Back to Journals » OncoTargets and Therapy » Volume 13
Toll-Like Receptor 9 Agonists in Cancer
Authors Karapetyan L, Luke JJ ![]() , Davar D
, Davar D ![]()
Received 26 May 2020
Accepted for publication 16 August 2020
Published 9 October 2020 Volume 2020:13 Pages 10039—10061
DOI https://doi.org/10.2147/OTT.S247050
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Leo Jen-Liang Su
Lilit Karapetyan,1 Jason J Luke1,2 *,* Diwakar Davar1,2 **
1University of Pittsburgh Medical Center (UPMC) Hillman Cancer Center (HCC), Pittsburgh, PA, USA; 2Department of Medicine, University of Pittsburgh, Pittsburgh, PA, USA
*These authors contributed equally to this work
Correspondence: Diwakar Davar
University of Pittsburgh Medical Center and Hillman Cancer Center, UPCI Research Pavilion, 5115 Centre Ave, Suite 1.32d, Pittsburgh, PA 15213, USA
Tel +1 412 623 7368
Fax +1 412 623 7704
Email [email protected]
Jason J Luke
University of Pittsburgh Medical Center and Hillman Cancer Center, 5115 Centre Ave, Pittsburgh, PA 15213, USA
Tel +1 412 623 7368
Fax +1 412 623 7704
Email [email protected]
Abstract: Toll-like receptor 9 (TLR9) is a pattern recognition receptor that is predominantly located intracellularly in immune cells, including dendritic cells, macrophages, natural killer cells, and other antigen-presenting cells (APC). The primary ligands for TLR9 receptors are unmethylated cytidine phosphate guanosine (CpG) oligodinucleotides (ODN). TLR9 agonists induce inflammatory processes that result in the enhanced uptake and killing of microorganisms and cancer cells as well as the generation of adaptive immune responses. Preclinical studies of TLR9 agonists suggested efficacy both as monotherapy and in combination with several agents, which led to clinical trials in patients with advanced cancer. In these studies, intravenous, intratumoral, and subcutaneous routes of administration have been tested; with anti-tumor responses in both treated and untreated metastatic sites. TLR9 agonist monotherapy is safe, although efficacy is minimal in advanced cancer patients; conversely, combinations appear to be more promising. Several ongoing phase I and II clinical trials are evaluating TLR9 agonists in combination with a variety of agents including chemotherapy, radiotherapy, targeted therapy, and immunotherapy agents. In this review article, we describe the distribution, structure and signaling of TLR9; discuss the results of preclinical studies of TLR9 agonists; and review ongoing clinical trials of TLR9 agonists singly and in combination in patients with advanced solid tumors.
Keywords: toll-like receptor, TLR, TLR9, CpG, ODN, innate immunity, innate agonist, cancer, cancer immunotherapy, dendritic cell
Introduction
The advent of monoclonal antibodies (mAb) targeting inhibitory immune checkpoints such as programmed cell death 1 (PD-1) or –ligand 1 (PD-L1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) have transformed the management of advanced cancers. Anti-PD(L)1 immune checkpoint inhibitors (ICI) singly or in combination with anti-CTLA-4 or other agents are approved across multiple indications in sixteen separate diseases including a histology-agnostic indication in patients with microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) solid tumors.1,2 The hallmark of ICI therapy is the durability of responses in a subset of patients as evidenced by progression-free survival (PFS) rates of 21–29% in melanoma and 22% in non-small cell lung cancer (NSCLC) with anti-PD(L)1 singly;3–7 and up to 36% with anti-PD-1/anti-CTLA-4 dual ICI in melanoma.8 However, the majority of patients do relapse and the question of how to improve outcomes in these patients remains a vexing problem for the field.
Biomarkers associated with an improved outcome to ICI therapy include CD8 T cell infiltrate (TIL),9,10 interferon (IFN)-γ gene expression signature,11,12 tumor mutation burden (TMB),12–14 and PD-L1 expression.15–18 However, not all T cell-inflamed tumors respond, and not all tumors are inflamed, underscoring the importance of therapeutic strategies directed at improving responses in T cell-uninflamed tumors.18–20 T cell-inflamed tumors are characterized by the presence of abundant CD8+ T cells, CXCR3-binding chemokines (such as CXCL9 and CXCL10) and to a lesser extent, B cells and plasma cells – collectively represented by IFN-γ gene expression signature.10,11,21,22 Conversely, the tumor microenvironment (TME) of T cell-uninflamed tumors is characterized by immuno-suppressive cells including regulatory T cells (T reg), myeloid-derived suppressor cells (MDSC), tumor‐associated macrophages (TAM), vascular endothelial cells and cancer-associated fibroblasts (CAF).23–26 It remains unclear in individual tumors whether the lack of anti-tumor immunity is related to defects at the level of T cell priming/trafficking, other mechanisms mediating tumor-intrinsic mechanisms of immune exclusion in TME, or a combination of both factors.
Adaptive immune recognition of a diverse array of antigens is mediated by the structural diversity of B and T cell receptors – attributable to somatic hypermutation in V-region genes, and junctional and combinatorial diversity generated during gene rearrangement in B and T cells, respectively.27 Conversely, the innate immune system comprising complement system, dendritic cells (DC), natural killer (NK) cells, macrophages, leucocytes and γδ T cells, is an older, evolutionarily conserved system that serves to prevent the spread and movement of foreign pathogens.
The concept of microbial pattern recognition was first proposed by Charles Janeway Jr. and describes the two cardinal features of innate immunity: the ability to distinguish non-self infectious molecules conserved across microbial species – termed pathogen-associated molecular patterns (PAMPs) - from self, and the ability to elicit adaptive immune responses against non-self PAMPs.28 Since the initial description in 1989, multiple pattern recognitional receptors (PRR) have been described that play critical roles in innate and adaptive immune responses including Toll-like receptors (TLR), retinoic acid-inducible gene I (RIG-I)-like receptors, nucleotide-binding oligomerization domain (NOD)-like receptors, and C-type lectin receptors (CLR) recognize and are critical to the activity of the innate immune system.29,30
The various PRRs recognize different PAMPs, are differentially expressed on immune cells and localize to different cellular localities (see Figure 1).31 TLRs mediate innate immune responses to a wide variety of distinct PAMPs derived from various microorganisms including viruses, intra and extra cellular bacteria, and fungi; and are expressed on a broad array of immune cells.31 RIG-I-like receptors (RLRs) RIG-I, MDA5, and LGP2 are broadly expressed in most tissues where they sense viral RNA ligands in the cytoplasm and trigger inflammatory and anti-viral innate immune responses.32,33 CLRs are expressed by multiple innate immune cells particularly DCs and myeloid cells and recognize a variety of glycans on pathogens and hence play an essential role in anti-fungal immune responses.34 NLRs including NOD1 and NOD2 are found in the cytoplasm of lymphocytes, macrophages and DCs where they recognize peptidoglycan motifs from bacterial cells and cooperate with TLR to activate nuclear factor-kappaB (NF-κB) signaling and mediate anti-bacterial immune responses.35,36 Stimulator of interferon genes (STING) is located in the endoplasmic reticulum of APCs, endothelial, and epithelial cells and is activated when plasma membrane-bound cGAS recognizes cytosolic double-stranded DNA. cGAS-STING signaling phosphorylates IRF3 leading to IFN-β transcription and type I IFN signaling.37,38
 |
Figure 1 Cellular distribution of various TLR and respective ligands in humansNotes: Created with BioRender.com. Abbreviations: CpG, cytidine phosphate guanosine; dsRNA, double stranded RNA; LPS, lipopolysaccharide; ODN, oligodinucleotide; ssRNA, single stranded RNA; TLR, Toll-like receptors. |
Type I and II IFNs are involved in innate and adaptive immunity and play an essential role in mediating anti-tumor immunity. Type I IFNs – IFN-α and IFN-β - are expressed in nucleated cells and enhance tumor antigen expression, stimulate dendritic cells (DCs), facilitate CD8+ cell expansion and differentiation to memory type cells, apoptosis, and regulate T cell activity resulting in tumor regression.39,40 Type II IFN (IFN-γ) is mainly expressed in Th1 cells, CD8+ T cells, and natural killer (NK) cells. IFN-γ decreases the activity of T reg cells, increases MHC I and II expression on DCs, enhances antigen presentation, activates proliferation of CD4+ and CD8+ T cells, and M1 macrophages.41–43 Through a downstream signaling cascade that begins with type I IFN induction, type I IFNs potently and rapidly translate signals from TLRs into effects on a broad array of tissues.44
Compelling preclinical data suggest that intra-tumoral administration of TLR9 agonists improves APC activation, in particular DCs in tumor-draining lymph nodes resulting in proinflammatory cytokine release, increased expression of type I IFN genes and T cell priming converting T cell uninflamed TMEs to T cell inflamed ones.45 Given the importance of type I IFN and CD8+ T cells in mediating response to ICI, there has been compelling interest in evaluating combinations of innate agonists, particularly TLR9 agonists, to augment response to ICI.10,46 The efficacy of TLR9 agonists with ICI therapy has been investigated in preclinical studies and is currently being tested in clinical trials; and represent a promising therapeutic strategy to overcome ICI resistance, particularly for T cell uninflamed tumors.47,48 In this review article, we describe TLR9 receptor distribution and function, describe TLR9 signaling and delineate the role of TLR9 agonists in cancer immunotherapy with a focus on ongoing clinical trials.
TLR9 Overview: Type and Distribution, Structure and Ligands, Activation and Signaling Pathways
TLR9 Type and Distribution
TLRs are homologous to the Toll receptor which was first identified in Drosophila melanogaster where it forms a complex with human nerve growth factor-like cystine knot protein Spätzle – the Toll-Spätzle complex – which is critical to both embryonic development and the generation of immune responses against fungi.49–51 Although structurally related to Drosophila TLR, molecular phylogenetic analyses have clarified that vertebrate TLR are highly conserved across various species and can be subclassified into six major families based on general class of PAMP recognized: TLR1 family which includes TLR1, TLR2, TLR6 and TLR10 (lipopeptide); TLR3 family [double stranded RNA (dsRNA)]; TLR4 family (lipopolysaccharide); TLR5 family (flagellin); TLR7–9 families [TLR7/8 – single stranded RNA (ssRNA) and TLR9 – double stranded DNA (dsDNA) or heme motifs]. The sixth remaining family – which includes TLR11-13 and TLR21-23 subfamilies – is represented in humans only as a pseudogene.52,53 Following synthesis within endoplasmic reticulum, traffic to Golgi, and proper folding, TLRs are either recruited to the cell surface (cell surface TLR) or intracellularly to endosomes (endosomal TLR) - a distribution that reflects the likelihood of exposure to the particular viral and/or bacterial nucleic acids that the TLRs sense as illustrated in Figure 1 (see Figure 1).
Cell surface TLRs include TLR1, TLR2, TLR4, TLR5, TLR6, whereas intracellular TLRs are localized in the endosome and include TLR3, TLR7, TLR8, TLR9, TLR10, TLR11, TLR12, and TLR13 – although TLR11, TLR12, and TLR13 are not expressed in human tissues.54 Of the endosomal TLRs, TLR3 recognizes dsRNA, TLR7 and TLR8 recognize ssRNA while TLR9 recognizes dsDNA.55–57 Expression patterns of the endosomal TLRs further determine likelihood of encountering and responding to various PAMPS: TLR3 is expressed ubiquitously; TLR7 is expressed in lung, placenta and spleen; TLR8 is preferentially expressed in peripheral immune cells and lung tissue.58 In humans, TLR9s are predominantly expressed by antigen presenting cells (APC) particularly B cells, T cells and DCs within immune-cell-rich tissues including spleen, lymph node, and bone marrow.59–62 In humans, TLR10 is primarily expressed within endosomes although its function remains puzzling. Compelling data suggest that TLR10 exerts both pro-inflammatory63,64 and anti-inflammatory65 effects – former observed in the setting of viral infection, while the latter is mediated through a B cell-intrinsic process through antibody-mediated crosslinking. Overall, the ligand specificity and function of TLR10 remain less well characterized.
TLR7, TLR8 and TLR9 are primarily located in intracellular vesicles within the endoplasmic reticulum (ER) and translocate to endosomes upon stimulation by ligands.66 The release of TLR9 from the ER is controlled by several mechanisms including tyrosine-based motifs in TLR9 cytoplasmic tail,67–70 and phosphorylation of TLR9.71 Key proteins required for TLR9 traffic from ER to endosomal compartments include glycoprotein 96 (gp96),72 UNC93B1,73,74 adapter protein 3 (AP-3),75,76 a protein associated with TLR4 (PRAT4A),77 and Slc15a4.75 The unique localization and trafficking requirements of the nucleic acid-sensing TLRs 7–9 serve as a regulatory mechanism to limit immune responses to host nucleic acids; and indeed, artificial localization of TLR9 to cell surface causes autoimmune manifestations.78,79 After leaving the ER, TLR9 and other nucleic acid-sensing TLRs traffic to the endosomal compartment where they are proteolytically processed.
