Back to Journals » Clinical Interventions in Aging » Volume 13
Tolerability of buprenorphine transdermal system in nursing home patients with advanced dementia: a randomized, placebo-controlled trial (DEP.PAIN.DEM)
Authors Erdal A ![]() , Flo E, Aarsland D, Selbaek G, Ballard C
, Flo E, Aarsland D, Selbaek G, Ballard C ![]() , Slettebo DD
, Slettebo DD ![]() , Husebo BS
, Husebo BS ![]()
Received 29 December 2017
Accepted for publication 17 March 2018
Published 16 May 2018 Volume 2018:13 Pages 935—946
DOI https://doi.org/10.2147/CIA.S161052
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Richard Walker
Ane Erdal,1 Elisabeth Flo,2 Dag Aarsland,3,4 Geir Selbaek,5–7 Clive Ballard,8 Dagrun D Slettebo,1 Bettina S Husebo1,9
1Department of Global Public Health and Primary Care, Centre for Elderly and Nursing Home Medicine, University of Bergen, Bergen, Norway; 2Department of Clinical Psychology, University of Bergen, Bergen, Norway; 3Department of Old Age Psychiatry, Institute of Psychiatry, Psychology and Neuroscience, King’s College London, London, UK; 4Centre for Age-Related Medicine, Stavanger University Hospital, Stavanger, Norway; 5Centre for Old Age Psychiatric Research, Innlandet Hospital Trust, Ottestad, Norway; 6National Advisory Unit on Aging and Health, Tønsberg, Norway; 7Institute of Health and Society, Faculty of Medicine, University of Oslo, Oslo, Norway; 8Exeter Medical School, University of Exeter, Exeter, UK; 9Municipality of Bergen, Bergen, Norway
Purpose: Buprenorphine transdermal system is increasingly prescribed in people with advanced dementia, but no clinical trial has investigated the safety and factors associated with discontinuation due to adverse events in this population.
Patients and methods: One hundred sixty-two people with advanced dementia and significant depression from 47 nursing homes were included and randomized to active analgesic treatment (acetaminophen/buprenorphine) or identical placebo for 13 weeks. In this secondary analysis, the main outcomes were time to and reasons for discontinuation of buprenorphine due to adverse events. Change in daytime activity as measured by actigraphy was a secondary outcome.
Results: Of the 44 patients who received active buprenorphine 5 μg/hour, 52.3% (n=23) discontinued treatment due to adverse events compared to 13.3% (6 of 45) in the placebo group (p<0.001). Psychiatric and neurological adverse events were the most frequently reported causes of discontinuation (69.6%, n=16). Concomitant use of antidepressants significantly increased the risk of discontinuation (HR 23.2, 95% CI: 2.95–182, p=0.003). Adjusted for age, sex, cognitive function, pain and depression at baseline, active buprenorphine was associated with 24.0 times increased risk of discontinuation (Cox model, 95% CI: 2.45–235, p=0.006). Daytime activity dropped significantly during the second day of active treatment (-21.4%, p=0.005) and decreased by 12.9% during the first week (p=0.053).
Conclusion: Active buprenorphine had significantly higher risk of discontinuation compared with placebo in people with advanced dementia and depression, mainly due to psychiatric and neurological adverse events. Daytime activity dropped significantly during the first week of treatment. Concomitant use of antidepressants further reduced the tolerability of buprenorphine.
