Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Toward a magic or imaginary bullet? Ligands for drug targeting to cancer cells: principles, hopes, and challenges
Authors Toporkiewicz M, Meissner J, Matusewicz L, Czogalla A, Sikorski AF
Received 18 September 2014
Accepted for publication 26 November 2014
Published 17 February 2015 Volume 2015:10(1) Pages 1399—1414
DOI https://doi.org/10.2147/IJN.S74514
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Monika Toporkiewicz, Justyna Meissner, Lucyna Matusewicz, Aleksander Czogalla, Aleksander F Sikorski
Laboratory of Cytobiochemistry, Faculty of Biotechnology, University of Wrocław, Wrocław, Poland
Abstract: There are many problems directly correlated with the systemic administration of drugs and how they reach their target site. Targeting promises to be a hopeful strategy as an improved means of drug delivery, with reduced toxicity and minimal adverse side effects. Targeting exploits the high affinity of cell-surface-targeted ligands, either directly or as carriers for a drug, for specific retention and uptake by the targeted diseased cells. One of the most important parameters which should be taken into consideration in the selection of an appropriate ligand for targeting is the binding affinity (KD). In this review we focus on the importance of binding affinities of monoclonal antibodies, antibody derivatives, peptides, aptamers, DARPins, and small targeting molecules in the process of selection of the most suitable ligand for targeting of nanoparticles. In order to provide a critical comparison between these various options, we have also assessed each technology format across a range of parameters such as molecular size, immunogenicity, costs of production, clinical profiles, and examples of the level of selectivity and toxicity of each. Wherever possible, we have also assessed how incorporating such a targeted approach compares with, or is superior to, original treatments.
Keywords: targeting, drug delivery, tumor, monoclonal antibody, EGFR, cancer
Introduction
In the past few decades, significant progress has been made in understanding the molecular principles of oncologic diseases. Based on the extensive knowledge base that has been developed regarding the “hallmarks” of cancers, many different strategies have been formulated and evaluated for cancer treatment and drug-targeting to tumor cells. Some of these systems exploit the overexpression of cancer-related surface-markers on diseased cells or the development of a dense, but leaky, vascular system within a tumor, forming the basis of a tumor targeting strategy.1
Many problems are currently associated with systemic drug administration. To reach the target site, the drug usually has to cross through several biological barriers in the organism, such as blood vessels, tissues, organs, cells, or even subcellular compartments within the target cell itself. Absence of specificity for the disease site, and the necessity to use very high doses of drugs to achieve sufficient local concentrations, promote the occurrence of nonspecific toxicity and other adverse side effects. A drug-targeting strategy could, potentially, solve the majority of these problems.
The concept of the “magic bullet” approach of drug-targeting was first proposed by Paul Ehrlich.2,3 It relies on the use of targeting-ligands, such as antibodies and their derivatives, peptides, or small molecules, which specifically bind to a receptor that is unique to, or overexpressed at the target site. The two key facilitators of an active delivery mechanism of molecules to a surface ligand are high binding-specificity and affinity. By targeting of nanoparticles carrying active pharmacological molecules, it is possible to achieve drug delivery to destination cells in vivo, maximizing the therapeutic efficacy of the drug and reducing its adverse side effects. The concept of directing drugs attached to “homing” molecules to sites of disease became possible due to recent advances on an interdisciplinary basis across the fields of tumor biology, chemistry, and bioengineered technologies.4,5
For many years the main driving force for drug delivery in cancer treatment was a nontargeted, or systemic drug administration. However, targeting is expected to increase intratumoral accumulation and, especially in the case of targeting by internalized ligands, to higher intracellular concentrations of the drug (Figure 1).6 Such approaches are focused on increasing the interactions between nanoparticles and cells by enhancing their internalization, but without altering the overall biodistribution.7 Two key benefits arise from such a targeting strategy, namely, that the specific antigen is accessible only on the targeted cells, and that antigen localization and expression remain as specific biomarkers of the target cell population throughout the treatment.8 These properties are now being actively exploited in biomedical research for targeting of drugs.
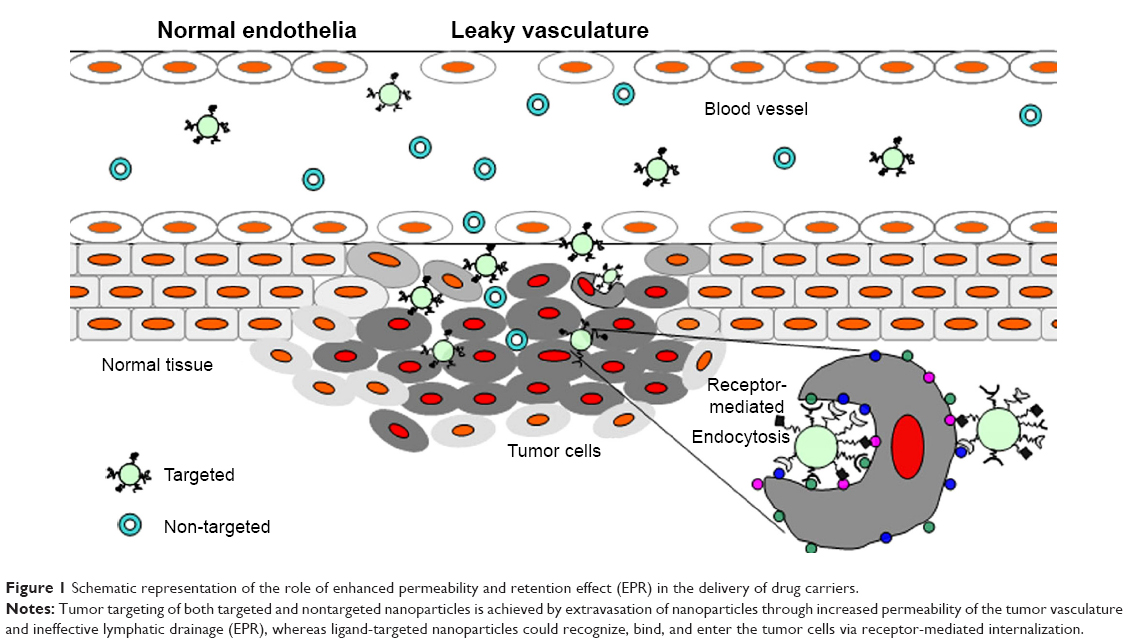 | Figure 1 Schematic representation of the role of enhanced permeability and retention effect (EPR) in the delivery of drug carriers. |
At present, many types of nanoparticles are under advanced studies as potential drug carriers. Nanoparticle-based drug formulations could potentially be more efficient and less toxic than conventional drug formulations. Some of the most widely used nanoparticles are liposomes, micelles, dendrimers, nanotubes, and polymers. For characteristics of these nanoparticles, see excellent reviews.9–14
Here, we focus on ligands which are used for targeting to cancer cells, such as monoclonal antibodies and their engineered fragments, peptides, aptamers, and proteins including DARPins, transferrin, lactoferrin, and lectins, as well as small molecules including folates and mannose derivatives. Their specificity, affinity and effectiveness, and their side effects, such as immunogenicity, are discussed.
Strategy for targeting
There are many different types of ligands which may be used as a basis for targeted delivery. Table 1 provides examples of ligands in terms of the most important parameters that should be taken into account when choosing a ligand for targeted drug delivery. We have focused on binding affinity as the most significant parameter, along with others, such as size, immunogenicity, clinical use, and cost of production. The ligand should be unique to the target cell and be characterized by the highest binding affinity and lowest immunogenicity. It should also enable penetration of the tumor. The best choice in this respect would be to use the most unique marker for a particular tumor or, even better, for its stem cells. However, at present, not many such markers are available, although substantial progress is being made in this field.15
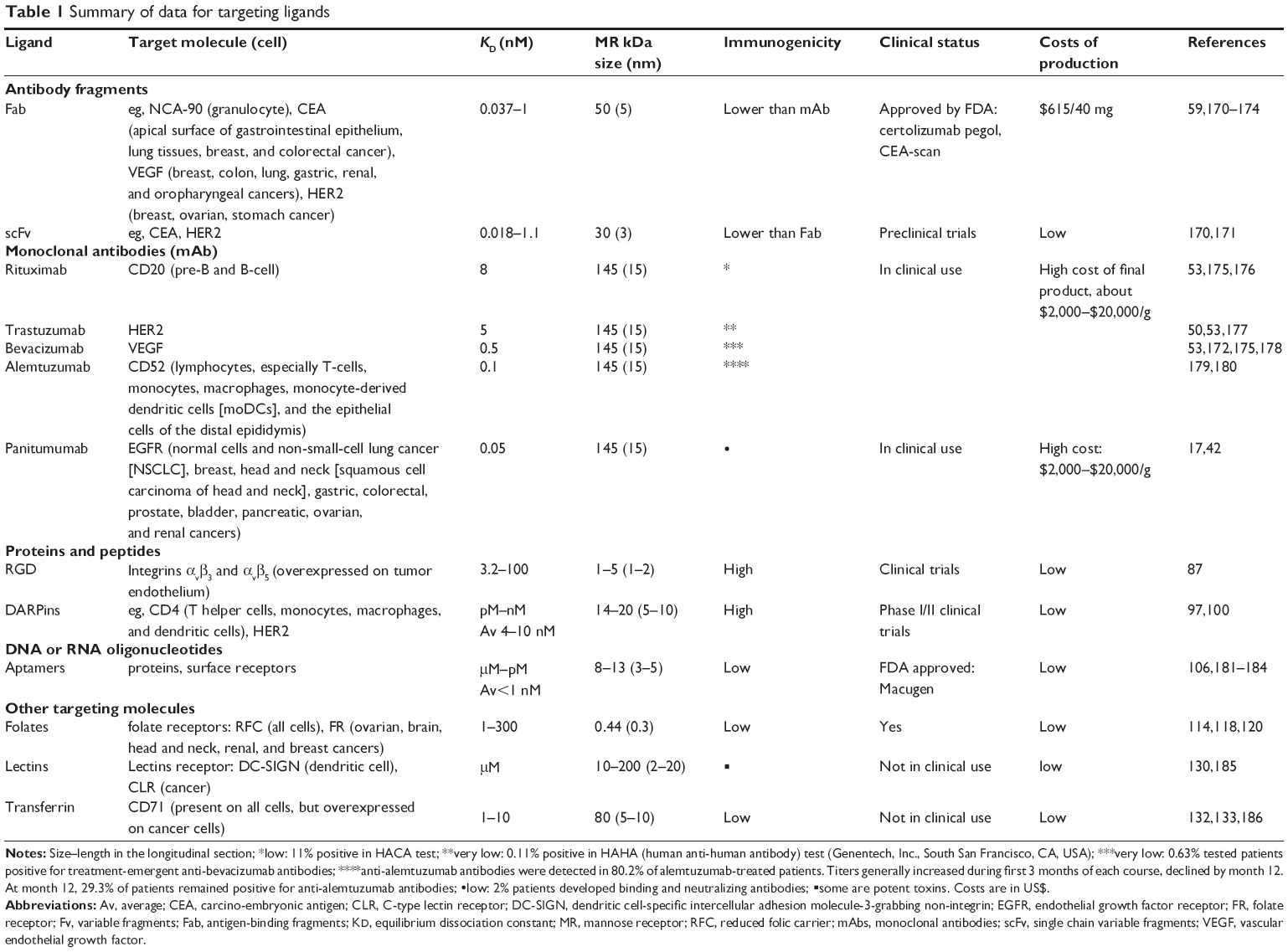 | Table 1 Summary of data for targeting ligands |
Monoclonal antibodies
Monoclonal antibodies (mAbs) possess many desirable technical attributes and advantages as tools for biomolecular targeting, including molecular homogeneity, specificity of interaction, high binding affinity in the nanomolar range, and ease of selection. mAbs represent a single molecular species that bind to antigens with the same affinity and promote the same effector functions. Therapeutic mAbs are usually completely humanized, or produced as chimeric proteins, in order to avoid unwanted immune reactions in patients.16,17 Chimeric antibodies contain human kappa and gamma constant regions (Fc domains) and murine light- and heavy-chain variable region (Fab domains). Chimeric mouse–human antibodies, such as rituximab, are genetically engineered. The protein sequences of humanized antibodies are essentially identical to that of their human variants, despite the nonhuman origin of some of the complementarity determining regions (CDR) of their variable chains. The production of humanized monoclonal antibodies can be accomplished using recombinant DNA technology followed by expression in mammalian cell culture.18 The main function of therapeutic mAbs is based on the induction of antibody-dependent cellular cytotoxicity (ADCC). However, in the case of a targeting approach, the antibody is typically used only as a “hook” that facilitates binding of the larger delivery particle with the site of interest on the target cell. Nevertheless, some mAbs also have direct pharmacological effects, mediated via a biological response within the target tissues.
