell migration and invasion. Flow cytometry assay showed that apatinib combined with APS increased apoptosis rate compared with apatinib monotherapy in ASPC-1 and PANC-1. In accord with the results of flow cytometry, we observed that decreased expression of the antiapoptotic protein (Bcl-2) in apatinib-treated group and its expression was further inhibited in apatinib + APS-treated group. Bax, a proapoptotic protein, was found to increase in apatinib-treated group, and its expression was further elevated in the combination group. The above results showed that apatinib induced cell apoptosis and that the apoptosis rate was increased with the addition of APS. It is well recognized that AKT and ERK are important kinases activated by many cellular stimuli, and both of them participate in the regulation of numerous cellular functions including proliferation, migration, and apoptosis. Disturbed activation of AKT and ERK pathway is associated with many human cancers.29 Over activation of p-AKT and p-ERK could activate Bcl-2 to prevent apoptosis and enhance proliferation and migration.29,30 In our study, we found that apatinib suppressed the expression of p-AKT and p-ERK and that the inhibitory effect was more evident after the addition of APS. The above findings clarified the mechanisms that APS enhanced the antitumor effects of apatinib. It has been identified that KRAS mutation is a well-known dominant driver signaling molecule in pancreatic cancer.31 In our study, APS and apatinib seem to inhibit the downstream effectors of RAS in the Ras/Raf/MEK/ERK signaling pathways. However, the mechanisms of how VEGFR-2 and KRAS interact remain to be elucidated in pancreatic cancer cells. A review showed that VEGF activated protein kinase C (PKC)-dependent, but Ras-independent, Raf-MEK-ERK pathway in endothelial cells.32 In the current study, we believe that VEGF binding to VEGFR-2 activated AKT and ERK signaling pathway to contribute in the survival of pancreatic cancer cells and that apatinib and APS inhibited the signaling pathways. However, the relationship between VEGFR-2 and KRAS in ASPC-1 and PANC-1 is unclear, and we will clarify it in the future. Moreover, we first investigated the changes in autophagy level induced by apatinib and APS in pancreatic cancer cells. Autophagy plays a crucial role in cell homeostasis, adaptation to stress situations, and the regulation of the inflammatory response. Blockade or uncontrolled activation of autophagy is associated with many diseases including cancers. In malignant cancer cells, autophagy was activated to combat adverse microenvironmental conditions such as nutritional deficiency and lack of oxygen to survive. Besides, autophagy could also be induced by chemotherapy and radiation therapy.33 The roles of autophagy investigated in pancreatic cancers have been controversial. Some research studies have considered autophagy as a tumor-promoting mechanism, whereas other studies showed its oncosuppressive functions.34,35 A growing body of evidence indicates that many different types of cancer cells protect themselves against therapeutic agents by inducing autophagy.36 Our study found that both apatinib and APS induced cellular autophagy in a dose-dependent manner. However, when apatinib was combined with APS, the expression of autophagy-related protein (LC3) was not elevated significantly compared with the apatinib-treated group; hence, we speculated that autophagy was not related to the synergetic effects of APS on apatinib in the present study, and the molecular mechanisms of the autophagy change remain to be elucidated in the future.
Conclusion
Our study first demonstrated that apatinib produced significant inhibition on cell proliferation, migration, and invasion in pancreatic cells. Meanwhile, we found that APS enhanced antitumor effects due to the further downregulation of p-AKT, p-ERK, and MMP-9 expression. Our data will provide an important reference for the application of targeted drugs toward pancreatic cancers. But one of the shortcomings in our study is that we observed the effectiveness of apatinib combined with APS in pancreatic cancer cells. The possible therapeutic benefit of apatinib and APS as well as other chemotherapy will be further evaluated in a multicenter clinical trial in the future.
Acknowledgments
We specially thank Zhaoyu Zhong for revising the manuscript. This work was supported by grants from the Beijing Natural Science Foundation (to Bangwei Cao, No 7172081; to Jing Wang, No QN2015-10), the Traditional Chinese Medicine Science and Technology Development Fund Project of Beijing (to Bangwei Cao, No JJ2016-16), and the Research Foundation of Beijing Friendship Hospital (to Jing Wang, No yyqdkt2014-10).
