Back to Journals » Cancer Management and Research » Volume 14
Towards Personalized Management of Ovarian Cancer
Authors Algethami M, Kulkarni S, Sadiq MT, Tang HKC, Brownlie J, Jeyapalan JN, Mongan NP, Rakha EA, Madhusudan S ![]()
Received 16 September 2022
Accepted for publication 10 December 2022
Published 15 December 2022 Volume 2022:14 Pages 3469—3483
DOI https://doi.org/10.2147/CMAR.S366681
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjeev K. Srivastava
Mashael Algethami,1 Sanat Kulkarni,2 Maaz T Sadiq,3 Hiu KC Tang,4 Juliette Brownlie,1 Jennie N Jeyapalan,1 Nigel P Mongan,1 Emad A Rakha,1 Srinivasan Madhusudan1,4
1Nottingham Biodiscovery Institute, School of Medicine, University of Nottingham, University Park, Nottingham, NG7 3RD, UK; 2Department of Medicine, City Hospital, Sandwell and West Birmingham Hospitals NHS Trust, Birmingham, B18 7QH, UK; 3Cancer Centre, Queen Elizabeth Hospital, University Hospitals Birmingham NHS Trust, Birmingham, B15 2GW, UK; 4Department of Oncology, Nottingham University Hospitals, Nottingham, NG51PB, UK
Correspondence: Srinivasan Madhusudan, Nottingham Biodiscovery Institute, School of Medicine, University Park, University of Nottingham, Nottingham, NG7 3RD, UK, Tel +44(0)115 823 1850, Fax +44(0)115 823 1849, Email [email protected]
Abstract: Despite advances in surgery and chemotherapy, the overall outcomes for patients with advanced ovarian cancer remain poor. Although initial response rates to platinum-based chemotherapy is about 60– 80%, most patients will have recurrence and succumb to the disease. However, a DNA repair–directed precision medicine strategy has recently generated real hope in improving survival. The clinical development of PARP inhibitors has transformed lives for many patients with BRCA germline-deficient and/or platinum-sensitive epithelial ovarian cancers. Antiangiogenic agents and intraperitoneal chemotherapy approaches may also improve outcomes in patients. Moreover, evolving immunotherapeutic opportunities could also positively impact patient outcomes. Here we review the current clinical state of PARP inhibitors and other clinically viable targeted approaches in ovarian cancer.
Keywords: ovarian cancer, poly-(ADP)-ribose polymerase, PARP, synthetic lethality, platinum chemotherapy
Introduction
Ovarian cancer is the fifth most common cancer in women in the UK with 7000 new cases diagnosed annually. The standard management approach in the treatment of epithelial ovarian cancer includes cytoreductive surgery and platinum-based chemotherapy.1–6 Cytoreductive surgery aims to remove cancerous tissue from the ovaries and adjacent tissues to achieve optimal debulking. Chemotherapy is usually employed either in the neoadjuvant setting (to downstage cancer) or after cytoreductive surgery (to treat microscopic seedling in optimally debulked tumors or reduce tumor burden in sub-optimally debulked cancer).1–6 Despite advances in these treatment modalities, the overall outcomes for patients with epithelial ovarian cancer remain poor with over 4000 patients dying from the condition each year in the UK. However, recent advances in ovarian cancer therapeutics have had a positive impact in improving survival outcomes. In this review, we will focus on emerging precision oncology strategies in treating advanced ovarian cancers.
Platinum Therapy in Ovarian Cancer
The initial response rate to platinum-based chemotherapy (carboplatin and paclitaxel or carboplatin monotherapy) in serous epithelial ovarian cancer (the most prevalent histological sub-type) is around 60–80%. However, most patients will develop platinum resistance (intrinsic or acquired) during the course of the disease which will adversely impact survival outcomes. Recent advances in biology have not only increased our understanding of the mechanism of action of platinum compounds7 but have also provided additional insights into the development of platinum resistance.8–14
A detailed discussion of the mechanism of action of platinum compounds is beyond the scope of this article, and several recent review articles are available.8–14 Briefly, cytotoxicity of platinating agents is primarily due to their ability to induce DNA damage in tumors. Platinum drugs, such as cisplatin and carboplatin, induce predominantly intra-strand cross-links adducts in DNA and to a lesser extent interstrand cross-links. These unrepaired lesions can block replication, eventually leading to an accumulation of double-strand break (DSBs) and subsequently apoptotic cell death. DNA intra-strand cross-links are repaired through the nucleotide excision repair (NER)10,15–18 pathway during the G1 phase of the cell cycle, whereas interstrand cross-links are processed through the Fanconi anemia (FA) pathway.19–21 DSB repair intermediates generated during NER or FA mediated repair can also be processed through DSB repair pathways. During the S/G2 phase of the cell cycle, DSBs are repaired through the high-fidelity homologous recombination (HR) pathway.22,23 On the other hand, error-prone non-homologous end joining (NHEJ) is involved in the repair of DSBs during G1 phase of the cell cycle.24,25 As cancers can possess impaired DNA repair pathways, platinum-induced DNA damage is more likely to accumulate in tumors and therefore leads to more selective killing. However, upregulation of DSB repair, as occurs in some tumors, can promote platinum resistance and as a result, recent efforts have also focused on the development of bioassays that can predict platinum sensitivity in tumors.
BRCA Germline Deficiency and Ovarian Cancer
BRCA1 and BRCA2 are tumor suppressor genes coding for proteins with critical roles in HR repair of DSBs. During HR, the use of a sister chromatid as a template, results in high-fidelity, error-free DNA repair.23 In HR deficiency states, such as due to BRCA1 or BRCA2 germline mutations, the error prone NHEJ pathway is required for DSB repair.26,27 NHEJ-mediated repair is associated with chromosomal instability, increased genome-wide loss of heterozygosity (LOH) and increased risk of development of cancers including ovarian cancers amongst others.28 A prospective epidemiological study of 9856 female BRCA 1 or 2 carriers reported a cumulative risk to 80 years of ovarian cancer at 44% for BRCA1 mutation group and 17% in the BRCA2 group.29 Importantly, the germline deficiency of BRCA1/2 or other HR components can also promote platinum sensitivity in ovarian cancers26,27 due to impaired repair of platinum-induced DSBs.
HR Deficiency (HRD) and Platinum Sensitivity
The cellular signaling pathway for identifying DSBs and subsequent HR repair require several effector components including BRCA1/2, ATM, ATR, CHK1, CHK2, PALB2. Deficiency or defective function of any of these components leads to an impairment in HR and interstrand cross-link repair, resulting in reliance on the error-prone NHEJ pathway. Somatic mutations in HR genes, although less common than germline, have been shown to occur in about 18% of the ovarian cancers. Phenotypically, these cells are therefore similar to BRCA-mutated cells – such HR-deficient (HRD) tumors are referred to as demonstrating ‘BRCAness’.30 In the presence of platinum agents, HR-deficient tumor cells will preferentially accumulate unrepaired cross-links and conduct error-prone NHEJ as compared to normal cells. The resulting mutational load and significant loss of genomic integrity will result in cell cycle arrest and apoptotic cell death. This therefore rationalizes the use of platinum agents in HR-deficient tumors.