TLR9 Structure and Ligands
Structurally, TLRs are type I transmembrane glycoproteins comprising an extracellular N-terminal ligand recognition domain, a single transmembrane helix, and an intracellular C-terminal cytoplasmic signaling domain.50 TLR extracellular domains (ECD) comprise repeated leucine-rich repeat (LRR) modules that bind PAMPs depending on TLR subtype as delineated in TLR9 Type and Distribution.28,80 Each LRR module is 20–43 amino acids long and comprises a variable part and a highly conserved “LxxLxLxxN” motif where “L” is leucine, isoleucine, valine or phenylalanine and “N” is asparagine, threonine, serine or cysteine.81,82 LRR modules form one or two horseshoe domains wherein the “LxxLxLxxN” motifs are located in inner concave surfaces, while the variable parts form the outer convex surface. TLR receptor structure is characterized by constituent LRR motifs, repeat numbers and is flanked by two cysteine clusters including 2–4 cysteine residues across each TLR subtype as described in TLR9 Type and Distribution.83 The TLR intracellular domain (ICD) comprises ~150 amino acids and shares sequence homology with the signaling domains of IL-1R super-family, and hence is termed Toll/interleukin-1 receptor (TIR).84,85 The tertiary structures of TLR1-6 have been determined and have previously been summarized in other reviews on the topic.83,86
Although TLRs 7–9 bear functional similarly to TLR3 in that they are localized intracellularly to endosomes and recognize nucleic acid PAMPs (see TLR9 Type and Distribution), the structures of TLRs 7–9 are markedly different from TLR3.83,87–90 The ECDs of TLRs 7–9 comprise 25 LRR modules, are heavily glycosylated, contain large insertions in LRRs 2, 5 and 8, and contain stretches of ~40 amino acid residues between LRRs 14 and 15.86 The insertions arise from the glycan-free ECD surface involved in dimerization and hence, likely give rise to structures involved in dimerization; while the ~40 residue stretches show a high degree of species variability and hence, are unlikely involved in dimerization. The TLR9 (and TLR7 and 8) ECD contains a Z-loop or hinge region between LRR14 and LRR15 where proteolytic cleavage by a cysteine lysosomal protease occurs to form proteolytically cleaved TLR9 (amino acids 471–1032) that maintains the horseshoe shape of the protomer.90–92 Both full length and proteolytically cleaved TLR9 (amino acids 471–1032) are predominantly monomeric in the absence of unmethylated cytidine phosphate guanosine (CpG). However, following ligand binding, proteolytically cleaved TLR9 dimerizes forming a homodimer.90,91,93
TLR9 preferentially detects unmethylated CpG oligodinucleotides (ODN) with a species-specific preference for hexamer CpG motifs (human 5′-GTCGTT-3 vs. murine 5′-GACGTT-3) that are less common in vertebrate DNA.94 There are three major classes of CpG ODNs based on different backbones and sequence motifs. Type A CpG are characterized by: poly G sequence at 5ʹ end, 3ʹ end or both; internal palindromic sequence containing GC nucleotides; and a partial phosphorothioate (PS) modified backbone.95 Type A CpG preferentially activate plasmacytoid DCs (pDC) and NK cells and induce significant IFN-α production by pDCs.96–98 Type B CpG are generally 18–28 nucleotides in length; have a complete PS-modified backbone; and contain one or more 6mer CpG motifs (5ʹ-PuPyCGPyPu-3ʹ) with the most potent ODNs in this class containing three 6mer sequences.61 Type B CpG preferentially activate B cells and less so NK cells with no effect on DCs.99,100 Type C CpG have features of both classes A and B: a complete PS-modified backbone and an internal palindromic motif.101 Consequently, the effects of type C CpG comprise features of both classes: strong direct B cell stimulation, IFN-α production by pDC, APC activation and maturation, and indirect NK cell activation.102
Besides species-specific ODN sequence preference for TLR9 activation, other factors including number and position of CpG motifs, nucleotides adjacent to CG dinucleotide, and ODN secondary structure influence ODN potency for TLR9 activation in humans.103–106 Separately, when considering the effects of TLR9 agonists in primates as compared to mice, it is important to consider the differential distribution and localization of TLR9 receptors in humans and mice.107 Synthetic TLR9 agonists, which are being used in clinical trials, have structural differences, which makes them nuclease resistant and also increases their half-life.108 A comprehensive list of synthetic TLR9 agonists categorized by type, structure and current status of clinical evaluation is provided in Table 1.
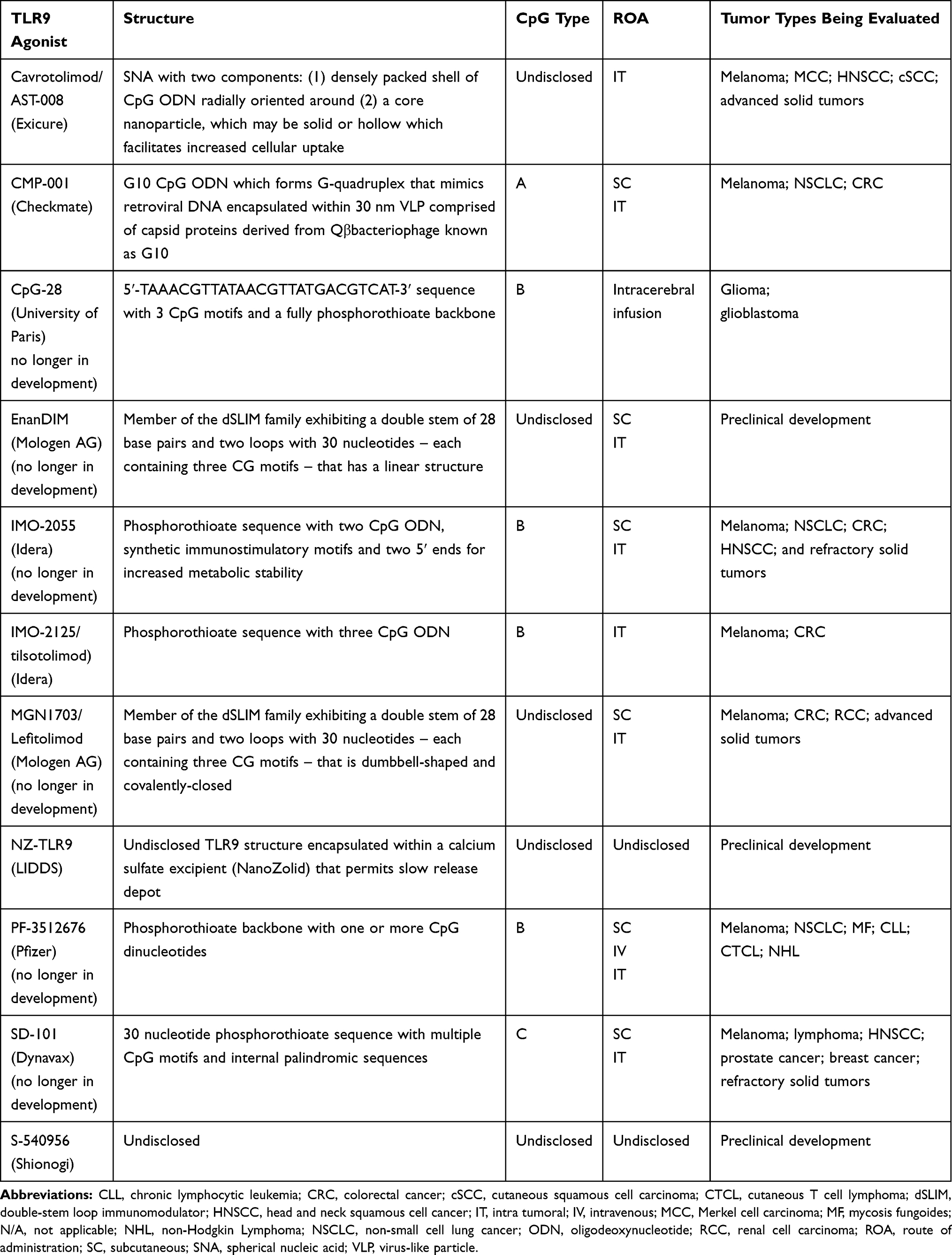 |
Table 1 Toll-Like Receptor 9 Agonists Recently or Currently in Development |
TLR9 Activation and Signaling Pathways
Activation of TLR9 signaling requires two CpG ODN to symmetrically bind to the C-terminal fragment of one TLR9 protomer and the CpG-binding groove in the N-terminal fragment of another, creating a homodimer.90,91,93 ODN that bind only to the N-terminal fragment are inhibitory;109 while methylated single stranded DNA (ssDNA) and dsDNA have a lower affinity for TLR9 and induce less TLR9 dimerization.110–112 Given the differential immunostimulatory activity of unmethylated and methylated CpG ODN, it was initially thought that methylation status of CpG ODN represented a means by which vertebrate TLR9 distinguished between pathogenic bacterial (unmethylated) and eukaryotic (methylated) DNA.112 However, it has since been shown that while TLR9 can recognize both unmethylated and methylated DNA; the greater immunostimulatory activity of unmethylated CpG ODN is secondary to an upstream process wherein unmethylated (but not methylated) CpG ODN induces TLR9 mobilization to the late endosomal compartment in a src-family kinase (SFK)-mediated signaling process, that permits co-localization irrespective of methylation status.113,114 Besides CpG ODN, TLR9 may be activated by endogenous ligands including heat shock protein (HSP), surfactant protein A (SP-A), fibronectin, high mobility group box 1 (HMGB1), in addition to numerous synthetic ligands.60
TLRs differentially recruit specific combinations of one or more among four TIR domain-containing adaptors that mediate downstream signaling: myeloid differentiation primary response gene 88 (MyD88), TIR domain-containing adaptor-inducing IFN-β (TRIF), TIR-containing adaptor protein/MyD88-adaptor-like (TIRAP/MAL), or TRIF-related adaptor molecule (TRAM).115 All TLR signaling pathways culminate in activation of the transcription factor NF-κB – either through MyD88-dependent or MyD88-independent (or TRIF-dependent) mechanisms.115 MyD88 consists of an N-terminal death domain (DD) and a C-terminal TIR domain.116 Following TLR activation, MyD88 DD interacts with DDs of IL-1 receptor-associated kinase (IRAK) family of protein kinases, particularly IRAK1 and IRAK4.117,118 Following sequential phosphorylation of IRAK1 and IRAK4, these dissociate from MyD88 and interact with TNF receptor-associated factor 6 (TRAF6), a RING-type ubiquitin E3 ligase.119 TRAF6 promotes ubiquitination of target proteins, including TRAF6 itself and NF-κB essential modifier (NEMO) along with the ubiquitin-conjugating enzyme complex Uev1A-Ubc13.120,121 Ubiquitinated NEMO and TRAF6 recruit transforming growth factor-β-activated kinase-1 (TAK1) and TAK1-binding proteins (TABs) to activate either mitogen-activated protein kinase (MAPK) (ERK, JNK, p38) pathway or the inhibitor of nuclear factor-κB (IκB) kinase (IKK) complex which leads to NF-κB signaling.122 Separate from the MyD88-dependent pathway, TLR3 and TLR4 in particular can activate NF-κB signaling through a TRIF-dependent pathway mediated through either TRAF6 or receptor interacting protein 1 (RIP1).123–125 TRIF directly binds to TRAF6 via its TRAF6-binding motifs.124,126 TRAF6 then activates NF-κB signaling via a TAK1-dependent mechanism similar to what occurs in the MyD88-dependent pathway.124 TAK1-deficient mice demonstrate impaired NF-κB and MAPK activation in response to multiple TLR ligands underscoring the importance of TAK1 in mediating NF-κB and MAPK activation through either MyD88-dependent or TRIF-dependent pathways.127,128
TLR signaling is homeostatically regulated by a number of mechanisms, failure of which results in autoimmunity and/or inflammatory diseases. The MyD88-dependent pathway can be inhibited by ST2825, SOCS1, and Cbl-b; while the TRIF-dependent pathway is suppressed by SARM and TAG.129,130 Aside from these, other molecules can directly inhibit binding of MyD88 or TRIF with TLRs or downstream molecules including TRAF3, TRAF6 and TAK1 – summarized in detail elsewhere.131 Finally, NF-κB signaling can directly be directly suppressed by cl-3, IκBNS, Nurr1, ATF3, and/or PDLIM2.132
B cells and pDC constitutively express TLR7 and TLR9 and produce large amounts of type I IFNs following exposure to cognate ligands ssRNA and CpG ODN, respectively. TLR9 (along with TLR7) signal via the MyD88-dependent pathway. While activation of MAPK and NF-κB signaling via a TAK1-dependent process, type I IFN production is mediated by IRF7 that is phosphorylated in the cytoplasm but subsequently translocates into the nucleus.133,134 The critical role played by IRF7 in response to TLR7/TLR9 signaling is underscored by the inability of IRF7-deficient (but not IRF3-deficient) mice to produce normal amounts of IFN-α in response to TLR7/TLR9 ligands.135 The TLR9 signaling pathway and pDC activation are illustrated in Figure 2.
 |
Figure 2 TLR and TLR9 cellular location and downstream signalingNotes: Created with BioRender.com. Abbreviations: AP-1, activator protein 1; IRF, interferon regulatory factor; IKK, IκB kinase; MAPK, mitogen activated protein kinase; MyD88, myeloid differentiation primary response 88; NFκB, nuclear factor kappa beta; SARM1, sterile α and TIR motif containing 1; TAK1, transforming growth factor-β-activated kinase-1; TLR, toll-like receptor; TRIF, TIR domain-containing adaptor-inducing IFN, beta; TRAF6, TNF receptor-associated factor 6 (TRAF6); TRAM, TRIF-related adaptor molecule; TIRAP/MAL, TIR-containing adaptor protein/MyD88-adaptor-like. |
TLR9 activation on DCs and pDCs, in particular, prompts the secretion of large quantities of type I IFNs, which has direct (tumor cell inhibition) and indirect (anti-tumor immune responses) effects on cancer cells, most pronounced in the early stages of the anti-tumor immune response. Type I IFN production in the tumor facilitates antigen cross-presentation by DC, which subsequently migrates to the tumor-draining lymph nodes where they cross-prime naive CD8+ T cells, further amplifying anti-tumor immune responses.136,137 This results in DC maturation (with expression of CD80 and CD86), increased MHC class I expression, and secretion of IL-12 leading to a Th1 skew.46,138,139
Preclinical and Clinical Studies of TLR9 Agonists
Preclinical Studies of TLR9 Agonists
The systemic and local use of TLR9 agonists as monotherapy and in combination with chemotherapy, radiation, targeted therapy, and immunotherapy have been explored in several preclinical models with promising therapeutic potential. In syngeneic models in multiple tumors including breast cancer, lung cancer, melanoma, colon, cervical, pancreatic cancer and lymphoma, TLR9 agonists inhibited tumor growth singly and in combination with chemotherapy, and targeted therapy including EGFR and Her-2/neu directed therapies.140–143 Across these various studies, TLR9 agonists were administered topically, subcutaneously, intratumorally or systemically; although subcutaneously and intra-tumoral administration resulted in greater tumor retention compared to intravenous administration. Intra-tumoral injections typically induced tumor rejection, and in some instances, potent abscopal effects in distant non-treated tumors were also observed.