Tumors and surrounding peritumoral cells can produce strongly immunosuppressive cytokines and growth factors, such as transforming growth factor-β (TGFβ). TGFβ has been shown to promote tumor escape from the control of the immune system. GC1008 is a fully humanized TGFβ-specific antibody that binds to all three isoforms of TGFβ.19 This antibody is currently being evaluated in clinical trials of patients with metastatic kidney cancer or malignant melanoma.20
One of the first molecules targeted by mAb therapy was the epidermal growth factor receptor (EGFR). EGFR belongs to the ErbB oncogene family, which consists of four members, namely, ErbB-1, -2, -3, and -4 (also known as HER-1, -2, -3, -4). EGFR plays a crucial pleotropic role in cell proliferation, differentiation, survival, angiogenesis, and metastasis.21 ErbB receptors consist of an N-terminal extracellular domain, a short transmembrane domain, an intracellular catalytic tyrosine kinase domain, and several intracellular tyrosine residues. EGFR exists in the plasma membrane as a monomer. Upon ligand binding, the receptor undergoes domain rearrangement that allows formation of a homo- or heterodimer with either HER2 or another EGFR molecule. This event brings two intracellular receptor domains together and triggers their intrinsic kinase activation.22–24 When activated, the EGFR kinase phosphorylates several tyrosine residues in the C-terminal tail of the EGFR which becomes a docking site for downstream signaling effectors that initiate signaling cascades and stimulate cell proliferation.25 Activation through homo- or heterodimerization underlies the combinatorial activation of the EGFR family of receptors, HER2, HER3, and HER4. HER2 is an atypical member of the ErbB family in that it is not directly activated by ligand, but, rather serves as a universal heterodimeric partner for each of the other ErbB family members.26
Normal cells express up to 1×105 EGF receptors per cell. However, tumor cells can express up to 200-fold more receptors per cell.27,28 Felder et al29 have showed the existence of two populations of EGFR: one of high affinity (1%–2% of the total number of EGFR) showing a twofold higher affinity, at 0.1 nM EGF, compared to the other of low affinity at saturating EGF concentrations. Overexpression of ErbB proteins is frequently found in many different human tumors of epithelial origin (approximately 30% of all tumors), such as non-small-cell lung cancer (NSCLC), breast, head and neck (squamous cell carcinoma of head and neck), gastric, colorectal, prostate, bladder, pancreatic, ovarian, and renal cancers.30–32 Overexpression, in association with a number of mutations in the ErbB family of genes have been implicated in malignant diseases, and their presence strongly correlates with aggressive pathological characteristics, drug resistance, and poor survival rates.33 Given the role of EGFR in contributing to the development of malignancy, the opportunity to target the EGFR pathway is considered as a potent strategy in medical oncology.27
Based on the canonical model of ligand-induced dimerization and activation of the EGFR,34,35 two general approaches involving mAbs have been proposed for its inhibition, namely, that the antibodies either recognize the ligand binding site (cetuximab, panitumubab) and inhibit ligand-mediated activation of the receptor, or they prevent the subsequent dimerization process of the receptor by inhibiting the structural changes occurring in the ectodomain upon ligand binding (matuzumab). Examples of anti-EGFR mAbs that have undergone or, currently are in clinical testing are cetuximab (IMC-C225),36 matuzumab (EMD 72000-monoclonal antibody 425),35,37 panitumumab, also known as ABX-EGF, VECTIBIX,38 and necitumumab (IMC-11F8).39
Cetuximab (IMC-C225) is a chimeric human–murine monoclonal anti-EGFR antibody that is currently in Phase II and Phase III testing. Cetuximab inhibits EGFR activation by competing directly with EGF for its binding site on domain III of the receptor, preventing ligand binding and receptor activation.40 Specifically, cetuximab binds to the EGFR and induces its internalization and degradation, with a concomitant upregulation of p27Kip1 and cell cycle arrest in G1, enhanced apoptosis, inhibition of angiogenesis, and induction of ADCC.41
Panitumumab is a fully humanized IgG2 mAb that binds with high affinity to the ligand-binding domain of EGFR.42 Panitumumab, similarly to cetuximab, competitively blocks the binding of EGF and TGFα to EGFR, thus inhibiting autophosphorylation induced by EGFR ligands.38 Cetuximab and panitumumab were recently launched and marketed for colon, head and neck, and/or lung cancers, covering a limited range of solid tumors.34,43 Necitumumab (IMC-11F8), designed to bind and block the ligand-binding site of EGFR, is another IgG1 antibody which is currently under investigation in clinical trials of patients with NSCLC.39,44
Matuzumab is a humanized murine IgG1 monoclonal antibody that binds at a site near to, but distinct from the EGF-binding site on the EGFR and displays a constellation of biochemical and inhibitory properties.45 The Fc receptor recognizes the Fc portion of the IgG1 protein. Hence, it is predicted that this antibody should be capable of triggering ADCC in human tumor cells that express the EGFR antigen. Phase I studies of this agent have demonstrated single-agent activity in colon cancer, with the predominant side effect being mild skin toxicity.46
HER2 is a more potent oncoprotein than the other ErbBs, and was first discovered as a rodent carcinogen-induced oncogene that encodes a variant of HER2 with a mutation that makes its tyrosine kinase constitutively active. Trastuzumab (Herceptin), which is a humanized monoclonal antibody to HER2, has been the first treatment to reach widespread clinical use, particularly for the treatment of metastatic breast cancer. Trastuzumab induces ADCC.47
Another monoclonal antibody directed to HER2, which has been clinically tested is pertuzumab, which binds directly to the dimerization arm of HER2 and blocks both its dimerization and activation in response to stimulation of an HER2 partner,48 such as HER3 (Table 1)49 (Figure 2).
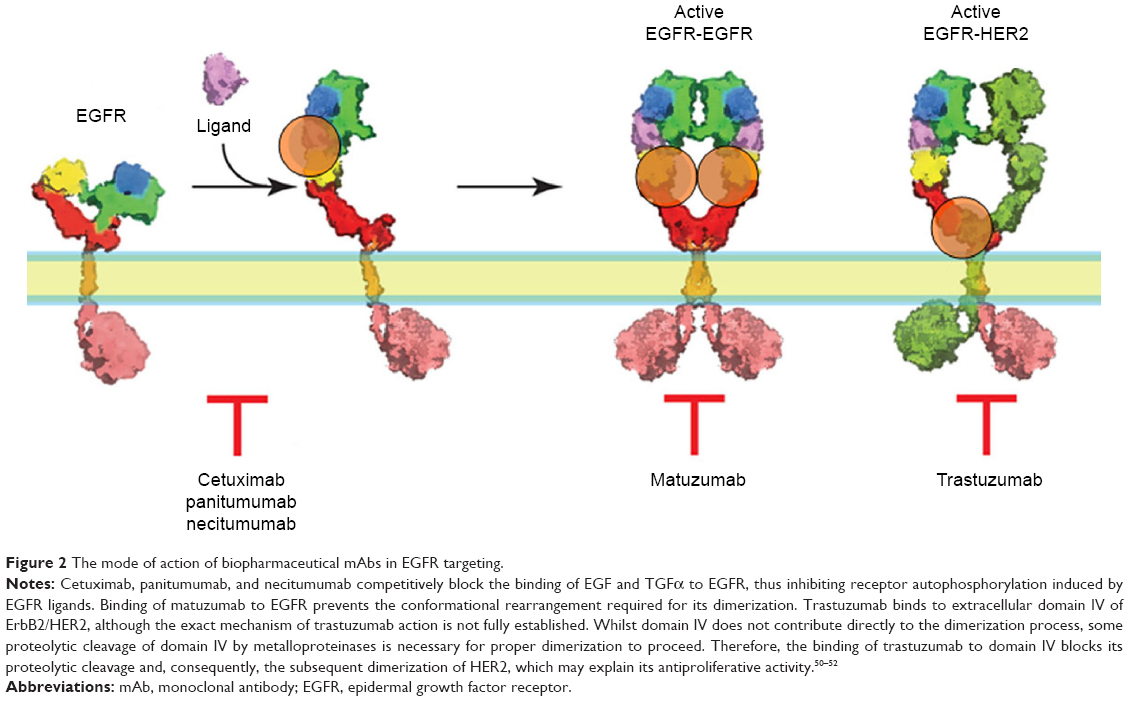 | Figure 2 The mode of action of biopharmaceutical mAbs in EGFR targeting. |
Alemtuzumab, a monoclonal IgG against CD52, is effective in patients with relapsing-multiple sclerosis and is currently in Phase III clinical trials. Alemtuzumab was more effective than interferon beta-1a in preventing relapses over two years of study, with a 59.9% improvement in previously untreated patients and a 49.4% improvement in patients who had had previous treatment with interferon beta.53
Mazar et al54 have developed a monoclonal antibody, ATN-291, that is specific for human urokinase plasminogen activator (uPA) (see “Peptides” section). The binding affinity of ATN-291 to uPA was about 0.5 nM. The ATN-291 and uPA complex was internalized in a manner specific to uPA. A novel stealth approach utilizing liposome-encapsulated arsenic trioxide, called nanobins (NB), utilized ATN-291 antibody as a targeting mechanism. ATN-291-NB has retained the binding affinity of free ATN-291. NBs were taken up by urokinase plasminogen activator receptor (uPAR)-expressing but not uPAR-negative cells. Several approaches have focused on using fully humanized antibodies to target uPAR. These antibodies exhibit antitumor effects, inhibiting tumor cell invasion in vitro, tumor growth by 25%–50% in vivo, and affected uPAR-dependent signaling in vivo.55 Approaches exploiting uPAR-targeted therapeutics have reached a stage where the targeting of uPAR has been validated as a promising strategy in cancer and the first uPAR-targeted molecule is expected to advance into the clinic in the near future.54
Based on our experience, targeting with BCL2 antisense oligonucleotides encapsulated within liposomes constituted with a therapeutic antibody against CD20 selectively and effectively reached their target site in vivo in a NOD-SCID mouse xenograft Daudi model. Moreover, a significant therapeutic effect was demonstrated via this treatment. CD20-BCL2 antisense targeted liposomes showed excellent therapeutic efficacy with 100% tumor growth inhibition compared with the mice treated with CD20-targeted liposomes containing scrambled BCL2 antisense sequence. The same formulation of BCL2-antisense encapsulated liposomes, but nonsurface-targeted, showed a much lower therapeutic effect with maximally about 30% tumor growth inhibition.56 Liposomes targeted with therapeutic antibody accumulated in the tumor more extensively when compared to nontargeted liposomes.