Identifying somatic HRD mutations poses a diagnostic challenge; somatic mutation screening requires analysis of tumor DNA and confirmation from germline mutation screening of normal cells (using a blood test). Furthermore, analysis of tumor tissue raises the issues of intra-tumoral heterogeneity and difficulty in identifying epigenetic modifications to HRD genes. A novel method of evaluating HRD is through a “functional” assessment of tumor DNA for a “mutational scar”. As HRD tumors are over reliant on the error-prone NHEJ pathway, they will typically develop a characteristic series of DNA changes including loss of heterozygosity (LOH), telomeric allelic imbalance and large-scale genetic transitions.31 A quantitative evaluation of these changes using next-generation sequencing can generate an HRD score, as performed in the NOVA trial.32 Alternatively, tumors may be categorized by the degree of LOH, as done in the ARIEL2 trial.33
The presence of HRD mutations, including both BRCA and non-BRCA mutations, has been shown to strongly correlate with greater sensitivity to platinum-based therapy as well as better overall survival in stage II to IV ovarian cancers (hazard ratio (HR) 0.6, 95% confidence interval (CI) 0.4–0.8; p=0.0006). Specifically, somatic HRD mutations were predictors of platinum sensitivity, although did not show a significant survival benefit, possibly due to a small sample size of this sub-type.34 These findings are further corroborated by Wen et al who found that higher HRD scores were significantly associated with platinum sensitivity and progression-free survival in ovarian cancers.35 In addition, there is now evidence to suggest that higher HRD scores are associated with greater response to platinum re-challenge in resistant patients.36 In summary, it is evident that HRD status of tumors can act as both a marker of platinum sensitivity (in primary and resistant disease) and as a prognostic indicator. However, platinum agents possess a significant side effect profile, including nephrotoxicity, neurotoxicity, and myelosuppression, which limit their utility in clinical practice. There is therefore a need for better tolerated, precision strategies that can also exploit HRD.37
Synthetic Lethality in BRCA Germline-Deficient or Platinum-Sensitive Ovarian Cancers
The enzyme poly-(ADP)-ribose polymerase 1 (PARP1) is essential during DNA repair. PARP1 binds to single-strand breaks (SSBs) that are generated as repair intermediates during DNA repair. On binding to SSBs, PARP1 is activated which in turn leads to the synthesis of PAR (poly-ADP-ribose) polymers. Auto-PARylation of PARP1 recruits several other DNA repair factors at sites of DNA damage which promotes coordination of efficient DNA repair. As discussed previously, BRCA1 and BRCA2 are essential for HR repair and germline mutations increase the risk of development of platinum-sensitive ovarian cancers.38,39
Synthetic lethality exploits inter-gene relationships in which loss of function of either of two related genes is non-lethal, but loss of both causes cell death. This offers the potential to specifically target cancer cells through pharmacological blockade of a protein known to be in a synthetic lethal relationship with a mutated tumor suppressor protein such as BRCA1 or BRCA2. Synthetic lethality targeting using PARP inhibitors in BRCA1/2 germline-deficient ovarian cancers is an exciting precision oncology strategy in ovarian cancers. The underlying molecular mechanism of the synthetically lethal relationship between BRCA and PARP is complex. Briefly, PARP inhibitors block PARP1 catalytic activity thereby preventing auto-PARylation and release of PARP from the SSB. The DNA-bound PARP1 is cytotoxic due to disruption of replication fork progression leading onto DSB accumulation and eventually apoptosis in BRCA1/2-deficient cancer cells. Tumors which demonstrate ‘BRCAness’ because of HRD phenotype can also show greater sensitivity to PARPi; this is thought to be due to the same synthetically lethal relationship. Following the pre-clinical discovery of PARPi-induced synthetic lethality, several clinical trials were rapidly initiated in BRCA1/2-deficient, HR-deficient or platinum-sensitive ovarian cancers leading to its clinical approval.38,39 Here we provide a summary of pivotal clinical trials in ovarian cancer patients.
Clinical Studies of PARP Inhibitors in Ovarian Cancers
PARP Inhibitors as Primary Maintenance Therapy
PARP inhibition has become an integral component of ovarian cancer management following FDA approval in December 2018 for olaparib in the first-line maintenance setting and subsequently EMA and NICE approval in 2019. The role of PARP inhibitors (PARPi) continues to expand rapidly with niraparib and rucaparib already enjoying widespread use globally and inclusion in various national guidelines.
The SOLO-I trial40 cemented olaparib’s place in the first-line maintenance setting in patients with BRCA-mutated advanced ovarian cancer. This international Phase III placebo-controlled trial included 391 patients with BRCA1/2 mutations and stage III/IV high-grade serous ovarian cancer (HGSOC) or endometrioid, fallopian tube or primary peritoneal cancer who had achieved a complete or partial response following first-line platinum-based chemotherapy. The initial analysis conducted after a median follow-up period of 41 months showed the rate of freedom from disease progression and death at 3 years to be 60% in the olaparib versus 27% in the placebo cohorts. On independent central review, this rate increased to 69% for olaparib and 35% for placebo, with a hazard ratio (HR) of 0.28. The SOLO-I trial reported an acceptable safety profile with most adverse effects being grade 1 or 2; however, rare cases of acute myeloid leukemia (1%) and interstitial lung disease (2%) were seen only in the olaparib cohort. The subsequent health-related quality of life analysis revealed no clinically significant difference in Trial Outcome Index scores.40
Preclinical data raised concerns that use of PARPi may compromise the efficacy of subsequent chemotherapy regimens in BRCA1/2 mutation carriers through development of secondary BRCA1/2 mutations.41,42 The SOLO-1 trial showed a significant increase in the time to second disease progression, suggesting that olaparib did not diminish the potential benefits patients had with second subsequent therapy.40
The PRIMA trial43 was a randomized, placebo-controlled phase III trial evaluating niraparib, another PARPi, in patients with advanced serous or endometrioid ovarian cancer who achieved a partial or complete response following first-line platinum-based therapy. The patient population in the PRIMA trial had several poor prognostic features when compared to the SOLO-1 population such as post-operative residual disease and over 63% receiving neoadjuvant chemotherapy. In the HRD-deficient group (according to the Mychoice test by Myriad Genetics), the reported median progression-free survival (mPFS) was 21.9 months with niraparib, versus 10.4 months in the placebo group (HR 0.43). In the overall population, mPFS in the niraparib and placebo groups was 13.8 and 8.2 months (HR 0.62). Interestingly, even in the HRD proficient group, niraparib resulted in a significant benefit (mPFS 8.1 vs 5.4 months, HR 0.68) supporting the hypothesis that niraparib may possess additional mechanisms of action beyond those involved in DNA damage repair.
In addition to success with olaparib and niraparib, the ATHENA-MONO phase III randomized placebo-controlled trial44 showed significantly beneficial effects for rucaparib monotherapy for primary maintenance therapy of high-grade ovarian cancer. Of the 538 patients included, 427 were allocated to rucaparib and 111 to placebo; mPFS for rucaparib was 20.2 months versus 9.2 months for placebo (HR 0.52, p<0.0001). Although hematological grade 3 or 4 treatment-related adverse events were common (28.7% with anemia and 14.2% with neutropenia), the trial highlights the effectiveness of rucaparib monotherapy for first-line maintenance.
Veliparib is generally considered to have a more favorable hematological toxicity profile than other PARPi, and in the Velia phase III trial45 was used in combination with carboplatin and paclitaxel followed by maintenance in the first-line setting for stage III/IV HGSOC, primary peritoneal or fallopian tube cancers. Whilst the trial did report favorable results (7 month mPFS improvement) in the veliparib group, due to patients continuing to receive veliparib as maintenance monotherapy it is unclear whether the benefit was derived from the veliparib–chemotherapy combination or the maintenance phase. The treatment interruption rate in the veliparib–chemotherapy combination group was almost 58% in comparison to 39% for chemotherapy alone, and even in the maintenance Phase 19% of the patients discontinued veliparib.45
PARPi have also been studied for use in the primary maintenance setting in combination with anti-angiogenic drugs such as bevacizumab and cediranib. Bevacizumab is extensively used in combination with chemotherapy in the neoadjuvant/adjuvant phases as well as in palliative and maintenance settings. Inhibition of angiogenesis causes a hypoxic state leading to downregulation of BRCA1 expression and consequently impaired HR, thereby rationalizing a combination with PARPi.
In the phase III PAOLA-1 trial,46 olaparib was combined with bevacizumab for maintenance after first-line chemotherapy, regardless of HRD status in patients with stage III/IV HGSOC, endometrioid, fallopian tube or primary peritoneal cancers. Reported mPFS in the overall population was 26.1 months versus 18.3 months (HR 0.63), and in the HRD-positive group (determined by the myChoice HRD plus assay and inclusive of mBRCA) mPFS was 37.2 compared to 17.7 months (HR 0.33). Importantly in the HRD-negative group, there was no significant difference in mPFS compared to placebo. The lack of an olaparib-only maintenance monotherapy arm within this trial was a limitation, and it is therefore unclear if, or the extent to which, the benefit in the HRD-positive group was due to synergistic effects between bevacizumab and olaparib or the sole effects of olaparib. The combination of olaparib with bevacizumab had an acceptable toxicity profile with no significant deterioration in quality-of-life analyses.46
Niraparib has also been combined with bevacizumab in the primary maintenance setting in a Phase II single-arm study47 including patients post-optimal debulking surgery. Analysis at 18 months showed PFS rates of 62% in the overall population, 76% in HRD and 47% in HR proficient groups.
PARP Inhibitors as Recurrent Maintenance Therapy
PARP inhibitors have also been evaluated as maintenance therapy for recurrent ovarian cancer. A second phase III trial, SOLO-2,48 with a similar design to SOLO-1, investigated a new tablet formulation of olaparib in patients who had achieved a complete or partial clinical response following two or more lines of chemotherapy. The mPFS was reported as 19.1 months in the olaparib group compared to 5.5 months in the placebo group, with a similar HR 0.30 to SOLO-1. Over 40% of the patients in this trial had received three or more lines of previous chemotherapy. Interestingly, exposure to bevacizumab as part of pre-maintenance chemotherapy did not alter the treatment effects for patients who received olaparib. The data from SOLO-2 also showed longer times to first and second subsequent therapies which generally would have included intravenous chemotherapy. As a result, these secondary end points hold particular clinical relevance given the advanced stage of disease for patients included in this trial.
