Back to Journals » International Journal of Nanomedicine » Volume 16
TPGS2000-DOX Prodrug Micelles for Improving Breast Cancer Therapy
Authors Tang L, Jiang W, Wu L, Yu X, He Z, Shan W, Fu L ![]() , Zhang Z, Zhao Y
, Zhang Z, Zhao Y ![]()
Received 21 August 2021
Accepted for publication 23 November 2021
Published 1 December 2021 Volume 2021:16 Pages 7875—7890
DOI https://doi.org/10.2147/IJN.S335405
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Mian Wang
Lan Tang, 1 Wenhui Jiang, 1 Lan Wu, 1 Xiaolan Yu, 1 Zheng He, 1 Weiguang Shan, 1 Lulu Fu, 1 Zhenhai Zhang, 2 Yunchun Zhao 3
1College of Pharmaceutical Science, Zhejiang University of Technology, Hangzhou, People’s Republic of China; 2Affiliated Hospital of Integrated Traditional Chinese and Western Medicine, Nanjing University of Chinese Medicine, Jiangsu Province Academy of Traditional Chinese Medicine, Nanjing, People’s Republic of China; 3Women’s Hospital, School of Medicine, Zhejiang University, Hangzhou, People’s Republic of China
Correspondence: Zhenhai Zhang
Affiliated Hospital of Integrated Traditional Chinese and Western Medicine, Nanjing University of Chinese Medicine, Nanjing, 210018, People’s Republic of China
Tel +86 13758190403
Email [email protected]
Yunchun Zhao
Women’s Hospital, School of Medicine, Zhejiang University, 1, Xueshi Road, Hangzhou, 310016, People’s Republic of China
Tel +86 15858178673
Email [email protected]
Background: Doxorubicin (DOX) is an anthracycline antibiotic that inhibits the growth of several solid and hematologic malignant tumors. Increasing the targeting ability of DOX and reducing the multi-drug resistance (MDR) of tumor cells to DOX are major aims for researchers.
Purpose: In this study, to increase therapeutic efficiency, reduce the side effects and the MDR of tumor cells to DOX, D-alpha-tocopheryl polyethylene glycol 2000 succinate monoester (TPGS2000)-DOX prodrug micelles were developed by grafting DOX to TPGS2000 via an amide bond that release DOX in the slightly acidic conditions in tumor tissue.
Materials and Methods: The TPGS2000-DOX micelles were constructed using polyethylene glycol 12-hydroxy stearate (Solutol HS15) as the carrier. The in vitro drug release profile and dilution stability of the nanomicelles were determined. The in vitro cytotoxicity and distribution of the nanomicelles in the tumor cells were also investigated. Moreover, we explored the therapeutic outcomes using the MCF-7/ADR tumor-bearing murine model.
Results: The average particle size was approximately 30 nm with a narrow distribution, which was conducive for solid tumor accumulation. The results of in vivo imaging and in vitro cellular uptake assays demonstrated that the TPGS2000-DOX micelles increased the tumor-targeting ability and cellular uptake of DOX. The anticancer potential of TPGS2000-DOX micelles was higher than that of DOX, as revealed by in vitro cytotoxic assays with MCF-7/ADR cells and in vivo antitumor assays with MCF-7 tumor-bearing nude mice.
Conclusion: TPGS2000-DOX prodrug micelles reverse the MDR of tumor cells, achieve passive targeting by forming nanomicelles, and subsequently enhance the efficacy and reduce the toxicity of DOX.
Keywords: doxorubicin, D-alpha-tocopheryl polyethylene glycol 2000 succinate monoester, breast cancer, multi-drug resistance, polymer prodrug nanomedicine
Introduction
Cancer is a serious threat to human health and compromises the quality of life of affected individuals. Breast cancer is the most common type of cancer among women, affecting one in eight women on average.1 It is a malignant tumor that originates from healthy mammary gland cells.2 Chemotherapy has become the standard treatment for malignant tumors.3 Doxorubicin (DOX) is an anthracycline antibiotic that inhibits the growth of several solid and hematologic malignant tumors and is presently one of the most effective antitumor drugs. However, DOX has numerous disadvantages, including a short half-life, cardiotoxicity, multi-drug resistance (MDR), and prolonged administration period.4–6
To increase therapeutic efficiency, reduce the side effects and the MDR of tumor cells to DOX, researchers have focused on nanometer drug delivery systems (NDDSs), including liposomes, nanoparticles, micelles, and dendrimers.7–16 Among these NDDSs, polymer micelles have emerged as a research hotspot in recent years to reverse MDR.17 Polymer micelles of small particle sizes and narrow distribution ranges achieve passive targeting by the enhanced permeability and retention effect (EPR).18,19 The hydrophilic shell in the core-shell structure of polymer micelles can escape recognition by the reticuloendothelial system, thereby prolonging the duration of circulation of the drug.20 Additionally, these micelles allow hydrophobic drugs to enter the core, which increases drug solubility and reduces the side effects of the drug. Furthermore, the P-glycoprotein (P-gp)-induced MDR of tumor cells can be reversibly transformed by polymer micelles prepared using amphiphilic block polymer surfactants, including D-alpha-tocopheryl polyethylene glycol succinate (TPGS), pluronic, and polyesters.21–26 However, traditional NDDSs generally carry drugs by physical embedding. The drugs easily leak from the carrier into the blood following intravenous injection, which leads to premature metabolism of the drug, decreasing the concentration of the drug at the tumor site.
To enhance the stability of NDDSs and prevent the premature release of drugs, several researchers have combined drugs and carriers via covalent bonds using various cross-linking methods. Using appropriate strategies, researchers have developed nano-preparations that utilize the differences between the in vivo environment and tumor environment to specifically release drugs at the tumor site.27,28 The combination of macromolecular precursor drugs and nano-drug delivery systems is a major topic of research. It has been reported that TPGS can be combined with mitoxantrone and gemcitabine to form prodrugs that can assemble into micelles in water.29 These polymer micelle prodrugs have the characteristics of both prodrugs and micelles. The polymer-drug conjugates can alter the biodistribution of the drug, increase its therapeutic effects, and reduce its side effects.30,31
TPGS, a water-soluble derivative of naturally occurring vitamin E, comprises a lipophilic alkyl tail and hydrophilic polar head. TPGS, which is an effective emulsifier owing to its bulky structure and large surface area, enhances the emulsification effect, drug encapsulation efficiency, and therapeutic effects. Thus, TPGS is useful to prepare nanoparticle formulations of anticancer drugs.32–35 Furthermore, TPGS can inhibit the drug efflux of P-gp by competitively inhibiting substrate binding, altering membrane fluidity, and inhibiting the ATPase activity of the outflow pump.36–41 In a previous study, DOX was coupled with TPGS1000 to develop the TPGS-CH=N-DOX prodrug.42 The study demonstrated that the resulting micelles have enhanced long-term stability in the physiological environment, improved tumor accumulation mediated by the EPR effect, and apparent cytotoxicity in DOX-resistant MCF-7/ADR cancer cells, which is mediated via the P-gp inhibitory activity of TPGS.
In this study, we chemically grafted DOX to TPGS2000 via an acid-sensitive amide bond to design a TPGS2000-DOX prodrug that can release DOX in the slightly acidic conditions in tumor tissue. TPGS2000-DOX was assembled into nanomicelles using Solutol HS15 (Figure 1A). We aimed to achieve passive targeting via the ERP effect of the TPGS2000-DOX nanomicelles and reversing the MDR of tumor cells by TPGS2000. We also determined the in vitro drug release profile and dilution stability of the nanomicelles. The in vitro cytotoxicity and distribution of the nanomicelles in the tumor cells were investigated using the DOX-sensitive MCF-7 human breast cancer cell line and DOX-resistant MCF-7 (MCF-7/ADR) cells. The therapeutic outcomes were further studied using the MCF-7/ADR tumor-bearing murine model.