Last, but not least, the generation of new or modified mAbs is both laborious and costly. Altogether, these aspects have prompted scientists to seek alternative antibody formats that provide the same binding specificity of mAbs, but with desired improvements in their performance (Table 1).
Antibody derivatives
To overcome the size restrictions of the full-length monoclonal antibody molecule, naturally-derived or fully synthetic antigen-binding fragments (Fab and Fab’; ~50 kDa), variable fragments (Fv; ~15 kDa), and single chain variable fragments (scFv; ~30 kDa) have been engineered and tested.57,58 Fab′ and scFv fragments can be selected by phage-display and are engineered more easily than mAbs, to control properties such as affinity (KD usually lower than 1 nM) or internalization capabilities.59 All of these antigen-binding fragments lack the Fc-antibody region, which is most immunogenic.
As mentioned above, IgGs have been dissected into their constituent domains as either monovalent Fab, scFv, single variable VH and VL domains, or bivalent fragments, such as (Fab′)2. Many such molecules are now in clinical or preclinical trials. A number of Fab fragments have already been approved by the FDA and a number of additional entities are currently in clinical trials.59 Antibody fragments seem set to join mAbs as powerful therapeutic and diagnostic agents, particularly for targeting cancer, inflammatory, autoimmune, and viral diseases. Antibody Fab and scFv fragments, containing VH and VL domains, usually retain the specific antigen-binding affinity of the parent antibody, but are usually characterized by improved pharmacokinetics with respect to tissue penetration. Antibody derivatives have a variety of applications, ranging from simple research tools as a diagnostic, or companion diagnostic, to highly refined biopharmaceutical drugs in their own right. Their huge selectivity and ease of engineering modulation has facilitated more sophisticated applications (delivery vehicles for gene therapy).59
CroFab® (Savage Laboratories, Melville, NY, USA) is a Fab (ovine) antibody fragment format approved by the FDA for clinical use in patients who have been bitten by venomous snakes, such as rattlesnakes, copperheads, and cottonmouths/water moccasins. Early use of CroFab®, within 6 hours of the snakebite, is advised to prevent clinical deterioration and the occurrence of systemic coagulation abnormalities.60 Sulesomab (LeukoScan®; Immunomedics, Inc., Morris Plains, NJ, USA) is a murine monoclonal IgG antibody Fab′ fragment labeled with the isotope technetium-99m. The fragment targets NCA-90, found on the cell membrane of granulocytes. Using a gamma camera, LeukoScan® can be used to detect osteomyelitis, a bone infection. LeukoScan® is not available in North America, but it has European-wide registration and it is also approved in Australia. A number of Fab fragments (Lucentis, Thromboview, CDP791, CDP870, MDX-H210) and scFv (pexelizumab, CC49, SGN-17, C6.5K-A), are currently in preclinical and clinical trials (Table 1).59
Single-domain antibodies consisting of a single monomeric variable antibody domain, also known as nanobodies, have progressed into the clinic, demonstrating the belief in the potential of this type of molecule as a valuable treatment option for patients suffering from Alzheimer’s complex disease, for which no adequate drugs are currently available.61
Peptides
The main strategy to select proper peptide ligands is to screen peptide libraries produced by phage-display62,63 or chemical synthesis.64 The phage-display method is more widely used and enables the selection of small peptides, a hundredth the mass of IgG antibodies.65 This method can be used to identify peptides that target a specific receptor with an affinity in the μM to nM range, or certain cell types, even if the receptors are unknown. A number of such peptides that home specifically to various organs under normal or pathological conditions have been identified.66 These peptides have been used for targeted delivery of oligonucleotides, drugs, imaging agents, nanoparticles, viruses, and liposomes.67
Peptide-based delivery has many advantages. The small size of peptides allows more efficient penetration to the tissue, compared to antibodies and proteins. Peptides can be chemically synthesized at a large scale relatively inexpensively and, unlike in the case of recombinant expression technologies, the removal of endotoxins or cell culture-derived contaminants is not necessary.67,68 Despite their small size, some peptides have binding affinities comparable to specific antibodies.69
In vivo phage-display technology makes use of peptide libraries composed of short, random, amino acid sequences expressed on the surface of the phage particles. Typically, these phage libraries are injected into the tail vein of a mouse and allowed to circulate for a short period of time, around 5–15 minutes, to allow for binding of phages displaying peptides that recognize surface epitopes on the target tissue. The bound phage can then be “rescued” from the target organ, amplified, and the whole process repeated a number of times in order to obtain a specific phage-peptide with high affinity for the target tissue.70 The principle behind the peptide homing strategy is that they should only recognize molecules which are upregulated in tumors and, therefore, would not recognize normal cells from the corresponding organ.67,70
Numerous peptide ligands have been isolated against various types of receptors or cells, such as RGD-containing peptides against integrin receptors in angiogenic tumor vasculature65,71–74 (Table 1), or specific for PDGFR-β receptor in pericytes and endothelial cells,75 KRK-containing peptides directed to angiogenic blood vessels and tumor cells,76 and a peptide recognizing thrombin receptor.77 Tissue-specific homing peptides have also been reported for pancreatic β cells,78 as well as specific peptides for tumor cells, especially lung tumor.65,79 There is also a known peptide sequence, designated as GE11, which recognizes the EGFR.80
Tumor-homing peptides have already entered clinical trials. Results from several Phase I and II trials have been reported, and a number of trials are currently ongoing or at the stage of recruiting patients for trials.63,81–84 The results of clinical trials so far have been very encouraging, with reports of improved outcomes in terms of therapeutic efficacy, such as the absence of any dose-limiting toxicity and good tolerance of all peptide-targeted therapy combinations.67
The most widely used peptides in the targeted-delivery applications are integrin-targeting RGD-peptides – the first tumor-targeting peptides discovered.68 Integrins αvβ3 and αvβ5 are overexpressed on tumor endothelium and some epithelial cells during tumor growth, angiogenesis, invasion, and metastasis. Therefore, they represent an interesting molecular target for a tumor-homing approach. RGD is a cell-adhesion motif present in many proteins of the extracellular matrix (ECM). This motif is recognized by αvβ3 and αvβ5 integrin receptors. The binding affinities of some of the RGD-containing derivatives for αvβ3 range from 3.2 to 100 nM.85,86 The addition of specific amino acid residues to peptide sequence motifs, such as RGD, that induce binding to cell-attachment proteins, strongly enhances the binding affinity of this peptide. The injection of the modified peptides induced antigen-specific serum antibodies.87
Among others targeting moieties, molecules such as small peptide LHRH (luteinizing hormone-releasing hormone) analogs or peptide analogs based on the uPAR binding region of uPA should also be mentioned. The receptor for LHRH is overexpressed in many tumors, including breast, ovarian, endometrial, prostate, hepatic, colorectal, and pancreatic cancers, renal cell carcinomas, and melanomas. Some small peptide LHRH analogs, such as DAla6 EA or DLeu6 EA, are characterized by high binding affinity to LHRH receptor (KD in nM range), and possess the ability to recognize a broad variety of tumors, but not normal cells.88 Use of these small peptides has certain advantages such as ease of preparation, low antigenicity, and increased stability when compared to conventional proteins.89 He et al89 showed that simple immunomodification of PEGylated mitoxantrone-loaded liposomes with LHRH analog, gonadorelin, against cancer-specific LHRH receptors results in development of universal tumor-targeted cytostatics delivery system.