Keywords: opioids, analgesics, dementia, drug safety, adverse drug reactions
Introduction
More than 80% of elderly people in long-term residential care have dementia.1 Approximately 50% of these individuals suffer from pain of clinically significant intensity.2 Cognitive impairment leads to difficulty in verbally expressing painful symptoms and complicates the assessment and treatment of pain.3 This may increase the risk of untreated chronic pain in people with dementia compared with cognitively intact patients.4 In the past few decades, systematic reviews have expressed concern that nursing home patients with dementia receive less analgesic treatment than those without dementia, despite comparable diagnoses of pain.5,6
Data from the entire population of Denmark in 2010 showed that 41% of the country’s 42,291 nursing home patients used opioids, and that patients without dementia received significantly more opioid analgesics compared with those with dementia (43% and 38%, respectively).7 A study including 425 patients from 12 nursing homes in Austria in 2011–2012 found that despite having more pain, fewer cognitively impaired patients received scheduled analgesic prescriptions compared with patients without cognitive impairment (36% and 58%, respectively).8 Several studies have reported similar rates of analgesic use in nursing home patients with and without dementia,9,10 with an overall increase in total analgesic use irrespective of cognitive state and a shift toward increased use of opioid analgesics.9 In Norway, the use of opioid analgesics in nursing home patients increased from 11% in 2000 to 24% in 2011, with a substantial increase in the use of strong opioids from 1.9% to 17.9%.9 In 2011, the odds ratio for the use of strong opioids in nursing home patients with dementia did not differ significantly compared with those without dementia.9
Buprenorphine transdermal system (TDS) has been recommended for elderly patients because of its favorable pharmacodynamic and pharmacokinetic profile, with low risk of serious adverse events.11 Buprenorphine, a strong opioid, is a partial mu receptor agonist and a kappa receptor antagonist. This pattern of activity gives a ceiling effect for respiratory depression, without a clinically relevant ceiling effect on analgesia.12 As one of few opioids, it does not require dose adjustment in renal insufficiency due to hepatic clearance.13 Buprenorphine TDS is prescribed to over 10% of nursing home patients in countries where it is marketed, with estimated use in people with dementia ranging from 10.5% to 14.8%.7,9,10 While buprenorphine TDS has shown high persistence rates in the general population, the rate of common adverse events such as nausea, dizziness, or sedation is higher than that of comparator opioids.14,15 Dementia, age-related physiological changes, multimorbidity, frailty, and interactions with psychotropic drugs may impact the safety and tolerability of buprenorphine TDS.16
There is a well-documented association between pain and increased depressive symptoms in people with dementia,17 and antidepressants have questionable efficacy for depression in these patients.18 In a recent study, we investigated whether analgesic treatment with acetaminophen or buprenorphine TDS could improve depression in people with dementia. Contrary to our hypothesis, we found that active treatment was associated with more persistent depressive symptoms, and 52% of patients who received active buprenorphine were withdrawn from the study due to adverse events during treatment.39 Few studies have assessed the tolerability and adverse effects of buprenorphine TDS in nursing home patients with dementia, and none with a placebo-controlled design. Furthermore, buprenorphine may have additive or synergistic interaction effects with other drugs that have sedative effects. Elderly patients and people with dementia are particularly vulnerable to adverse effects such as sedation, but interactions between opioids and other commonly prescribed psychotropic drugs such as antidepressants have not been studied in this population. Similarly, anticholinergic drugs may negatively impact cognition in people with dementia, but we do not know whether high anticholinergic drug burden is associated with poorer tolerability of buprenorphine. There is a need to investigate clinically significant interactions between opioids and anticholinergic and psychotropic drugs in people with dementia.
In these secondary analyses of our study, the primary aim was to assess the tolerability of buprenorphine TDS in nursing home patients with moderate to severe dementia, controlling for pain intensity, depressive symptoms, cognitive state, and concomitant use of psychotropic and anticholinergic drugs. Secondary aims were to assess which adverse effects most frequently caused discontinuation and to determine how daytime activity changed during the first week of treatment.
Patients and methods
Study design and population
The current study comprises secondary analyses of data collected in the randomized, placebo-controlled trial “Efficacy of analgesic treatment for depression in nursing home patients with dementia (DEP.PAIN.DEM),” which was conducted in 47 nursing homes in 10 municipalities of Norway, including people with dementia (Mini-Mental State Examination [MMSE] ≤20) and depression (Cornell Scale for Depression in Dementia [CSDD] ≥8; full inclusion and exclusion criteria are presented in Table 1). The intervention consisted of a stepwise increase in analgesic treatment, and patients who did not use scheduled analgesics at baseline or used acetaminophen ≤1 g/day were prescribed acetaminophen in a total dose of 1 g three times daily. Patients who already used acetaminophen >1 g daily, nonsteroidal antiinflammatory drugs (except low-dose acetylsalicylic acid), or buprenorphine 5 μg/hour, or who had difficulty swallowing tablets, were prescribed buprenorphine TDS 5 μg/hour in addition to their regular treatment and randomized to receive active treatment or placebo for 13 weeks with no further dose adjustment.39 Patients who received buprenorphine/placebo TDS are included in the current analyses.