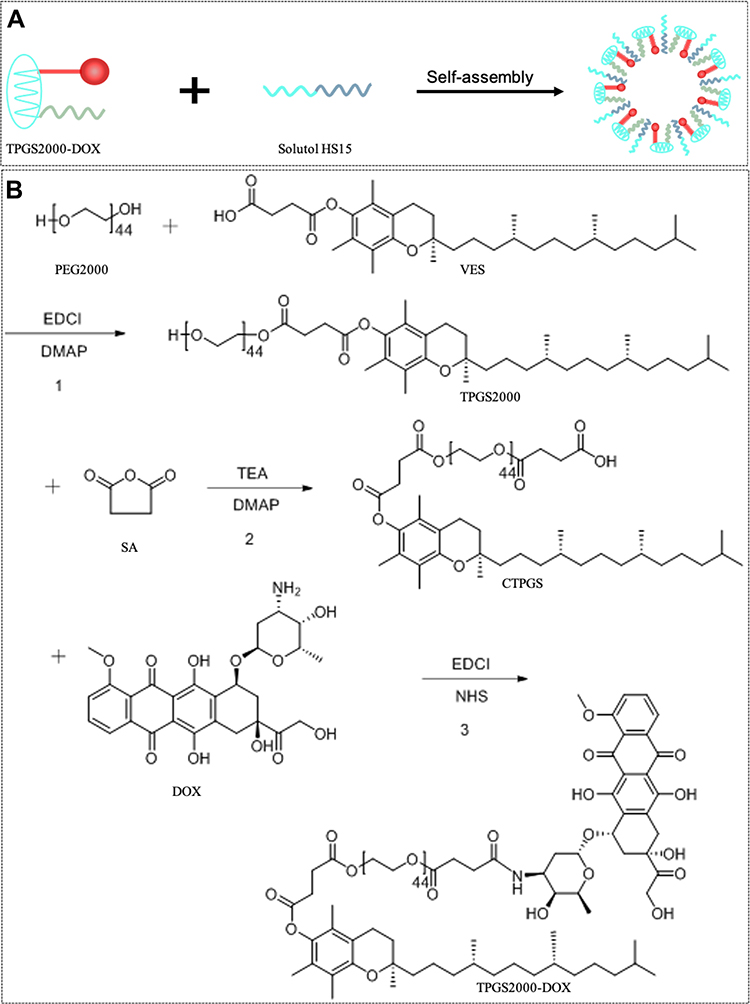 |
Figure 1 (A) The schematic illustration of TPGS2000-DOX micelles. (B) Synthetic pathways in formation of TPGS2000-DOX. |
Materials and Methods
Materials
Chemicals
DOX·HCl was purchased from Dalian Meilun Biological Technology Co., Ltd. (Dalian, China). TPGS2000 was synthesized in our laboratory using Vitamin E succinate (VES) and polyethylene glycol (PEG) 2000 and 1-ethyl-3-(3-dimethyl aminopropyl)-1-carbodiimide hydrochloride (EDCI), 4-dimethylpyridine (DMAP), and N-hydroxysuccinimide (NHS) were purchased from Shanghai Covalent Chemistry (Shanghai, China). Succinic anhydride (SA) was procured from Aladdin Reagent Co., Ltd. (Shanghai, China). Solutol HS15 was purchased from BASF Corporation (Ludwigshafen, Germany). Triethylamine (TEA), methylene chloride, chloroform, 1,4-dioxane, and anhydrous ethers were procured from Shanghai Ling Feng Chemical Reagent Co., Ltd. (Shanghai, China). RPMI 1640 medium (with penicillin-streptomycin), trypsin, and phosphate-buffered saline (PBS) buffer were purchased from Genome Biomedical Technology Co., Ltd. (Hangzhou, China). Fetal bovine serum (FBS) was procured from Gibco (NY, USA) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was purchased from Sigma (USA). All other reagents were of analytical grade or higher purity.
Cell Lines and Experimental Animals
A549 and MCF-7 cells were obtained from the Cell Bank of the Chinese Academy of Sciences. MCF-7/ADR cells were purchased from Nanjing Keygen Biotechnology Co., Ltd. (Nanjing, China). These cells were cultured with RPMI 1640 medium supplemented with 10% FBS, penicillin (100 U/mL), and streptomycin (100 U/mL) at 37 °C in an atmosphere of 5% CO2.
Specific pathogen-free BALB/c female nude mice, aged 4–6 weeks and weighing 18–22 g, were purchased from Shanghai Silaike Experimental Animal Co., Ltd. (Shanghai, China). All animal studies were approved by the Animal Care and Ethics Committee of Zhejiang Technology University (Permit Number: 20180301019). All animal experiments complied with the Guide for Care and Use of Laboratory Animals by the Institute of Laboratory Animal Research.
Synthesis of TPGS2000-DOX
TPGS2000 is synthesized by esterification of VES and PEG2000. The reaction process was that 5.3 g VES, 3.8 g EDCI and 1.2 g DMAP were mixed and stirred in anhydrous DCM for 1 h in an ice bath. Then 20 g PEG2000 was added and reacted at 30 °C for 48 hours under nitrogen protection. The reaction product was purified by silica gel column chromatography and eluted with chloroform/methanol (20:1) to obtain TPGS 2000. TPGS2000 was activated by SA via a ring-opening reaction in the presence of DMAP to obtain the CTPGS reagent.43 Then, 1 g of CTPGS was allowed to react with 457.2 mg of EDCI and 268.8 mg of NHS in DMF at 0 °C under nitrogen protection for 12 h. Subsequently, 236 mg of DOX·HCl and 200 μL of TEA were allowed to react in DMF at 25 °C under nitrogen protection for 1 h to remove the hydrochloride. The two solutions were mixed and reacted for 48 h at 25 °C under nitrogen protection. The product was initially dialyzed in DMF for 24 h to remove the unconjugated DOX and dialyzed again with distilled water to remove excess reagents (MWCO = 2000 Da). The solution was freeze-dried to obtain 721 mg of red powder. The synthetic pathway was shown in Figure 1B.
Preparation of TPGS2000-DOX Micelles
TPGS2000-DOX micelles were prepared by the thin-film hydration method. Briefly, the TPGS2000-DOX conjugate and Solutol HS15 were dissolved together in ethanol. A rotary evaporator (BUCHI, Switzerland) was used to evaporate the ethanol to form a thin film of TPGS2000-DOX and HS15. Nitrogen gas was subsequently blown over the thin film for 30 min to remove the residual reagents. The film was hydrated with deionized water and stirred for 1 h at 60 °C to obtain a micellar solution. Finally, the micellar solution was centrifuged at 10,000 rpm for 10 min and filtered using cellulose acetate filters (pore size 0.22 μm) to remove the precipitate and obtain the TPGS2000-DOX micellar solution.
Characterization of TPGS2000-DOX Micelles
Morphology and Size
The particle size and zeta potential of the TPGS2000-DOX micelles were analyzed using Malvern Zetasizer Instruments (DelsaNano, Beckman Coulter). The shape and surface morphology of the micelles were observed using a transmission electron microscope (TEM; Japan) following negative staining with a 2% solution of phosphotungstic acid.
In vitro Drug Release Behavior
The dialysis bag diffusion method was employed to study the in vitro DOX release behavior of the TPGS2000-DOX micelles. Briefly, 3 mL of the micelles was sealed into a dialysis bag (MWCO = 2000 Da) and dialyzed with 50 mL of PBS (pH 5.0 or pH 7.4). The experiment was conducted at 37 °C with rotating at 100 rpm. Then, 3 mL of the release medium samples were withdrawn after 0.5, 1, 2, 4, 8, 12, 24, 48, and 72 h and replenished with the same volume of fresh medium. Each measurement was obtained in triplicate. The samples were finally analyzed by a fluorophotometer at λex of 468 nm and λem of 597 nm.
Cellular Studies
Cytotoxicity
In vitro cytotoxicity assays were performed with A549, MCF-7, and MCF-7/ADR cells, to compare the cytotoxicity of the various DOX formulations, using the standard MTT assay. Briefly, A549, MCF-7, and MCF-7/ADR cells were seeded into 96-well microplates individually at a density of 5×104 viable cells/well and incubated overnight with RPMI 1640 culture medium to facilitate cell adherence. The cells were incubated with a series of DOX formulations at different concentrations for 48 h. Then, 20 μL of 5 mg/mL MTT in PBS solution was added to each well, following which the cells were incubated for an additional 4 h. Cells that had been cultured under the same conditions but in the absence of DOX represented the blank control. Following incubation, the MTT solution was replaced with 150 μL of dimethyl sulfoxide and shaken for 10 min to dissolve the crystals. The absorbance of the cell solution was finally measured at 490 nm using a microplate reader (Bio-Rad iMark, USA). The cell survival rate is graphically represented by a histogram.