uPAR is selectively overexpressed in most solid tumors and several hematological malignancies. uPAR mediates various signaling events essential for the differentiation and migration of cells within the tumor environment.90 The internalization of uPAR requires formation of the uPA-PAI-1-uPAR complex (complex composed of urokinase plasminogen activator, plasminogen activator inhibitor 1, and urokinase plasminogen activator receptor). Several studies have attempted to exploit the internalization of uPAR to deliver cytotoxic therapeutics to tumors. Peptide inhibitors of uPA binding to uPAR, based on the growth factor domain (GFD) of uPA, have been described. Those peptides usually bind to human uPAR with high affinity (KD <1 nM).91 Disulfide cyclized GFD-derived peptide (amino acids 19–31)-DOTA conjugates bound to 213Bi were cytotoxic to OV-MZ-6 ovarian cancer cells in vitro.92
DARPins (designed ankyrin repeat proteins)
DARPins are derived from natural ankyrin repeat proteins, which are the most abundant proteins identified in the human genome.93 DARPins libraries have been engineered by a consensus-design approach to generate designed ankyrin repeat proteins as an alternative to antibody-based scaffolds.94 DARPins are composed of four to six modules and their molecular weight ranges from 14 to 21 kDa, which is almost one-tenth of the size of the conventional IgG, and one-third the size of Fab fragments.95 Usually, with only a few rounds of selection, many different target-specific DARPins can be obtained, with affinities ranging from pM to low nM.96 Due to the small size of DARPins, they offer a much higher tissue penetration compared to antibodies and, therefore, are able to reach targets far outside of the bloodstream. The lack of an Fc domain in DARPin structures makes them less immunogenic than many of the alternative formats.97 The thermal stability of DARPins is usually very high, with the midpoint of denaturation between 65°C and 95°C,98 and with biological half-lives exceeding 60 days.97 Recent studies with full-consensus DARPins revealed that these proteins belong to the most stable proteins described to date. This stability, in association with the fact that the DARPin fold shows no flexible peptide domains, might explain why no proteolytic digestion has been detected in any experiment with their use.97,99
An investigation of the in vivo accumulation of HER2-specific DARPins in mouse xenografts, demonstrated a strong and direct correlation between the total amount located in the tumor and the respective affinity of the DARPin.96 In a separate study, DARPins interacting with human CD4 with very high affinity (KD value of 8.9 nM) were selected. This affinity is comparable with the range of KDs of high affinity antibodies (Table 1).100
Some DARPins are particularly suited to deliver active pharmacological moieties to sites of disease. This could be a benefit both in oncology, where DARPins are used to deliver toxins to tumors, and in treatment of inflammatory diseases, where DARPins could be designed to inhibit cytokines at sites of inflammation. At present, it is still unclear whether ankyrin-repeat proteins can be delivered in vivo at a level and in a format that are suited for controlling and affecting intracellular mechanisms and interactions. It has been shown, however, that PEG-modified DARPins show an increased serum half-life and still accumulate well in tumor tissue.97 Meanwhile, DARPins for ocular indications are currently in Phase I/II clinical trials.101
Aptamers
Aptamers are single-stranded DNA or RNA oligonucleotides, between 25–50 bases in length, with a molecular mass generally less than 20 kDa, and are derived from combinatorial libraries through an in vitro selection process called Systematic Evolution of Ligands through Exponential enrichment (SELEX).102 Aptamers are a novel and particularly interesting class of targeting ligands with a unique ability to bind a variety of targets including peptides, enzymes, antibodies, various cell surface receptors, and even small organic molecules with nanomolar or even picomolar affinities.103 In general, compared to other targeting agents, aptamers exploit unique benefits, such as a relatively small size, low immunogenicity, high affinity and selectivity, and the ease of their in vitro synthesis, which makes them attractive alternatives to antibodies and peptides.104 The SELEX protocol allows in vitro selection of aptamers capable of binding to a specific ligand with high selectivity and sensitivity. To make them more resistant to nucleolytic degradation, aptamers are typically chemically modified.105 Since the protocol for the in vitro selection of aptamers does not depend on a binding reaction in a biological system, the real affinity for a ligand in vivo could be completely different. To solve these drawbacks, aptamers are sometimes selected directly using whole living cells, pathogens, or even animal models.106 Some preclinical toxicological studies conducted on selected aptamers did not reveal any toxicity, either in rats (dosing of up to 100 mg/kg) or in dogs (96-hour continuous infusion at doses up to 10 mg/kg/day).107
Macugen® (Valeant Pharmaceuticals North America LLC, Bridgewater, NJ, USA) is the first and, so far the only aptamer approved by the FDA for clinical use,108 although there are eight other aptamers reportedly enrolled in clinical trials (Table 1).109
Other targeting molecules
Folic acid (or folate), other vitamins, transferrin, growth factors, hormones, and carbohydrates (hyaluronic acid) are naturally occurring ligands to cell surface receptors. The binding affinity values vary in range from low μM for lectins to high nM for folates. They have a huge advantage over other targeting approaches, due to lower molecular weights than antibodies, lower immunogenic index, relatively cheap production and, ease of handling and storage. Receptors for these ligands are often overexpressed on tumor cells, providing a rationale for selective drug delivery. However, receptor expression is generally not specific to tumor cells and normal cells may suffer some toxicity (Table 1).110
Folates
Folates are low molecular-weight vitamins required by eukaryotic cells for single-carbon metabolism and de novo nucleotide synthesis. There are two different mechanisms for the cellular uptake of folic acid (FA): reduced folate carrier (RFC) and folate receptor (FR). The carrier is a low affinity membrane-spanning protein that transports reduced folate directly into the cytosol.111 RFC is present on virtually all cells, whereas the high affinity FR (KD =1 nM) is expressed at high levels mainly on cancer cells eg, ovarian, brain, head and neck, renal, and breast cancers.112–114 Because animal cells lack key enzymes of the folate biosynthetic pathway, their survival and proliferation are dependent on their ability to acquire and utilize this vitamin.115 It has been demonstrated that receptor-mediated uptake of FA could also be successfully exploited to facilitate entry of an FA-attached molecule, macromolecule, or liposomes into cells.116–119 Gabizon et al120 showed that folate-targeted liposomes bind to the FR of J6456 lymphoma cells in vivo and play a significant role in liposome biodistribution in solid tumors. On the other hand, the experiments carried out by Leamon et al121 revealed that folate-targeted liposomes accumulated mainly in the mouse liver, because of activated liver-derived macrophages (Kupffer cells), which express FR.122 These studies indicate that liposome-targeting to the FR receptor has the potential to alter liposome biodistribution.120 This methodology is currently being investigated for the selective delivery of imaging and therapeutic agents to tumor tissue. Phase I and II clinical studies for the first folate-containing imaging agents have been initiated,118 but so far no folate-targeting particles are in clinical use.
Lectins
Lectins are multidomain proteins that can recognize and bind specifically to sugar complexes attached to proteins and lipids. Characteristic features of most interactions involving carbohydrates (either protein–carbohydrate or carbohydrate–carbohydrate interactions) are high specificity and low affinity. Nature overcomes this low affinity by clustering ligands and receptors at the cell surface, using avidity as a means of achieving a sufficient binding strength.123 Different cell types express various glycan arrays and transformed or cancer cells often express different glycans compared with their normal counterparts.124 A large number of different approaches have been used for C-type lectin receptor (CLR) for targeting glyconanoparticles, glycodendrimers, glycofullerenes, glycoclusters, and glycolipids.125 Fluorescent gold nanoparticles were used to display multiple copies of structural motifs of the N-linked high-mannose glycan of HIV gp120 as efficient ligands of dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN), also known as CD209.126 Mannose-terminated glycodendrons, were shown to inhibit Ebola virus infection of DCs by DC-SIGN.127 Glycofullerenes presenting mannose residues were used to inhibit DC-SIGN-dependent cell infection by pseudotyped viral particles.128 Multivalent polycationic glyco-amphiphilic cyclodextrins were prepared and used for targeting the mannose receptor (MR) on macrophages.129 Multibranched mannosylated lipids were prepared and incorporated into liposomes to allow for MR-mediated endocytosis by monocyte-derived dendritic cells.125 Barrientos et al130 have examined the interaction of lactose-functionalized gold nanoparticles (GNPs) with two different galactose-specific carbohydrate-binding proteins: an enzyme – β galactosidase from Escherichia coli and a lectin – the agglutinin from Viscum album. The carbohydrate binding site architecture and mode of recognition of these two representative proteins are different. The observed stability, together with the lack of toxicity against several cell lines, makes the GNP technology a promising strategy for the development of potential drugs to intervene in carbohydrate-mediated processes in vivo.
Transferrin
Transferrin (Tf) is an 80 kDa glycoprotein secreted by the liver. Iron-loaded Tf (monoferric or diferric) binds with its receptor, TfR, with affinity ranging from 1 to 10 nM.131,132 Due to their high rate of proliferation, cancer cells have dramatically increased iron requirements, in comparison with their normal cell counterparts. This phenomenon is associated with an increased expression of TfR, which can be correlated with tumor stage and cancer progression.133 A wide variety of Tf conjugates have been examined to deliver chemotherapeutic drugs, toxic protein (ricin A chain, saponin), PEG-protein conjugates, RNases, and nucleic acid conjugates.133 Adriamycin (ADR), which is an antineoplastic drug, has been chemically conjugated to transferrin (Tf-ADR) in an effort to deliver it directly to cancer cells overexpressing TfR.134 It has been shown that the Tf-ADR conjugate had a lower IC50 in HL60 and K562 cells, in comparison to the free drug.135 The same conjugate was also used in a different study, where it exhibited effective tissue biodistribution, a prolonged half-life of adriamycin in murine blood, and controlled release from transferrin conjugates. In nude mice bearing xenografts of H-MESO-1 tumor cells, IV-administered Tf-ADR increased the life span of mice by 69%, in comparison to 30% in the case of mice treated with ADR alone.131,134 Additionally, doxorubicin targeted with transferrin DOX–TRF was able to overcome MDR in leukemia cell lines, while having only a very limited effect on normal tissue cells.136 Conjugation of transferrin with ricin A-chain-toxic protein (RTA), allows TfR-mediated delivery of RTA into cells that can restore its toxicity, since RTA itself lacks any binding activity to the cells. The IC50 of RTA–transferrin conjugates in human leukemia CEM cells was between ten- and 10,000-fold lower when compared to the nonlinked combination of Tf and RTA.137 Mann et al138 have used transferrin as the targeting component conjugated to a trimeric HIV gp140 model vaccine antigen-cargo via a biotin–streptavidin linkage (Tf-gp140). Tf targets the highly efficient transcytotic and recycling transferrin receptor (CD71) that is expressed on both nasal and vaginal mucosal epithelium and is actively transcytosed. This conjugate has been successfully utilized as a delivery system for drugs, when associated with microparticles or emulsion formulations, for the delivery of anticancer agents and in gene therapy.
Lactoferrin
Lactoferrin (Lf) is a mammalian iron-binding glycoprotein, which belongs to the transferrin family. Previously, Lf was successfully exploited as a targeting ligand for delivery in the brain, as the Lf receptor (LfR) is expressed in the endothelial cells forming the blood–brain barrier (BBB).139,140 Moreover, many recent studies have revealed that lactoferrin can bind to multiple receptors on hepatocytes, including low-density lipoprotein receptor-related protein receptors (LRP-R),141 and the asialoglycoprotein receptors (ASGP-R),142 which also belong to the LfR family. It has been demonstrated that Lf binds ASGP-R with high affinity (a KD of aproximately 80 nM) in a galactose-independent manner,143 implying that lactoferrin is a good ligand for binding to ASGP-R. Lf–PEG-modified liposomes have been demonstrated to be a promising targeted drug delivery system for liver cancer chemotherapy, exhibiting a remarkable binding affinity and specificity toward hepatoma cells, and improved accumulation within hepatic tumor cells, but displaying low cellular toxicity against normal liver cells.144
Mannose derivates
The mannose-6-phosphate receptors (MPRs) are type I transmembrane glycoproteins that bind their specific oligosaccharide through a mannose-6-phosphate (M6P) recognition site at pH 6.5–6.7 in the trans-Golgi network and release it inside late-endosomes, where the pH is around 6.0.145 M6P residues are exclusively added to the N-linked, high mannose oligosaccharide residue of soluble lysosomal proteins.