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. | ||
Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. | ||
Kooby DA, Gillespie TW, Liu Y, et al. Impact of adjuvant radiotherapy on survival after pancreatic cancer resection: an appraisal of data from the national cancer data base. Ann Surg Oncol. 2013;20(11):3634–3642. | ||
Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol. 2005;23(5):1011–1027. | ||
Büchler P, Reber HA, Büchler MW, Friess H, Hines OJ. VEGF-RII influences the prognosis of pancreatic cancer. Ann Surg. 2002;236(6):738–749. | ||
Luo J, Guo P, Matsuda K, et al. Pancreatic cancer cell-derived vascular endothelial growth factor is biologically active in vitro and enhances tumorigenicity in vivo. Int J Cancer. 2001;92(3):361–369. | ||
Tian S, Quan H, Xie C, et al. YN968D1 is a novel and selective inhibitor of vascular endothelial growth factor receptor-2 tyrosine kinase with potent activity in vitro and in vivo. Cancer Sci. 2011;102(7):1374–1380. | ||
Li J, Qin S, Xu J, et al. Randomized, double-blind, placebo-controlled phase III trial of apatinib in patients with chemotherapy-refractory advanced or metastatic adenocarcinoma of the stomach or gastroesophageal junction. J Clin Oncol. 2016;34(13):1448–1454. | ||
Zhang H. Apatinib for molecular targeted therapy in tumor. Drug Des Devel Ther. 2015;9:6075–6081. | ||
Peng H, Zhang Q, Li J, et al. Apatinib inhibits VEGF signaling and promotes apoptosis in intrahepatic cholangiocarcinoma. Oncotarget. 2016;7(13):17220–17229. | ||
Peng S, Zhang Y, Peng H, et al. Intracellular autocrine VEGF signaling promotes EBDC cell proliferation, which can be inhibited by apatinib. Cancer Lett. 2016;373(2):193–202. | ||