 | Table 1 Inclusion and exclusion criteria |
Procedures
Clinicians were advised to keep doses of psychotropic and analgesic drugs unchanged during the study period, if possible. If lasting changes were made to regular analgesic treatment or antidepressants, the patient was withdrawn from the study. The study treatment was prescribed in addition to any regular or as-needed analgesics. In mild to moderate acute pain, patients were given as-needed analgesics in addition to study treatment, and the number of doses given during the study period was recorded. Patients with severe pain at baseline were excluded because it would be unethical to risk treating them with a placebo. We therefore ensured that the included patients would not suffer from prolonged or unnecessary untreated pain because of the study protocol. Furthermore, the physician who was responsible for the patient had full authority to discontinue study treatment promptly if clinical changes necessitated treatment with a known dose of active analgesic. Written informed consent was obtained from patients with medical decision-making capacity, or written presumed consent was obtained from a legally authorized representative in those with reduced capacity to consent in accordance with ethics committee requirements and current Norwegian legislation. The trial was approved by the Regional Committee for Medical and Health Research Ethics (REC-West 2013/1474) and the Norwegian Medicines Agency (EudraCT 2013-002226-23), and registered at ClinicalTrials.gov (NCT02267057).
Randomization and masking
The trial was double blinded, and participants were randomly allocated to each arm in a 1:1 ratio according to computer-generated random numbers in blocks of 12 with no stratification factors. Buprenorphine TDS and identical, inert placebo (Mundipharma Research Limited, Cambridge, UK) were packed and marked indiscernibly, identifiable only by pack number. Patients, nursing home staff, physicians, pharmacy, researchers, and statisticians were all masked to group identity until completion of the analyses.
Assessments
Assessments were made by the researchers in collaboration with the nursing home staff and included scheduled assessments at baseline, 6 and 13 weeks in addition to any spontaneous reports during the whole 13-week period. The tolerability of buprenorphine TDS was operationalized by assessing how many patients discontinued treatment due to adverse events (defined as suspected adverse event, clinical deterioration, or death) and how long treatment lasted before such discontinuation. Discontinuation for other reasons, such as protocol violation, was not included in the analysis, and in the following, “discontinuation” refers only to those cases defined here as caused by adverse events. To ensure that all suspected adverse events were reported, the proxy rater received standardized detailed verbal and written information about known possible adverse effects of buprenorphine. They were instructed to pay attention to and report changes in any of the symptoms listed as potential adverse events, as well as any other clinical changes that occurred during treatment. If any clinical changes were observed during treatment, the raters were instructed to contact the researchers by phone immediately to report the symptoms. This information was distributed to other staff members, along with instructions to contact the researchers by phone immediately upon suspicion of any adverse event. In addition, the researchers asked specifically whether any adverse events were suspected at other contacts with the nursing home staff and during scheduled follow-up at 6 and 13 weeks of treatment. All suspected adverse events, irrespective of whether the patient discontinued treatment, were recorded verbatim as reported by nursing home staff, in as much detail as possible, including information about time from initiation to presenting symptoms and discontinuation of treatment.
Demographic information and a complete list of scheduled drug prescriptions (excluding prescriptions given pro re nata, ie, “as needed”) were extracted from the patients’ medical records at baseline. The total number of scheduled drug prescriptions was counted. Analgesic use was assessed by counting the number of prescriptions for drugs classified as systemic analgesics (Anatomical Therapeutic Chemical [ATC] code N02 or M01A). In addition, the individual and total numbers of scheduled psychotropic drugs were counted (antidepressants [N06A], anti-dementia [N06D], antipsychotic [N05A], anxiolytic [N05B], hypnotic and sedative [N05C], and antiepileptic [N03A] drugs). The total anticholinergic cognitive burden (ACB) was calculated by assigning 1 point for each prescribed drug with mild anticholinergic properties, 2 points for each drug with moderate anticholinergic effects, and 3 points for each drug with strong anticholinergic properties.19,20 Between-group differences in drug use and morbidity at baseline were assessed by counting the number of prescriptions for drugs within each ATC group (A–V).
Activity was assessed by actigraphy registration using the Philips Actiwatch Spectrum, which was worn on the patients’ dominant or mobile wrist continuously for 14 days (7 days before and 7 days after treatment was started).21,22 Total activity counts per day (Total AC) and mean intensity of activity per minute (AC/minute) for daily 12-hour intervals (09:00–21:00) were extracted from the Respironics Actiware 6.0.9 software. Mean activity counts for Total AC and AC/minute were calculated for both 7-day periods in all patients with at least 5 valid days of actigraphy recording per week.