The cation-independent mannose 6-phosphate receptor (CIM6PR) plays important roles in various biological processes. Its main role is transporting and sorting those lysosomal enzymes that contain an M6P-recognition marker in their structure from the trans-Golgi network to the lysosomes. CIM6PR also mediates the endocytosis of extracellular ligands such as insulin-like growth factor 2 (IGF2), retinoic acid and M6P-containing proteins.146–148
An endothelial cell monolayer associated with pericytes and astrocytes, known as the BBB, separates the blood from the cerebral parenchyma and prevents the penetration of drugs into the central nervous system. This barrier is characterized by tight intercellular junctions. The BBB prevents the uptake of all large molecules, with only small (<5 kDa), lipid-soluble and electrically-neutral molecules able to passively diffuse across this barrier. In the case of brain tumors, minor local disruptions of the BBB take place.149 Targeting of the BBB, therefore, represents a promising strategy for improving drug delivery to brain tumors. Carrier-mediated transport systems mediate the passage of nutrients through the BBB. Over twenty transporters have so far been identified, such as carriers for D-glucose (GLUT1), monocarboxylic acids (MCT1), large neutral amino acids (LAT1), cationic amino acids, or organic cations.150 The GLUT1 transporter promotes the transport of D-glucose from the blood to the brain. It mediates the passage of substances exhibiting similar structures to d-glucose through the BBB, including 2-deoxyglucose, galactose, mannose, and glucose analogs. Mannose-modified liposomes prepared from p-aminophenyl-α-mannoside were able to cross through the BBB using the glucose transporter with high affinity toward M6P receptor.151
Conclusion: hopes and challenges
Four key requirements seem to be essential for effective targeted drug delivery: retention, evasion, targeting (recognition), and release.152 Our current understanding of drug targeting to tumor cells is based on the combination of a few independent phenomena involving events associated with the enhanced permeability and retention (EPR) effect, properties of nanoparticles, increased retention in the circulation, and the type of ligand–receptor interaction.153 Through the EPR effect, macromolecular therapeutics tend to accumulate within tumors after systemic delivery.154 As such, nanoparticles represent an approach for the delivery of large drug payloads specifically to tumors. However, because many drugs require cellular internalization for efficacy, accumulation within the tumor microenvironment by the EPR effect may not correlate with therapeutic outcome. The specific ligand–receptor interaction for intracellular localization could occur only after blood circulation and extravasation steps, and the extending circulation time is a strategy commonly taken to increase the fraction of nanoparticles reaching a target tumor. Efficient ligand–receptor interaction is dependent upon various factors, including the extent of expression of specific receptors on target cells relative to nontarget cells, receptor availability on the surface of the target cells, the rate of internalization, and recycling of receptor after binding with ligand etc. Moreover, it is not known what fraction of cells express a specific receptor at a given time point and what is the expression pattern of that receptor in the whole tumor. Emanuel et al155 showed that successful delivery of immunoliposomes against fibrocarcinoma antigen was highly dependent on the stage of tumor development, with superior delivery by nontargeted liposomes at all stages except the micrometastases stage. The complexities related to identifying the effective ligand–receptor interaction may help explain the observations of Call et al156 on the lack of improved uptake of folate-targeted liposomes by target cells. Despite the success of in vitro experiments, poor accumulation of folate-targeted liposomes in KB (cells derived via HeLa contamination) tumor tissue was observed and the delivery efficiency was comparable with nontargeted liposomes.121
Tumor heterogeneity should also be considered for effective tumor treatment. A tumor is not a monoculture or collective mass of a single cell type. In fact, aiming at cancer cells with a single surface marker results in focusing only at a single population among mixed ones which are constantly changing and adapting. Detection and diagnosis of a particular cell type by a single surface marker can result in the overestimation of a cancer, due to commonly shared features with normal cells within the tumor. Generally, the approach involving only one surface marker could be regarded as “outdated”. The multiple surface marker approach is considered as a better alternative of cancer cell detection and elimination. There is significant research activity based on the use of a primary tumor sample as a template to explore new targeting moieties with advanced techniques, including phage-display and aptamers screening approaches. These cell-specific approaches are expected to result in the targeting of tumors with greater selectivity.153
One key strategy in the pursuit of the vision of Ehrlich, that “antibodies are in a way magic bullets that identify their target themselves without harming the organism” was the development of the hybridoma technology.157 Interest in antibody therapeutics has increased over the last decade due to the number of approved agents for the treatment of cancer and other diseases validating this strategy.54 Several mAbs have been already approved for clinical use, with more than 150 additional mAbs in clinical trials worldwide.16,158 So far, 23 different mAbs have been approved by the FDA for clinical use. Among these, seven products are directed to cancer, namely: rituximab (anti-CD20), bevacizumab (anti-VEGF), alemtuzumab (anti-CD52) and targeted to EGFR: cetuximab, penitumumab, trastuzumab (anti-HER2), and matuzumab.159–161 Therapeutic antibodies provide clinical benefit to patients with cancer and have been established as “standard of care” agents for several highly prevalent human cancers.
Immunoconjugates, where mAbs are covalently linked to drugs, toxins, or radioisotopes, have also been successfully commercialized, eg, Zevalin (ibritumomab tiuxetan), Bexxar (131I-tositumomab), and Mylotarg (gemtuzumab ozogamicin).162–164 There is also growing interest in the range of antibody derivatives and peptides which could be used not only for drug delivery but, also, as diagnostic tools. Because the first generation of liposome therapeutics was focused on reducing systemic toxicity, the current clinically available therapeutic liposome formulations do not exhibit active targeting at the cellular level.165 Kirpotin et al166 demonstrated that both HER2 antibody-targeted and nontargeted liposomes reached tumors in vivo in a human breast cancer xenograft model. However, the uptake of immunoliposomes targeted with antibody against HER2 was approximately sixfold higher in tumor cells than nontargeted particles. This observation suggests that active targeting can be successfully exploited to promote cellular binding and internalization.
Whether the mechanism of action of immunoliposomes involves cellular binding or internalization, there are other barriers that liposomes may encounter within the tumor tissue including disordered vasculature, increased hydrostatic pressure, and the “binding-site barrier”. The latter may be generated by a fraction of immunoliposomes, which is bound to the first line of tumor cells and may hinder diffusion through the tumor parenchyma.167 Because the binding-site barrier prevents homogeneous drug activity throughout the tumor, scFvs have often proved more efficacious in improving tumor penetration.168 Achieving enhanced cellular uptake of immunoliposomes and maintaining high bioavailability is most important, but functionalized liposomes can suffer from increased recognition by the immune system. This can result in high in vitro activity that does not translate into in vivo efficacy. A potential solution to this problem is to improve the stealth capacity of immunoliposomes by masking the targeting ligand with polyethylene glycol while in the bloodstream, and exposing that ligand when the liposomes reach the tumor site.165 Kuai et al169 used cleavable disulfide PEG5000 lipid to mask TATs (trans activating transcriptional activators) covalently attached to PEG2000 lipids at the surface of liposome. That strategy resulted in higher tumor accumulation of the liposome preparations and lower capture by the reticuloendothelial system (RES) system.
Tumor-homing peptides have recently entered clinical trials, with a number of other trials currently ongoing or at the stage of recruiting patients.63,81–84 Interestingly, no dose-limiting toxicity has been reported so far in these trials and all peptide-targeted therapy combinations have been well tolerated.67 The future challenge for this emerging clinical therapeutic approach will be to find novel ways to exploit them, alternatively after chemical manipulation, such as conjugation to active pharmacological molecules or nanoparticles, in order to deliver them with an even greater efficacy and selectivity to the target tissue.
In summary, an increasing range of different classes of ligands have become available for targeted delivery to cancer cell targets, which are characterized by high binding affinity, low toxicity, high stability in blood, and low immunogenicity. However, the “magic bullet” should refer to a system that delivers all of the drug load to the target, without any side effect on nontarget tissues. Based upon what has been mentioned in the literature to date, we could conclude that this field is still in its infancy, although several promising avenues have the potential of delivering novel therapeutic strategies with promising results.
Acknowledgments
This work was supported by Wroclaw Research Centre EIT+ within the project “Biotechnologies and advanced medical technologies” – BioMed (POIG.01.01.02-02-003/08). Publication cost supported by Wroclaw Centre of Biotechnology, programme The Leading National Research Centre (KNOW) for years 2014–2018. The authors are grateful to Dr Michał Grzybek from the Paul Langerhans Institute, Medical Faculty, TU Dresden, for careful reading and discussions of the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Hanahan D, Weinberg RA. The Hallmarks of Cancer. Cell. 2000;100(1):57–70. | ||