Over 75% of the patients with ovarian cancer do not harbor BRCA1/2 mutations (varying by subtype) and with the addition of non-HGSOC, primary peritoneal and fallopian tube cancers, a large proportion of patients are not eligible for olaparib-based maintenance treatment. The NOVA trial32 investigated niraparib and included patients with platinum-sensitive, ovarian cancer following two or more lines of chemotherapy (aside from one patient included within the trial) who achieved a partial or complete response. A total of 553 patients were included in the trial and stratified according to BRCA and HRD status. The niraparib group had a significantly longer mPFS compared to placebo (21 and 5.5 months, respectively, HR 0.27). HRD-positive patients had better outcomes (mPFS 12.8 versus 3.9 months, HR 0.38) and even in the non-mBRCA cohort, niraparib showed a clinically significant benefit (9.3 months versus 3.9 months HR 0.45) with 20% of patients continuing treatment post 18 months. Almost 50% of the patients in the NOVA trial were heavily pre-treated having received three or more lines of chemotherapy. The side effect profile of niraparib was broadly comparable to that of olaparib with the most common grade 3 and 4 adverse effects (AE) being hematological in nature (thrombocytopenia 33.8%, anemia 25.3% and neutropenia 19.6%). There were five cases (1.4%) of myelodysplastic syndrome reported in the niraparib group which is widely recognized as a class-specific adverse effect.
Rucaparib has also been evaluated in the ARIEL3 trial in the maintenance setting following second- or later-line chemotherapy. In ARIEL3, patients were grouped according to BRCA status and presence of HRD, the latter determined by the extent of genomic loss of heterozygosity (LOH) as described earlier. This large Phase III trial randomized 564 patients in a 2:1 ratio to rucaparib or placebo. In the cohort of patients with BRCA mutations, the mPFS was 16.6 months for rucaparib (n=130) versus placebo (n=66) (HR 0.23, 95% CI 0.16–0.34, P<0.0001).49
In patients with HRD (inclusive of mBRCA and non mBRCA with “high” LOH) the mPFS was 13.6 months in the rucaparib group (n=236) versus 5.4 months in the placebo group (n=118). In the intention-to-treat population, the mPFS was 10.8 months versus 5.4 months (HR 0.32) in the rucaparib and placebo groups, respectively. Notably, 37% of the patients had previous exposure to three or more lines of chemotherapy. Furthermore, 19% of the patients had >2cm of disease at baseline in the rucaparib group and achieved significant benefit, with the “bulky” disease group trending towards greater benefit. The utility of LOH as a biomarker to predict response to rucaparib is drawn into question however, given the observed benefits of rucaparib in tumors without an identified HRD; within the LOH “low” and BRCA wild-type group approximately 30% of the patients achieved benefit from rucaparib at 1 year, compared to 5% in the placebo group. These findings support those of the PRIMA trial suggesting alternative, non-HRD-based synthetic lethality mechanisms of actions of PARP inhibitors may exist.
Talazoparib has significantly greater PARP trapping capabilities than other clinically available PARP inhibitors.50 However, whilst there is a general paucity of evidence for talazoparib in ovarian cancer, a recent review provides an update on ongoing ovarian cancer studies.50 Consequently, talazoparib is not currently approved for therapeutic use in ovarian cancers.
PARP Inhibitor Monotherapy for Relapsed Advanced Ovarian Cancer (Table 1)
In addition to these promising findings in the maintenance setting, PARPi have been investigated in the setting of relapsed disease. The ICEBERG-3 trial (NCT00628251) showed benefit with olaparib as a single agent versus pegylated liposomal doxorubicin (PLD) in patients with relapsed, platinum-resistant disease and BRCA mutation. This study rationalized the larger phase III SOLO-3 trial51 comparing olaparib single agent to physicians choice non-platinum chemotherapy following at least two lines of platinum-based chemotherapy in patients with platinum-sensitive or partially sensitive (defined as progression 6–12 months after platinum-based chemotherapy) relapsed disease. The trial included 266 patients randomized 2:1 to olaparib or chemotherapy (PLD/gemcitabine/topotecan). Approximately 50% of the patients had received three or more lines of chemotherapy.
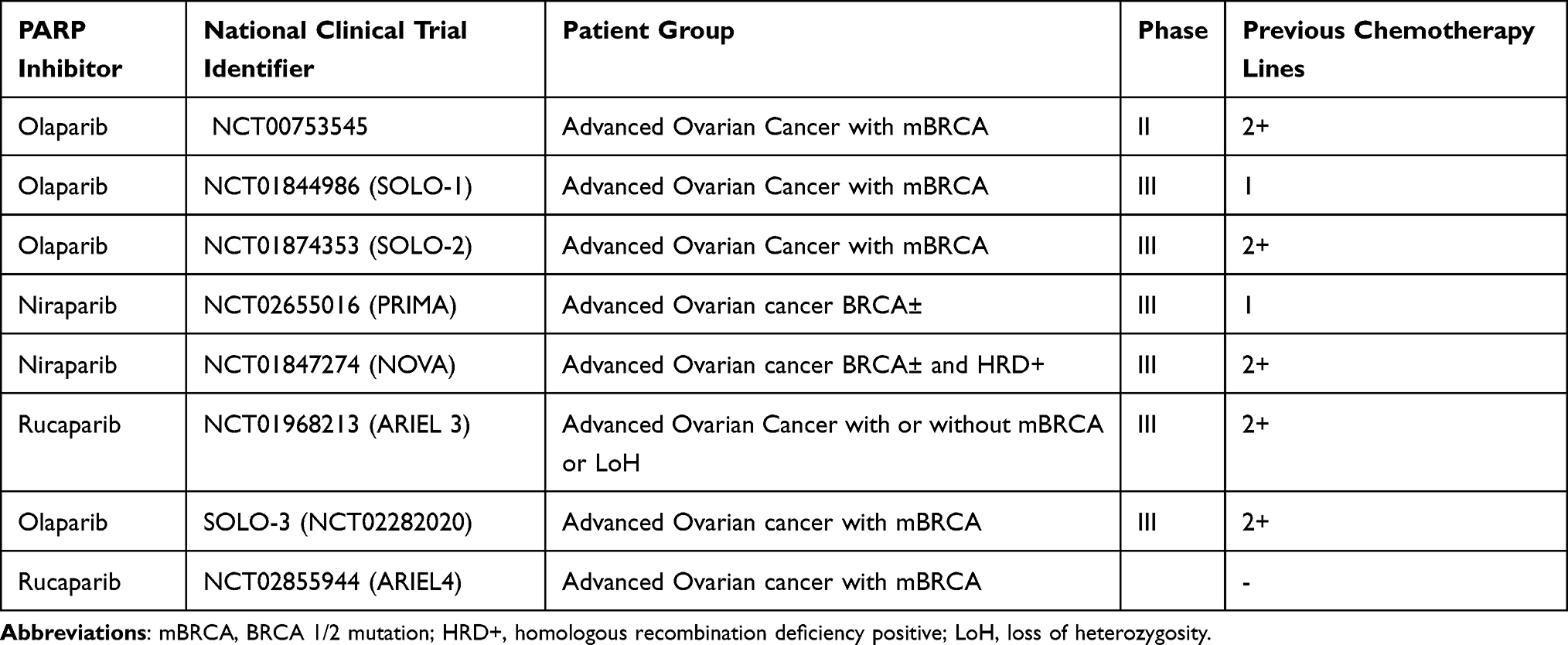 |
Table 1 Clinical Trials Beyond Phase 1 Evaluating PARP Inhibitors as Monotherapy |
The reported overall response rate (ORR) was 72% with olaparib and 52% with chemotherapy with earlier use of olaparib correlating with higher response rates. MPFS was 13.2 versus 8.5 months for olaparib and chemotherapy, respectively, with durations of treatment with olaparib over double those of various chemotherapy agents. Olaparib was associated with an inferior toxicity profile with higher rates of fatigue, nausea and diarrhea reported; however, with dose modifications, the discontinuation rate of olaparib was lower than that of chemotherapy. These findings are further supported by a randomized phase II trial of maintenance olaparib compared to placebo in 265 women with platinum-sensitive relapsed ovarian cancer.52 The study showed significantly greater mPFS in the olaparib cohort (8.4 months versus 4.8 months, HR 0.35, p<0.001) and significant improvements in overall survival with no new safety concerns over the two-year follow-up.52
More recently, the ARIEL4 trial53 compared rucaparib with chemotherapy in patients with mBRCA who had progressed on at least two previous lines of treatment. In the chemotherapy arm, platinum-sensitive ovarian cancer was treated with platinum-based regimens whilst paclitaxel was used for platinum-resistant disease. An mPFS of 7.4 months in the rucaparib arm was reported compared to 5.7 months with chemotherapy (HR 0.64, p=0.001), further demonstrating the benefits of PARPi monotherapy for relapsed mBRCA advanced ovarian cancer.