Given the immunomodulatory role of radiation therapy (RT), several preclinical studies have evaluated the combination of TLR9 agonists and RT and demonstrated synergy in immunocompetent murine tumor models.144,145 In these studies, mice were able to reject tumor rechallenge demonstrating evidence of immunological memory. The anti-tumor effects of RT and TLR9 agonist combination therapy were abrogated in nude mice suggest that immune cells mediated these effects.146,147 Using a LLC model, Zhang et al demonstrated that RT/TLR9 combination led to a more potent tumor-specific humoral response compared to either TLR9 or RT singly.146 Further, the treatment was still effective in B cell-deficient mice although less so than in non-deficient mice, suggesting that although not critical, B cells were at least partly responsible for the anti-tumor effects observed.146
A major barrier limiting the effect of anti-PD(L)1 and/or anti-CTLA-4 ICI in tumors is the lack of spontaneous tumor-infiltrating T cells and defective IFN-α production in the tumor microenvironment (TME) in non-inflamed tumors.12,46 Preclinically, TLR9 agonists are potent adjuvants of cancer vaccines with strong immunostimulatory effects. TLR9 agonists in combination with peptide vaccines increase antigen-specific T cells in multiple tumors including melanoma, NSCLC, breast cancer, and sarcoma.148–151 These antigen-specific T cells are detectable ex vivo, produce IFN-γ, and are lytic.148 Several lines of evidence suggest that TLR9 agonists in combination with ICI may enhance anti-tumor T cell responses and augment clinical benefits as compared with either agent singly. Firstly, TLR9 agonists expand antigen-specific CD8+ T cells that upregulate PD-1 expression.152,153 Secondly, TLR9 agonists in combination with anti-PD-1 ICI transform the TME, increasing CD8+ T cells, NK cells, DCs, and B cells even in PD-1 non-responding tumors.153,154 Thus, TLR9 agonists, particularly in combination with ICI, offer a promising therapeutic agent to circumvent the lack of IFN-α production observed in uninflamed tumors, which are poorly T cell-infiltrated and often fail to respond to immune checkpoint blockade.
TLR9 agonists have demonstrated potent synergy with anti-PD-1 and/or anti-CTLA-4 ICI in multiple models including melanoma,153,155 lung cancer,156 HPV positive oropharyngeal cancer,157 head and neck squamous cell carcinoma,158 breast cancer,153 colorectal cancer,153 and lymphoma.159 Using two separate melanoma models of diametric immunogenicity, Reilley et al demonstrated the importance of intra-tumoral therapy and TME-centric immune activation in mediating responses to TLR9 agonist and anti-PD-1/anti-CTLA-4 ICI.155 In immunogenic B16/OVA melanoma, TLR9 agonist monotherapy resulted in tumor regression in treated and untreated tumors associated with increased infiltration of tumor antigen-specific T cells, reduced T regs and proinflammatory cytokines.155 Conversely, in non-immunogenic B16/F10 melanoma, while intra-tumoral TLR9 agonist combined with anti-PD-1 or anti-CTLA-4 ICI resulted in regression of treated tumors, untreated tumors did not respond; although this was overcome partially by a more potent anti-CTLA-4 antibody and TLR9 agonist, underscoring the importance of the relative potency of these agents in mediating their effects. The antitumor effects of CpG/IL-10 combination were abrogated in germ-free (GF) or antibiotic-treated mice as a result of the failure of tumor-infiltrating myeloid cells to produce inflammatory cytokines such as TNF and IL12 – linking intestinal microbiota composition to the outcome of TLR9 agonism, an adding the composition of the gut microbiome as another dimension affecting the outcome of TLR9 agonism and ICI.160,161
In summary, TLR9 agonists result in secretion of proinflammatory cytokines, chemokines such as monocyte chemoattractant protein-1 (MCP-1), IFN-γ-inducible 10 protein (IP-10), activation of pDCs, NK cells and Th1 polarization. TLR9 agonists have demonstrated single agent activity and immunological memory in immunocompetent murine models, primarily with intra-tumoral rather than systemic/subcutaneous drug administration. Synergies observed with RT and with immunotherapies in particular anti-PD-1 and/or anti-CTLA-4 ICI argued for further studies in humans. Below we summarize the safety and efficacy data for TLR9 agonists in advanced solid malignancies.
Clinical Trials Evaluating TLR9 Agonists in Solid Tumors: Safety
TLR9 agonists have been evaluated in a plethora of clinical trials singly and in combination with chemotherapy, radiotherapy, and immunotherapy. Select monotherapy and combination trials are summarized in Tables 2 and 3 respectively (see Tables 2 and 3). Of note, in dose-escalation studies of TLR9 agonists, incidence of adverse events (AE) did not increase in a dose-dependent fashion; and maximal tolerated dose (MTD) was typically not achieved. Class-related AEs included flu-like symptoms, were generally of low-severity, with a low incidence (3–10%) of grade 3/4 events and tended to peak between third and sixth doses.
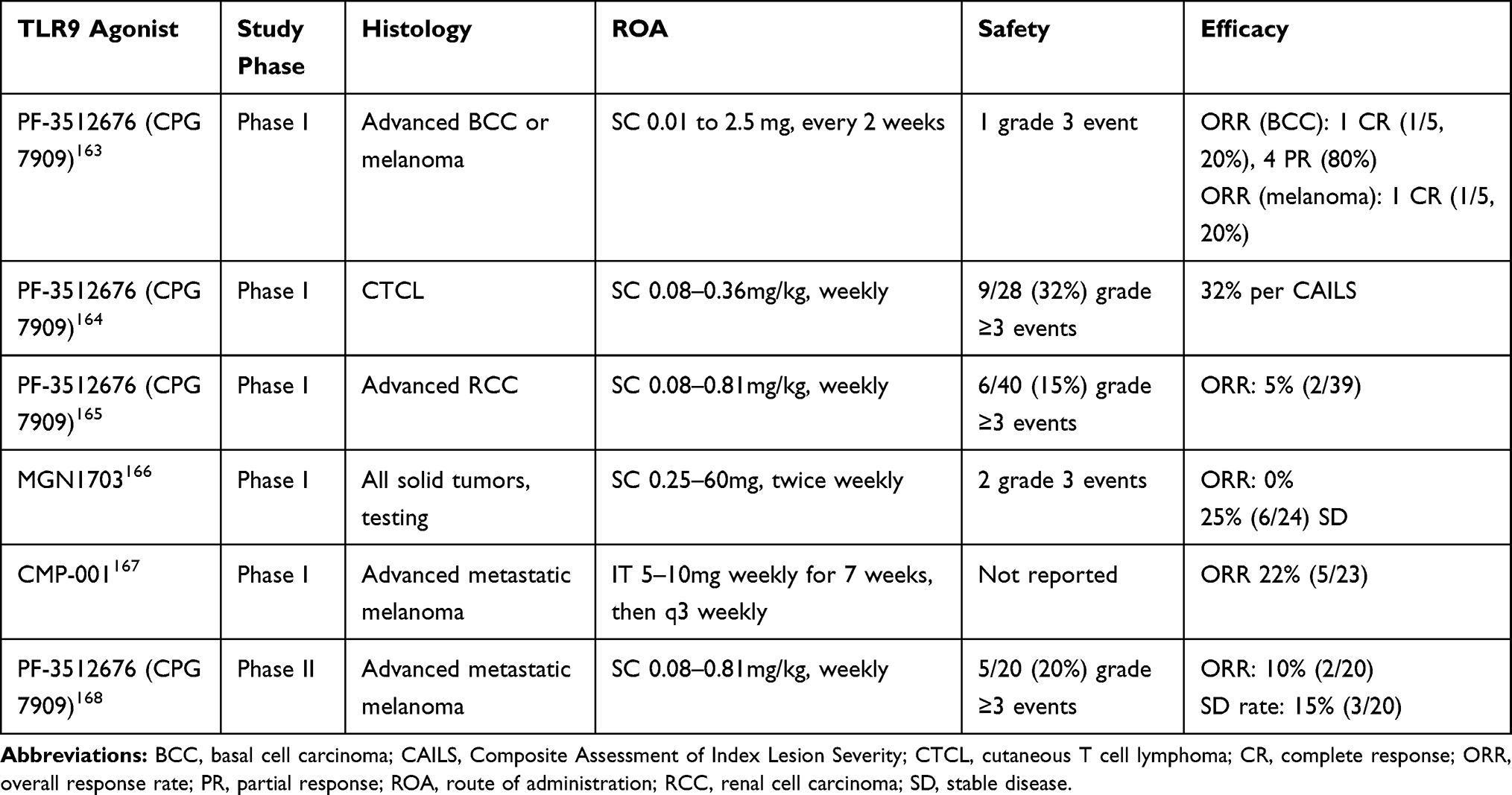 |
Table 2 Clinical Trials of TLR9 Agonists as Monotherapy |
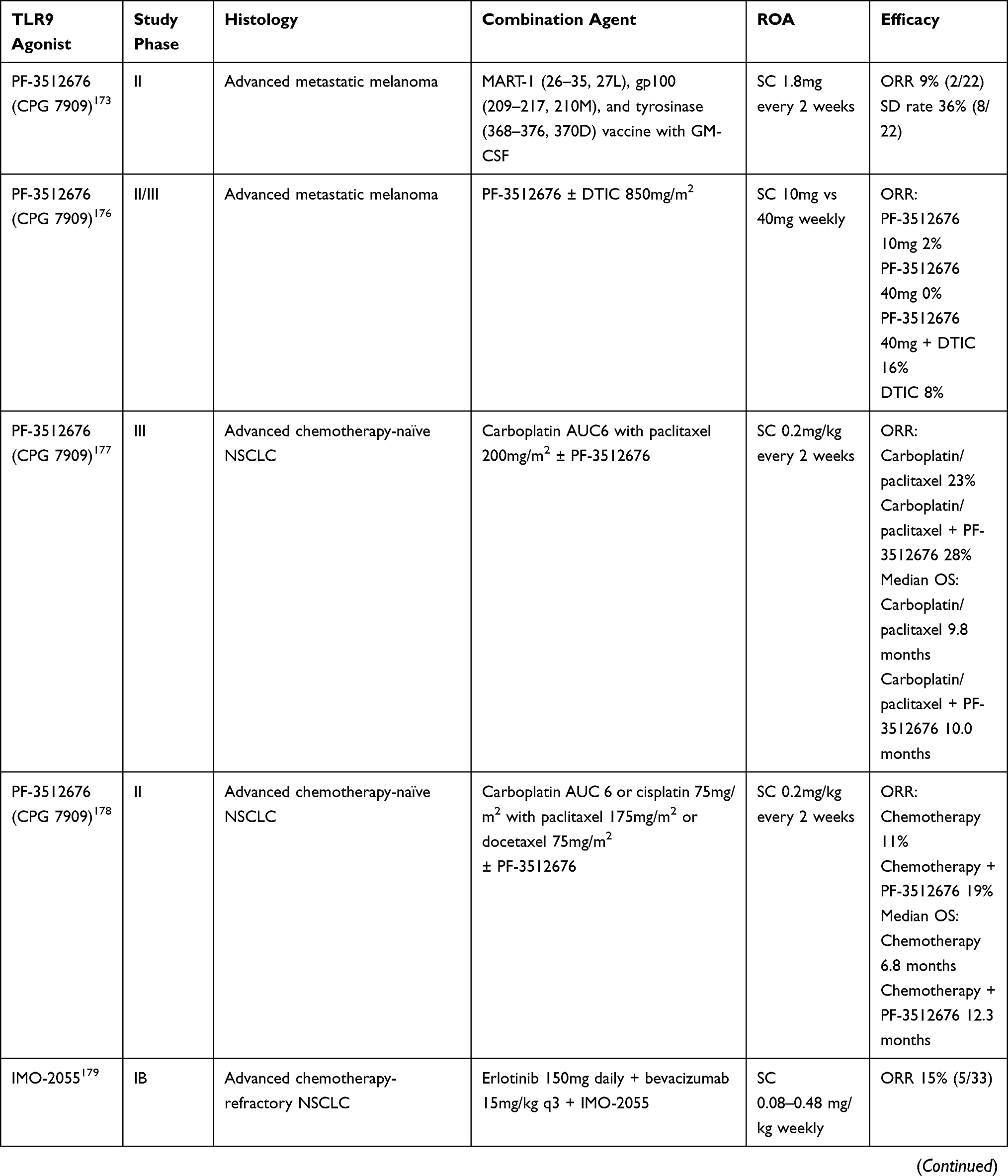 | 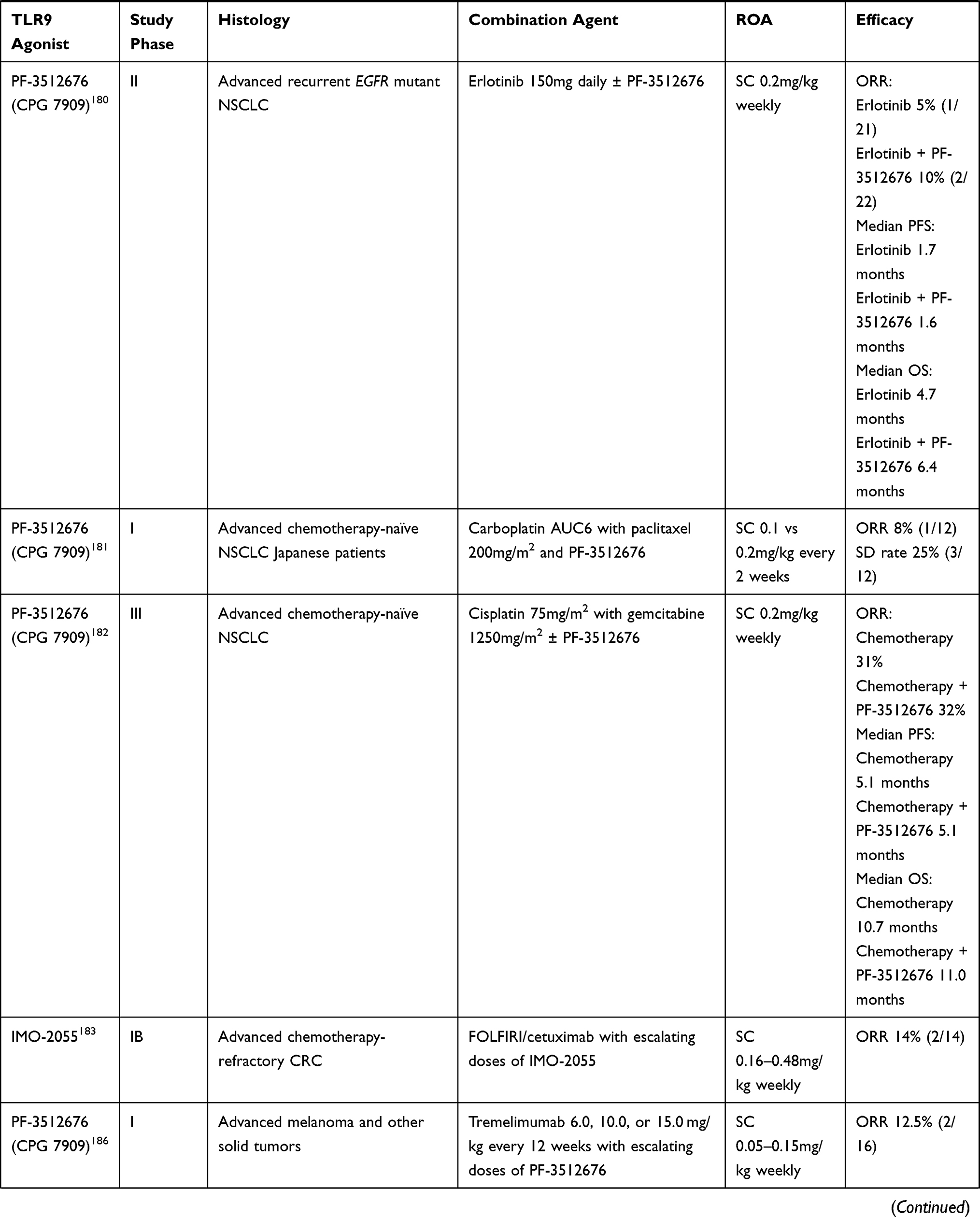 |  |
Table 3 Clinical Trials of TLR9 Agonists in Combination Trials |
Cytokine release syndrome (CRS) is a severe AE that can occur after TLR9 agonist injections and is related to IFN-induced cytokine and chemokine release. CRS, as observed with TLR9 agonists, is graded and managed similarly as chimeric antigen receptor-T cell therapy-related CRS. As CRS symptoms can appear rapidly, patients treated in TLR9 agonist studies are typically monitored for 4–6 hours following TLR9 agonist administration. CRS prophylaxis with low-dose corticosteroids is not typically mandated; although corticosteroid prophylaxis is recommended for patients who experienced Grade 3 and 4 toxicity with prior injections or with concomitant adrenal insufficiency. Other class-specific AEs include injection site reactions with subcutaneous and intra-tumoral injection of drugs.