In vitro Cellular Uptake
The cellular uptake was evaluated by flow cytometry and confocal laser scanning microscopy (CLSM). DOX was detected at an excitation wavelength of 488 nm and an emission wavelength of 593 nm. The MCF-7 and MCF-7/ADR cells were seeded into 6-well plates at a density of 2×105 cells/well and incubated overnight at 37 °C to facilitate adherence. The culture medium was subsequently removed and the cells were washed twice with serum-free RPMI 1640 medium. The MCF-7 and MCF-7/ADR cells were treated with different formulations of DOX containing 2 μg/mL or 4 μg/mL DOX. Following incubation for 1 h or 4 h, the culture medium was removed and the cells were rinsed twice or thrice with PBS (pH = 7.4, 4 °C). After trypsinization, the cells were collected by centrifugation at 1,200 rpm for 5 min. The cellular uptake was finally determined using a flow cytometer (Becton Dickinson, USA).
Cellular Distribution
The MCF-7 and MCF-7/ADR cells were seeded into 6-well plates at a density of 3×105 cells/well and incubated overnight at 37 °C in an incubator with an atmosphere of 5% CO2 to facilitate cell adherence. The cells were rinsed twice with a serum-free RPMI 1640 culture medium and stabilized for 30 min in the incubator. After removing the culture medium, the cells were treated with 5 μg/mL DOX or the TPGS2000-DOX micelles. Following incubation for 4 h, the cells were rinsed twice or thrice with PBS (pH = 7.4, 4 °C) and incubated and stained with 4ʹ,6-diamidino-2-phenylindole (DAPI) (50 μL, 500 ng/mL) for 5 min. The cells were then washed and fixed with 4% paraformaldehyde for 20 min. The stationary liquid was finally removed and the cells were observed by CLSM.
Determination of Apoptosis
The MCF-7/ADR cells were seeded into 6-well plates at a density of 2×105 cells/well and incubated overnight at 37 °C in a humidified atmosphere containing 5% CO2. The cells were subsequently treated with 0.744 μg/mL, 3.72 μg/mL, and 18.6 μg/mL of DOX or 0.2906 μg/mL, 1.453 μg/mL, and 7.265 μg/mL of DOX-loaded micelles for 48 h. After washing twice with ice-cold PBS, the cells were detached using trypsin and resuspended at a density of 5×105 cells/mL. The cell suspensions were kept in the dark during fluorescein isothiocyanate (FITC)-Annexin V and 7-AAD staining for 15 min at 37 °C. Apoptosis was finally determined by flow cytometry. Based on the fluorescence intensity of FITC-Annexin V and 7-amino-actinomycin D (7-AAD), the cells were divided into four quadrants, representing living cells, early apoptotic cells, late apoptotic cells, and necrotic cells.
Cell Cycle Assay
The MCF-7/ADR cells were grown in 6-well plates at a density of 2×105 cells/well and treated with different concentrations of DOX or DOX-loaded micelles for 48 h. The cells were subsequently collected and washed twice with ice-cold PBS. After fixing with 70% (v/v) cold ethanol, the cells were stored at 4 °C for 1 h. The cell suspensions were centrifuged before propidium iodide (PI) staining to remove the ethanol solution. The cell pellets were re-suspended in 1.0 mL of PI staining solution comprising 10 mg/mL PI and 100 mg/mL RNase A and incubated at 37 °C in an incubator for 30 min before analysis by flow cytometry.
In vivo Antitumor Effects
Female nude mice (4–6 weeks) were selected to establish MCF-7 breast cancer xenograft models for investigating the potential in vivo anticancer effects of the TPGS2000-DOX micelles. 1×108 MCF-7 cells suspended in 200 μL PBS were subcutaneously injected at the left flank of each nude mouse. When the tumor volume (V) reached 100–150 mm3 (V =0.5 × length × width2), the mice were randomly assigned into one of three groups (n = 6 per group): saline, DOX, and TPGS2000-DOX micelle (equivalent dose of DOX, 10 mg/kg). The corresponding treatments were administered to each group by tail vein injection on days 1, 3, 5, 7, and 9. The tumor volumes and body weights were measured daily until day 21. The mice were euthanized at the end of the experiment and their tumor weights were recorded.
In vivo Imaging
When the tumor volumes reached 100–150 mm3, the MCF-7 tumor-bearing nude mice were randomly divided into two groups (n = 3 per group). The anesthetized nude mice were then individually administered DOX or the solution of DOX-loaded micelles by tail vein injection at a dose equivalent to 10 mg/kg DOX. The nude mice were optically visualized using an IVIS Lumina XRMS Series III imaging system (PerkinElmer, USA) at 0.5, 1, 2, 4, 6, 8, 12, and 24 h. After 24 h, the mice were sacrificed and the tumors and organs, including the heart, liver, spleen, lungs, and kidneys, were dissected. The in vivo biodistribution, which was related to the fluorescence intensity, was determined by in vitro imaging.
Statistical Analyses
All data are presented as the mean ± standard deviation (SD). The t-test was performed to compare two groups, whereas multiple groups were compared by one-way analysis of variance (ANOVA). P values < 0.05 were statistically significant.
Results
Synthesis and Characterization of the TPGS2000-DOX Conjugate
To prepare the TPGS2000-DOX micelle, the TPGS2000-DOX conjugate was first synthesized by linking TPGS2000 and DOX via an amide bond. In this study, the formation of the conjugate structures was confirmed by hydrogen nuclear magnetic resonance spectroscopy (1H-NMR), Fourier-transform infrared spectroscopy (FT-IR), and mass spectroscopy. As shown in Figure 2D and E, the hydrogen spectrum of CTPGS had multi-peak signals in δH 2.58~2.72, which was the hydrogen signal of succinic anhydride. In addition, it could be found from the mass spectrum that CTPGS had an m/z 2200~3200, which was 100 lager than TPGS2000, which also proved that the succinic aside and TPGS2000 were successfully conjugated. The hydrogen spectrum of the synthesized TPGS2000-DOX conjugate had several characteristic signals of DOX, namely, the δH 7.90 (d, 2H), 7.64 (d, 1H), 5.50 (s, 1H), and 3.99 (s, H) signals. Additionally, the synthesized conjugate had the characteristic PEG δH 3.51 (brs, 176H, OCH2CH2O) peak, indicating that DOX had been successfully linked to TPGS2000 (Figure 2A). The FT-IR spectrum of the TPGS2000-DOX conjugate showed characteristic peaks of an amide-linked structure at 1655.2 cm−1 and 1578.3 cm−1 (Figure 2B). The results confirmed that DOX was successfully linked to the carboxylic group (-COOH) of TPGS2000 via the formation of an amide bond. Furthermore, the analysis of the mass spectra showed that the molecular weight distribution of the TPGS2000-DOX conjugate ranged between 2400 and 3800 Da, with an average relative molecular mass of approximately 3131.1868 Da (Figure 2C). The data obtained by the chemical analyses were in good agreement with the theoretical values, which indicates that the coupling of DOX to TGPS2000 was successful.
 |
Figure 2 (A) 1H-NMR spectrum of TPGS2000-DOX conjugate. (B) FT-IR spectrum of TPGS2000-DOX. (C) Mass spectrum of TPGS2000-DOX. (D) 1H-NMR spectrum of CTPGS. (E) Mass spectrum of CTPGS. |
Preparation and Characterization of TPGS2000-DOX Micelles
TPGS2000-DOX has poor water solubility and cannot spontaneously form micelles. In this study, the low toxicity surfactant, Solutol HS15, was used to prepare the TPGS2000-DOX mixed nanomicelles using the thin-film dispersion method. The size of the particles and the results obtained from the zeta potentiometer analyzer are depicted in Figure 3A and B. The particle size of the drug-loaded micelles was 24.9 ± 0.72 nm and the polydispersity index was 0.259. The zeta potential of the micelles was −2.86 + 0.12 mV.
 |
Figure 3 (A) Zeta potential distribution of TPGS2000-DOX micelles. (B) Size distribution of TPGS2000-DOX micelles. (C) Transmission electron micrographs of TPGS2000-DOX micelles. (D) Drug release of TPGS2000-DOX micelles at pH = 7.4 and pH = 5.0. |
The results of the morphological characterization by TEM (Figure 3C) revealed that the prepared micelles were generally spherical, with a uniform size distribution. The drug-loaded micelles appeared to be small and had a narrow size distribution.