PARP Inhibitor-Based Combination Strategies for Relapsed Advanced Ovarian Cancer
Platinum Agents
A Phase I/Ib trial investigating the combination of olaparib with platinum-based treatment54 demonstrated encouraging efficacy; however due to significant hematological toxicity, notably 42% developing grade 3 or 4 neutropenia, treatment intensity could not be maintained. In a larger international randomized phase II controlled trial,55 162 patients with recurrent, platinum-sensitive high-grade ovarian cancer with less than three previous lines of chemotherapy were enrolled. 81 patients were randomized to the olaparib group in combination with carboplatin and paclitaxel followed by maintenance olaparib monotherapy whilst 75 patients were randomized to chemotherapy alone. A significantly superior mPFS was reported with olaparib (12.2 months compared to 9.6 months) and subgroup analyses confirmed better outcomes in mBRCA patients as expected. Increased Grade 3/4 neutropenia was observed in the combination group (43% vs 35%) whilst other hematological toxicities were similar across the groups.55 Other adverse effects were on average 10% more commonly reported with Olaparib + chemotherapy, grade 3+ toxicities were 7% more frequent; however, discontinuation rates were broadly similar (19%).
Cediranib
Cediranib is an oral inhibitor of the vascular endothelial growth factor (VEGF) receptor tyrosine kinase 1, 2 and 3 and is known to have activity against ovarian cancer.56 A Phase II clinical trial to investigate the benefit of using cediranib alongside olaparib versus olaparib alone57 randomized 44 patients to the olaparib/cediranib group and 46 to the olaparib alone group. There was a significantly better mPFS reported in the combination group (17.7 months) compared to olaparib alone (9 months) (HR 0.42). The results from these trials provided the basis for two currently ongoing phase III trials comparing the oral cediranib/olaparib combination to monotherapy (ICON9 study, NCT03278717) and to investigator-choice chemotherapy in patients with recurrent ovarian cancer (COCOS study, NCT02502266).
Pegylated Liposomal Doxorubicin (PLD)
PLD is already a heavily used agent as monotherapy or in combination with platinum agents. The ROLANDO study combined Olaparib with PLD in heavily pretreated (including platinum resistant) patients. An impressive 77% disease control rate was reported (29% PR); however, grade 3+ toxicities were common when PLD was dosed at 40mg/m2 (47%). The lower PLD dose of 30mg/m2 as per with platinum combination was much more tolerable (21% Grade 3+ toxicities). Other trials have explored PLD in combination with veliparib and quadruple regimens including platinum agents and bevacizumab (Table 2).
 |
Table 2 Examples of Clinical Trials Evaluating PARPi in Combination with Chemotherapy and Targeted Treatment |
Cyclophosphamide
PARP inhibitors have been shown to potentiate DNA damage resulting from alkylating agents in preclinical models.58 In advanced ovarian cancer, cyclophosphamide has been used in long term, low-dose regimens (metronomic dosing), especially for patients where fitness to tolerate toxicity is a concern. A Phase I trial aimed to assess a veliparib in combination with metronomic cyclophosphamide in 35 patients with refractory solid tumors and lymphomas.59 A maximum tolerated dose of 60mg was established to be given orally once a day for 7, 14 or 21 days alongside daily cyclophosphamide 50mg. This data led to a phase II trial which compared combination cyclophosphamide/veliparib versus cyclophosphamide alone in mBRCA patients with high-grade ovarian, primary peritoneal or fallopian tube cancer. This study did not show a significant improvement in outcomes through the addition of veliparib to metronomic cyclophosphamide.60
Mitomycin C (MMC)
Veliparib was combined with MMC in a dose escalation study61 and compared to veliparib alone. Six of 61 patients were reported to have a response to treatment, 5 of whom were in the combination arm. The trial concluded that veliparib alone or with MMC could be safely administered to patients and can provide clinical benefit in some.
Immunotherapy Strategies
PARPi have been shown to promote anti-tumor activity by upregulating PDL-1 expression in animal model cancer cells. The blockade of PD-1/PDL-1 interaction was theorized to resensitize PARPi treated cells to T-cell mediated cytotoxicity.62,63 The combination of immunotherapy with PARPi is therefore being explored in early-phase trials with Phase II/III trials on the horizon. A Phase I/II trial (NCT02571725) is currently recruiting mBRCA patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancers after one line of platinum-based treatment. This trial is designed to combine daily olaparib with a CTLA-4 inhibitor (tremelimumab) given on day 1 of a 28-day cycle for the first six cycles followed by 12-weekly thereafter until disease progression or unacceptable toxicity. This combination was reported to be tolerable in ASCO 2017 and a following phase II trial is underway.
Another phase I/II trial64 assessed the combination of a PARPi (niraparib 200 or 300mg OD) in combination with an anti-PD-1 monoclonal antibody (pembrolizumab 200mg IV 3 weekly) for the treatment of recurrent ovarian cancer (irrespective of BRCA status). Of the cohort of 62 patients, the objective response rate (ORR) was 18% (90% CI 11–29%) with complete response seen in 3 patients (5%), partial response in 8 (13%) and 28 (47%) had stable disease. Common treatment-related adverse events included fatigue (53%), nausea (42%) and anemia (36%); notably 10 (19%) patients had immune-related adverse effects (thought to be due to pembrolizumab) with 3 (6%) of these having grade 3 severity. In summary, this study demonstrates that PARPi and anti-PD-1 immunotherapy combinations can be effective and well tolerated in this patient population.64
There are multiple ongoing phase II/III trials of immunotherapy-PARPi combinations for recurrent ovarian cancer, including with other targeted agents such as bevacizumab (Table 3).
 |
Table 3 Examples of Currently Ongoing Trials Investigating PARP Inhibitors Combined with Immunotherapy |
Mechanisms of Resistance to PARP Inhibitors
Although PARP inhibitors therapy has improved survival outcome, the response rate to PARPi monotherapy is around 40–50% and progression-free survival is about 7 months. The clinical data clearly indicate that intrinsic or acquired resistance can limit the benefit of PARPi therapy. Delineating the mechanisms of resistance is therefore an area of intense research.65–68 Pre-clinical investigations of PARP inhibitor resistance in BRCA-deficient cells have highlighted several mechanisms of resistance. This includes restoration of HR (via reactivation of BRCA function through BRCA reversion, loss of BRCA1 promoter methylation, HSP90 activation, inactivation of 53BP1, reduced levels of REV7 or down regulation of PARP1), restoration of stalled replication fork protection (RAD51 overexpression) and reduced cellular uptake of PARP inhibitors (overexpression of ABCB1).65–68 Whether these pre-clinical mechanisms can be translated to a real term clinical application is yet to be established.