Studies of TLR9 agonists in combination with chemotherapy, targeted therapy, radiation, vaccines and other immunotherapy agents have generally not been associated with higher than expected AEs with one exception. IMO-2055 was studied in combination with 5FU/Cisplatin/Cetuximab in recurrent/metastatic HNSCC in a phase IB trial that was terminated early due to the higher rate of observed toxicities.162 All patients in the study experienced at least one adverse effect at any grade, and 92% had Grade 3 and greater AEs, with 31% considered related to IMO-2055 including injection site reactions, QT prolongation, bacteremia, and sepsis. TLR9 agonists have been studied in combination with various ICI including anti-PD-(L)1 and/or anti-CTLA-4 inhibitors with no significant additional AEs aside from class-specific flu-like symptoms and injection site reactions. Of note, TLR9/ICI combinations do not appear to be associated with increased frequency of immune-related adverse events (irAEs) in comparison to ICI monotherapy.
Clinical Trials Evaluating TLR9 Agonists in Solid Tumors: Efficacy
When used as monotherapy in advanced solid tumors, TLR9 agonists including PF-3512676 (CPG 7909) and MGN1703 did not reveal any clinically meaningful efficacy results in advanced solid tumors and chronic leukemias with CMP-001 being a rare exception albeit in a small number of patients (see Table 2).163–168 CpG-28 is a CpG ODN administered via intra-cerebral infusion to facilitate maximal brain penetration that was evaluated in treatment-refractory high-grade gliomas and glioblastomas (GBM) with minimal toxicity and preliminary evidence of efficacy in a difficult patient population in phase I and II trials.169,170 However, in a randomized trial of standard chemoradiotherapy with or without CpG-28 injected into surgical cavity post tumor removal, the addition of CpG-28 did not improve survival.171 Phase I single agent data for motolimod (VTX2337), lefitolimod (MGN1703), tilsotolimod (IMO-2125), and SD-101 have not been published.
Intra-tumoral vaccination with CpG in combination with peptide vaccination resulted in increased tumor-antigen specific CD8+ T cells in multiple tumors including melanoma, NSCLC, breast cancer and sarcoma.172–174 These tumor-antigen specific CD8+ T cells were detectable ex vivo, produced IFN-γ and were lytic; and CpG administration expanded tumor-antigen specific clonotypes and primed non-TA-reactive CD8+ T cells resulting in expansion of novel clonotypes.148,175 However, while results of local CpG in combination with peptide vaccination treatments were promising in melanoma,173 systemic administration of CpG in combination with chemotherapy did not result in clinical responses in melanoma (PF-3512676),176 NSCLC (PF-3512676 and IMO-2055),177–182 and colorectal cancer (IMO-2055).183
Combinations with radiation have been evaluated in indolent lymphomas and mycosis fungoides.172,184,185 In mycosis fungoides, PF-3512676 combined with in situ vaccination with RT produced meaningful responses in distant lesions along with significant reductions in T regs intratumorally.184 In patients with treatment-naive indolent lymphomas, intra-tumoral SD-101 in combination with low-dose RT was well tolerated. Responses were observed in both treated and untreated lesions, and were associated with increases in CD8+ and CD4+ effector T cells.185
While it is clear that CpG administration strongly induces tumor-specific CD8+ T cell responses, objective responses are rare and T cell responses are not sustained – suggesting that augmentation of T cell trafficking with anti-PD-1 and/or anti-CTLA-4 ICI may be additive. As delineated above, in preclinical models, intra-tumoral (rather than systemic) TLR9/ICI combinations are efficacious with evidence of increased tumor-specific CD8+ T cells and reduced T regs. A phase I study of PF-3512676 with CTLA-4 inhibitor tremelimumab demonstrated few responses (2/17, 6%) in heavily pre-treated melanoma albeit with a high rate of toxicity that precluded further development.186
Below we describe the current state of development for some of the TLR9 agonists in advanced testing, with a focus on combinations with ICIs in various solid tumors, summarized in Table 3 (see Table 3).
Lefitolimod (MGN1703)
Lefitolimod (MGN1703) is a synthetic type C TLR9 agonist of the double-stem loop immunomodulators (dSLIM) family comprising a covalently closed dumbbell-shaped structure with a middle 28 base pair section flanked by two 30 nucleotides single-stranded loops at either end.187,188 Following a promising phase I study in advanced malignancies, lefitolimod (MGN1703) was rapidly advanced into the maintenance setting in two phase II trials in extensive-stage small cell lung cancer (IMPULSE) and metastatic colorectal cancer after front line chemotherapy.189,190 While primary endpoint of improved overall/progression-free survival was not met in the intent-to-treat arm in either study, improved pharmacodynamic and efficacy signals in subgroup analyses prompted a phase III trial of subcutaneous lefitolimod (MGN1703) as maintenance therapy compared with standard maintenance regimens in patients with metastatic CRC who had achieved tumor reduction with induction therapy (IMPALA, NCT02077868). Negative results have been reported and the further development of this agent is uncertain at this time.
Tilsotolimod (IMO-2125)
Tilsotolimod (IMO-2125) is a type B TLR9 agonist containing a PS backbone with the sequence 5′-TCG*AACG*TTCG*-X-G*CT TG*CAAG*CT-5′ where G* represents 2′-deoxy-7-deaza-guanosine and X is a glycerol linker.191 Tilsotolimod (IMO-2125) is being evaluated in multiple solid tumors with the development in advanced melanoma being furthest along. ILLUMINATE-204 is a multi-center, phase 1/2 trial in patients with anti-PD-1 refractory advanced melanoma wherein escalating doses of tilsotolimod (IMO-2125) was studied with either ipilimumab or pembrolizumab in the phase I portion; after which tilsotolimod (IMO-2125) 8mg in combination with ipilimumab was studied in phase II portion. In 49 evaluable anti-PD1 refractory melanoma patients, investigators reported objective response rates (ORR) of 22% with 71% disease control rate and 48% grade 3/4 treatment-related adverse events (TRAE), although the frequency of irAE was not significantly increased.192 The combination of tilsotolimod (IMO-2125) and ipilimumab is being evaluated in a pivotal phase III trial (ILLUMINATE-301) in anti-PD-1 refractory advanced melanoma compared to ipilimumab; while a separate study (ILLUMINATE-206) is studying tilsotolimod (IMO-2125) with ipilimumab/nivolumab in microsatellite-stable colorectal cancer.
SD-101
SD-101 is a type C TLR9 agonist that induces large quantities of type I IFNs.153 In combination with RT in treatment-naive B cell lymphoma, SD-101 was well tolerated and demonstrated abscopal responses.185 When studied in PD-1 naïve and PD-1 relapsed/refractory melanoma in combination with anti-PD-1 ICI pembrolizumab, ORR with SD-101/pembrolizumab was 78% in treatment-naïve and 15% in PD-1 relapsed/refractory melanoma; while induction of IFN-responsive genes and significant increases of inflammatory cells including CD8+ T cells were observed in circulating leucocytes and in tumors, respectively.154
CMP-001
CMP-001 is a type A CpG composed of (i) a virus-like particle (VLP) comprising capsid proteins derived from bacteriophage Qb, which encapsulate (ii) CpG ODN G10 which I a 30-nucleotide strand, flanked by 10 guanines on either end designed to induce high levels of type I IFN production. CMP-001 was designed to be taken up by pDC via anti-Qb antibodies that bind to FCR-γ on pDC facilitating uptake and enabling antigen cross-presentation to T cells and other effector cells. Following CMP-001 injection intratumorally, anti-Qb antibodies are rapidly generated. These anti-Qb antibodies facilitate opsonization of CMP-001 by pDC in a FcRγ-dependent fashion that promotes pDC uptake, IFN-α induction and are critical to anti-tumor efficacy.159 Although subcutaneous dosing of CMP-001 is being explored in several studies, it is unclear whether this mode of drug delivery results in anti-Qb antibody generation and CMP-001 opsonization to the degree observed with intra-tumoral therapy. In the CMP-001-001 study in PD-1 relapsed/refractory melanoma, intra-tumoral CMP-001 with pembrolizumab resulted in ORR of 25%. Side effects were class-specific including flu-like symptoms (7%), hypotension (6%), hypertension (5%), AST/ALT elevation (2–3%), injection site reactions (27%); and majority were grade 1–2. Median duration of response was not reached (>17 months) with similar responses in treated and non-treated sites.193 The preliminary results of CMP-001 combination with atezolizumab and radiation therapy showed no responses in advanced NSCLC.194
Pre-clinically, compelling data suggest that neoadjuvant immunotherapy (compared with adjuvant immunotherapy) results in greater tumor-specific immune responses and improved eradication of distant metastases following primary tumor resection in orthotopic murine models of melanoma and breast carcinoma.195,196 Separately, in a sub-analysis of patients treated in CMP-001-001 study, high fraction of baseline lymph node tumor burden was a favorable feature associated with longer PFS.197 Compared to post-operative adjuvant therapy, neoadjuvant therapy permits an in vivo assessment of tumor biology, and represents the ideal scenario for studying predictive biomarkers and intermediate end points that might predict long-term clinical outcomes. These observations prompted evaluation of neoadjuvant CMP-001 with nivolumab in high-risk stage III melanoma prior to definitive surgery; wherein the primary endpoint of the study was the rate of major pathologic response (MPR) as assessed using recently developed criteria.198–200 In this study, patients received intra-tumoral CMP-001 and nivolumab for 7 weeks pre-operatively. Following definitive surgery, patients continued nivolumab and subcutaneous CMP-001 every 4 weeks post-operatively for 12 months. In a cohort of 20 patients, 75% MPR rate was observed – significantly better than what has been reported with neoadjuvant pembrolizumab monotherapy and similar to what has been reported with neoadjuvant ipilimumab/nivolumab.201–205 This has prompted a randomized phase II study of the combination compared to pembrolizumab monotherapy (EA6194) and a phase II/III study of the combination against anti-PD(L)1 ICI in PD-1 naïve metastatic melanoma (CMP-001-010).
NZ-TLR
NanoZolid® technology represents a novel approach of creating controlled-release products using cold isostatic pressing to encapsulate active substances in a highly dense calcium sulfate microstructure. This technology has been used to generate depot-formulations of anti-androgens for use in prostate cancer.206 NZ-TLR is a TLR9 agonist with an undisclosed TLR9 structure encapsulated within a calcium sulfate excipient (NanoZolid) that permits slow release depot formulation following intra-tumoral dosing obviating the need for frequent dosing. Preclinical data in syngeneic tumor models have been reported but not published with plans for first-in-human clinical trials in 2021.