Xu J, Liu X, Yang S, Zhang X, Shi Y. Apatinib plus icotinib in treating advanced non-small cell lung cancer after icotinib treatment failure: a retrospective study. Onco Targets Ther. 2017;10:4989–4995. | ||
Luo C, Shen J, Ying J, et al. Case report of a KIT-mutated melanoma patient with an excellent response to apatinib and temozolomide combination therapy. Onco Targets Ther. 2017;10:4553–4557. | ||
Huang WH, Liao WR, Sun RX. Astragalus polysaccharide induces the apoptosis of Human hepatocellular carcinoma cells by decreasing the expression of Notch1. Int J Mol Med. 2016;38(2):551–557. | ||
Li J, Bao Y, Lam W, et al. Immunoregulatory and anti-tumor effects of polysaccharopeptide and Astragalus polysaccharides on tumor-bearing mice. Immunopharmacol Immunotoxicol. 2008;30(4):771–782. | ||
Guo L, Bai SP, Zhao L, Wang XH. Astragalus polysaccharide injection integrated with vinorelbine and cisplatin for patients with advanced non-small cell lung cancer: effects on quality of life and survival. Med Oncol. 2012;29(3):1656–1662. | ||
Tian QE, De Li H, Yan M, Cai HL, Tan QY, Zhang WY. Effects of Astragalus polysaccharides on P-glycoprotein efflux pump function and protein expression in H22 hepatoma cells in vitro. BMC Complement Altern Med. 2012;12:94. | ||
Chiaravalli M, Reni M, O’Reilly EM. Pancreatic ductal adenocarcinoma: state-of-the-art 2017 and new therapeutic strategies. Cancer Treat Rev. 2017;60:32–43. | ||
Saridaki Z, Androulakis N, Vardakis N, et al. A triplet combination with irinotecan (CPT-11), oxaliplatin (LOHP), continuous infusion 5-fluorouracil and leucovorin (FOLFOXIRI) plus cetuximab as first-line treatment in KRAS wt, metastatic colorectal cancer: a pilot phase II trial. Br J Cancer. 2012;107(12):1932–1937. | ||