Targeting Angiogenesis
Bevacizumab is a monoclonal antibody that sequesters and neutralizes all isoforms of the VEGF-receptor ligand VEGF-A, and there is clinical evidence of efficacy in metastatic colorectal cancer, non-small cell lung cancer, advanced renal cell carcinoma and in hepatocellular carcinoma.69 Phase II trials of bevacizumab in women with ovarian cancer have shown evidence of objective tumor responses in a small proportion (16%) of patients with advanced, platinum-resistant disease who generally have a dismal prognosis.70
Antiangiogenic Agents as Adjuvant Therapy and Primary Maintenance
The ICON7 study was a pivotal, large randomized clinical trial that enrolled patients with high-risk early stage (I and IIa with clear-cell histology or poorly differentiated tumors) and advanced stage (IIB-IV) epithelial ovarian cancer, after upfront surgery, to standard-of-care combination chemotherapy with carboplatin and paclitaxel or the same chemotherapy backbone alongside bevacizumab (7.5mg/kg intravenously every 3 weeks) with maintenance bevacizumab until progression.71 The likelihood of achieving an objective radiologic response was statistically significantly higher in the patients receiving bevacizumab alongside chemotherapy compared with those receiving chemotherapy alone – 67% versus 48% (p<0.001). MPFS was 17.3 months with chemotherapy alone and 19 months with chemotherapy and bevacizumab (HR 0.81, p=0.004). In terms of progression-free survival, the maximal effect of bevacizumab was seen at 12 months and diminished by 24 months. In the updated analyses, progression-free survival with 42 months of follow-up was 22.4 months without bevacizumab versus 24.1 months with bevacizumab (P=0.04 by Log rank test); the PFS benefit was greater with bevacizumab in patients at high risk of progression, with progression-free survival with 42 months of follow-up of 14.5 months with standard therapy alone and 18.1 months with bevacizumab added. Despite these modest improvements in PFS with the addition of bevacizumab, the final overall survival results from the ICON-7 trial showed that overall survival was equivalent with chemotherapy alone versus chemotherapy with bevacizumab – 44.6 months versus 45.5 months (p=0.85).72 In a pre-planned exploratory analysis, however, patients with poor prognostic factors achieved a statistically significant overall survival benefit with the addition of bevacizumab – 34.5 months versus 39.3 months (p=0.03). Of key importance, in view of the fact that the majority of patients in the trial had advanced disease that was incurable, is the effect of treatments on quality of life and global quality-of-life (assessed by a validated questionnaire) was poorer in patients receiving bevacizumab with chemotherapy.73
The other pertinent study Is the GOG clinical trial74 comparing standard chemotherapy with carboplatin-paclitaxel to carboplatin-paclitaxel-bevacizumab (6 cycles) and carboplatin-paclitaxel-bevacizumab followed by maintenance bevacizumab for patients with advanced epithelial ovarian cancer. MPFS was 10.3 months with chemotherapy alone compared with 14.1 months with chemotherapy, bevacizumab, and maintenance bevacizumab (p<0.001). When the progression-free survival analysis was restricted to patients with radiologic (RECIST) progression and those with serologic progression only (Ca125) were excluded, mPFS was 12 months versus 18 months in the chemotherapy alone and chemotherapy-bevacizumab maintenance group (p<0.001). However, despite impressive gains in progression-free survival, this did not translate into any overall survival benefit with median overall survival of 39.3 months and 39.7 months in the chemotherapy and chemotherapy-bevacizumab groups, respectively. The final protocol-specified analysis of overall survival from the GOG study confirmed no overall survival benefit for the total population; however, an exploratory subset analysis did find that for patients with Stage IV disease median overall survival was 42.8 with chemotherapy, bevacizumab and maintenance bevacizumab versus 32.6 months with chemotherapy alone.74
Anti-Angiogenic Agents for Relapsed Advanced Ovarian Cancer
Patients were systemic-therapy naïve in the ICON-7 and GOG studies, and anti-angiogenic treatment has also been studied in the second and subsequent line setting. The key study was AURELIA, a randomized Phase III trial comparing investigator’s choice of chemotherapy with chemotherapy and concurrent bevacizumab in advanced, platinum-refractory (progression within 6 months) epithelial ovarian cancer.75 Patients could have received a maximum of two prior chemotherapy regimens, and those with platinum-refractory disease were excluded. Chemotherapy was chosen from liposomal doxorubicin, paclitaxel and topotecan. About 85% of the patients had papillary serous histology, and just over half of patients had poorly differentiated tumors. This study met its primary endpoint with an improvement in mPFS from 3.4 to 6.7 months with the addition of bevacizumab. The rate of objective response (RECIST and/or Ca125) was 12.6% with chemotherapy versus 30.9% with chemotherapy and bevacizumab. There was no statistically significant different in overall survival, however (13.3 versus 16.6 months), in keeping with the results in the treatment-naïve setting.
The results of the OCEANS trial76 are informative in terms of the role of anti-angiogenic therapy alongside chemotherapy in the platinum-sensitive, relapsed setting). This was a randomized, placebo-controlled comparison of carboplatin-gemcitabine with or without concurrent bevacizumab followed by maintenance bevacizumab. Thirty-five percent of the patients were 65 years of age or older, 75% had a performance status of 0, 78% had papillary serous histology and 59% had a platinum-free interval of greater than 1 year. The addition of concurrent and then maintenance bevacizumab to chemotherapy led to an increase in median progression-free survival of 4 months (p=0.0001) and in improvement in objective response rate from 57.4% to 78.5%, but there was no significant overall survival benefit (approximately 33 months in both arms).77
As discussed previously, PARPi and bevacizumab combination therapy has been shown to have beneficial effects in the primary maintenance setting. Further to this, the AVANOVA2 phase II randomized trial78 compared bevacizumab and niraparib to niraparib alone (1:1 allocation) in 97 patients with high-grade serous or endometrioid platinum-sensitive recurrent ovarian cancer. The trial demonstrated significantly improved mPFS in the combination group (11.9 months versus 5.5 months, HR 0.35, p<0.0001) and similar incidence of adverse effects across groups except for greater incidence of hypertension with anti-VEGF therapy.78 These results rationalize further phase III trials of the niraparib–bevacizumab combination for recurrent, platinum-sensitive ovarian cancer.
Trebananib is an anti-angiogenic treatment whose mechanism of action is to inhibit the interaction between pro-angiogenic factors angiopoietins 1 and 2 to the Tie2 receptor. It was clinically evaluated in the setting of relapsed, advanced epithelial ovarian cancer in the TRINOVA-1 trial.79 There was significantly improved progression-free survival when trebananib was given alongside weekly paclitaxel chemotherapy (versus placebo) – 7.2 versus 5.4 months (p<0.0001). Trebananib was well-tolerated with the main toxicity being peripheral oedema. The final analysis of the TRINOVA-1 study showed no significant difference in overall survival in the intention-to-treat population, however patients with ascites at baseline had a statistically significant two-month improvement in overall survival with trebananib.80
Cediranib is an oral small-molecule inhibitor VEGFR-2, PDGFR and c-Kit and has a predominantly anti-angiogenic mode of action. In the randomized phase II trial discussed earlier, cediranib demonstrated significantly greater mPFS in combination with olaparib in patients with relapsed high-grade serous or high-grade endometrioid or germline BRCA mutant ovarian cancer.57 Median overall survival was numerically superior in the combination therapy group (44.2 months) compared to the olaparib monotherapy group (33.3 months) although this was not statistically significant (p=0.11). Cediranib was associated with fatigue, hypertension and diarrhea.
Aflibercept is a fusion protein that comprises an immunoglobulin G backbone fused to the extracellular sequences of human VEGFR1 and 2. It functions as a soluble decoy receptor binding the key pro-angiogenic factor VEGF-A with higher affinity than its natural receptor. It was evaluated in patients with platinum-resistant/refractory advanced epithelial ovarian cancer at two different doses in a Phase II study.81 There was a hint of clinical activity in these heavily pre-treated patients with approximately 12% of the patients achieving clinical benefit (objective response or disease stabilization for 6 months or more).
Other Targeted Approaches
The rationale behind intraperitoneal chemotherapy as opposed to that delivered via the systemic route is that far higher concentrations of chemotherapy on the peritoneal surfaces can be achieved. Intraperitoneal delivery of chemotherapy may improve outcomes by eliminating residual microscopic peritoneal disease more efficiently than intravenous administration. A randomized Phase III trial, which recruited patients in the late 1980s and early 1990s, compared intravenous or intra-peritoneal cisplatin alongside maximal surgical debulking and intravenous cyclophosphamide chemotherapy in patients with stage III epithelial ovarian cancer.82 Patients were newly diagnosed with stage III disease and underwent full surgical staging and maximal debulking with a maximum diameter of 2cm for any residual lesions. Six cycles of chemotherapy were delivered using cyclophosphamide 600mg/m2 and intravenous cisplatin 100mg/m2. Intraperitoneal cisplatin was given at the same dose in 2 liters of fluid as quickly as possible at body temperature. In terms of efficacy, the pathological complete response rate (at second-look laparotomy) was 36% with intravenous and 47% with intraperitoneal cisplatin. Median overall survival was 41 months with intravenous and 49 months with intraperitoneal cisplatin. Peripheral neuropathy, tinnitus, leucopenia and hearing loss were far less common with intraperitoneal cisplatin. This study was however performed in the pre-taxane era and cyclophosphamide chemotherapy would be considered sub-optimal in contemporary practice.
A much more recent Phase III randomized study provided an important update as to the true role of intraperitoneal chemotherapy in this setting.83 Approximately 90% of the patients had high-grade serous papillary histology and one-third of patients had disease involving 6–8 anatomical regions of the abdomen. Optimal cytoreductive surgery (no macroscopic disease) was achieved in two-thirds of patients. In contrast to the 1996 study, where upfront surgical debulking was mandated, almost all included patients (90%) had advanced disease that was not felt to be amenable to upfront optimal cytoreductive surgery. These patients received neoadjuvant chemotherapy with carboplatin-paclitaxel for three cycles followed by randomization to surgery alone or surgery and hyperthermic intraperitoneal chemotherapy (HIPEC, 100mg/m2 cisplatin) at a temperature of 40°C with the entire abdomen being perfused for a 90-minute period. Completion of the planned three cycles of post-operative chemotherapy was achieved in over 90% of the patients. Median recurrence-free survival was 3.5 months longer in the HIPEC arm (14.2 versus 10.7 months) and 3-year recurrence-free survival was achieved in 17% of the surgery-HIPEC treated patients versus 8% in the surgery only group. Three-year overall survival probability was 62% with surgery-HIPEC and 48% with surgery alone. Inpatient stay was only 2 days longer with the addition of HIPEC, and it was feasible for post-operative chemotherapy to commence within 4 weeks in both treatment groups.