Cavrotolimod (AST-008)
Spherical nucleic acids (SNAs) are a unique class of nanomaterial comprised of an inorganic nanoparticle core that acts as a scaffold for the assembly and orientation of an outer nucleic acid shell. SNA technology confers properties that distinguish SNAs from linear DNA or RNA counterparts including increased cellular uptake, improved pharmacokinetics, and biodistribution.207 Cavrotolimod (AST-008) is a novel SNA configuration of TLR9 with undisclosed structure that is currently being tested singly and in combination with pembrolizumab in patients with advanced Merkel cell carcinoma and cutaneous squamous cell carcinoma. Preclinical data suggested that intra-tumoral cavrotolimod (AST-008) activated immune cells and elicited a Th1 type cytokine response. Early data from a phase I study suggested that cavrotolimod (AST-008) was safe and well tolerated with no dose-limiting AEs noted. Dose-dependent increases in NK cells and CD8 T cells peripherally were observed intra-tumoral cavrotolimod (AST-008) singly with pembrolizumab.207
Conclusions
TLR9 agonists are well tolerated as monotherapy and do not appear to increase the toxicity of chemotherapeutic, targeted, radiation, and other immunotherapy agents as part of combination therapy. The main adverse effects are injection site reactions and flu-like symptoms, of mild to moderate severity and typically well managed with symptomatic treatment – and have resulted in treatment discontinuation in only a small number of patients in clinical trials.
In preclinical studies, TLR9 agonists demonstrated efficacy singly and in combination with a variety of agents. Intra-tumoral and subcutaneous routes of administration showed better local and distant responses in comparison to intravenous routes. In clinical trials, single-agent TLR9 agonist therapy for advanced solid tumors did not demonstrate significant efficacy, especially with an intravenous route of administration. While combinations with chemotherapy and targeted therapy yielded disappointing results, combinations with RT in indolent lymphomas have been promising. However, the greatest excitement has been reserved for the combinations of TLR9 agonists with ICI. Tilsotolimod (IMO-2125), SD-101 and CMP-001 all demonstrated efficacy in PD-1 relapsed/refractory melanoma in small non-randomized phase II studies. Pivotal trials testing these agents against active comparators in advanced PD-1 relapsed/refractory melanoma are either ongoing (tilsotolimod/IMO-2125 – ILLUMINATE-301) or planned (CMP-001 - CMP-001-010).
Separately, the unusual response rate observed with CMP-001 in combination with nivolumab in a small phase II neoadjuvant trial raises several questions: do neoadjuvant pathological responses translate into durable relapse-free survival; is the response rate in this setting predictive of what may be seen in PD-1 naïve metastatic melanoma; and is the neoadjuvant setting with its limited tumor burden and nodal involvement the ideal setting wherein to evaluate innate agonists in melanoma and other cancers?
TLR9 agonists augment antigen presentation to APC, and promote T cell trafficking to create a T cell inflamed TME resulting in an increased response rate of ICI therapy. However, several questions remain. What is the ideal ICI partner for augmenting the benefit of innate agonists in general and TLR9 agonists in particular: anti-PD-1, anti-CTLA-4, combination of anti-PD-1/anti-CTLA-4 or other? Are TLR9 agonists enough to convert T cell uninflamed tumors to inflamed tumors; or are combinations with other innate immunostimulants such as RIG-I, STING agonists required? While accessibility of tumors has limited the scope of intra-tumoral injections, could recent advancements in image-guided procedures provide an opportunity to target visceral metastatic lesions? Could advances in structural chemistry permit depot formulations that maintain efficacy with fewer injections? How best can TLR9 agonists be combined with RT, and possibly newer modalities including alpha-emitters?208 Ongoing clinical trials will help to answer some questions while elucidating biomarkers of resistance and response with the eventual goal of broadening the use of TLR9 agonists in solid tumors.
Funding
This work was supported by grants from Harry J Lloyd Trust and Melanoma Research Foundation Breakthrough Consortium (DD); DOD W81XWH-17-1-0265, Sy Holzer Endowed Immunotherapy Research Fund Award and the Hillman Fellows for Innovative Cancer Research Program (JJL).
Disclosure
JJL declares Consultant fees: TTC Oncology, 7 Hills, Alphamab Oncology, Pyxis, Spring bank, Tempest, Abbvie, Akrevia, Algios, Array, Astellas, Bayer, Bristol-Myers Squibb, Eisai, EMD Serono, Ideaya, Incyte, Janssen, Merck, Mersana, Novartis, PTx, RefleXion, Regeneron, Rubius, Silicon, Tesaro, Vividion; Equity: Actym, Alphamab Oncology, Arch Oncology, Kanaph, Mavu (now part of AbbVie), Onc.AI, Pyxis, Tempest; Research Support: (all to institution for clinical trials unless noted) AbbVie, Agios (IIT), Array (IIT), Astellas, Bristol-Myers Squibb, CheckMate (SRA), Compugen, Corvus, EMD Serono, Evelo (SRA), Five Prime, FLX Bio, Genentech, Immatics, Immunocore, Incyte, Leap, MedImmune, Macrogenics, Necktar, Novartis, Palleon (SRA), Merck, Spring bank, Tesaro, Tizona, Xencor; Travel: Akrevia, Bayer, Bristol-Myers Squibb, EMD Serono, Incyte, Janssen, Merck, Mersana, Novartis, Pyxis, RefleXion; Patents: (both provisional) Serial #15/612,657 (Cancer Immunotherapy), PCT/US18/36,052 (Microbiome Biomarkers for Anti-PD-1/PD-L1 Responsiveness: Diagnostic, Prognostic and Therapeutic Uses Thereof).
DD reports Research support from BMS, CellSight Technologies, Checkmate Pharmaceutics, GlaxoSmithKline, Merck; and received consultancy fees from Array Biopharma, Merck, Incyte, Immunocore; also Speakers’ Bureau: Medical Education Network. The authors report no other conflicts of interest in this work.
References
1. Marabelle A, Le DT, Ascierto PA, et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/mismatch repair-deficient cancer: results from the phase II KEYNOTE-158 study. J Clin Oncol. 2020;38(1):1–10. doi:10.1200/JCO.19.02105
2. Zhao B, Zhao H, Zhao J. Efficacy of PD-1/PD-L1 blockade monotherapy in clinical trials. Ther Adv Med Oncol. 2020;12:1758835920937612. doi:10.1177/1758835920937612
3. Hamid O, Robert C, Daud A, et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann Oncol. 2019;30(4):582–588. doi:10.1093/annonc/mdz011
4. Herbst RS, Garon EB, Kim DW, et al. Long-term outcomes and retreatment among patients with previously treated, programmed death-Ligand 1Positive, advanced nonsmall-cell lung cancer in the KEYNOTE-010 study. J Clin Oncol. 2020;38(14):1580–1590. doi:10.1200/JCO.19.02446
5. Larkin J, Lao CD, Urba WJ, et al. Efficacy and safety of nivolumab in patients with BRAF V600 mutant and BRAF wild-type advanced melanoma: a pooled analysis of 4 clinical trials. JAMA Oncol. 2015;1(4):433–440. doi:10.1001/jamaoncol.2015.1184
6. Ribas A, Hamid O, Daud A, et al. Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA. 2016;315(15):1600–1609. doi:10.1001/jama.2016.4059
7. Weber JS, Hodi FS, Wolchok JD, et al. Safety profile of nivolumab monotherapy: a pooled analysis of patients with advanced melanoma. J Clin Oncol. 2017;35(7):785–792. doi:10.1200/JCO.2015.66.1389
8. Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2019;381(16):1535–1546. doi:10.1056/NEJMoa1910836
9. Rohatgi A, Hartman D, Karunamurthy A, Burkette J. Evaluation of CD8 score by automated quantitative image analysis in metastatic melanoma treated with PD1 blockade: preliminary results. J Immunother Cancer. 2019;7(282).
10. Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568–571. doi:10.1038/nature13954
11. Ayers M, Lunceford J, Nebozhyn M, et al. IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017;127(8):2930–2940. doi:10.1172/JCI91190
12. Cristescu R, Mogg R, Ayers M, et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science. 2018;362(6411):eaar3593. doi:10.1126/science.aar3593
13. Cao D, Xu H, Xu X, Guo T, Ge W. High tumor mutation burden predicts better efficacy of immunotherapy: a pooled analysis of 103078 cancer patients. Oncoimmunology. 2019;8(9):e1629258. doi:10.1080/2162402X.2019.1629258
14. Wu HX, Wang ZX, Zhao Q, et al. Tumor mutational and indel burden: a systematic pan-cancer evaluation as prognostic biomarkers. Ann Transl Med. 2019;7(22):640. doi:10.21037/atm.2019.10.116
15. Daud AI, Wolchok JD, Robert C, et al. Programmed death-ligand 1 expression and response to the anti-programmed death 1 antibody pembrolizumab in melanoma. J Clin Oncol. 2016;34(34):4102–4109. doi:10.1200/JCO.2016.67.2477
16. Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515(7528):563–567. doi:10.1038/nature14011
17. Taube JM, Klein A, Brahmer JR, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res. 2014;20(19):5064–5074. doi:10.1158/1078-0432.CCR-13-3271
18. Trujillo JA, Sweis RF, Bao R, Luke JJ. T cell-inflamed versus non-T cell-inflamed tumors: a conceptual framework for cancer immunotherapy drug development and combination therapy selection. Cancer Immunol Res. 2018;6(9):990–1000. doi:10.1158/2326-6066.CIR-18-0277
19. Olson DJ, Luke JJ. The T-cell-inflamed tumor microenvironment as a paradigm for immunotherapy drug development. Immunotherapy. 2019;11(3):155–159. doi:10.2217/imt-2018-0171
20. Gajewski TF, Corrales L, Williams J, Horton B, Sivan A, Spranger S. Cancer immunotherapy targets based on understanding the T cell-inflamed versus non-T cell-inflamed tumor microenvironment. Adv Exp Med Biol. 2017;1036:19–31.
21. Harlin H, Meng Y, Peterson AC, et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009;69(7):3077–3085.
22. Mikucki ME, Fisher DT, Matsuzaki J, et al. Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat Commun. 2015;6(1):7458. doi:10.1038/ncomms8458
23. Kraman M, Bambrough PJ, Arnold JN, et al. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science. 2010;330(6005):827–830. doi:10.1126/science.1195300
24. Salmon H, Franciszkiewicz K, Damotte D, et al. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J Clin Invest. 2012;122(3):899–910. doi:10.1172/JCI45817
25. Buckanovich RJ, Facciabene A, Kim S, et al. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat Med. 2008;14(1):28–36. doi:10.1038/nm1699
26. Fridman WH, Pagès F, Sautès-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012;12(4):298–306. doi:10.1038/nrc3245
27. Minervina A, Pogorelyy M, Mamedov I. T-cell receptor and B-cell receptor repertoire profiling in adaptive immunity. Transpl Int. 2019;32(11):1111–1123. doi:10.1111/tri.13475
28. Janeway CA
29. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805–820. doi:10.1016/j.cell.2010.01.022
30. Medzhitov R. Approaching the asymptote: 20 years later. Immunity. 2009;30(6):766–775. doi:10.1016/j.immuni.2009.06.004
31. Walsh D, McCarthy J, O’Driscoll C, Melgar S. Pattern recognition receptors–molecular orchestrators of inflammation in inflammatory bowel disease. Cytokine Growth Factor Rev. 2013;24(2):91–104. doi:10.1016/j.cytogfr.2012.09.003
32. Cagliani R, Sironi M. Pathogen-driven selection in the human genome. Int J Evol Biol. 2013;2013:204240. doi:10.1155/2013/204240
33. Loo YM, Gale M
34. Tang J, Lin G, Langdon WY, Tao L, Zhang J. Regulation of C-type lectin receptor-mediated antifungal immunity. Front Immunol. 2018;9(123). doi:10.3389/fimmu.2018.00123
35. Martínez-Torres RJ, Chamaillard M. The ubiquitin code of NODs signaling pathways in health and disease. Front Immunol. 2019;10(2648). doi:10.3389/fimmu.2019.02648
36. Mukherjee T, Hovingh ES, Foerster EG, Abdel-Nour M, Philpott DJ, Girardin SE. NOD1 and NOD2 in inflammation, immunity and disease. Arch Biochem Biophys. 2019;670:69–81. doi:10.1016/j.abb.2018.12.022
37. Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol. 2015;15(12):760–770. doi:10.1038/nri3921
38. Flood BA, Higgs EF, Li S, Luke JJ, Gajewski TF. STING pathway agonism as a cancer therapeutic. Immunol Rev. 2019;290(1):24–38. doi:10.1111/imr.12765
39. Sikora AG, Jaffarzad N, Hailemichael Y, et al. IFN-alpha enhances peptide vaccine-induced CD8+ T cell numbers, effector function, and antitumor activity. J Immunol. 2009;182(12):7398–7407. doi:10.4049/jimmunol.0802982
40. Hervas-Stubbs S, Perez-Gracia JL, Rouzaut A, Sanmamed MF, Le Bon A, Melero I. Direct effects of type I interferons on cells of the immune system. Clin Cancer Res. 2011;17(9):2619–2627. doi:10.1158/1078-0432.CCR-10-1114
41. Lu C, Klement JD, Ibrahim ML, et al. Type I interferon suppresses tumor growth through activating the STAT3-granzyme B pathway in tumor-infiltrating cytotoxic T lymphocytes. J Immunother Cancer. 2019;7(1):157. doi:10.1186/s40425-019-0635-8
42. Castro F, Cardoso AP, Gonçalves RM, Serre K, Oliveira MJ. Interferon-gamma at the crossroads of tumor immune surveillance or evasion. Front Immunol. 2018;9:847.