In vitro Drug Release
The conditions of drug release in the blood circulation and the intracellular compartment of the tumor cells were simulated in the in vitro drug release experiments. The drug release of the TPGS2000-DOX micelles was determined using buffer solutions at pH 7.4 and pH 5.0 (Figure 3D). Under the condition of physiological environment pH 7.4, after 96 h, the cumulative release of DOX in micelles is only 20%, indicating that micelles have a slow-release effect and can exist stably in blood circulation. In the pH 5.0 system, red precipitation was observed gradually. As time went by, the precipitation increased, indicating that the micelles could not exist stably for a long time in the pH 5.0 system, and were destroyed to release DOX. The release of DOX could be attributed to the fact that the amide bonds break easily in an acidic condition.
Cellular Uptake of DOX-Loaded Micelles
The cellular uptake of DOX and TPGS2000-DOX micelles was studied using MCF-7 and MCF-7/ADR cells. The relative fluorescence intensity of DOX uptake was studied qualitatively and quantitatively by flow cytometry and CLSM.
Flow cytometry was used to quantitatively evaluate the cellular uptake of DOX and TPGS2000-DOX micelles (Figure 4A and B). Following incubation with MCF-7 cells for 1 h, the fluorescence intensity of DOX was lower than that of the TPGS2000-DOX micelles (P < 0.01). In contrast, the fluorescence intensity of DOX was significantly higher than that of the TPGS2000-DOX micelles following incubation with MCF-7 cells for 4 h (P<0.05). The fluorescence intensity of the DOX group was approximately 1.11-fold higher than that of the TPGS2000-DOX group following incubation with MCF-7/ADR cells for 1 h (P < 0.05). When incubated with MCF-7/ADR cells for 4 h, there was no significant difference in the fluorescence intensity between the DOX group and the TPGS2000-DOX group. The alterations in fluorescence intensity could have been induced by the efflux of DOX by P-gp.
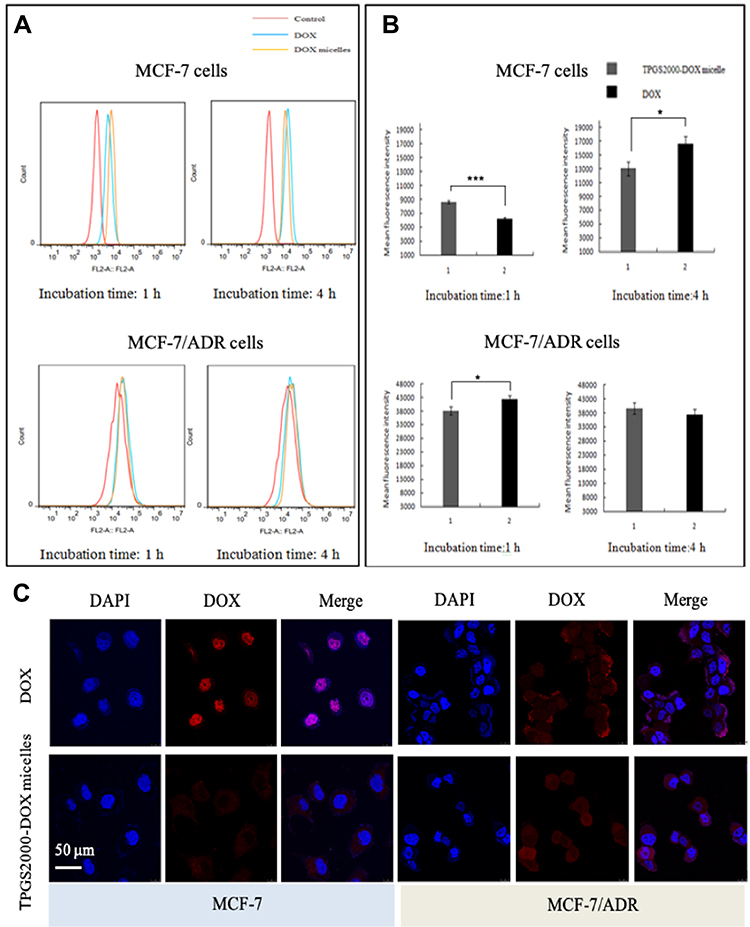 |
Figure 4 In vitro cell uptake studies. (A) CLSM micro images. (B) Flow cytometry analyses (*P < 0.05; ***P < 0.01). (C) The mean fluorescence intensity for cell internalization of free DOX and TPGS2000-DOX micelles after incubation with MCF-7 and MCF-7/ADR cells for 4h. |
To further explore the mechanism of internalization of the drug-loaded micelles, all cells were stained blue with DAPI and studied by CLSM. In MCF-7 cells, the intracellular red fluorescence of the DOX group was high in the DOX channel and the overlapping channels showed distinct purple fluorescence following 4 h of incubation. This indicated that DOX was distributed in the entire intracellular compartment and primarily localized in the nucleus. However, the TPGS2000-DOX micelles were primarily localized in the cytoplasmic compartment, under the same conditions. In the MCF-7/ADR cells, strong red fluorescence was observed on the cell membrane of the DOX group (Figure 4C). It showed that DOX is mainly located on the cell membrane after being effluxed. And in the TPGS2000-DOX micelles, there was no obvious red fluorescence on the cell membrane in the DOX channel, and the overlapping channel showed that the micelles were mainly located in the cytoplasm and were not recognized and discharged by P-gp.
In vitro Cytotoxicity
The in vitro cytotoxicity of the TPGS2000-DOX micelles, TPGS2000-DOX conjugates, DOX, and the blank micelle carriers was determined (Figure 5A–C). The results demonstrated that these substances had inhibitory effects on the growth of the MCF-7, MCF-7/ADR, and A549 cell lines.
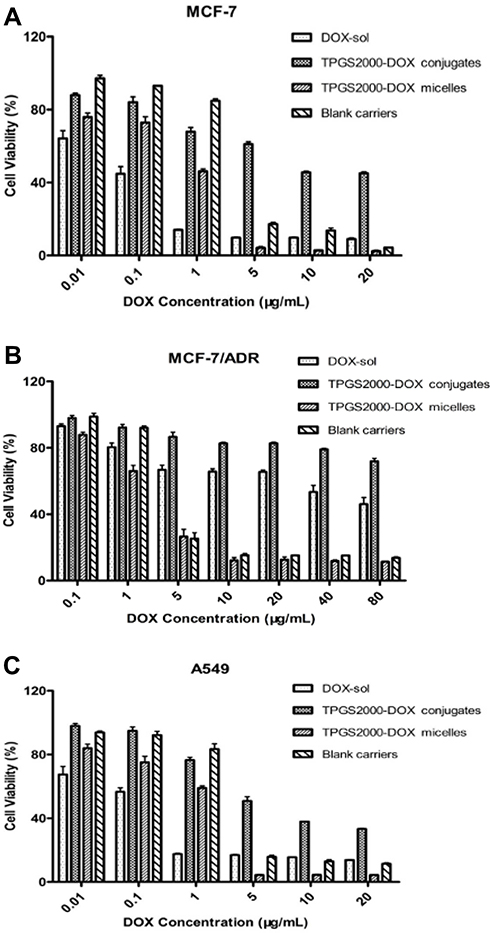 |
Figure 5 The in vitro cell toxicity of DOX-Sol, TPGS2000-DOX conjugates, TPGS2000-DOX micelles and blank carriers against MCF-7 (A), MCF-7/ADR (B) and A549 cells (C). |
The cytotoxicity of the TPGS2000-DOX micelles was higher than that of DOX and conjugated DOX, over a concentration range of 5 to 20 μg/mL, in the MCF-7 (P < 0.05) and A549 cell lines (P < 0.01) (Figure 5A and C). Although the blank carrier control showed some inhibitory effects, the inhibitory effect of the TPGS2000-DOX micelles was significantly greater than that of the blank carrier (concentration of 0.01 to 10 μg/mL: P < 0.01, the concentration of 20 μg/mL: P < 0.05). As shown in Figure 5, the changes in the concentration of blank micelle carrier from 1 μg/mL to 5 μg/mL (concentration of DOX equivalent) had led to a significant reduction in cell viability. The micelle vectors used to prepare the TPGS2000-DOX micelles significantly showed cell cytotoxicity, which might be due to the concentration of vectors, which caused the change of cell osmosis pressure. The TPGS2000-DOX conjugates were less cytotoxic than TPGS2000-DOX micelles because they were administered in the form of a mixed suspension. This led to the conjugates being mostly deposited at the bottom of the plate, which made intracellular entry difficult and subsequently resulted in reduced cytotoxicity.