Within the caveats of a small study (120 patients), where the method of randomization was not explicitly described, performed over an 8-year period at a single centre, HIPEC in addition to cytoreductive surgery and chemotherapy for first recurrence of advanced (FIGO stage IIIC and IV) epithelial ovarian cancer appeared to be effective. Median overall survival was 26.7 months compared with 13.4 months with HIPEC versus no HIPEC and the 3-year overall survival was 75% versus 18%, respectively.84
Other Histological Subtypes
Epithelial ovarian cancer can be further classified by histological subtype into serous, endometrioid, clear cell and mucinous. Notably, trials for PARPi in high-grade ovarian cancers discussed above typically excluded clear cell and mucinous cancers. Although the pivotal trials for antiangiogenic and immunotherapy agents included these subtypes, this was in small numbers due to their rarity. Ovarian clear cell carcinoma (OCCC) is rare, with higher prevalence in Asian populations, and characterized by chemoresistance and poor prognosis.85 However, pre-clinical evidence suggests PARPi may act as potent chemosensitizers in OCCC86 and is reviewed in more detail in.85 Due to its low prevalence, it is challenging to extrapolate data from the small numbers of OCCC patients included in clinical trials of antiangiogenic and immunotherapy agents, although the current evidence is reviewed by Ogasawara et al.87 The same issue plagues the mucinous subtype, with a paucity of cases present in large clinical trials limiting the reliability of conclusions from subtype-specific analysis. Despite not reaching statistical significance; however, evidence from GOG-0241 and ICON-6 suggest potentially beneficial effects of bevacizumab and cediranib respectively and is reviewed in more detail in.88 Furthermore, the scarcity of HRD in mucinous ovarian cancers implies these tumors are unlikely to respond to platinum-based chemotherapy or PARPi.89
Conclusion
Metastatic ovarian cancer remains a devastating disease. Over the last decade, significant advances have transformed the lives of many ovarian cancer patients. This is largely due to 1) increasingly patients are being treated in highly specialized high volume gynaecologic-oncology centres and 2) well-conducted clinical trials investigating precision oncology strategies in ovarian cancer. A major advance has been the use of PARP inhibitor therapy in the maintenance setting that has substantially improved progression-free survival in BRCA germline-deficient as well as platinum-sensitive ovarian cancers. However, several challenges remain including intrinsic or acquired resistance to PARPi therapy. The development of actionable predictive biomarkers of resistance will provide opportunities for further fine-tuning of precision oncology therapeutics. Moreover, the emergence of new DNA repair inhibitors in clinical trials (including those targeting ATM, ATR, WEE1 and other evolving targets) in solid tumors will likely impact on ovarian cancer therapeutics. The evolution of newer antiangiogenic agents and the emergence of immunotherapeutic options also provide an opportunity for improvements in survival.
With advancements in these precision strategies, there arises a need to identify clinically relevant predictive biomarkers within tumors, and therefore optimal candidates for these therapies. Essential in aiding this transition towards “personalized oncology” is the development of validated, tumor-based companion diagnostic testing such as the FDA-approved Foundation OneⓇ test90 or the NCC OncopanelⓇ test.91 The former test can assess the tumor mutational burden and PD-L1 expression (predictive of immunotherapy response) as well as degree of LOH, HRD and BRCA1/2 expression (predictive of PARPi response).90 Increasing the availability of companion testing and access to stratified precision therapies has the potential to change the face of ovarian cancer therapeutics.
Acknowledgments
Research in Madhusudan lab is funded by Naaz-Coker Ovarian cancer fellowship, University of Nottingham.
Disclosure
Mashael Algethami and Sanat Kulkarni are joint first authors. The authors declare no conflict of interest.
References
1. Baert T, Ferrero A, Sehouli J., et al. The systemic treatment of recurrent ovarian cancer revisited. Ann Oncol. 2021;32(6):710–725. doi:10.1016/j.annonc.2021.02.015
2. DiSilvestro P, Colombo N, Harter P, Gonzalez-Martin A, Ray-Coquard I, Coleman RL. Maintenance treatment of newly diagnosed advanced ovarian cancer: time for a paradigm shift? Cancers. 2021;13(22):5756. doi:10.3390/cancers13225756
3. Elyashiv O, Wong YNS, Ledermann JA. Frontline maintenance treatment for ovarian cancer. Curr Oncol Rep. 2021;23(8):97. doi:10.1007/s11912-021-01088-w
4. Nikolaidi A, Fountzilas E, Fostira F, Psyrri A, Gogas H, Papadimitriou C. Neoadjuvant treatment in ovarian cancer: new perspectives, new challenges. Front Oncol. 2022;12:820128. doi:10.3389/fonc.2022.820128
5. Schoutrop E, Moyano-Galceran L, Lheureux S, et al. Molecular, cellular and systemic aspects of epithelial ovarian cancer and its tumor microenvironment. Semin Cancer Biol. 2022;86(Pt 3):207–223. doi:10.1016/j.semcancer.2022.03.027
6. Shaik B, Zafar T, Balasubramanian K, Gupta SP. An overview of ovarian cancer: molecular processes involved and development of target-based chemotherapeutics. Curr Top Med Chem. 2021;21(4):329–346. doi:10.2174/1568026620999201111155426
7. Ali R, Aouida M, Alhaj Sulaiman A, Madhusudan S, Ramotar D. Can cisplatin therapy be improved? Pathways that can be targeted. Int J Mol Sci. 2022;23(13):7241. doi:10.3390/ijms23137241
8. Chen SH, Chang JY. New insights into mechanisms of cisplatin resistance: from tumor cell to microenvironment. Int J Mol Sci. 2019;20(17):85.
9. Devarajan N, Manjunathan R, Ganesan SK. Tumor hypoxia: the major culprit behind cisplatin resistance in cancer patients. Crit Rev Oncol Hematol. 2021;162:103327. doi:10.1016/j.critrevonc.2021.103327
10. Duan M, Ulibarri J, Liu KJ, Mao P. Role of nucleotide excision repair in cisplatin resistance. Int J Mol Sci. 2020;21(23):9248. doi:10.3390/ijms21239248
11. Skowron MA, Oing C, Bremmer F, et al. The developmental origin of cancers defines basic principles of cisplatin resistance. Cancer Lett. 2021;519:199–210. doi:10.1016/j.canlet.2021.07.037
12. Wang L, Zhao X, Fu J, Xu W, Yuan J. The role of tumour metabolism in cisplatin resistance. Front Mol Biosci. 2021;8:691795. doi:10.3389/fmolb.2021.691795
13. Makovec T. Cisplatin and beyond: molecular mechanisms of action and drug resistance development in cancer chemotherapy. Radiol Oncol. 2019;53(2):148–158. doi:10.2478/raon-2019-0018
14. Marchetti C, De Felice F, Romito A, et al. Chemotherapy resistance in epithelial ovarian cancer: mechanisms and emerging treatments. Semin Cancer Biol. 2021;77:144–166. doi:10.1016/j.semcancer.2021.08.011
15. D’Souza A, Blee AM, Chazin WJ. Mechanism of action of nucleotide excision repair machinery. Biochem Soc Trans. 2022;50(1):375–386. doi:10.1042/BST20210246
16. Duan M, Speer RM, Ulibarri J, Liu KJ, Mao P. Transcription-coupled nucleotide excision repair: new insights revealed by genomic approaches. DNA Repair (Amst). 2021;103:103126. doi:10.1016/j.dnarep.2021.103126
17. Liakos A, Lavigne MD, Fousteri M. Nucleotide excision repair: from neurodegeneration to cancer. Adv Exp Med Biol. 2017;1007:17–39.