Torchilin VP. Drug targeting. Eur J Pharm Sci. 2000;11(Suppl 2):S81–S91. | ||
Muro S. Challenges in design and characterization of ligand-targeted drug delivery systems. J Control Release. 2012;164(2):125–137. | ||
Lammers T, Kiessling F, Hennink WE, Storm G. Drug targeting to tumors: principles, pitfalls and (pre-) clinical progress. J Control Release. 2012;161(2):175–187. | ||
Kiessling A, Wehner R, Fussel S, Bachmann M, Wirth MP, Schmitz M. Tumor-associated antigens for specific immunotherapy of prostate cancer. Cancers. 2012;4(1):193–217. | ||
Zhang L, Li Y, Yu JC. Chemical modification of inorganic nanostructures for targeted and controlled drug delivery in cancer treatment. J Mater Chem B. 2014;2(5):452–470. | ||
Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2(12):751–760. | ||
Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliv Rev. 2014;66:2–25. | ||
Parnham MJ, Wetzig H. Toxicity screening of liposomes. Chem Phys Lipids. 1993;64(1–3):263–274. | ||
Markman JL, Rekechenetskiy A, Holler E, Ljubimova JY. Nanomedicine therapeutic approaches to overcome cancer drug resistance. Adv Drug Deliv Rev. 2013;65(13–14):1866–1879. | ||
Kopecek J. Polymer-drug conjugates: origins, progress to date and future directions. Adv Drug Deliv Rev. 2013;65(1):49–59. | ||
Wu J, Huang W, He Z. Dendrimers as carriers for siRNA delivery and gene silencing: a review. Scientific World Journal. 2013;2013:630–654. | ||
Kumar N, Ravikumar MN, Domb AJ. Biodegradable block copolymers. Adv Drug Deliv Rev. 2001;53(1):23–44. | ||
Sosnik A, Carcaboso AM. Nanomedicines in the future of pediatric therapy. Adv Drug Deliv Rev. 2014;73:140–161. | ||
Angeloni V, Tiberio P, Appierto V, Daidone MG. Implications of stemness-related signaling pathways in breast cancer response to therapy. Semin Cancer Biol. Epub Aug 18, 2014. | ||
Reichert JM, Rosensweig CJ, Faden LB, Dewitz MC. Monoclonal antibody successes in the clinic. Nat Biotechnol. 2005;23(9):1073–1078. | ||
Weeraratne D, Chen A, Pennucci JJ, et al. Immunogenicity of panitumumab in combination chemotherapy clinical trials. BMC Clin Pharmacol. 2011;11:17. | ||
Riechmann L, Clark M, Waldmann H, Winter G. Reshaping human antibodies for therapy. Nature. 1988;332(6162):323–327. | ||
Weiner LM, Surana R, Wang S. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat Rev Immunol. 2010;10(5):317–327. | ||
ClinicalTrials.gov. Part 2 of Phase 1 Study of GC1008 to Treat Advanced Melanoma (Part 2 Will Only Accept and Treat Patients With Advanced Malignant Melanoma). Available from: http://clinicaltrials.gov/ct2/show/results/NCT00899444. Accessed November 14, 2014. | ||
Pines G, Kostler WJ, Yarden Y. Oncogenic mutant forms of EGFR: lessons in signal transduction and targets for cancer therapy. FEBS Lett. 2010;584(12):2699–2706. | ||
Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103(2):211–225. | ||
Schlessinger J. Signal transduction. Autoinhibition control. Science. 2003;300(5620):750–752. | ||
Dawson JP, Berger MB, Lin CC, Schlessinger J, Lemmon MA, Ferguson KM. Epidermal growth factor receptor dimerization and activation require ligand-induced conformational changes in the dimer interface. Mol Cell Biol. 2005;25(17):7734–7742. | ||
Leahy DJ. A molecular view of anti-ErbB monoclonal antibody therapy. Cancer Cell. 2008;13(4):291–293. | ||
Holbro T, Hynes NE. ErbB receptors: directing key signaling networks throughout life. Annu Rev Pharmacol Toxicol. 2004;44:195–217. | ||
Herbst RS, Shin DM. Monoclonal antibodies to target epidermal growth factor receptor-positive tumors: a new paradigm for cancer therapy. Cancer. 2002;94(5):1593–1611. | ||
Schmitz KR, Bagchi A, Roovers RC, van Bergen en Henegouwen PM, Ferguson KM. Structural evaluation of EGFR inhibition mechanisms for nanobodies/VHH domains. Structure. 2013;21(7):1214–1224. | ||
Felder S, LaVin J, Ullrich A, Schlessinger J. Kinetics of binding, endocytosis, and recycling of EGF receptor mutants. J Cell Biol. 1992;117(1):203–212. | ||
Low K, Wacker M, Wagner S, Langer K, von Briesen H. Targeted human serum albumin nanoparticles for specific uptake in EGFR-Expressing colon carcinoma cells. Nanomedicine. 2011;7(4):454–463. | ||
Benhabbour SR, Luft JC, Kim D, et al. In vitro and in vivo assessment of targeting lipid-based nanoparticles to the epidermal growth factor-receptor (EGFR) using a novel Heptameric ZEGFR domain. J Control Release. 2012;158(1):63–71. | ||
Mickler FM, Mockl L, Ruthardt N, Ogris M, Wagner E, Brauchle C. Tuning nanoparticle uptake: live-cell imaging reveals two distinct endocytosis mechanisms mediated by natural and artificial EGFR targeting ligand. Nano Lett. 2012;12(7):3417–3423. | ||
Dancey JE. Predictive factors for epidermal growth factor receptor inhibitors–the bull’s-eye hits the arrow. Cancer Cell. 2004;5(5):411–415. | ||
Burgess AW. EGFR family: structure physiology signalling and therapeutic targets. Growth Factors. 2008;26(5):263–274. | ||
Wikstrand CJ, Hale LP, Batra SK, et al. Monoclonal antibodies against EGFRvIII are tumor specific and react with breast and lung carcinomas and malignant gliomas. Cancer Res. 1995;55(14):3140–3148. | ||
Bardelli A, Janne PA. The road to resistance: EGFR mutation and cetuximab. Nat Med. 2012;18(2):199–200. | ||
Bier H, Reiffen KA, Haas I, Stasiecki P. Dose-dependent access of murine anti-epidermal growth factor receptor monoclonal antibody to tumor cells in patients with advanced laryngeal and hypopharyngeal carcinoma. Eur Arch Otorhinolaryngol. 1995;252(7):433–439. | ||
Yang XD, Jia XC, Corvalan JR, Wang P, Davis CG. Development of ABX-EGF, a fully human anti-EGF receptor monoclonal antibody, for cancer therapy. Crit Rev Oncol Hematol. 2001;38(1):17–23. | ||
Dienstmann R, Felip E. Necitumumab in the treatment of advanced non-small cell lung cancer: translation from preclinical to clinical development. Expert Opin Biol Ther. 2011;11(9):1223–1231. | ||
Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell. 2005;7(4):301–311. | ||
Meira DD, Nobrega I, de Almeida VH, et al. Different antiproliferative effects of matuzumab and cetuximab in A431 cells are associated with persistent activity of the MAPK pathway. Eur J Cancer. 2009;45(7):1265–1273. | ||
Saltz L, Easley C, Kirkpatrick P. Panitumumab. Nat Rev Drug Discov. 2006;5(12):987–988. | ||
Imai K, Takaoka A. Comparing antibody and small-molecule therapies for cancer. Nat Rev Cancer. 2006;6(9):714–727. | ||
Li S, Kussie P, Ferguson KM. Structural basis for EGF receptor inhibition by the therapeutic antibody IMC-11F8. Structure. 2008;16(2):216–227. | ||
Schmiedel J, Blaukat A, Li S, Knochel T, Ferguson KM. Matuzumab binding to EGFR prevents the conformational rearrangement required for dimerization. Cancer Cell. 2008;13(4):365–373. | ||
Vanhoefer U, Tewes M, Rojo F, et al. Phase I study of the humanized antiepidermal growth factor receptor monoclonal antibody EMD72000 in patients with advanced solid tumors that express the epidermal growth factor receptor. J Clin Oncol. 2004;22(1):175–184. | ||
Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med. 2000;6(4):443–446. | ||
Agus DB, Akita RW, Fox WD, et al. Targeting ligand-activated ErbB2 signaling inhibits breast and prostate tumor growth. Cancer Cell. 2002;2(2):127–137. | ||
Capelan M, Pugliano L, De Azambuja E, et al. Pertuzumab: new hope for patients with HER2-positive breast cancer. Ann Oncol. 2013;24(2):273–282. | ||
Baselga J, Albanell J. Mechanism of action of anti-HER2 monoclonal antibodies. Ann Oncol. 2001;12(Suppl 1):S35–S41. | ||
Roskoski R Jr. The ErbB/HER receptor protein-tyrosine kinases and cancer. Biochem Biophys Res Commun. 2004;319(1):1–11. | ||
Molina MA, Codony-Servat J, Albanell J, Rojo F, Arribas J, Baselga J. Trastuzumab (herceptin), a humanized anti-Her2 receptor monoclonal antibody, inhibits basal and activated Her2 ectodomain cleavage in breast cancer cells. Cancer Res. 2001;61(12):4744–4749. | ||
Shaughnessy AF. Monoclonal antibodies: magic bullets with a hefty price tag. BMJ. 2012;345:e8346. | ||
Mazar AP, Ahn RW, O’Halloran TV. Development of novel therapeutics targeting the urokinase plasminogen activator receptor (uPAR) and their translation toward the clinic. Curr Pharm Des. 2011;17(19):1970–1978. | ||
Nykjaer A, Conese M, Christensen EI, et al. Recycling of the urokinase receptor upon internalization of the uPA:serpin complexes. EMBO J. 1997;16(10):2610–2620. | ||
Wyrozumska PM, Toporkiewicz J, Szarawarska M, et al. Liposome-coated lipoplex–based carrier for antisense oligonucleotides. Cancer Biol Ther. Epub Nov 20, 2014. | ||
Gong R, Chen W, Dimitrov DS. Expression, purification, and characterization of engineered antibody CH2 and VH domains. Methods Mol Biol. 2012;899:85–102. | ||
Miller KR, Koide A, Leung B, et al. T cell receptor-like recognition of tumor in vivo by synthetic antibody fragment. PLoS One. 2012;7(8):e43746. | ||
Holliger P, Hudson PJ. Engineered antibody fragments and the rise of single domains. Nat Biotechnol. 2005;23(9):1126–1136. | ||
Savage Laboratories. CroFab®-The primary pit viper antivenom treatment in the United States for more than 13 years. Available from: http://www.savagelabs.com/index.htm. Accessed November 14, 2014. | ||
Drugs.com. Ablynx’s Partner Boehringer Ingelheim Initiates Phase I Study With Nanobody to Treat Alzheimer’s Disease. Available from: http://www.drugs.com/clinical_trials/ablynx-s-partner-boehringer-ingelheim-initiates-phase-study-nanobody-alzheimer-s-16281.html. Accessed October, 2013. | ||
Smith GP, Petrenko VA. Phage Display. Chem Rev. 1997;97(2):391–410. | ||
Koivunen E, Arap W, Rajotte D, Lahdenranta J, Pasqualini R. Identification of receptor ligands with phage display peptide libraries. J Nucl Med. 1999;40(5):883–888. | ||
Marasco D, Perretta G, Sabatella M, Ruvo M. Past and future perspectives of synthetic peptide libraries. Curr Protein Pept Sci. 2008;9(5):447–467. | ||
Lee TY, Lin CT, Kuo SY, Chang DK, Wu HC. Peptide-mediated targeting to tumor blood vessels of lung cancer for drug delivery. Cancer Res. 2007;67(22):10958–10965. | ||
Brown KC. New approaches for cell-specific targeting: identification of cell-selective peptides from combinatorial libraries. Curr Opin Chem Biol. 2000;4(1):16–21. | ||
Laakkonen P, Vuorinen K. Homing peptides as targeted delivery vehicles. Integr Biol (Camb). 2010;2(7–8):326–337. | ||
Zhang XX, Eden HS, Chen X. Peptides in cancer nanomedicine: drug carriers, targeting ligands and protease substrates. J Control Release. 2012;159(1):2–13. | ||