The resistance of the MCF-7/ADR cell line to DOX was considerably apparent, and cell viability was approximately 50% even when the concentration of DOX was nearly 80 μg/mL (Figure 5B). However, the TPGS2000-DOX micelles were more cytotoxic than DOX, at the same concentration (concentration of 0.1 μg/mL: P < 0.05, concentration of 1 to 80 μg/mL: P < 0.01). Furthermore, the cytotoxicity of the TPGS2000-DOX conjugates to the ADR cell line was significantly lower than that of the DOX (P < 0.01). This could be presumably attributed to the slow rate of release of DOX, and the efficacy of DOX was further reduced owing to efflux by P-gp. In general, although the DOX-loaded micelles are less toxic to cancer cells than DOX, they have better selectivity to protect normal cells from DOX-induced damage.
The half-maximal inhibitory concentration (IC50) values of the different formulations of DOX and the blank micelle carriers were calculated to determine the in vitro antitumor effect in Table 1. The IC50 value of DOX was lower than that of TPGS2000-DOX micelles in normal MCF-7 and A549 cell lines. One possible reason for this observation was that the TPGS2000-DOX micelles primarily entered the cells via endocytosis, which was slower than the passive diffusion of DOX. However, the release rate of the prototype drug, DOX, from the DOX-loaded micelles was slow owing to the mechanism by which DOX was linked during loading. The effective intracellular concentration of the DOX-loaded micelles remained relatively low. The IC50 of the TPGS2000-DOX micelles was significantly lower than that of DOX in the MCF-7/ADR cells. These results, combined with the results provided in Table 2, revealed that the drug resistance index of the TPGS2000-DOX micelles to MCF-7/ADR cells was 4.78, and the reverse drug resistance factor of DOX was 35.76. This indicated that the DOX-loaded micelles effectively reversed the MDR of tumor cells to DOX. Additionally, the IC50 value of the blank micelle carriers was low, which was consistent with the results of the cellular activity inhibition assays.
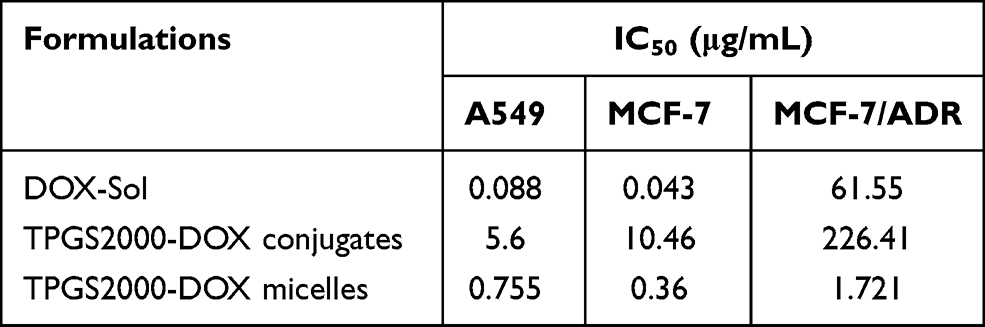 |
Table 1 IC50 Values of Different DOX Formulations and Blank Carriers Against A549, MCF-7 and MCF-7/ADR |
 |
Table 2 The Resistant Index and the Reversal Factor of TPGS2000-DOX Micelles |
TPGS2000-DOX Micelles Induced Apoptosis
The percentages of living cells, early apoptotic cells, late apoptotic cells, and necrotic cells following treatment with different concentrations of DOX and TPGS2000-DOX micelles were different (Figure 6A and B). The results demonstrated that, as the concentration of DOX and DOX-loaded micelles increased, the sum of the percentages of apoptotic and necrotic cells increased accordingly. These observations, together with the inhibitory activity of DOX on MCF-7/ADR cells, indicated that the cytotoxic effect of DOX and DOX-loaded micelles on MCF-7 cells was primarily caused by the induction of apoptosis and necrosis. Additionally, owing to the high expression of P-gp in MCF-7/ADR cells, the TPGS2000-DOX micelles had a greater effect on the induction of apoptosis than DOX (P < 0.01). Moreover, the increase in the rate of apoptosis induced by the TPGS2000-DOX micelles was primarily attributed to late apoptosis. This indicated that the TPGS2000-DOX micelles induced the forward transition from early apoptosis to late apoptosis. As early apoptosis is reversible and late apoptosis is irreversible, it is beneficial to induce apoptosis in tumor cells by promoting the transition from early apoptosis to late apoptosis. The effect was greater when DOX was modified by TGPS and loaded into micelles.
 |
Figure 6 (A) Apoptosis induction detected by flow cytometry with FITC-Annexin V conjugated with 7ADD staining. (B) Graphical representation of apoptosis in MCF-7/ADR cells. (C) Cell cycle distribution. |
Effect on Cell Cycle Arrest
The results of cell cycle analysis by flow cytometry are depicted in Figure 6C. The population distribution of cells in the G0/G1, S, and G2/M phases of the cell cycle was (35.08 ± 3.21) %, (17.26 ± 1.75) %, and (47.66 ± 3.53) %, respectively, in the control group. When these cells were treated with the maximum concentration of DOX, the proportion of cells in the G0/G1 phase decreased significantly ((35.08 ± 3.21) % to (26.51 ± 3.60) %, P < 0.05), whereas that in the G2/M phase increased concomitantly ((47.66 ± 3.53) % to (56.91 ± 2.95) %, P < 0.05). In contrast, when the MCF-7/ADR cells were treated with the maximum concentration of DOX-loaded micelles, the G0/G1 phase was significantly arrested compared with that in the control ((35.08 ± 3.21) % vs (60.46 ± 2.74) %, P < 0.01). These results demonstrated that the proliferation of MCF-7/ADR resistant cells was significantly inhibited at different stages in the groups that were treated with DOX and TPGS2000-DOX micelles. The MCF-7/ADR cells that had been treated with DOX were arrested at the G2/M phase, which indicated that DOX promoted cell differentiation and induced DNA damage in the differentiation phase of the cell cycle. The TPGS2000-DOX micelles prevented the cells from entering the S phases of the cell cycle, where DNA is replicated and the quantity of DNA is doubled. This subsequently resulted in the slower growth rate and decreased proliferative activity of the MCF-7/ADR tumor cells.
In vivo Antitumor Effects
The therapeutic efficacy of the TPGS2000-DOX micelles following intravenous administration was evaluated in nude mice bearing subcutaneous MCF-7 xenografts. The body weights of the saline group and the group that was treated with the TPGS2000-DOX micelles remained relatively stable (P > 0.1) (Figure 7A). However, the bodyweight of the nude mice that were treated with DOX reduced abruptly. Additionally, the quality of the fur was poor and not glossy during treatment, which could be attributed to the severe toxicity and side effects of DOX. The tumor volumes were altered in each group during treatment (Figure 7B). The tumor volumes in the saline control group increased rapidly, whereas tumor growth was inhibited in the other two groups. The average tumor volumes of the groups that were treated with DOX and TPGS2000-DOX micelles were 78.03% and 66.56% (78.03% vs 66.56%, P < 0.05), respectively, at the end of experimentation compared with those of the saline group. The tumor inhibition rates of DOX group (the average tumor weight was 0.84 g) and TPGS2000-DOX micelles (the average tumor weight was 0.65 g) were 27.60% and 43.88% (27.60% vs 43.88%, P < 0.01), respectively. The actual tumor photograph was seen in Figure 7C. The results suggested that the TPGS2000-DOX micelles effectively reduced drug toxicity and significantly improved the antitumor activity.