18. Sugasawa K. Molecular mechanisms of DNA damage recognition for mammalian nucleotide excision repair. DNA Repair (Amst). 2016;44:110–117. doi:10.1016/j.dnarep.2016.05.015
19. Niraj J, Farkkila A, D’Andrea AD. The Fanconi Anemia Pathway in Cancer. Annu Rev Cancer Biol. 2019;3:457–478. doi:10.1146/annurev-cancerbio-030617-050422
20. Shukla P, Solanki A, Ghosh K, Vundinti BR. DNA interstrand cross-link repair: understanding role of Fanconi anemia pathway and therapeutic implications. Eur J Haematol. 2013;91(5):381–393. doi:10.1111/ejh.12169
21. Su X, Huang J. The Fanconi anemia pathway and DNA interstrand cross-link repair. Protein Cell. 2011;2(9):704–711. doi:10.1007/s13238-011-1098-y
22. Wright WD, Shah SS, Heyer WD. Homologous recombination and the repair of DNA double-strand breaks. J Biol Chem. 2018;293(27):10524–10535. doi:10.1074/jbc.TM118.000372
23. Zhao W, Wiese C, Kwon Y, Hromas R, Sung P. The BRCA tumor suppressor network in chromosome damage repair by homologous recombination. Annu Rev Biochem. 2019;88:221–245. doi:10.1146/annurev-biochem-013118-111058
24. Hnizda A, Blundell TL. Multicomponent assemblies in DNA-double-strand break repair by NHEJ. Curr Opin Struct Biol. 2019;55:154–160. doi:10.1016/j.sbi.2019.03.026
25. Williams GJ, Hammel M, Radhakrishnan SK, Ramsden D, Lees-Miller SP, Tainer JA. Structural insights into NHEJ: building up an integrated picture of the dynamic DSB repair super complex, one component and interaction at a time. DNA Repair (Amst). 2014;17:110–120. doi:10.1016/j.dnarep.2014.02.009
26. Neff RT, Senter L, Salani R. BRCA mutation in ovarian cancer: testing, implications and treatment considerations. Ther Adv Med Oncol. 2017;9(8):519–531. doi:10.1177/1758834017714993
27. Nelson HD, Huffman LH, Fu R, Harris EL, Force USPST. Genetic risk assessment and BRCA mutation testing for breast and ovarian cancer susceptibility: systematic evidence review for the U.S. Preventive Services Task Force. Ann Intern Med. 2005;143(5):362–379. doi:10.7326/0003-4819-143-5-200509060-00012
28. Powell SN, Kachnic LA. Roles of BRCA1 and BRCA2 in homologous recombination, DNA replication fidelity and the cellular response to ionizing radiation. Oncogene. 2003;22(37):5784–5791. doi:10.1038/sj.onc.1206678
29. Kuchenbaecker KB, Hopper JL, Barnes DR, et al. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA. 2017;317(23):2402–2416. doi:10.1001/jama.2017.7112
30. Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer. 2016;16(2):110–120. doi:10.1038/nrc.2015.21
31. da Cunha Colombo Bonadio RR, Fogace RN, Miranda VC, Diz M. Homologous recombination deficiency in ovarian cancer: a review of its epidemiology and management. Clinics. 2018;73(suppl1):e450s. doi:10.6061/clinics/2018/e450s
32. Mirza MR, Monk BJ, Herrstedt J, et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med. 2016;375(22):2154–2164. doi:10.1056/NEJMoa1611310
33. Swisher EM, Lin KK, Oza AM, et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, Phase 2 trial. Lancet Oncol. 2017;18(1):75–87. doi:10.1016/S1470-2045(16)30559-9
34. Pennington KP, Walsh T, Harrell MI, et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin Cancer Res. 2014;20(3):764–775. doi:10.1158/1078-0432.CCR-13-2287
35. Wen H, Feng Z, Ma Y, et al. Homologous recombination deficiency in diverse cancer types and its correlation with platinum chemotherapy efficiency in ovarian cancer. BMC Cancer. 2022;22(1):550. doi:10.1186/s12885-022-09602-4
36. da Costa A, Do Canto LM, Larsen SJ, et al. Genomic profiling in ovarian cancer retreated with platinum based chemotherapy presented homologous recombination deficiency and copy number imbalances of CCNE1 and RB1 genes. BMC Cancer. 2019;19(1):422. doi:10.1186/s12885-019-5622-4
37. Oun R, Moussa YE, Wheate NJ. The side effects of platinum-based chemotherapy drugs: a review for chemists. Dalton Trans. 2018;47(19):6645–6653. doi:10.1039/C8DT00838H
38. Lord CJ, Ashworth A. PARP inhibitors: synthetic lethality in the clinic. Science. 2017;355(6330):1152–1158. doi:10.1126/science.aam7344
39. Lord CJ, Tutt AN, Ashworth A. Synthetic lethality and cancer therapy: lessons learned from the development of PARP inhibitors. Annu Rev Med. 2015;66:455–470. doi:10.1146/annurev-med-050913-022545
40. Moore K, Colombo N, Scambia G, et al. Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. 2018;379(26):2495–2505. doi:10.1056/NEJMoa1810858
41. Sakai W, Swisher EM, Karlan BY, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451(7182):1116–1120. doi:10.1038/nature06633
42. Swisher EM, Sakai W, Karlan BY, Wurz K, Urban N, Taniguchi T. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res. 2008;68(8):2581–2586. doi:10.1158/0008-5472.CAN-08-0088
43. Gonzalez-Martin A, Pothuri B, Vergote I, et al. Niraparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. 2019;381(25):2391–2402. doi:10.1056/NEJMoa1910962
44. Monk BJ, Parkinson C, Lim MC, et al. A randomized, phase iii trial to evaluate rucaparib monotherapy as maintenance treatment in patients with newly diagnosed ovarian cancer (ATHENA-MONO/GOG-3020/ENGOT-ov45). J Clin Oncol. 2022;JCO2201003.
45. Coleman RL, Fleming GF, Brady MF, et al. Veliparib with first-line chemotherapy and as maintenance therapy in ovarian cancer. N Engl J Med. 2019;381(25):2403–2415. doi:10.1056/NEJMoa1909707
46. Ray-Coquard I, Pautier P, Pignata S, et al. Olaparib plus bevacizumab as first-line maintenance in ovarian cancer. N Engl J Med. 2019;381(25):2416–2428. doi:10.1056/NEJMoa1911361
47. Hardesty MM, Krivak TC, Wright GS, et al. OVARIO phase II trial of combination niraparib plus bevacizumab maintenance therapy in advanced ovarian cancer following first-line platinum-based chemotherapy with bevacizumab. Gynecol Oncol. 2022;166(2):219–229. doi:10.1016/j.ygyno.2022.05.020
48. Pujade-Lauraine E, Ledermann JA, Selle F, et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a double-blind, randomised, placebo-controlled, Phase 3 trial. Lancet Oncol. 2017;18(9):1274–1284. doi:10.1016/S1470-2045(17)30469-2
49. Coleman RL, Oza AM, Lorusso D, et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390(10106):1949–1961. doi:10.1016/S0140-6736(17)32440-6
50. Boussios S, Abson C, Moschetta M, et al. Poly (ADP-Ribose) polymerase inhibitors: talazoparib in ovarian cancer and beyond. Drugs R D. 2020;20(2):55–73. doi:10.1007/s40268-020-00301-8
51. Penson RT, Valencia RV, Cibula D, et al. Olaparib Versus Nonplatinum Chemotherapy in Patients With Platinum-Sensitive Relapsed Ovarian Cancer and a Germline BRCA1/2 Mutation (SOLO3): a Randomized Phase III Trial. J Clin Oncol. 2020;38(11):1164–1174. doi:10.1200/JCO.19.02745
52. Ledermann JA, Harter P, Gourley C, et al. Overall survival in patients with platinum-sensitive recurrent serous ovarian cancer receiving olaparib maintenance monotherapy: an updated analysis from a randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Oncol. 2016;17(11):1579–1589. doi:10.1016/S1470-2045(16)30376-X
53. Kristeleit R, Lisyanskaya A, Fedenko A, et al. Rucaparib versus standard-of-care chemotherapy in patients with relapsed ovarian cancer and a deleterious BRCA1 or BRCA2 mutation (ARIEL4): an international, open-label, randomised, phase 3 trial. Lancet Oncol. 2022;23(4):465–478. doi:10.1016/S1470-2045(22)00122-X
54. Lampert EJ, Hays JL, Kohn EC, et al. Phase I/Ib study of olaparib and carboplatin in heavily pretreated recurrent high-grade serous ovarian cancer at low genetic risk. Oncotarget. 2019;10(30):2855–2868. doi:10.18632/oncotarget.26869
55. Oza AM, Cibula D, Benzaquen AO, et al. Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: a randomised phase 2 trial. Lancet Oncol. 2015;16(1):87–97. doi:10.1016/S1470-2045(14)71135-0