43. Musella M, Manic G, De Maria R, Vitale I, Sistigu A. Type-I-interferons in infection and cancer: unanticipated dynamics with therapeutic implications. Oncoimmunology. 2017;6(5):e1314424–e1314424. doi:10.1080/2162402X.2017.1314424
44. Perkins DJ, Vogel SN. Space and time: new considerations about the relationship between Toll-like receptors (TLRs) and type I interferons (IFNs). Cytokine. 2015;74(2):171–174. doi:10.1016/j.cyto.2015.03.001
45. Moynihan KD, Irvine DJ. Roles for innate immunity in combination immunotherapies. Cancer Res. 2017;77(19):5215–5221. doi:10.1158/0008-5472.CAN-17-1340
46. Corrales L, Matson V, Flood B, Spranger S, Gajewski TF. Innate immune signaling and regulation in cancer immunotherapy. Cell Res. 2017;27(1):96–108. doi:10.1038/cr.2016.149
47. Agrawal S, Kandimalla ER. Intratumoural immunotherapy: activation of nucleic acid sensing pattern recognition receptors. Immuno Oncol Tech. 2019;3:15–23. doi:10.1016/j.iotech.2019.10.001
48. Hamid O, Ismail R, Puzanov I. Intratumoral immunotherapy—update 2019. Oncologist. 2020;25(3):e423–e438. doi:10.1634/theoncologist.2019-0438
49. Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA. The dorsoventral regulatory gene cassette spatzle/toll/cactus controls the potent antifungal response in drosophila adults. Cell. 1996;86(6):973–983. doi:10.1016/S0092-8674(00)80172-5
50. Gay NJ, Gangloff M. Structure and function of toll receptors and their ligands. Annu Rev Biochem. 2007;76(1):141–165. doi:10.1146/annurev.biochem.76.060305.151318
51. Weber AN, Tauszig-Delamasure S, Hoffmann JA, et al. Binding of the drosophila cytokine spatzle to toll is direct and establishes signaling. Nat Immunol. 2003;4(8):794–800. doi:10.1038/ni955
52. Roach JC, Glusman G, Rowen L, et al. The evolution of vertebrate toll-like receptors. Proc Natl Acad Sci U S A. 2005;102(27):9577–9582. doi:10.1073/pnas.0502272102
53. Coban C, Ishii KJ, Kawai T, et al. Toll-like receptor 9 mediates innate immune activation by the malaria pigment hemozoin. J Exp Med. 2005;201(1):19–25. doi:10.1084/jem.20041836
54. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol. 2010;11(5):373–384. doi:10.1038/ni.1863
55. Gantier MP, Tong S, Behlke MA, et al. TLR7 is involved in sequence-specific sensing of single-stranded RNAs in human macrophages. J Immunol. 2008;180(4):2117–2124. doi:10.4049/jimmunol.180.4.2117
56. Hemmi H, Kaisho T, Takeuchi O, et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol. 2002;3(2):196–200. doi:10.1038/ni758
57. Mancuso G, Gambuzza M, Midiri A, et al. Bacterial recognition by TLR7 in the lysosomes of conventional dendritic cells. Nat Immunol. 2009;10(6):587–594. doi:10.1038/ni.1733
58. Chuang TH, Ulevitch RJ. Cloning and characterization of a sub-family of human toll-like receptors: hTLR7, hTLR8 and hTLR9. Eur Cytokine Netw. 2000;11(3):372–378.
59. Krieg AM. Therapeutic potential of toll-like receptor 9 activation. Nat Rev Drug Discov. 2006;5(6):471–484. doi:10.1038/nrd2059
60. Krieg AM. Toll-like receptor 9 (TLR9) agonists in the treatment of cancer. Oncogene. 2008;27(2):161–167. doi:10.1038/sj.onc.1210911
61. Krieg AM, Yi AK, Matson S, et al. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature. 1995;374(6522):546–549. doi:10.1038/374546a0
62. Nishimura M, Naito S. Tissue-specific mRNA expression profiles of human toll-like receptors and related genes. Biol Pharm Bull. 2005;28(5):886–892. doi:10.1248/bpb.28.886
63. Lee SM, Kok KH, Jaume M, et al. Toll-like receptor 10 is involved in induction of innate immune responses to influenza virus infection. Proc Natl Acad Sci U S A. 2014;111(10):3793–3798. doi:10.1073/pnas.1324266111
64. Regan T, Nally K, Carmody R, et al. Identification of TLR10 as a key mediator of the inflammatory response to listeria monocytogenes in intestinal epithelial cells and macrophages. J Immunol. 2013;191(12):6084–6092. doi:10.4049/jimmunol.1203245
65. Hess NJ, Jiang S, Li X, Guan Y, Tapping RI. TLR10 is a B cell intrinsic suppressor of adaptive immune responses. J Immunol. 2017;198(2):699–707. doi:10.4049/jimmunol.1601335
66. Iavarone C, Ramsauer K, Kubarenko AV, et al. A point mutation in the amino terminus of TLR7 abolishes signaling without affecting ligand binding. J Immunol. 2011;186(7):4213–4222. doi:10.4049/jimmunol.1003585
67. Chockalingam A, Brooks JC, Cameron JL, Blum LK, Leifer CA. TLR9 traffics through the golgi complex to localize to endolysosomes and respond to CpG DNA. Immunol Cell Biol. 2009;87(3):209–217. doi:10.1038/icb.2008.101
68. Chockalingam A, Rose WA
69. Leifer CA, Brooks JC, Hoelzer K, et al. Cytoplasmic targeting motifs control localization of toll-like receptor 9. J Biol Chem. 2006;281(46):35585–35592. doi:10.1074/jbc.M607511200
70. Leifer CA, Kennedy MN, Mazzoni A, Lee C, Kruhlak MJ, Segal DM. TLR9 is localized in the endoplasmic reticulum prior to stimulation. J Immunol. 2004;173(2):1179–1183. doi:10.4049/jimmunol.173.2.1179
71. Hasan M, Gruber E, Cameron J, Leifer CA. TLR9 stability and signaling are regulated by phosphorylation and cell stress. J Leukoc Biol. 2016;100(3):525–533. doi:10.1189/jlb.2A0815-337R
72. Brooks JC, Sun W, Chiosis G, Leifer CA. Heat shock protein gp96 regulates toll-like receptor 9 proteolytic processing and conformational stability. Biochem Biophys Res Commun. 2012;421(4):780–784. doi:10.1016/j.bbrc.2012.04.083
73. Majer O, Liu B, Woo BJ, Kreuk LSM, Van Dis E, Barton GM. Release from UNC93B1 reinforces the compartmentalized activation of select TLRs. Nature. 2019;575(7782):371–374. doi:10.1038/s41586-019-1611-7
74. Pelka K, Bertheloot D, Reimer E, et al. The chaperone UNC93B1 regulates toll-like receptor stability independently of endosomal TLR transport. Immunity. 2018;48(5):911–922e917. doi:10.1016/j.immuni.2018.04.011
75. Blasius AL, Arnold CN, Georgel P, et al. Slc15a4, AP-3, and hermansky-pudlak syndrome proteins are required for toll-like receptor signaling in plasmacytoid dendritic cells. Proc Natl Acad Sci U S A. 2010;107(46):19973–19978. doi:10.1073/pnas.1014051107
76. Sasai M, Linehan MM, Iwasaki A. Bifurcation of toll-like receptor 9 signaling by adaptor protein 3. Science. 2010;329(5998):1530–1534. doi:10.1126/science.1187029
77. Takahashi K, Shibata T, Akashi-Takamura S, et al. A protein associated with toll-like receptor (TLR) 4 (PRAT4A) is required for TLR-dependent immune responses. J Exp Med. 2007;204(12):2963–2976. doi:10.1084/jem.20071132
78. Barton GM, Kagan JC, Medzhitov R. Intracellular localization of Toll-like receptor 9 prevents recognition of self DNA but facilitates access to viral DNA. Nat Immunol. 2006;7(1):49–56. doi:10.1038/ni1280
79. Mouchess ML, Arpaia N, Souza G, et al. Transmembrane mutations in toll-like receptor 9 bypass the requirement for ectodomain proteolysis and induce fatal inflammation. Immunity. 2011;35(5):721–732. doi:10.1016/j.immuni.2011.10.009
80. Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1(2):135–145. doi:10.1038/35100529
81. Kajava AV. Structural diversity of leucine-rich repeat proteins. J Mol Biol. 1998;277(3):519–527. doi:10.1006/jmbi.1998.1643
82. Kobe B, Kajava AV. The leucine-rich repeat as a protein recognition motif. Curr Opin Struct Biol. 2001;11(6):725–732. doi:10.1016/S0959-440X(01)00266-4
83. Matsushima N, Tanaka T, Enkhbayar P, et al. Comparative sequence analysis of leucine-rich repeats (LRRs) within vertebrate toll-like receptors. BMC Genom. 2007;8(1):124. doi:10.1186/1471-2164-8-124
84. Xu Y, Tao X, Shen B, et al. Structural basis for signal transduction by the toll/interleukin-1 receptor domains. Nature. 2000;408(6808):111–115. doi:10.1038/35040600
85. Nyman T, Stenmark P, Flodin S, Johansson I, Hammarstrom M, Nordlund P. The crystal structure of the human toll-like receptor 10 cytoplasmic domain reveals a putative signaling dimer. J Biol Chem. 2008;283(18):11861–11865. doi:10.1074/jbc.C800001200
86. Botos I, Segal DM, Davies DR. The structural biology of Toll-like receptors. Structure. 2011;19(4):447–459. doi:10.1016/j.str.2011.02.004
87. Zhang Z, Ohto U, Shibata T, et al. Structural analysis reveals that toll-like receptor 7 is a dual receptor for guanosine and single-stranded RNA. Immunity. 2016;45(4):737–748. doi:10.1016/j.immuni.2016.09.011
88. Yoo E, Salunke DB, Sil D, et al. Determinants of activity at human toll-like receptors 7 and 8: quantitative structure-activity relationship (QSAR) of diverse heterocyclic scaffolds. J Med Chem. 2014;57(19):7955–7970. doi:10.1021/jm500744f
89. Tanji H, Ohto U, Shibata T, et al. Toll-like receptor 8 senses degradation products of single-stranded RNA. Nat Struct Mol Biol. 2015;22(2):109–115. doi:10.1038/nsmb.2943
90. Ohto U, Shibata T, Tanji H, et al. Structural basis of CpG and inhibitory DNA recognition by toll-like receptor 9. Nature. 2015;520(7549):702–705. doi:10.1038/nature14138
91. Ewald SE, Lee BL, Lau L, et al. The ectodomain of Toll-like receptor 9 is cleaved to generate a functional receptor. Nature. 2008;456(7222):658–662. doi:10.1038/nature07405
92. Sepulveda FE, Maschalidi S, Colisson R, et al. Critical role for asparagine endopeptidase in endocytic toll-like receptor signaling in dendritic cells. Immunity. 2009;31(5):737–748. doi:10.1016/j.immuni.2009.09.013
93. Park B, Brinkmann MM, Spooner E, Lee CC, Kim YM, Ploegh HL. Proteolytic cleavage in an endolysosomal compartment is required for activation of toll-like receptor 9. Nat Immunol. 2008;9(12):1407–1414. doi:10.1038/ni.1669
94. Bauer S, Kirschning CJ, Hacker H, et al. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc Natl Acad Sci U S A. 2001;98(16):9237–9242. doi:10.1073/pnas.161293498
95. Krug A, Rothenfusser S, Hornung V, et al. Identification of CpG oligonucleotide sequences with high induction of IFN-α/β in plasmacytoid dendritic cells. Eur J Immunol. 2001;31(7):2154–2163. doi:10.1002/1521-4141(200107)31:7<2154::AID-IMMU2154>3.0.CO;2-U
96. Verthelyi D, Ishii KJ, Gursel M, Takeshita F, Klinman DM. Human peripheral blood cells differentially recognize and respond to two distinct CpG motifs. J Immunol. 2001;166(4):2372–2377. doi:10.4049/jimmunol.166.4.2372
97. Hartmann G, Weeratna RD, Ballas ZK, et al. Delineation of a CpG phosphorothioate oligodeoxynucleotide for activating primate immune responses in vitro and in vivo. J Immunol. 2000;164(3):1617–1624. doi:10.4049/jimmunol.164.3.1617
98. Sivori S, Carlomagno S, Moretta L, Moretta A. Comparison of different CpG oligodeoxynucleotide classes for their capability to stimulate human NK cells. Eur J Immunol. 2006;36(4):961–967. doi:10.1002/eji.200535781
99. Janeway’s Immunobiology. Ninth edition by Kenneth Murphy, Casey Weaver, Allan Mowat, Leslie Berg, David Chaplin, Charles A. Janeway, Paul Travers, and Mark Walport. Q Rev Biol. 2018;93(1):59.
100. Gürsel M, Verthelyi D, Gürsel I, Ishii KJ, Klinman DM. Differential and competitive activation of human immune cells by distinct classes of CpG oligodeoxynucleotide. J Leukoc Biol. 2002;71(5):813–820.