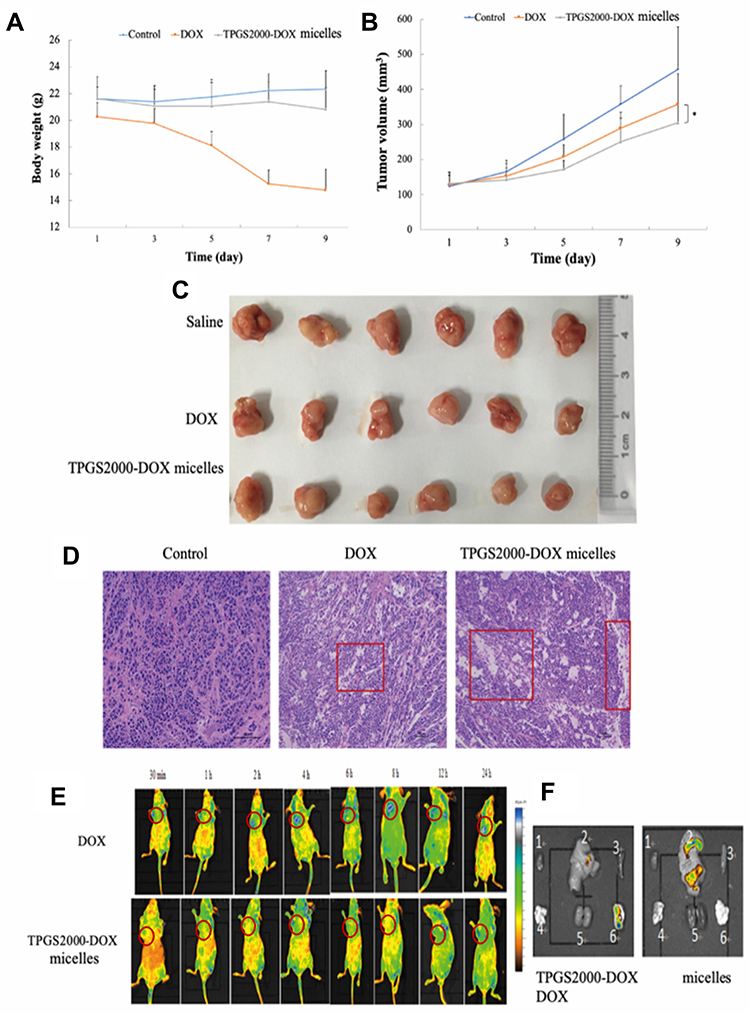 |
Figure 7 The body weight (A) and the tumor volume (B) change of the mice treated with Saline, free DOX and TPGS2000 -DOX micelles over time (*P < 0.05). (C) Photographs of excised tumor weight at the end of the experiment. (D) Images of H&E-stained sections of the tumor collected from mice in the different treatment groups with 100-micron scale rod. (E) In vivo fluorescence image of nude mice after administration of TPGS2000-DOX micelles at 30min, 1h, 2 h, 4 h, 6h, 8 h, 12h and 24 h. (F) Ex vivo DOX fluorescence images of isolated different organs (1- heart, 2-liver, 3-spleen, 4-lung, 5-kidney and 6-tumor). |
Hematoxylin and eosin staining images of the tissue sections showed that the number of tumor cells and tumor histology were significantly altered following treatment. The histological images revealed that the tumor cells in the saline group proliferated more actively than those in the DOX and TPGS2000-DOX groups (Figure 7D). The area of apoptotic and necrotic cells in the tumor sections of the groups that were treated with DOX and TPGS2000-DOX micelles was markedly greater than that of the saline group and was particularly pronounced in the group that was treated with the TPGS2000-DOX micelles. These results were consistent with those of the tumor suppression studies. The results of histological analyses further confirmed that the TPGS2000-DOX micelles could improve the antitumor activity of the drug.
In vivo and in vitro Fluorescence Imaging
An in vivo fluorescence imaging system was used to study the in vivo distribution of DOX and TPGS2000-DOX micelles and the in vitro retention of tissues. In the group that received DOX, fluorescence was observed at the tumor site after 30 min of injection and was distributed throughout the body of the nude mice 8 h after injection (Figure 7E). It was also observed that the fluorescence intensity altered irregularly over time and DOX was either easily dispersed to the other tissues or excreted following metabolism, which confirmed that the tumor-targeting activity of DOX was weak. The fluorescence of the TPGS2000-DOX micelles was localized at the tumor site 2 h after injection and the intensity reached a peak of 4 h after injection. This phenomenon was attributed to the fact that nano-sized micelles can easily permeate into leaky blood vessels and are retained in the tumor site via the EPR effect.6,7 As TPGS2000-DOX micelles are gradually metabolized, the intensity of fluorescence decreases at a corresponding rate. The results of in vivo fluorescence imaging demonstrated that the tumor localization of the TPGS2000-DOX micelles was more pronounced than that of DOX, indicating a certain degree of tumor-targeting of the TPGS2000-DOX micelles.
To further investigate the distribution of DOX and TPGS2000-DOX micelles in the organs and tumors, all tumors and major organs were excised 24 h after injection for further in vitro experiments and fluorescence imaging (Figure 7F). The fluorescence intensity indicated that the accumulation of TPGS2000-DOX micelles in the tumors was greater than that of DOX. The results further confirmed that the duration of the circulation of the TPGS2000-DOX micelles was prolonged, and their accumulation at the tumor site was augmented, compared with DOX. In addition to the tumors, the hepatic tissues exhibited fluorescence in both the DOX and TPGS2000-DOX groups, and the liver tissues of the DOX group exhibited a stronger fluorescence intensity than those of the TPGS2000-DOX group. This could be attributed to the centralization and metabolism of drugs at high concentrations in the liver. This result also demonstrated that the TPGS2000-DOX micelles could target tumor sites and reduce the concentration of the drug in the normal tissues, indicating that the TPGS2000-DOX micelles maximize therapeutic efficacy and reduce the toxic side effects of DOX.
Discussion
DOX has a strong antitumor activity, however, the MDR of tumor cells induced by the long-term administration of DOX affects its efficacy. The preparation of DOX prodrug micelles can reverse the MDR of tumors via the use of appropriate carrier materials. Although the TPGS2000-DOX prodrug can ameliorate the MDR of tumor cells to DOX, it has poor water solubility and is difficult to administer clinically. In this work, in addition to enhancing the water solubility of TPGS2000-DOX, Solutol HS15 also can reverse the MDR of tumors.44 According to reports, mixed nanomicelles that carried a positive charge on the surface could easily interact with the proteins in the blood serum, thereby reducing the efficacy of the drug.45 The TPGS2000-DOX micelle carries a negative charge, which ensures its stability in the blood. Additionally, particles with sizes of 10–200 nm can gather in solid tumors using the strong penetration and interception effect of tumor tissues and play a passive targeting role.46–48 In the in vitro drug release experiments of TPGS2000-DOX micelles, the DOX prototype drug dissociates from the micelles at pH 5.0. This indicated that TPGS2000-DOX was prone to hydrolysis and cleavage under acidic conditions owing to the presence of an amide bond. This implied that the DOX-loaded micelles became unstable in the intracellular compartment of tumor cells, where the loaded DOX could be effectively released. In summary, the results suggested that the TPGS2000-DOX conjugate existed stably in the weakly alkaline environment of normal tissues and could accumulate in the tumor site via the EPR effect, where the loaded DOX was effectively released in the acidic environment of tumor cells as pH-sensitive micelles reported.49,50 The mechanisms of internalization of the drug-loaded micelles had been explored by flow cytometry and CLSM. First, both assays show that the tumor cells MCF-7 and MCF-7/ADR had the efflux of DOX. It was also observed that the intracellular accumulation of DOX was high, which explained the corresponding increase in fluorescence intensity when the duration of incubation was increased. However, in the MCF-7/ADR cell line, with high P-gp expression, the fluorescence intensity treated with DOX decrease significantly as the duration of incubation was increased, owing to the efflux of DOX by P-gp. In contrast, the fluorescence intensity of the group that was treated with TPGS2000-DOX micelles remained considerably high over time, as the TPGS2000-DOX micelles could bypass recognition by P-gp and enter the tumor cells. As the micelles were primarily internalized into the cells by endocytosis, the TPGS2000-DOX micelles were largely distributed in the cytoplasm, which was consistent with the results observed in CLSM herein. These results, combined with those of in vitro drug release, revealed that the release of DOX continued at a low rate after 4 h of incubation. The accumulation of DOX on the cell membrane could be attributed to the efflux of DOX by P-gp before nuclear entry. DOX was able to enter the ADR tumor cells that were treated with the TPGS2000-DOX micelles and were well-dispersed throughout the intracellular compartment owing to its inhibitory effect on P-gp. These results confirm that the micelles are primarily internalized into the tumor cells by endocytosis. These results reveal that the TPGS2000-DOX micelles can effectively evade efflux by P-gp and reverse the MDR of tumor cells to DOX.