McGuire MJ, Samli KN, Chang YC, Brown KC. Novel ligands for cancer diagnosis: selection of peptide ligands for identification and isolation of B-cell lymphomas. Exp Hematol. 2006;34(4):443–452. | ||
Laakkonen P, Akerman ME, Biliran H, et al. Antitumor activity of a homing peptide that targets tumor lymphatics and tumor cells. Proc Natl Acad Sci U S A. 2004;101(25):9381–9386. | ||
Koivunen E, Wang B, Ruoslahti E. Phage libraries displaying cyclic peptides with different ring sizes: ligand specificities of the RGD-directed integrins. Biotechnology (N Y). 1995;13(3):265–270. | ||
Pasqualini R, Koivunen E, Ruoslahti E. A peptide isolated from phage display libraries is a structural and functional mimic of an RGD-binding site on integrins. J Cell Biol. 1995;130(5):1189–1196. | ||
Sugahara KN, Teesalu T, Karmali PP, et al. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell. 2009;16(6):510–520. | ||
Wang K, Zhang X, Zhang L, et al. Development of biodegradable polymeric implants of RGD-modified PEG-PAMAM-DOX conjugates for long-term intratumoral release. Drug Deliv. Epub March 27, 2014. | ||
Joyce JA, Laakkonen P, Bernasconi M, Bergers G, Ruoslahti E, Hanahan D. Stage-specific vascular markers revealed by phage display in a mouse model of pancreatic islet tumorigenesis. Cancer Cell. 2003;4(5):393–403. | ||
Hoffman JA, Giraudo E, Singh M, et al. Progressive vascular changes in a transgenic mouse model of squamous cell carcinoma. Cancer Cell. 2003;4(5):383–391. | ||
Meiring MS, Litthauer D, Harsfalvi J, van Wyk V, Badenhorst PN, Kotze HF. In vitro effect of a thrombin inhibition peptide selected by phage display technology. Thromb Res. 2002;107(6):365–371. | ||
Samli KN, McGuire MJ, Newgard CB, Johnston SA, Brown KC. Peptide-mediated targeting of the islets of Langerhans. Diabetes. 2005;54(7):2103–2108. | ||
Oyama T, Sykes KF, Samli KN, Minna JD, Johnston SA, Brown KC. Isolation of lung tumor specific peptides from a random peptide library: generation of diagnostic and cell-targeting reagents. Cancer Lett. 2003;202(2):219–230. | ||
Li Z, Zhao R, Wu X, et al. Identification and characterization of a novel peptide ligand of epidermal growth factor receptor for targeted delivery of therapeutics. FASEB J. 2005;19(14):1978–1985. | ||
Eskens FA, Dumez H, Hoekstra R, et al. Phase I and pharmacokinetic study of continuous twice weekly intravenous administration of Cilengitide (EMD 121974), a novel inhibitor of the integrins alphavbeta3 and alphavbeta5 in patients with advanced solid tumours. Eur J Cancer. 2003;39(7):917–926. | ||
Krag DN, Shukla GS, Shen GP, et al. Selection of tumor-binding ligands in cancer patients with phage display libraries. Cancer Res. 2006;66(15):7724–7733. | ||
MacDonald TJ, Stewart CF, Kocak M, et al. Phase I clinical trial of cilengitide in children with refractory brain tumors: Pediatric Brain Tumor Consortium Study PBTC-012. J Clin Oncol. 2008;26(6):919–924. | ||
Gregorc V, Santoro A, Bennicelli E, et al. Phase Ib study of NGR-hTNF, a selective vascular targeting agent, administered at low doses in combination with doxorubicin to patients with advanced solid tumours. Br J Cancer. 2009;101(2):219–224. | ||
Huang G, Zhou Z, Srinivasan R, et al. Affinity manipulation of surface-conjugated RGD peptide to modulate binding of liposomes to activated platelets. Biomaterials. 2008;29(11):1676–1685. | ||
Alloatti D, Giannini G, Vesci L, et al. Camptothecins in tumor homing via an RGD sequence mimetic. Bioorg Med Chem Lett. 2012;22(20):6509–6512. | ||
Yano A, Onozuka A, Matin K, Imai S, Hanada N, Nisizawa T. RGD motif enhances immunogenicity and adjuvanicity of peptide antigens following intranasal immunization. Vaccine. 2003;22(2):237–243. | ||
YoungLai EV, Todoroff EC. The pituitary gonadotropin-releasing hormone (GnRH) receptor of the female rabbit: characterization and developmental aspects. Can J Physiol Pharmacol. 1992;70(12):1639–1646. | ||
He Y, Zhang L, Song C. Luteinizing hormone-releasing hormone receptor-mediated delivery of mitoxantrone using LHRH analogs modified with PEGylated liposomes. Int J Nanomedicine. 2010;5:697–705. | ||
Li Y, Cozzi PJ. Targeting uPA/uPAR in prostate cancer. Cancer Treat Rev. 2007;33(6):521–527. | ||
Ploug M, Ostergaard S, Gardsvoll H, et al. Peptide-derived antagonists of the urokinase receptor. Affinity maturation by combinatorial chemistry, identification of functional epitopes, and inhibitory effect on cancer cell intravasation. Biochemistry. 2001;40(40):12157–12168. | ||
Knor S, Sato S, Huber T, et al. Development and evaluation of peptidic ligands targeting tumour-associated urokinase plasminogen activator receptor (uPAR) for use in alpha-emitter therapy for disseminated ovarian cancer. Eur J Nucl Med Mol Imaging. 2008;35(1):53–64. | ||
Lander ES, Linton LM, Birren B, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. | ||
Kajander T, Cortajarena AL, Regan L. Consensus design as a tool for engineering repeat proteins. Methods Mol Biol. 2006;340:151–170. | ||
Leader B, Baca QJ, Golan DE. Protein therapeutics: a summary and pharmacological classification. Nat Rev Drug Discov. 2008;7(1):21–39. | ||
Zahnd C, Wyler E, Schwenk JM, et al. A designed ankyrin repeat protein evolved to picomolar affinity to Her2. J Mol Biol. 2007;369(4):1015–1028. | ||
Stumpp MT, Binz HK, Amstutz P. DARPins: a new generation of protein therapeutics. Drug Discov Today. 2008;13(15–16):695–701. | ||
Wetzel SK, Settanni G, Kenig M, Binz HK, Pluckthun A. Folding and unfolding mechanism of highly stable full-consensus ankyrin repeat proteins. J Mol Biol. 2008;376(1):241–257. | ||
Binz HK, Amstutz P, Kohl A, et al. High-affinity binders selected from designed ankyrin repeat protein libraries. Nat Biotechnol. 2004;22(5):575–582. | ||
Schweizer A, Rusert P, Berlinger L, et al. CD4-specific designed ankyrin repeat proteins are novel potent HIV entry inhibitors with unique characteristics. PLoS Pathog. 2008;4(7):e1000109. | ||
ClinicalTrials.gov. A Phase I/II, Open-label, Single Ascending Dose Study Evaluating the Safety, Preliminary Efficacy, and Pharmacokinetics of Intravitreal MP0112 in Patients With Diabetic Macular Edema (DME). Available from: http://clinicaltrials.gov/show/NCT01042678. Accessed January 1, 2010. | ||
Janas T, Janas T. The selection of aptamers specific for membrane molecular targets. Cell Mol Biol Lett. 2011;16(1):25–39. | ||
Jayasena SD. Aptamers: an emerging class of molecules that rival antibodies in diagnostics. Clin Chem. 1999;45(9):1628–1650. | ||
Missailidis S, Thomaidou D, Borbas KE, Price MR. Selection of aptamers with high affinity and high specificity against C595, an anti-MUC1 IgG3 monoclonal antibody, for antibody targeting. J Immunol Methods. 2005;296(1–2):45–62. | ||
Keefe AD, Cload ST. SELEX with modified nucleotides. Curr Opin Chem Biol. 2008;12(4):448–456. | ||
Dua P, Kim S, Lee DK. Nucleic acid aptamers targeting cell-surface proteins. Methods. 2011;54(2):215–225. | ||
Ireson CR, Kelland LR. Discovery and development of anticancer aptamers. Mol Cancer Ther. 2006;5(12):2957–2962. | ||
Sundaram P, Kurniawan H, Byrne ME, Wower J. Therapeutic RNA aptamers in clinical trials. Eur J Pharm Sci. 2013;48(1–2):259–271. | ||
Cibiel A, Pestourie C, Duconge F. In vivo uses of aptamers selected against cell surface biomarkers for therapy and molecular imaging. Biochimie. 2012;94(7):1595–1606. | ||
Sapra P, Allen TM. Ligand-targeted liposomal anticancer drugs. Prog Lipid Res. 2003;42(5):439–462. | ||
Antony AC. The biological chemistry of folate receptors. Blood. 1992;79(11):2807–2820. | ||
Weitman SD, Lark RH, Coney LR, et al. Distribution of the folate receptor GP38 in normal and malignant cell lines and tissues. Cancer Res. 1992;52(12):3396–3401. | ||
Ai J, Xu Y, Li D, Liu Z, Wang E. Folic acid as delivery vehicles: targeting folate conjugated fluorescent nanoparticles to tumors imaging. Talanta. 2012;101:32–37. | ||
Chen C, Ke J, Zhou XE, et al. Structural basis for molecular recognition of folic acid by folate receptors. Nature. 2013;500(7463):486–489. | ||
McHugh M, Cheng YC. Demonstration of a high affinity folate binder in human cell membranes and its characterization in cultured human KB cells. J Biol Chem. 1979;254(22):11312–11318. | ||
Lee RJ, Low PS. Delivery of liposomes into cultured KB cells via folate receptor-mediated endocytosis. J Biol Chem. 1994;269(5):3198–3204. | ||
Wang S, Lee RJ, Cauchon G, Gorenstein DG, Low PS. Delivery of antisense oligodeoxyribonucleotides against the human epidermal growth factor receptor into cultured KB cells with liposomes conjugated to folate via polyethylene glycol. Proc Natl Acad Sci U S A. 1995;92(8):3318–3322. | ||
Leamon CP, Low PS. Delivery of macromolecules into living cells: a method that exploits folate receptor endocytosis. Proc Natl Acad Sci U S A. 1991;88(13):5572–5576. | ||
Gabizon A, Horowitz AT, Goren D, et al. Targeting folate receptor with folate linked to extremities of poly(ethylene glycol)-grafted liposomes: in vitro studies. Bioconjug Chem. 1999;10(2):289–298. | ||
Gabizon A, Horowitz AT, Goren D, Tzemach D, Shmeeda H, Zalipsky S. In vivo fate of folate-targeted polyethylene-glycol liposomes in tumor-bearing mice. Clin Cancer Res. 2003;9(17):6551–6559. | ||
Leamon CP, Cooper SR, Hardee GE. Folate-liposome-mediated antisense oligodeoxynucleotide targeting to cancer cells: evaluation in vitro and in vivo. Bioconjug Chem. 2003;14(4):738–747. | ||
Turk MJ, Breur GJ, Widmer WR, et al. Folate-targeted imaging of activated macrophages in rats with adjuvant-induced arthritis. Arthritis Rheum. 2002;46(7):1947–1955. | ||
Varki A. Biological roles of oligosaccharides: all of the theories are correct. Glycobiology. 1993;3(2):97–130. | ||
Bies C, Lehr CM, Woodley JF. Lectin-mediated drug targeting: history and applications. Adv Drug Deliv Rev. 2004;56(4):425–435. | ||
Lepenies B, Lee J, Sonkaria S. Targeting C-type lectin receptors with multivalent carbohydrate ligands. Adv Drug Deliv Rev. 2013;65(9):1271–1281. | ||
Arnaiz B, Martinez-Avila O, Falcon-Perez JM, Penades S. Cellular uptake of gold nanoparticles bearing HIV gp120 oligomannosides. Bioconjug Chem. 2012;23(4):814–825. | ||
Ribeiro-Viana R, Sanchez-Navarro M, Luczkowiak J, et al. Virus-like glycodendrinanoparticles displaying quasi-equivalent nested polyvalency upon glycoprotein platforms potently block viral infection. Nat Commun. 2012;3:1303. | ||
Luczkowiak J, Munoz A, Sanchez-Navarro M, et al. Glycofullerenes inhibit viral infection. Biomacromolecules. 2013;14(2):431–437. | ||