Ottaiano A, Capozzi M, De Divitiis C, et al. Gemcitabine mono-therapy versus gemcitabine plus targeted therapy in advanced pancreatic cancer: a meta-analysis of randomized phase III trials. Acta Oncol. 2017;56(3):377–383. | ||
Kindler HL, Niedzwiecki D, Hollis D, et al. Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: phase III trial of the cancer and leukemia group B (CALGB 80303). J Clin Oncol. 2010;28(22):3617–3622. | ||
Li CM, Liu ZC, Bao YT, Sun XD, Wang L. Extraordinary response of metastatic pancreatic cancer to apatinib after failed chemotherapy: a case report and literature review. World J Gastroenterol. 2017;23(41):7478–7488. | ||
Liang L, Wang L, Zhu P, et al. Apatinib concurrent gemcitabine for controlling malignant ascites in advanced pancreatic cancer patient: a case report. Medicine. 2017;96(47):e8725. | ||
Li Q, Bao JM, Li XL, Zhang T, Shen XH. Inhibiting effect of Astragalus polysaccharides on the functions of CD4+CD25 highTreg cells in the tumor microenvironment of human hepatocellular carcinoma. Chin Med J (Engl). 2012;125(5):786–793. | ||
Zhang D, Zheng J, Ni M, et al. Comparative efficacy and safety of Chinese herbal injections combined with the FOLFOX regimen for treating gastric cancer in China: a network meta-analysis. Oncotarget. 2017;8(40):68873–68889. | ||
Wu J, Yu J, Wang J, et al. Astragalus polysaccharide enhanced antitumor effects of apatinib in gastric cancer AGS cells by inhibiting AKT signalling pathway. Biomed Pharmacother. 2018;100:176–183. | ||
Xu Y, Li Z, Jiang P, et al. The co-expression of MMP-9 and tenascin-C is significantly associated with the progression and prognosis of pancreatic cancer. Diagn Pathol. 2015;10:211. | ||
Candido S, Abrams SL, Steelman LS, et al. Roles of NGAL and MMP-9 in the tumor microenvironment and sensitivity to targeted therapy. Biochim Biophys Acta. 2016;1863(3):438–448. | ||