It is worth pondering that the blank carriers also show a certain degree of toxicity to cells. Previous studies have demonstrated that Solutol HS15 is not merely an inert compound, and may reverse ADR and alter the permeability of the cell membrane.51,52 The cytotoxicity of the blank vectors could be attributed to the fact that high concentrations of lipids alter the permeability of the cell membrane, which can subsequently lead to cell death. Therefore, the carrier materials used to prepare the micelles might confer better biosecurity at low concentrations than that at high concentrations.
Conclusion
In this study, we designed and synthesized a DOX prodrug, TPGS2000-DOX, by directly conjugating the hydrophobic DOX to a very short chain PEG, TPGS2000, which was synthesized using PEG2000 and VES. The activated TPGS2000 was subsequently conjugated to DOX via an amide bond to form the TPGS2000-DOX prodrug. A mixed micelle, comprising TPGS2000-DOX and Solutol HS15, was developed to prepare particles of appropriate size, good uniformity, and stability. The results of in vivo imaging demonstrated that the TPGS2000-DOX micelles could effectively target DOX to the tumor site. The TPGS2000-DOX micelles were relatively stable in PBS at pH 7.4 without any burst release. It was observed that the TPGS2000-DOX micelles bypassed the P-gp-mediated efflux of DOX, leading to the accumulation of DOX in the cytoplasm of resistant MCF-7/ADR cells. Additionally, the results of in vitro antitumor assays demonstrated that the cytotoxicity of the TPGS2000-DOX micelles was significantly higher than that of DOX. The results of in vivo tumor suppression in nude mice bearing MCF-7 tumors suggested that the therapeutic efficacy of the TPGS2000-DOX micelles was greater than that of DOX and could achieve better safety based on the changes in body weights. However, there was no significant difference between the tumor volumes of TPGS2000-DOX micelles and DOX groups. This could be primarily attributed to the fact that TPGS2000 was directly coupled to DOX to form the TPGS2000-DOX micelle, which might have suppressed the effective release of DOX at the tumor site.
Acknowledgments
This research was funded by the National Natural Science Foundation of China [Grant No. 81403117 and 81802630] and the Public welfare project of Zhejiang Science and Technology Department [Grant No.GF20H280010].
Disclosure
The authors report no conflicts of interest in this work.
References
1. Alphandéry E. Perspectives of breast cancer thermotherapies. J Cancer. 2014;5(6):472–479. doi:10.7150/jca.8693
2. Avazzadeh R, Vasheghani-Farahani E, Soleimani M, Amanpour S, Sadeghi M. Synthesis and application of magnetite dextran-spermine nanoparticles in breast cancer hyperthermia. Prog Biomater. 2017;6(3):75–84. doi:10.1007/s40204-017-0068-8
3. O’Brien ME, Borthwick A, Rigg A, et al. Mortality within 30 days of chemotherapy: a clinical governance benchmarking issue for oncology patients. Br J Cancer. 2006;95(12):1632–1636. doi:10.1038/sj.bjc.6603498
4. Thorn CF, Oshiro C, Marsh S, et al. Doxorubicin pathways: pharmacodynamics and adverse effects. Pharmacogenet Genomics. 2011;21(7):440–446. doi:10.1097/FPC.0b013e32833ffb56
5. Eom YW, Kim MA, Park SS, et al. Two distinct modes of cell death induced by doxorubicin: apoptosis and cell death through mitotic catastrophe accompanied by senescence-like phenotype. Oncogene. 2005;24(30):4765–4777. doi:10.1038/sj.onc.1208627
6. Broxterman HJ, Gotink KJ, Verheul HM. Understanding the causes of multidrug resistance in cancer: a comparison of doxorubicin and sunitinib. Drug Resist Updat. 2009;12(4–5):114–126. doi:10.1016/j.drup.2009.07.001
7. Schilt Y, Berman T, Wei X, Barenholz Y, Raviv U. Using solution X-ray scattering to determine the high-resolution structure and morphology of PEGylated liposomal doxorubicin nanodrugs. Biochim Biophys Acta. 2016;1860(1):108–119. doi:10.1016/j.bbagen.2015.09.012
8. Kubo T, Sugita T, Shimose S, et al. Targeted systemic chemotherapy using magnetic liposomes with incorporated Adriamycin for osteosarcoma in hamsters. Int J Oncol. 2001;18(1):121–125. doi:10.3892/ijo.18.1.121
9. Wu J, Lu Y, Lee A, et al. Reversal of multidrug resistance by transferrin-conjugated liposomes co-encapsulating doxorubicin and verapamil. J Pharm Pharm Sci. 2007;10(3):350–357.
10. Prabaharan M, Grailer JJ, Pilla S, Steeber DA, Gong S. Gold nanoparticles with a monolayer of doxorubicin-conjugated amphiphilic block copolymer for tumor-targeted drug delivery. Biomaterials. 2009;30(30):6065–6075. doi:10.1016/j.biomaterials.2009.07.048
11. Wang F, Wang YC, Dou S, et al. Doxorubicin-tethered responsive gold nanoparticles facilitate intracellular drug delivery for overcoming multidrug resistance in cancer cells. ACS Nano. 2011;5(5):3679–3692. doi:10.1021/nn200007z
12. Tian Y, Guo R, Jiao Y, et al. Redox stimuli-responsive hollow mesoporous silica nanocarriers for targeted drug delivery in cancer therapy. Nanoscale Horiz. 2016;1(6):480–487. doi:10.1039/C6NH00139D
13. Shi C, Guo X, Qu Q, et al. Actively targeted delivery of anticancer drug to tumor cells by redox-responsive star-shaped micelles. Biomaterials. 2014;35(30):8711–8722. doi:10.1016/j.biomaterials.2014.06.036
14. Guo X, Shi C, Wang J, Di S, Zhou S. pH-triggered intracellular release from actively targeting polymer micelles. Biomaterials. 2013;34(18):4544–4554. doi:10.1016/j.biomaterials.2013.02.071
15. Wei T, Chen C, Liu J, et al. Anticancer drug nanomicelles formed by self-assembling amphiphilic dendrimer to combat cancer drug resistance. Proc Natl Acad Sci USA. 2015;112(10):2978–2983. doi:10.1073/pnas.1418494112
16. Chen K, Liao S, Guo S, et al. Multistimuli-responsive PEGylated polymeric bioconjugate-based nano-aggregate for cancer therapy. Chem Eng J. 2020;391:123543. doi:10.1016/j.cej.2019.123543
17. Chen Z, Zhang W, Yuping LI, Wang X. Progress on nano-sized drug delivery system reversing tumor and multidrug resistance of osteosarcoma. J Pharmaceut Pract. 2016;34:103–105.
18. Mikhail AS, Allen C. Block copolymer micelles for delivery of cancer therapy: transport at the whole body, tissue and cellular levels. J Control Release. 2009;138(3):214–223. doi:10.1016/j.jconrel.2009.04.010
19. Nishiyama N, Kataoka K. Current state, achievements, and future prospects of polymeric micelles as nanocarriers for drug and gene delivery. Pharmacol Ther. 2006;112(3):630–648. doi:10.1016/j.pharmthera.2006.05.006
20. Wu Y, Li F, Zhang X, et al. Tumor microenvironment-responsive PEGylated heparin-pyropheophorbide-a nanoconjugates for photodynamic therapy. Carbohydr Polym. 2021;255:117490. doi:10.1016/j.carbpol.2020.117490
21. Chavanpatil MD, Khdair A, Gerard B, et al. Surfactant-polymer nanoparticles overcome P-glycoprotein-mediated drug efflux. Mol Pharm. 2007;4(5):730–738. doi:10.1021/mp070024d
22. Qiu L, Qiao M, Chen Q, et al. Enhanced effect of pH-sensitive mixed copolymer micelles for overcoming multidrug resistance of doxorubicin. Biomaterials. 2014;35:9877–9887.