Diaz-Moscoso A, Guilloteau N, Bienvenu C, et al. Mannosyl-coated nanocomplexes from amphiphilic cyclodextrins and pDNA for site-specific gene delivery. Biomaterials. 2011;32(29):7263–7273. | ||
Barrientos AG, de la Fuente JM, Jimenez M, et al. Modulating glycosidase degradation and lectin recognition of gold glyconanoparticles. Carbohydr Res. 2009;344(12):1474–1478. | ||
Kratz F, Beyer U, Roth T, et al. Transferrin conjugates of doxorubicin: synthesis, characterization, cellular uptake, and in vitro efficacy. J Pharm Sci. 1998;87(3):338–346. | ||
Bou-Abdallah F, Terpstra TR. The thermodynamic and binding properties of the transferrins as studied by isothermal titration calorimetry. Biochim Biophys Acta. 2012;1820(3):318–325. | ||
Daniels TR, Bernabeu E, Rodriguez JA, et al. The transferrin receptor and the targeted delivery of therapeutic agents against cancer. Biochim Biophys Acta. 2012;1820(3):291–317. | ||
Singh M, Atwal H, Micetich R. Transferrin directed delivery of adriamycin to human cells. Anticancer Res. 1998;18(3A):1423–1427. | ||
Berczi A, Barabas K, Sizensky JA, Faulk WP. Adriamycin conjugates of human transferrin bind transferrin receptors and kill K562 and HL60 cells. Arch Biochem Biophys. 1993;300(1):356–363. | ||
Lubgan D, Jozwiak Z, Grabenbauer GG, Distel LV. Doxorubicin-transferrin conjugate selectively overcomes multidrug resistance in leukaemia cells. Cell Mol Biol Lett. 2009;14(1):113–127. | ||
Raso V, Basala M. A highly cytotoxic human transferrin-ricin A chain conjugate used to select receptor-modified cells. J Biol Chem. 1984;259(2):1143–1149. | ||
Mann JF, Stieh D, Klein K, et al. Transferrin conjugation confers mucosal molecular targeting to a model HIV-1 trimeric gp140 vaccine antigen. J Control Release. 2012;158(2):240–249. | ||
Huang R, Ke W, Liu Y, Jiang C, Pei Y. The use of lactoferrin as a ligand for targeting the polyamidoamine-based gene delivery system to the brain. Biomaterials. 2008;29(2):238–246. | ||
Ye Y, Sun Y, Zhao H, et al. A novel lactoferrin-modified beta-cyclodextrin nanocarrier for brain-targeting drug delivery. Int J Pharm. 2013;458(1):110–117. | ||
Meilinger M, Haumer M, Szakmary KA, et al. Removal of lactoferrin from plasma is mediated by binding to low density lipoprotein receptor-related protein/alpha 2-macroglobulin receptor and transport to endosomes. FEBS Lett. 1995;360(1):70–74. | ||
Bennatt DJ, McAbee DD. Identification and isolation of a 45-kDa calcium-dependent lactoferrin receptor from rat hepatocytes. Biochemistry. 1997;36(27):8359–8366. | ||
McAbee DD, Jiang X, Walsh KB. Lactoferrin binding to the rat asialoglycoprotein receptor requires the receptor’s lectin properties. Biochem J. 2000;348(Pt 1):113–117. | ||
Wei M, Xu Y, Zou Q, et al. Hepatocellular carcinoma targeting effect of PEGylated liposomes modified with lactoferrin. Eur J Pharm Sci. 2012;46(3):131–141. | ||
Coutinho MF, Prata MJ, Alves S. Mannose-6-phosphate pathway: a review on its role in lysosomal function and dysfunction. Mol Genet Metab. 2012;105(4):542–550. | ||
Tong PY, Gregory W, Kornfeld S. Ligand interactions of the cation-independent mannose 6-phosphate receptor. The stoichiometry of mannose 6-phosphate binding. J Biol Chem. 1989;264(14):7962–7969. | ||
Jeanjean A, Gary-Bobo M, Nirde P, Leiris S, Garcia M, Morere A. Synthesis of new sulfonate and phosphonate derivatives for cation-independent mannose 6-phosphate receptor targeting. Bioorg Med Chem Lett. 2008;18(23):6240–6243. | ||
Zaccheo OJ, Prince SN, Miller DM, et al. Kinetics of insulin-like growth factor II (IGF-II) interaction with domain 11 of the human IGF-II/mannose 6-phosphate receptor: function of CD and AB loop solvent-exposed residues. J Mol Biol. 2006;359(2):403–421. | ||
Gururangan S, Friedman HS. Innovations in design and delivery of chemotherapy for brain tumors. Neuroimaging Clin N Am. 2002;12(4):583–597. | ||
Allen DD, Geldenhuys WJ. Molecular modeling of blood-brain barrier nutrient transporters: in silico basis for evaluation of potential drug delivery to the central nervous system. Life Sci. 2006;78(10):1029–1033. | ||
Adrian JE, Kamps JA, Poelstra K, Scherphof GL, Meijer DK, Kaneda Y. Delivery of viral vectors to hepatic stellate cells in fibrotic livers using HVJ envelopes fused with targeted liposomes. J Drug Target. 2007;15(1):75–82. | ||
Mills J, Needham D. Targeted drug delivery. Expert Opin Ther Patents. 1999;9:1499–1513. | ||
Bae YH, Park K. Targeted drug delivery to tumors: myths, reality and possibility. J Control Release. 2011;153(3):198–205. | ||
Matsumura Y, Oda T, Maeda H. [General mechanism of intratumor accumulation of macromolecules: advantage of macromolecular therapeutics]. Gan To Kagaku Ryoho. 1987;14(3 Pt 2):821–829. Japanese. | ||
Emanuel N, Kedar E, Bolotin EM, Smorodinsky NI, Barenholz Y. Targeted delivery of doxorubicin via sterically stabilized immunoliposomes: pharmacokinetics and biodistribution in tumor-bearing mice. Pharm Res. 1996;13(6):861–868. | ||
Call GB, Olson JM, Chen J, et al. Genomewide clonal analysis of lethal mutations in the Drosophila melanogaster eye: comparison of the X chromosome and autosomes. Genetics. 2007;177(2):689–697. | ||
Strebhardt K, Ullrich A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat Rev Cancer. 2008;8(6):473–480. | ||
Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev. 2013;65(1):36–48. | ||
Oldham RK, Dillman RO. Monoclonal antibodies in cancer therapy: 25 years of progress. J Clin Oncol. 2008;26(11):1774–1777. | ||
Elbakri A, Nelson PN, Abu Odeh RO. The state of antibody therapy. Hum Immunol. 2010;71(12):1243–1250. | ||
Oliveira S, Heukers R, Sornkom J, Kok RJ, van Bergen En Henegouwen PM. Targeting tumors with nanobodies for cancer imaging and therapy. J Control Release. 2013;172(3):607–617. | ||
Kaminski MS, Zelenetz AD, Press OW, et al. Pivotal study of iodine I 131 tositumomab for chemotherapy-refractory low-grade or transformed low-grade B-cell non-Hodgkin’s lymphomas. J Clin Oncol. 2001;19(19):3918–3928. | ||
Sievers EL, Larson RA, Stadtmauer EA, et al. Efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukemia in first relapse. J Clin Oncol. 2001;19(13):3244–3254. | ||
Witzig TE, Gordon LI, Cabanillas F, et al. Randomized controlled trial of yttrium-90-labeled ibritumomab tiuxetan radioimmunotherapy versus rituximab immunotherapy for patients with relapsed or refractory low-grade, follicular, or transformed B-cell non-Hodgkin’s lymphoma. J Clin Oncol. 2002;20(10):2453–2463. | ||
Noble GT, Stefanick JF, Ashley JD, Kiziltepe T, Bilgicer B. Ligand-targeted liposome design: challenges and fundamental considerations. Trends Biotechnol. 2014;32(1):32–45. | ||
Kirpotin DB, Drummond DC, Shao Y, et al. Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res. 2006;66(13):6732–6740. | ||
Sawant RR, Torchilin VP. Challenges in development of targeted liposomal therapeutics. AAPS J. 2012;14(2):303–315. | ||
Yokota T, Milenic DE, Whitlow M, Schlom J. Rapid tumor penetration of a single-chain Fv and comparison with other immunoglobulin forms. Cancer Res. 1992;52(12):3402–3408. | ||
Kuai R, Yuan W, Li W, et al. Targeted delivery of cargoes into a murine solid tumor by a cell-penetrating peptide and cleavable poly(ethylene glycol) comodified liposomal delivery system via systemic administration. Mol Pharm. 2011;8(6):2151–2161. | ||
Quintero-Hernandez V, Juarez-Gonzalez VR, Ortiz-Leon M, Sanchez R, Possani LD, Becerril B. The change of the scFv into the Fab format improves the stability and in vivo toxin neutralization capacity of recombinant antibodies. Mol Immunol. 2007;44(6):1307–1315. | ||
Ahmad ZA, Yeap SK, Ali AM, Ho WY, Alitheen NB, Hamid M. scFv antibody: principles and clinical application. Clin Dev Immunol. 2012;2012:980250. | ||
Padro T, Bieker R, Ruiz S, et al. Overexpression of vascular endothelial growth factor (VEGF) and its cellular receptor KDR (VEGFR-2) in the bone marrow of patients with acute myeloid leukemia. Leukemia. 2002;16(7):1302–1310. | ||
Kokkonen N, Ulibarri IF, Kauppila A, et al. Hypoxia upregulates carcinoembryonic antigen expression in cancer cells. Int J Cancer. 2007;121(11):2443–2450. | ||
Savage Laboratories. DigiFab digoxin immune Fab (ovine). Available from: http://www.savagelabs.com/index.htm. Accessed December 23, 2014. | ||
Kelley B. Industrialization of mAb production technology: the bioprocessing industry at a crossroads. MAbs. 2009;1(5):443–452. | ||
Li M, Xiao X, Zhang W, Liu L, Xi N, Wang Y. Nanoscale distribution of CD20 on B-cell lymphoma tumour cells and its potential role in the clinical efficacy of rituximab. J Microsc. 2014;254(1):19–30. | ||
Carrasco-Triguero M, Yi JH, Dere R, et al. Immunogenicity assays for antibody-drug conjugates: case study with ado-trastuzumab emtansine. Bioanalysis. 2013;5(9):1007–1023. | ||
Ferrara N, Hillan KJ, Gerber HP, Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004;3(5):391–400. | ||
Lin TS, Flinn IW, Modali R, et al. FCGR3A and FCGR2A polymorphisms may not correlate with response to alemtuzumab in chronic lymphocytic leukemia. Blood. 2005;105(1):289–291. | ||
Ratzinger G, Reagan JL, Heller G, Busam KJ, Young JW. Differential CD52 expression by distinct myeloid dendritic cell subsets: implications for alemtuzumab activity at the level of antigen presentation in allogeneic graft-host interactions in transplantation. Blood. 2003;101(4):1422–1429. | ||
White RR, Sullenger BA, Rusconi CP. Developing aptamers into therapeutics. J Clin Invest. 2000;106(8):929–934. | ||
Cheng WW, Das D, Suresh M, Allen TM. Expression and purification of two anti-CD19 single chain Fv fragments for targeting of liposomes to CD19-expressing cells. Biochim Biophys Acta. 2007;1768(1):21–29. | ||
Radom F, Jurek PM, Mazurek MP, Otlewski J, Jelen F. Aptamers: molecules of great potential. Biotechnol Adv. 2013;31(8):1260–1274. | ||
Javier DJ, Nitin N, Levy M, Ellington A, Richards-Kortum R. Aptamer-targeted gold nanoparticles as molecular-specific contrast agents for reflectance imaging. Bioconjug Chem. 2008;19(6):1309–1312. | ||
Jimenez-Barbero J, Dragoni E, Venturi C, et al. Alpha-O-linked glycopeptide mimetics: synthesis, conformation analysis, and interactions with viscumin, a galactoside-binding model lectin. Chemistry. 2009;15(40):10423–10431. | ||
Bennett MJ, Lebron JA, Bjorkman PJ. Crystal structure of the hereditary haemochromatosis protein HFE complexed with transferrin receptor. Nature. 2000;403(6765):46–53. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.