101. Hanagata N. Structure-dependent immunostimulatory effect of CpG oligodeoxynucleotides and their delivery system. Int J Nanomedicine. 2012;7:2181–2195. doi:10.2147/IJN.S30197
102. Marshall JD, Fearon K, Abbate C, et al. Identification of a novel CpG DNA class and motif that optimally stimulate B cell and plasmacytoid dendritic cell functions. J Leukoc Biol. 2003;73(6):781–792. doi:10.1189/jlb.1202630
103. Hartmann G, Battiany J, Poeck H, et al. Rational design of new CpG oligonucleotides that combine B cell activation with high IFN‐α induction in plasmacytoid dendritic cells. Eur J Immunol. 2003;33(6):1633–1641. doi:10.1002/eji.200323813
104. Kandimalla ER, Bhagat L, Cong YP, et al. Secondary structures in CpG oligonucleotides affect immunostimulatory activity. Biochem Biophys Res Commun. 2003;306(4):948–953. doi:10.1016/S0006-291X(03)01080-5
105. Agrawal S, Kandimalla ER. Synthetic agonists of toll-like receptors 7, 8 and 9. Biochem Soc Trans. 2007;35(Pt 6):1461–1467. doi:10.1042/BST0351461
106. Klinman DM, Currie D. Hierarchical recognition of CpG motifs expressed by immunostimulatory oligodeoxynucleotides. Clin Exp Immunol. 2003;133(2):227–232. doi:10.1046/j.1365-2249.2003.02216.x
107. Miyazato A, Nakamura K, Yamamoto N, et al. Toll-like receptor 9-dependent activation of myeloid dendritic cells by deoxynucleic acids from candida albicans. Infect Immun. 2009;77(7):3056–3064. doi:10.1128/IAI.00840-08
108. Adamus T, Kortylewski M. The revival of CpG oligonucleotide-based cancer immunotherapies. Contemp Oncol (Pozn). 2018;22(1A):56–60. doi:10.5114/wo.2018.73887
109. Lee S, Kang D, Ra EA, Lee TA, Ploegh HL, Park B. Negative self-regulation of TLR9 signaling by its N-terminal proteolytic cleavage product. J Immunol. 2014;193(7):3726–3735. doi:10.4049/jimmunol.1400210
110. Bird AP, Taggart MH, Nicholls RD, Higgs DR. Non-methylated CpG-rich islands at the human alpha-globin locus: implications for evolution of the alpha-globin pseudogene. EMBO J. 1987;6(4):999–1004. doi:10.1002/j.1460-2075.1987.tb04851.x
111. Cornelie S, Hoebeke J, Schacht AM, et al. Direct evidence that toll-like receptor 9 (TLR9) functionally binds plasmid DNA by specific cytosine-phosphate-guanine motif recognition. J Biol Chem. 2004;279(15):15124–15129. doi:10.1074/jbc.M313406200
112. Rutz M, Metzger J, Gellert T, et al. Toll-like receptor 9 binds single-stranded CpG-DNA in a sequence- and pH-dependent manner. Eur J Immunol. 2004;34(9):2541–2550. doi:10.1002/eji.200425218
113. de Jong SD, Basha G, Wilson KD, et al. The immunostimulatory activity of unmethylated and methylated CpG oligodeoxynucleotide is dependent on their ability to colocalize with TLR9 in late endosomes. J Immunol. 2010;184(11):6092–6102. doi:10.4049/jimmunol.0802442
114. de Jong S, Chikh G, Sekirov L, et al. Encapsulation in liposomal nanoparticles enhances the immunostimulatory, adjuvant and anti-tumor activity of subcutaneously administered CpG ODN. Cancer Immunol Immunother. 2007;56(8):1251–1264. doi:10.1007/s00262-006-0276-x
115. Kawai T, Akira S. Signaling to NF-kappaB by toll-like receptors. Trends Mol Med. 2007;13(11):460–469. doi:10.1016/j.molmed.2007.09.002
116. Bonnert TP, Garka KE, Parnet P, Sonoda G, Testa JR, Sims JE. The cloning and characterization of human MyD88: a member of an IL-1 receptor related family. FEBS Lett. 1997;402(1):81–84. doi:10.1016/S0014-5793(96)01506-2
117. Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4(7):499–511. doi:10.1038/nri1391
118. West AP, Koblansky AA, Ghosh S. Recognition and signaling by toll-like receptors. Annu Rev Cell Dev Biol. 2006;22(1):409–437. doi:10.1146/annurev.cellbio.21.122303.115827
119. Muroi M, Tanamoto K. TRAF6 distinctively mediates MyD88- and IRAK-1-induced activation of NF-kappaB. J Leukoc Biol. 2008;83(3):702–707. doi:10.1189/jlb.0907629
120. Adhikari A, Xu M, Chen ZJ. Ubiquitin-mediated activation of TAK1 and IKK. Oncogene. 2007;26(22):3214–3226. doi:10.1038/sj.onc.1210413
121. Chen F, Bhatia D, Chang Q, Castranova V. Finding NEMO by K63-linked polyubiquitin chain. Cell Death Differ. 2006;13(11):1835–1838. doi:10.1038/sj.cdd.4402014
122. Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412(6844):346–351. doi:10.1038/35085597
123. Meylan E, Burns K, Hofmann K, et al. RIP1 is an essential mediator of toll-like receptor 3-induced NF-kappa B activation. Nat Immunol. 2004;5(5):503–507. doi:10.1038/ni1061
124. Sato S, Sugiyama M, Yamamoto M, et al. Toll/IL-1 receptor domain-containing adaptor inducing IFN-beta (TRIF) associates with TNF receptor-associated factor 6 and TANK-binding kinase 1, and activates two distinct transcription factors, NF-kappa B and IFN-regulatory factor-3, in the toll-like receptor signaling. J Immunol. 2003;171(8):4304–4310. doi:10.4049/jimmunol.171.8.4304
125. Yamamoto M, Sato S, Mori K, et al. Cutting edge: a novel toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-β promoter in the toll-like receptor signaling. J Immunol. 2002;169(12):6668–6672. doi:10.4049/jimmunol.169.12.6668
126. Jiang Z, Mak TW, Sen G, Li X. Toll-like receptor 3-mediated activation of NF- B and IRF3 diverges at toll-IL-1 receptor domain-containing adapter inducing IFN-. Proc Natl Acad Sci U S A. 2004;101(10):3533–3538. doi:10.1073/pnas.0308496101
127. Sanjo H, Takeda K, Tsujimura T, Ninomiya-Tsuji J, Matsumoto K, Akira S. TAB2 is essential for prevention of apoptosis in fetal liver but not for interleukin-1 signaling. Mol Cell Biol. 2003;23(4):1231–1238. doi:10.1128/MCB.23.4.1231-1238.2003
128. Shim JH, Xiao C, Paschal AE, et al. TAK1, but not TAB1 or TAB2, plays an essential role in multiple signaling pathways in vivo. Genes Dev. 2005;19(22):2668–2681. doi:10.1101/gad.1360605
129. Han C, Jin J, Xu S, Liu H, Li N, Cao X. Integrin CD11b negatively regulates TLR-triggered inflammatory responses by activating Syk and promoting degradation of MyD88 and TRIF via Cbl-b. Nat Immunol. 2010;11(8):734–742. doi:10.1038/ni.1908
130. Palsson-McDermott EM, Doyle SL, McGettrick AF, et al. TAG, a splice variant of the adaptor TRAM, negatively regulates the adaptor MyD88-independent TLR4 pathway. Nat Immunol. 2009;10(6):579–586. doi:10.1038/ni.1727
131. Abe T, Shapira SD. Negative regulation of cytosolic sensing of DNA. Int Rev Cell Mol Biol. 2019;344:91–115.
132. Saitoh T, Tun-Kyi A, Ryo A, et al. Negative regulation of interferon-regulatory factor 3-dependent innate antiviral response by the prolyl isomerase Pin1. Nat Immunol. 2006;7(6):598–605. doi:10.1038/ni1347
133. Honda K, Yanai H, Mizutani T, et al. Role of a transductional-transcriptional processor complex involving MyD88 and IRF-7 in toll-like receptor signaling. Proc Natl Acad Sci U S A. 2004;101(43):15416–15421. doi:10.1073/pnas.0406933101
134. Kawai T, Sato S, Ishii KJ, et al. Interferon-alpha induction through toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat Immunol. 2004;5(10):1061–1068. doi:10.1038/ni1118
135. Honda K, Yanai H, Negishi H, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434(7034):772–777. doi:10.1038/nature03464
136. Diamond MS, Kinder M, Matsushita H, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med. 2011;208(10):1989–2003. doi:10.1084/jem.20101158
137. Fuertes MB, Kacha AK, Kline J, et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med. 2011;208(10):2005–2016. doi:10.1084/jem.20101159
138. Maldonado-Lopez R, De Smedt T, Michel P, et al. CD8alpha+ and CD8alpha- subclasses of dendritic cells direct the development of distinct T helper cells in vivo. J Exp Med. 1999;189(3):587–592. doi:10.1084/jem.189.3.587
139. Mashayekhi M, Sandau MM, Dunay IR, et al. CD8alpha(+) dendritic cells are the critical source of interleukin-12 that controls acute infection by toxoplasma gondii tachyzoites. Immunity. 2011;35(2):249–259. doi:10.1016/j.immuni.2011.08.008
140. Wang H, Rayburn ER, Wang W, Kandimalla ER, Agrawal S, Zhang R. Immunomodulatory oligonucleotides as novel therapy for breast cancer: pharmacokinetics, in vitro and in vivo anticancer activity, and potentiation of antibody therapy. Mol Cancer Ther. 2006;5(8):2106–2114. doi:10.1158/1535-7163.MCT-06-0158
141. Damiano V, Caputo R, Bianco R, et al. Novel toll-like receptor 9 agonist induces epidermal growth factor receptor (EGFR) inhibition and synergistic antitumor activity with EGFR inhibitors. Clin Cancer Res. 2006;12(2):577–583. doi:10.1158/1078-0432.CCR-05-1943
142. Yuan S, Qiao T, Li X, et al. Toll-like receptor 9 activation by CpG oligodeoxynucleotide 7909 enhances the radiosensitivity of A549 lung cancer cells via the p53 signaling pathway. Oncol Lett. 2018;15(4):5271–5279. doi:10.3892/ol.2018.7916
143. Jiang W, Wang D, Zhu F, Bhagat L, DiMuzio J, Agrawal S. Abstract B159: modulation of checkpoint expression in tumor microenvironment by intratumoral administration of a novel TLR9 agonist: rationale for combination therapy. Cancer Immunol Res. 2016;4(1 Supplement):B159–B159.
144. Mason KA, Ariga H, Neal R, et al. Targeting toll-like receptor 9 with CpG oligodeoxynucleotides enhances tumor response to fractionated radiotherapy. Clin Cancer Res. 2005;11(1):361–369.
145. Milas L, Mason KA, Ariga H, et al. CpG oligodeoxynucleotide enhances tumor response to radiation. Cancer Res. 2004;64(15):5074–5077. doi:10.1158/0008-5472.CAN-04-0926
146. Zhang H, Liu L, Yu D, et al. An in situ autologous tumor vaccination with combined radiation therapy and TLR9 agonist therapy. PLoS One. 2012;7(5).