23. Fan Z, Chen C, Pang X, et al. Adding vitamin E-TPGS to the formulation of Genexol-PM: specially mixed micelles improve drug-loading ability and cytotoxicity against multidrug-resistant tumors significantly. PLoS One. 2015;10:120–129.
24. Danson S, Ferry D, Alakhov V, et al. Phase I dose escalation and pharmacokinetic study of pluronic polymer-bound doxorubicin (SP1049C) in patients with advanced cancer. Br J Cancer. 2004;90(11):2085–2091. doi:10.1038/sj.bjc.6601856
25. Wu W, Guan Z. Docetaxel-loaded mixed micelles composed of Solutol HS15 and Pluronic F127 or folate-conjugated F127: preparation, optimization and in vitro comparative characterization. J Chin Pharm Sci. 2015;24(2):95–103. doi:10.5246/jcps.2015.02.011
26. Xiao L, Xiong X, Sun X, et al. Role of cellular uptake in the reversal of multidrug resistance by PEG-b-PLA polymeric micelles. Biomaterials. 2011;32(22):5148–5157. doi:10.1016/j.biomaterials.2011.03.071
27. O’Reilly RK, Hawker CJ, Wooley KL. Cross-linked block copolymer micelles: functional nanostructures of great potential and versatility. Chem Soc Rev. 2006;35(11):1068–1083. doi:10.1039/b514858h
28. Cai H, Xiang Y, Zeng Y, et al. Cathepsin B-responsive and gadolinium-labeled branched glycopolymer-PTX conjugate-derived nanotheranostics for cancer treatment. Acta Pharm Sin B. 2021;11(2):544–559. doi:10.1016/j.apsb.2020.07.023
29. Gao Y, Ping Q, Zong L. Preparation and antitumor activity of mitoxantrone conjugated D-α-tocopherylpolyethylene glycol 1000 succinate prodrug micelle. J China Pharm Univ. 2016;47:311–316.
30. Duncan R, Vicent MJ. Polymer therapeutics-prospects for 21st century: the end of the beginning. Adv Drug Deliv Rev. 2013;65(1):60–70. doi:10.1016/j.addr.2012.08.012
31. Wang W, Zhang X, Li Z, et al. Dendronized hyaluronic acid-docetaxel conjugate as a stimuli-responsive nano-agent for breast cancer therapy. Carbohydr Polym. 2021;267:118160. doi:10.1016/j.carbpol.2021.118160
32. Traber MG, Schiano TD, Steephen AC, Kayden HJ, Shike M. Efficacy of water-soluble vitamin E in the treatment of vitamin E malabsorption in short-bowel syndrome. Am J Clin Nutr. 1994;59(6):1270–1274. doi:10.1093/ajcn/59.6.1270
33. Win KY, Feng SS. In vitro and in vivo studies on vitamin E TPGS-emulsified poly(D,L-lactic-co-glycolic acid) nanoparticles for paclitaxel formulation. Biomaterials. 2006;27(10):2285–2291. doi:10.1016/j.biomaterials.2005.11.008
34. Gill KK, Kaddoumi A, Nazzal S. Mixed micelles of PEG(2000)-DSPE and vitamin-E TPGS for concurrent delivery of paclitaxel and parthenolide: enhanced chemosenstization and antitumor efficacy against non-small cell lung cancer (NSCLC) cell lines. Eur J Pharm Sci. 2012;46(1–2):64–71. doi:10.1016/j.ejps.2012.02.010
35. Guo Y, Luo J, Tan S, Otieno BO, Zhang Z. The applications of Vitamin E TPGS in drug delivery. Eur J Pharm Sci. 2013;49(2):175–186. doi:10.1016/j.ejps.2013.02.006
36. Wempe MF, Wright C, Little JL, et al. Inhibiting efflux with novel non-ionic surfactants: rational design based on vitamin E TPGS. Int J Pharm. 2009;370(1–2):93–102. doi:10.1016/j.ijpharm.2008.11.021
37. Zhang Z, Tan S, Feng SS. Vitamin E TPGS as a molecular biomaterial for drug delivery. Biomaterials. 2012;33(19):4889–4906. doi:10.1016/j.biomaterials.2012.03.046
38. Zhang Z, Chen Y, Deng J, et al. Solid dispersion of berberine-phospholipid complex/TPGS 1000/SiO2: preparation, characterization and in vivo studies. Int J Pharm. 2014;465(1–2):306–316. doi:10.1016/j.ijpharm.2014.01.023
39. Vijayakumar MR, Kumari L, Patel KK, et al. Intravenous administration of trans-resveratrol-loaded TPGS-coated solid lipid nanoparticles for prolonged systemic circulation, passive brain targeting and improved in vitro cytotoxicity against C6 glioma cell lines. RSC Adv. 2016;6(55):50336–50348. doi:10.1039/C6RA10777J
40. Dintaman JM, Silverman JA. Inhibition of P-glycoprotein by D-alpha-tocopheryl polyethylene glycol 1000 succinate (TPGS). Pharm Res. 1999;16(10):1550–1556. doi:10.1023/A:1015000503629
41. Zhao HZ, Tan EC, Yung LY. Potential use of cholecalciferol polyethylene glycol succinate as a novel pharmaceutical additive. J Biomed Mater Res A. 2008;84(4):954–964. doi:10.1002/jbm.a.31402
42. Bao Y, Yin M, Hu X, et al. A safe, simple and efficient doxorubicin prodrug hybrid micelle for overcoming tumor multidrug resistance and targeting delivery. J Control Release. 2016;235:182–194. doi:10.1016/j.jconrel.2016.06.003
43. Cao N, Feng SS. Doxorubicin conjugated to D-alpha-tocopheryl polyethylene glycol 1000 succinate (TPGS): conjugation chemistry, characterization, in vitro and in vivo evaluation. Biomaterials. 2008;29(28):3856–3865. doi:10.1016/j.biomaterials.2008.05.016
44. Coon JS, Knudson W, Clodfelter K, Lu B, Weinstein RS. Solutol HS 15, nontoxic polyoxyethylene esters of 12-hydroxystearic acid, reverses multidrug resistance. Cancer Res. 1991;51(3):897–902.
45. Zhang XZ. Fabrication and application of reduction-degradable poly (L-succinimide) nanomicelles prodrugs carriers [Master thesis]. East China Normal University; 2012.
46. Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7(9):771–782. doi:10.1038/nrd2614
47. Wang AZ, Langer R, Farokhzad OC. Nanoparticle delivery of cancer drugs. Annu Rev Med. 2012;63(1):185–198. doi:10.1146/annurev-med-040210-162544
48. Farokhzad OC, Langer R. Impact of nanotechnology on drug delivery. ACS Nano. 2009;3(1):16–20. doi:10.1021/nn900002m
49. Wang Z, Deng X, Ding J, et al. Mechanisms of drug release in pH-sensitive micelles for tumour targeted drug delivery system: a review. Int J Pharm. 2018;535(1–2):253–260. doi:10.1016/j.ijpharm.2017.11.003
50. Sun D, Lv X, Wang X, Yu A, Wang Y. Mixed micelles based on a pH-sensitive prodrug and TPGS for enhancing drug efficacy against multidrug-resistant cancer cells. Colloids Surf B Biointerfaces. 2017;159:419–426. doi:10.1016/j.colsurfb.2017.07.082
51. Shubber S, Vllasaliu D, Rauch C, et al. Mechanism of mucosal permeability enhancement of CriticalSorb® (Solutol® HS15) investigated in vitro in cell cultures. Pharm Res. 2015;32(2):516–527. doi:10.1007/s11095-014-1481-5
52. Chauvet S, Barras A, Boukherroub R, Bouron A. Lipid nanocapsules containing the non-ionic surfactant Solutol HS15 inhibit the transport of calcium through hyperforin-activated channels in neuronal cells. Neuropharmacology. 2015;99:726–734. doi:10.1016/j.neuropharm.2015.08.043
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.