56. Matulonis UA, Berlin S, Ivy P, et al. Cediranib, an oral inhibitor of vascular endothelial growth factor receptor kinases, is an active drug in recurrent epithelial ovarian, fallopian tube, and peritoneal cancer. J Clin Oncol. 2009;27(33):5601–5606. doi:10.1200/JCO.2009.23.2777
57. Liu JF, Barry WT, Birrer M, et al. Combination cediranib and olaparib versus olaparib alone for women with recurrent platinum-sensitive ovarian cancer: a randomised phase 2 study. Lancet Oncol. 2014;15(11):1207–1214. doi:10.1016/S1470-2045(14)70391-2
58. Donawho CK, Luo Y, Luo Y, et al. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin Cancer Res. 2007;13(9):2728–2737. doi:10.1158/1078-0432.CCR-06-3039
59. Kummar S, Ji J, Morgan R, et al. A phase I study of veliparib in combination with metronomic cyclophosphamide in adults with refractory solid tumors and lymphomas. Clin Cancer Res. 2012;18(6):1726–1734. doi:10.1158/1078-0432.CCR-11-2821
60. Kummar S, Oza AM, Fleming GF, et al. Randomized trial of oral cyclophosphamide and veliparib in high-grade serous ovarian, primary peritoneal, or fallopian tube cancers, or BRCA-mutant ovarian cancer. Clin Cancer Res. 2015;21(7):1574–1582. doi:10.1158/1078-0432.CCR-14-2565
61. Villalona-Calero MA, Duan W, Zhao W, et al. Veliparib alone or in combination with mitomycin C in patients with solid tumors with functional deficiency in homologous recombination repair. J Natl Cancer Inst. 2016;108(7):djv437. doi:10.1093/jnci/djv437
62. Jiao S, Xia W, Yamaguchi H, et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin Cancer Res. 2017;23(14):3711–3720. doi:10.1158/1078-0432.CCR-16-3215
63. Xue C, Xu Y, Ye W, et al. Expression of PD-L1 in ovarian cancer and its synergistic antitumor effect with PARP inhibitor. Gynecol Oncol. 2020;157(1):222–233. doi:10.1016/j.ygyno.2019.12.012
64. Konstantinopoulos PA, Waggoner S, Vidal GA, et al. Single-arm Phases 1 and 2 trial of niraparib in combination with pembrolizumab in patients with recurrent platinum-resistant ovarian carcinoma. JAMA Oncol. 2019;5(8):1141–1149. doi:10.1001/jamaoncol.2019.1048
65. Chiappa M, Guffanti F, Bertoni F, Colombo I, Damia G. Overcoming PARPi resistance: preclinical and clinical evidence in ovarian cancer. Drug Resist Updat. 2021;55:100744. doi:10.1016/j.drup.2021.100744
66. Dias MP, Moser SC, Ganesan S, Jonkers J. Understanding and overcoming resistance to PARP inhibitors in cancer therapy. Nat Rev Clin Oncol. 2021;18(12):773–791. doi:10.1038/s41571-021-00532-x
67. Kyo S, Kanno K, Takakura M, et al. Clinical landscape of PARP inhibitors in ovarian cancer: molecular mechanisms and clues to overcome resistance. Cancers. 2022;14(10):2504. doi:10.3390/cancers14102504
68. Piombino C, Cortesi L. Insights into the possible molecular mechanisms of resistance to PARP inhibitors. Cancers. 2022;14(11):2804. doi:10.3390/cancers14112804
69. Keating GM. Bevacizumab: a review of its use in advanced cancer. Drugs. 2014;74(16):1891–1925. doi:10.1007/s40265-014-0302-9
70. Cannistra SA, Matulonis UA, Penson RT, et al. Phase II study of bevacizumab in patients with platinum-resistant ovarian cancer or peritoneal serous cancer. J Clin Oncol. 2007;25(33):5180–5186. doi:10.1200/JCO.2007.12.0782
71. Perren TJ, Swart AM, Pfisterer J, et al. A phase 3 trial of bevacizumab in ovarian cancer. N Engl J Med. 2011;365(26):2484–2496. doi:10.1056/NEJMoa1103799
72. Oza AM, Cook AD, Pfisterer J, et al. Standard chemotherapy with or without bevacizumab for women with newly diagnosed ovarian cancer (ICON7): overall survival results of a phase 3 randomised trial. Lancet Oncol. 2015;16(8):928–936. doi:10.1016/S1470-2045(15)00086-8
73. Stark D, Nankivell M, Pujade-Lauraine E, et al. Standard chemotherapy with or without bevacizumab in advanced ovarian cancer: quality-of-life outcomes from the International Collaboration on Ovarian Neoplasms (ICON7) phase 3 randomised trial. Lancet Oncol. 2013;14(3):236–243. doi:10.1016/S1470-2045(12)70567-3
74. Burger RA, Brady MF, Bookman MA, et al. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N Engl J Med. 2011;365(26):2473–2483. doi:10.1056/NEJMoa1104390
75. Pujade-Lauraine E, Hilpert F, Weber B, et al. Bevacizumab combined with chemotherapy for platinum-resistant recurrent ovarian cancer: the AURELIA open-label randomized phase III trial. J Clin Oncol. 2014;32(13):1302–1308. doi:10.1200/JCO.2013.51.4489
76. Aghajanian C, Blank SV, Goff BA, et al. OCEANS: a randomized, double-blind, placebo-controlled phase III trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. J Clin Oncol. 2012;30(17):2039–2045. doi:10.1200/JCO.2012.42.0505
77. Aghajanian C, Goff B, Nycum LR, Wang YV, Husain A, Blank SV. Final overall survival and safety analysis of OCEANS, a phase 3 trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent ovarian cancer. Gynecol Oncol. 2015;139(1):10–16. doi:10.1016/j.ygyno.2015.08.004
78. Mirza MR, Avall Lundqvist E, Birrer MJ, et al. Niraparib plus bevacizumab versus niraparib alone for platinum-sensitive recurrent ovarian cancer (NSGO-AVANOVA2/ENGOT-ov24): a randomised, phase 2, superiority trial. Lancet Oncol. 2019;20(10):1409–1419. doi:10.1016/S1470-2045(19)30515-7
79. Monk BJ, Poveda A, Vergote I, et al. Anti-angiopoietin therapy with trebananib for recurrent ovarian cancer (TRINOVA-1): a randomised, multicentre, double-blind, placebo-controlled phase 3 trial. Lancet Oncol. 2014;15(8):799–808. doi:10.1016/S1470-2045(14)70244-X
80. Monk BJ, Poveda A, Vergote I, et al. Final results of a phase 3 study of trebananib plus weekly paclitaxel in recurrent ovarian cancer (TRINOVA-1): long-term survival, impact of ascites, and progression-free survival-2. Gynecol Oncol. 2016;143(1):27–34. doi:10.1016/j.ygyno.2016.07.112
81. Tew WP, Colombo N, Ray-Coquard I, et al. Intravenous aflibercept in patients with platinum-resistant, advanced ovarian cancer: results of a randomized, double-blind, phase 2, parallel-arm study. Cancer. 2014;120(3):335–343. doi:10.1002/cncr.28406
82. Alberts DS, Liu PY, Hannigan EV, et al. Intraperitoneal cisplatin plus intravenous cyclophosphamide versus intravenous cisplatin plus intravenous cyclophosphamide for stage III ovarian cancer. N Engl J Med. 1996;335(26):1950–1955. doi:10.1056/NEJM199612263352603
83. van Driel WJ, Koole SN, Sikorska K, et al. Hyperthermic intraperitoneal chemotherapy in ovarian cancer. N Engl J Med. 2018;378(3):230–240. doi:10.1056/NEJMoa1708618
84. Spiliotis J, Halkia E, Lianos E, et al. Cytoreductive surgery and HIPEC in recurrent epithelial ovarian cancer: a prospective randomized phase III study. Ann Surg Oncol. 2015;22(5):1570–1575. doi:10.1245/s10434-014-4157-9
85. Wong OGW, Li J, Cheung ANY. Targeting DNA damage response pathway in ovarian clear cell carcinoma. Front Oncol. 2021;11. doi:10.3389/fonc.2021.666815
86. Soto J. The treatment of clear cell ovarian cancer with the poly(ADP-ribose) polymerase (PARP1) inhibitors (AG14361, veliparib, olaparib) as chemosensitizers. bioRxiv. 2020.
87. Ogasawara A, Sato S, Hasegawa K. Current and future strategies for treatment of ovarian clear cell carcinoma. J Obstet Gynaecol Res. 2020;46(9):1678–1689. doi:10.1111/jog.14350
88. Nugawela D, Gorringe KL. Targeted therapy for mucinous ovarian carcinoma: evidence from clinical trials. Int J Gynecol Cancer. 2022;ijgc-2022–003658.
89. Gorringe KL, Cheasley D, Wakefield MJ, et al. Therapeutic options for mucinous ovarian carcinoma. Gynecol Oncol. 2020;156(3):552–560. doi:10.1016/j.ygyno.2019.12.015
90. Foundation Medicine. FoundationOne®Cdx. FoundationOne CDx | foundation Medicine. Available from: https://www.foundationmedicine.com/test/foundationone-cdx.
91. Yoshii Y, Okazaki S, Takeda M. Current status of next-generation sequencing-based cancer genome profiling tests in japan and prospects for liquid biopsy. Life. 2021;11(8):5148.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.