147. Meng Y, Carpentier AF, Chen L, et al. Successful combination of local CpG-ODN and radiotherapy in malignant glioma. Int J Cancer. 2005;116(6):992–997. doi:10.1002/ijc.21131
148. Fourcade J, Kudela P, Andrade Filho PA, et al. Immunization with analog peptide in combination with CpG and montanide expands tumor antigen-specific CD8+ T cells in melanoma patients. J Immunother. 2008;31(8):781–791. doi:10.1097/CJI.0b013e318183af0b
149. Karbach J, Gnjatic S, Bender A, et al. Tumor-reactive CD8+ T-cell responses after vaccination with NY-ESO-1 peptide, CpG 7909 and montanide ISA-51: association with survival. Int J Cancer. 2010;126(4):909–918. doi:10.1002/ijc.24850
150. Speiser DE, Liénard D, Rufer N, et al. Rapid and strong human CD8+ T cell responses to vaccination with peptide, IFA, and CpG oligodeoxynucleotide 7909. J Clin Invest. 2005;115(3):739–746. doi:10.1172/JCI23373
151. Valmori D, Souleimanian NE, Tosello V, et al. Vaccination with NY-ESO-1 protein and CpG in montanide induces integrated antibody/Th1 responses and CD8 T cells through cross-priming. Proc Natl Acad Sci U S A. 2007;104(21):8947–8952. doi:10.1073/pnas.0703395104
152. Humbert M, Guery L, Brighouse D, Lemeille S, Hugues S. Intratumoral CpG-B promotes antitumoral neutrophil, cDC, and T-cell cooperation without reprograming tolerogenic pDC. Cancer Res. 2018;78(12):3280–3292. doi:10.1158/0008-5472.CAN-17-2549
153. Wang S, Campos J, Gallotta M, et al. Intratumoral injection of a CpG oligonucleotide reverts resistance to PD-1 blockade by expanding multifunctional CD8+ T cells. Proc Natl Acad Sci. 2016;113(46):E7240–E7249. doi:10.1073/pnas.1608555113
154. Ribas A, Medina T, Kummar S, et al. SD-101 in combination with pembrolizumab in advanced melanoma: results of a phase Ib, multicenter study. Cancer Discov. 2018;8(10):1250–1257. doi:10.1158/2159-8290.CD-18-0280
155. Reilley MJ, Morrow B, Ager CR, Liu A, Hong DS, Curran MA. TLR9 activation cooperates with T cell checkpoint blockade to regress poorly immunogenic melanoma. J Immunother Cancer. 2019;7(1):323. doi:10.1186/s40425-019-0811-x
156. Gallotta M, Assi H, Degagne E, Kannan SK, Coffman RL, Guiducci C. Inhaled TLR9 agonist renders lung tumors permissive to pd-1 blockade by promoting optimal CD4(+) and CD8(+) T-cell interplay. Cancer Res. 2018;78(17):4943–4956. doi:10.1158/0008-5472.CAN-18-0729
157. Dorta-Estremera S, Hegde VL, Slay RB, et al. Targeting interferon signaling and CTLA-4 enhance the therapeutic efficacy of anti-PD-1 immunotherapy in preclinical model of HPV+ oral cancer. J Immunother Cancer. 2019;7(1):252. doi:10.1186/s40425-019-0728-4
158. Sato-Kaneko F, Yao S, Ahmadi A, et al. Combination immunotherapy with TLR agonists and checkpoint inhibitors suppresses head and neck cancer. JCI Insight. 2017;2(18):18. doi:10.1172/jci.insight.93397
159. Lemke-Miltner CD, Blackwell SE, Yin C, et al. Antibody opsonization of a TLR9 agonist-containing virus-like particle enhances in situ immunization. J Immunol. 2020;204(5):1386–1394. doi:10.4049/jimmunol.1900742
160. Goldszmid RS, Dzutsev A, Viaud S, Zitvogel L, Restifo NP, Trinchieri G. Microbiota modulation of myeloid cells in cancer therapy. Cancer Immunol Res. 2015;3(2):103–109. doi:10.1158/2326-6066.CIR-14-0225
161. Iida N, Dzutsev A, Stewart CA, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science. 2013;342(6161):967–970. doi:10.1126/science.1240527
162. Machiels J-P, Kaminsky M-C, Keller U, et al. Phase Ib trial of the toll-like receptor 9 agonist IMO-2055 in combination with 5-fluorouracil, cisplatin, and cetuximab as first-line palliative treatment in patients with recurrent/metastatic squamous cell carcinoma of the head and neck. Invest New Drugs. 2013;31(5):1207–1216. doi:10.1007/s10637-013-9933-z
163. Hofmann MA, Kors C, Audring H, Walden P, Sterry W, Trefzer U. Phase 1 evaluation of intralesionally injected TLR9-agonist PF-3512676 in patients with basal cell carcinoma or metastatic melanoma. J Immunother. 2008;31(5):520–527. doi:10.1097/CJI.0b013e318174a4df
164. Kim YH, Girardi M, Duvic M, et al. Phase I trial of a Toll-like receptor 9 agonist, PF-3512676 (CPG 7909), in patients with treatment-refractory, cutaneous T-cell lymphoma. J Am Acad Dermatol. 2010;63(6):975–983. doi:10.1016/j.jaad.2009.12.052
165. Thompson JA, Kuzel T, Drucker BJ, Urba WJ, Bukowski RM. Safety and efficacy of PF-3512676 for the treatment of stage IV renal cell carcinoma: an open-label, multicenter phase I/II study. Clin Genitourin Cancer. 2009;7(3):E58–65. doi:10.3816/CGC.2009.n.025
166. Weihrauch MR, Richly H, von Bergwelt-baildon MS, et al. Phase I clinical study of the toll-like receptor 9 agonist MGN1703 in patients with metastatic solid tumours. Eur J Cancer. 2015;51(2):146–156. doi:10.1016/j.ejca.2014.11.002
167. Milhem MM. Durable responses in anti-PD-1 refractory melanoma following intratumoral injection of a toll-like receptor 9 (TLR9) agonist, CMP-001, in combination with pembrolizumab. J Immunother Cancer. 2020;8:A1–A12. doi:10.1136/LBA2019.4
168. Pashenkov M, Goëss G, Wagner C, et al. Phase II trial of a toll-like receptor 9–activating oligonucleotide in patients with metastatic melanoma. J Clin Oncol. 2006;24(36):5716–5724. doi:10.1200/JCO.2006.07.9129
169. Carpentier A, Laigle-Donadey F, Zohar S, et al. Phase 1 trial of a CpG oligodeoxynucleotide for patients with recurrent glioblastoma. Neuro Oncol. 2006;8(1):60–66. doi:10.1215/S1522851705000475
170. Carpentier A, Metellus P, Ursu R, et al. Intracerebral administration of CpG oligonucleotide for patients with recurrent glioblastoma: a phase II study. Neuro Oncol. 2010;12(4):401–408. doi:10.1093/neuonc/nop047
171. Ursu R, Carpentier A, Metellus P, et al. Intracerebral injection of CpG oligonucleotide for patients with de novo glioblastoma – a phase II multicentric, randomised study. Eur J Cancer. 2017;73:30–37. doi:10.1016/j.ejca.2016.12.003
172. Brody JD, Ai WZ, Czerwinski DK, et al. In situ vaccination with a TLR9 agonist induces systemic lymphoma regression: a phase I/II study. J Clin Oncol. 2010;28(28):4324–4332. doi:10.1200/JCO.2010.28.9793
173. Tarhini AA, Leng S, Moschos SJ, et al. Safety and immunogenicity of vaccination with MART-1 (26-35, 27L), gp100 (209-217, 210M), and tyrosinase (368-376, 370D) in adjuvant with PF-3512676 and GM-CSF in metastatic melanoma. J Immunother. 2012;35(4):359–366. doi:10.1097/CJI.0b013e31825481fe
174. Goldinger SM, Dummer R, Baumgaertner P, et al. Nano-particle vaccination combined with TLR-7 and −9 ligands triggers memory and effector CD8⁺ T-cell responses in melanoma patients. Eur J Immunol. 2012;42(11):3049–3061. doi:10.1002/eji.201142361
175. Fourcade J, Sun Z, Pagliano O, et al. PD-1 and Tim-3 regulate the expansion of tumor antigen-specific CD8⁺ T cells induced by melanoma vaccines. Cancer Res. 2014;74(4):1045–1055. doi:10.1158/0008-5472.CAN-13-2908
176. Weber JS, Zarour H, Redman B, et al. Randomized Phase 2/3 trial of CpG oligodeoxynucleotide PF‐3512676 alone or with dacarbazine for patients with unresectable stage III and IV melanoma. Cancer. 2009;115(17):3944–3954. doi:10.1002/cncr.24473
177. Hirsh V, Paz-Ares L, Boyer M, et al. Randomized phase III trial of paclitaxel/carboplatin with or without PF-3512676 (toll-like receptor 9 agonist) as first-line treatment for advanced non–small-cell lung cancer. J Clin Oncol. 2011;29(19):2667–2674. doi:10.1200/JCO.2010.32.8971
178. Manegold C, Gravenor D, Woytowitz D, et al. Randomized phase II trial of a toll-like receptor 9 agonist oligodeoxynucleotide, PF-3512676, in combination with first-line taxane plus platinum chemotherapy for advanced-stage non-small-cell lung cancer. J Clin Oncol. 2008;26(24):3979–3986. doi:10.1200/JCO.2007.12.5807
179. Smith DA, Conkling P, Richards DA, et al. Antitumor activity and safety of combination therapy with the toll-like receptor 9 agonist IMO-2055, erlotinib, and bevacizumab in advanced or metastatic non-small cell lung cancer patients who have progressed following chemotherapy. Cancer Immunol Immunother. 2014;63(8):787–796. doi:10.1007/s00262-014-1547-6
180. Belani CP, Nemunaitis JJ, Chachoua A, et al. Phase 2 trial of erlotinib with or without PF-3512676 (CPG 7909, a toll-like receptor 9 agonist) in patients with advanced recurrent EGFR-positive non-small cell lung cancer. Cancer Biol Ther. 2013;14(7):557–563. doi:10.4161/cbt.24598
181. Yamada K, Nakao M, Fukuyama C, et al. Phase I study of TLR9 agonist PF-3512676 in combination with carboplatin and paclitaxel in patients with advanced non-small-cell lung cancer. Cancer Sci. 2010;101(1):188–195. doi:10.1111/j.1349-7006.2009.01361.x
182. Manegold C, Van Zandwijk N, Szczesna A, et al. A phase III randomized study of gemcitabine and cisplatin with or without PF-3512676 (TLR9 agonist) as first-line treatment of advanced non-small-cell lung cancer. Ann Oncol. 2012;23(1):72–77. doi:10.1093/annonc/mdr030
183. Chan E, Kwak EL, Hwang J, Heiskala M, de La Bourdonnaye G, Mita M. Open-label phase 1b study of FOLFIRI plus cetuximab plus IMO-2055 in patients with colorectal cancer who have progressed following chemotherapy for advanced or metastatic disease. Cancer Chemother Pharmacol. 2015;75(4):701–709. doi:10.1007/s00280-015-2682-2
184. Frank MJ, Reagan PM, Bartlett NL, et al. In situ vaccination with a TLR9 agonist and local low-dose radiation induces systemic responses in untreated indolent lymphoma. Cancer Discov. 2018;8(10):1258–1269. doi:10.1158/2159-8290.CD-18-0743
185. Levy R, Reagan PM, Friedberg JW, et al. SD-101, a novel class C CpG-oligodeoxynucleotide (ODN) toll-like receptor 9 (TLR9) agonist, given with low dose radiation for untreated low grade B-cell lymphoma: interim results of a phase 1/2 trial. Blood. 2016;128(22):2974. doi:10.1182/blood.V128.22.2974.2974
186. Millward M, Underhill C, Lobb S, et al. Phase I study of tremelimumab (CP-675 206) plus PF-3512676 (CPG 7909) in patients with melanoma or advanced solid tumours. Br J Cancer. 2013;108(10):1998–2004. doi:10.1038/bjc.2013.227
187. Wittig B, Märten A, Dorbic T, et al. Therapeutic vaccination against metastatic carcinoma by expression-modulated and immunomodified autologous tumor cells: a first clinical phase I/II trial. Hum Gene Ther. 2001;12(3):267–278. doi:10.1089/10430340150218404
188. Wittig B, Schmidt M, Scheithauer W, Schmoll H-J. MGN1703, an immunomodulator and toll-like receptor 9 (TLR-9) agonist: from bench to bedside. Crit Rev Oncol Hematol. 2015;94(1):31–44. doi:10.1016/j.critrevonc.2014.12.002
189. Thomas M, Ponce-Aix S, Navarro A, et al. Immunotherapeutic maintenance treatment with toll-like receptor 9 agonist lefitolimod in patients with extensive-stage small-cell lung cancer: results from the exploratory, controlled, randomized, international phase II IMPULSE study. Ann Oncol. 2018;29(10):2076–2084. doi:10.1093/annonc/mdy326
190. Schmoll H-J, Wittig B, Arnold D, et al. Maintenance treatment with the immunomodulator MGN1703, a toll-like receptor 9 (TLR9) agonist, in patients with metastatic colorectal carcinoma and disease control after chemotherapy: a randomised, double-blind, placebo-controlled trial. J Cancer Res Clin Oncol. 2014;140(9):1615–1624. doi:10.1007/s00432-014-1682-7
191. Wang D, Jiang W, Zhu F, Mao X, Agrawal S. Modulation of the tumor microenvironment by intratumoral administration of IMO-2125, a novel TLR9 agonist, for cancer immunotherapy. Int J Oncol. 2018;53(3):1193–1203. doi:10.3892/ijo.2018.4456
192. Idera Pharmaceuticals announces final clinical safety and efficacy data from illuminate-204 trial in advanced melanoma [news release] [press release]. April 21 2020.
193. Kirkwood J. Durable Responses in Anti-Pd-1 Refractory Melanoma Following Intra-Tumoral Injection of a Toll-Like Receptor 9 (TLR9) Agonist, CMP-001, in Combination with Pembrolizumab Abstract #: 11548/o87. sitc 2019. 2019.
194. Pharmaceuticals C. CMP-001 in combination with atezolizumab in participants with non-small cell lung cancer. In: Checkmate Pharmaceuticals Investigator’s Brochure.
195. Liu J, Blake SJ, Yong MCR, et al. Improved efficacy of neoadjuvant compared to adjuvant immunotherapy to eradicate metastatic disease. Cancer Discov. 2016;
196. Liu J, O’Donnell JS, Yan J, et al. Timing of neoadjuvant immunotherapy in relation to surgery is crucial for outcome. Oncoimmunology. 2019;8(5):e1581530. doi:10.1080/2162402X.2019.1581530
197. Houshmand S. Evaluation of Treatment Response in Phase IB of the Study of Patients with Advanced Melanoma Treated by CMP-001 in Combination with Pembrolizumab: Relationship with Pre-Treatment Tumor Burden (TB) Using Volumetric Computed Tomography (CT) Biomarkers RSNA. 2019.
198. Cottrell TR, Thompson ED, Forde PM, et al. Pathologic features of response to neoadjuvant anti-PD-1 in resected non-small-cell lung carcinoma: a proposal for quantitative immune-related pathologic response criteria (irPRC). Ann Oncol. 2018;29(8):1853–1860. doi:10.1093/annonc/mdy218
199. Tetzlaff MT, Messina JL, Stein JE, et al. Pathological assessment of resection specimens after neoadjuvant therapy for metastatic melanoma. Ann Oncol. 2018;29(8):1861–1868. doi:10.1093/annonc/mdy226
200. Stein JE, Soni A, Danilova L, et al. Major pathologic response on biopsy (MPRbx) in patients with advanced melanoma treated with anti-PD-1: evidence for an early, on-therapy biomarker of response. Ann Oncol. 2019;30(4):589–596. doi:10.1093/annonc/mdz019
201. Amaria RN, Reddy SM, Tawbi HA, et al. Neoadjuvant immune checkpoint blockade in high-risk resectable melanoma. Nat Med. 2018;24(11):1649–1654. doi:10.1038/s41591-018-0197-1
202. Menzies AM, Rozeman EA, Amaria RN, et al. Pathological response and survival with neoadjuvant therapy in melanoma: a pooled analysis from the international neoadjuvant melanoma consortium (INMC). J Clin Oncol. 2019;37(15_suppl):9503. doi:10.1200/JCO.2019.37.15_suppl.9503
203. Huang AC, Orlowski RJ, Xu X, et al. A single dose of neoadjuvant PD-1 blockade predicts clinical outcomes in resectable melanoma. Nat Med. 2019;25(3):454–461. doi:10.1038/s41591-019-0357-y
204. Rozeman EA, Menzies AM, ACJ VA, et al. Identification of the optimal combination dosing schedule of neoadjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma (OpACIN-neo): a multicentre, phase 2, randomised, controlled trial. Lancet Oncol. 2019;20(7):948–960. doi:10.1016/S1470-2045(19)30151-2
205. Blank CU, Rozeman EA, Fanchi LF, et al. Neoadjuvant versus adjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma. Nat Med. 2018;24(11):1655–1661. doi:10.1038/s41591-018-0198-0
206. Tammela TL, Haggman M, Ladjevardi S, et al. An intraprostatic modified release formulation of antiandrogen 2-hydroxyflutamide for localized prostate cancer. J Urol. 2017;198(6):1333–1339. doi:10.1016/j.juro.2017.07.072
207. Daniel WLLU, Coates S, Bexon AS, Mix S. Abstract CT044: AST-008, a TLR9 agonist spherical nucleic acid, activated NK cells, T cells, and cytokines in healthy subjects in a phase I clinical trial. Clin Cancer Res. 2019;79(13).
208. Domankevich V, Cohen A, Efrati M, et al. Combining alpha radiation-based brachytherapy with immunomodulators promotes complete tumor regression in mice via tumor-specific long-term immune response. Cancer Immunol Immunother. 2019;68(12):1949–1958. doi:10.1007/s00262-019-02418-5
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.