Asati V, Mahapatra DK, Bharti SK. PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as anticancer agents: structural and pharmacological perspectives. Eur J Med Chem. 2016;109:314–341. | ||
McCubrey JA, Steelman LS, Chappell WH, et al. Role of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007;1773(8):1263–1284. | ||
Mann KM, Ying H, Juan J, Jenkins NA, Copeland NG. KRAS-related proteins in pancreatic cancer. Pharmacol Ther. 2016;168:29–42. | ||
Simons M, Gordon E, Claesson-Welsh L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat Rev Mol Cell Biol. 2016;17(10):611–625. | ||
Galluzzi L, Bravo-San Pedro JM, Demaria S, Formenti SC, Kroemer G. Activating autophagy to potentiate immunogenic chemotherapy and radiation therapy. Nat Rev Clin Oncol. 2017;14(4):247–258. | ||
Gómez VE, Giovannetti E, Peters GJ. Unraveling the complexity of autophagy: potential therapeutic applications in pancreatic ductal adenocarcinoma. Semin Cancer Biol. 2015;35:11–19. | ||
Alderton GK. Autophagy: surviving stress in pancreatic cancer. Nat Rev Cancer. 2015;15(9):513. | ||
Dalby KN, Tekedereli I, Lopez-Berestein G, Ozpolat B. Targeting the prodeath and prosurvival functions of autophagy as novel therapeutic strategies in cancer. Autophagy. 2010;6(3):322–329. |
Supplementary materials
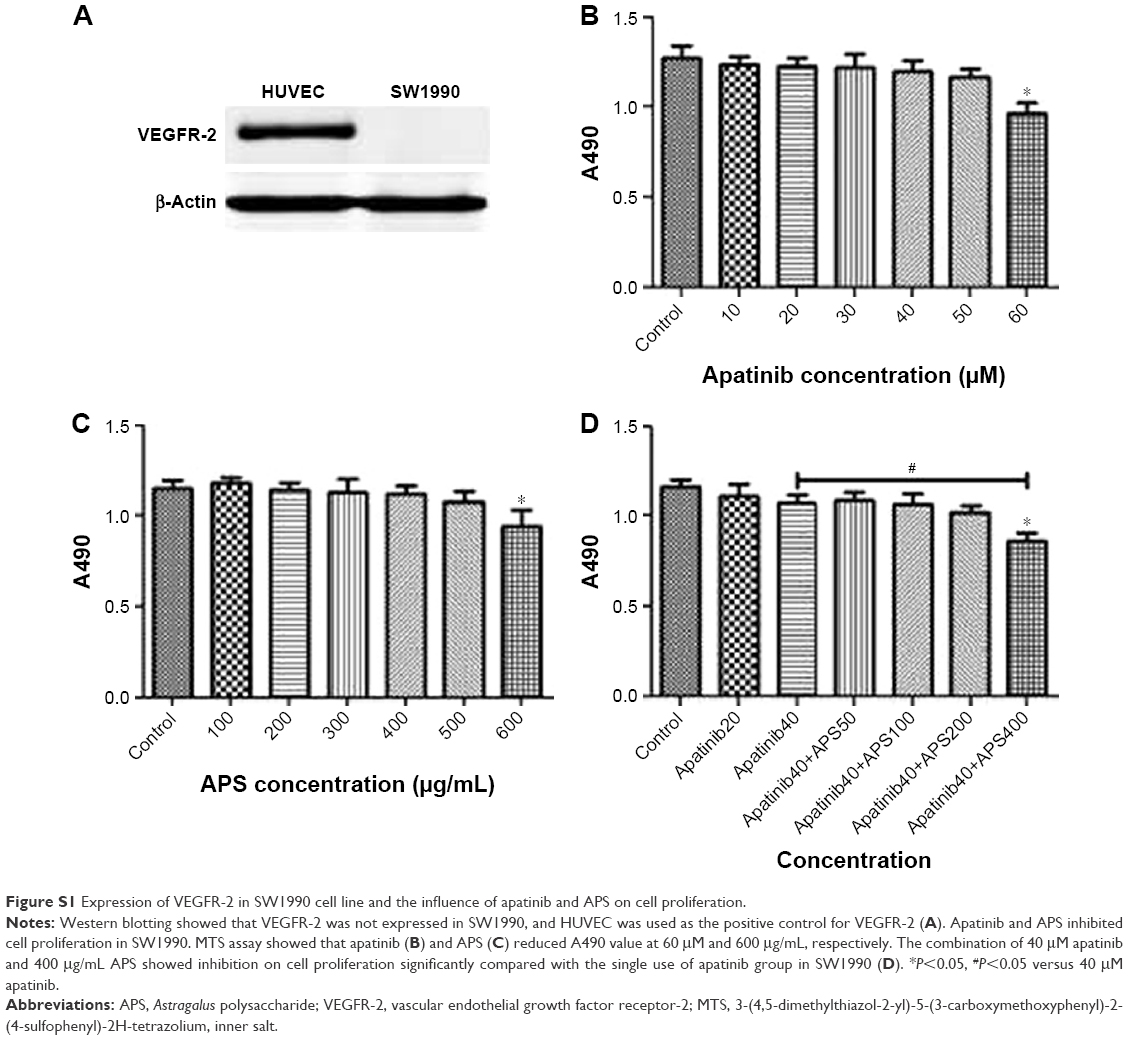 | Figure S1 Expression of VEGFR-2 in SW1990 cell line and the influence of apatinib and APS on cell proliferation. |
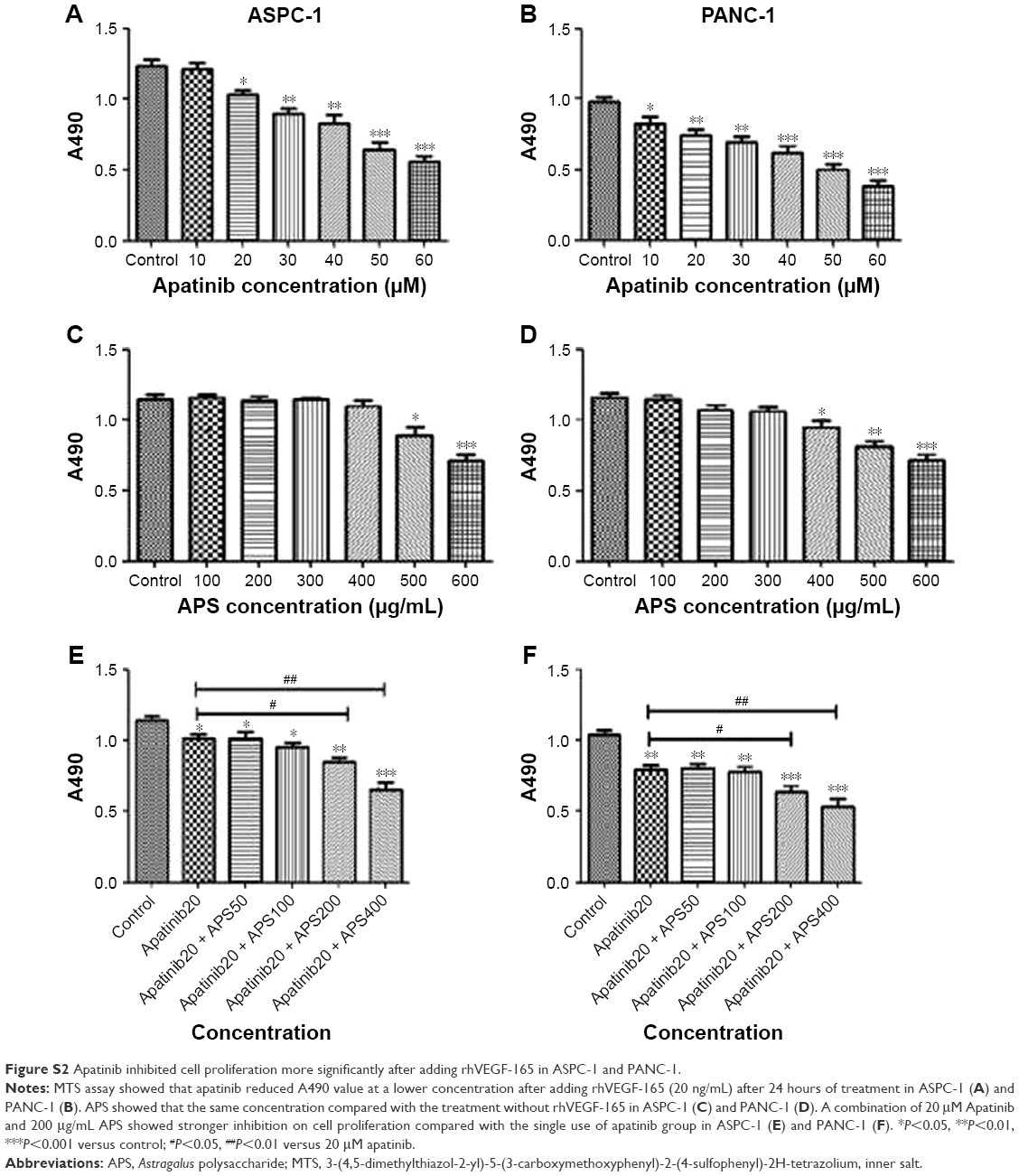 | Figure S2 Apatinib inhibited cell proliferation more significantly after adding rhVEGF-165 in ASPC-1 and PANC-1. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.