Cognitive function was assessed using the MMSE, a 30-item questionnaire administered directly to the patient covering 11 domains (registration, orientation to time and place, short-term recall, attention, calculation, long-term recall, naming, repetition, comprehension [verbal and written], writing, and visuospatial construction) to yield a sum score from 0 (most severe impairment) to 30 (no impairment).23,24
Pain was assessed using the Mobilization-Observation-Behavior-Intensity-Dementia-2 Pain Scale (MOBID-2), a two-part staff-administered instrument to assess pain in people with advanced dementia.25 The proxy evaluation of inferred pain intensity is based on the patient’s pain behaviors during standardized, guided movements of different body parts (Part 1), and pain behaviors that might be related to internal organs, head, and skin are recorded on an anatomical figure along with the inferred pain intensity for each region to allow monitoring over time (Part 2). The scale yields a final score from 0 (no pain) to 10 (worst pain imaginable). Good interrater and test–retest reliability, internal consistency, and validity have been shown, and the MOBID-2 scale has also demonstrated responsiveness to change.25
Depressive symptoms were assessed using the CSDD, which is a validated and widely used screening tool for depression in people with dementia.26 It is administered by an interview with a proxy rater who is familiar with the patient, and it contains 19 items in five domains (mood-related signs, behavioral disturbance, physical signs, cyclic functions, and ideational disturbance). Each item is rated from 0 (no symptom) to 2 (severe symptoms) to yield a sum score of between 0 (no depression) and 38 (most severe depression).26
The main outcome measure was time to discontinuation of treatment due to adverse events. Secondary outcome measures were reasons for discontinuation, and change in total daytime activity and mean intensity of daily activity as measured by actigraphy recording.
Sample size calculation
The DEP.PAIN.DEM trial was designed to obtain 90% power to detect a 2-point CSDD difference between active treatment (acetaminophen or buprenorphine) and identical placebo, with an SD of 5, a standardized effect size of 0.4, p<0.05. The sample size was calculated using a sample size formula for longitudinal continuous response, adjusted for within-subject correlation between repeated measurements which was estimated to be 0.25 using data from the first 113 patients. One hundred thirty-two participants (66 in each group) were required, and adjusting for 20% dropouts, our final aim was to include 165 participants in total.39
Statistical analysis
Baseline characteristics were described as mean and standard deviation for continuous variables, and with the number of patients and percentages of the sample size for categorical variables. Between-group differences were tested using independent-samples t-test for continuous variables with normal distribution; Mann-Whitney U-test for continuous variables with non-normal distribution; and Pearson’s χ2 test for categorical variables. We used a Kaplan–Meier survival plot and Cox regression models to determine whether patients who were randomized to receive active buprenorphine had a higher risk of discontinuation compared to those who received placebo. Cox regression analyses were repeated with age, sex, and MOBID-2, CSDD, and MMSE as covariates to determine which variables should be included in the adjusted analyses. To further assess whether the risk of discontinuation of active buprenorphine was modified by drug use, we tested the interaction between the treatment effect and each of the drug variables (total number of prescribed drugs, ACB score, total number of psychotropic drugs, and use of each class of psychotropic drugs [N06A, N06D, N05A, N05B, N05C, N03A]) on discontinuation, both unadjusted and adjusted for age, sex, and MOBID-2, CSDD, and/or MMSE at baseline if these covariates impacted discontinuation risk. We used cluster-robust variance estimates to account for dependence within nursing homes. To assess immediate changes in daytime activity during the early days of treatment, we used linear mixed-effects models for Total AC and AC/minute/day using the mean recording from the 7 days before treatment was initiated as baseline. Time was included as a categorical variable, with fixed effects for time, intervention, and their interaction in the models. The models were fitted with random intercepts for patients to account for correlation between longitudinal measurements, random slope for time, and residual error structure specified as independent by day. We regarded p<0.05 as significant. All statistical analyses were conducted with STATA/IC 15 (Stata Corp LP, College Station, TX, USA).
Results
In total, 162 patients were included in the DEP.PAIN.DEM trial: 73 were prescribed acetaminophen/placebo tablets, and 89 patients were prescribed buprenorphine/placebo TDS and included in the current study. In the latter group, 44 were allocated to active treatment (hereafter, “active group”), and 45 to placebo (hereafter, “placebo group”; see Figure 1). Characteristics of the included patients at baseline are shown in Table 2. The groups were comparable at baseline on all tested variables except that the active group received more drugs in ATC group M (seven patients in active treatment and one patient in placebo; p=0.025), and the placebo group received more drugs in ATC group N (mean number of prescriptions 2.2 [SD 1.6] in the active group and 3.2 [SD 1.7] in the placebo group; p=0.001). From the latter ATC group, use of antidepressants and total number of psychotropic drugs were significantly higher in the placebo group; we also found significantly higher ACB in the placebo group (Table 2). Fifteen patients used buprenorphine TDS 5 μg/hour prior to inclusion, eight of whom were allocated to receive active treatment.
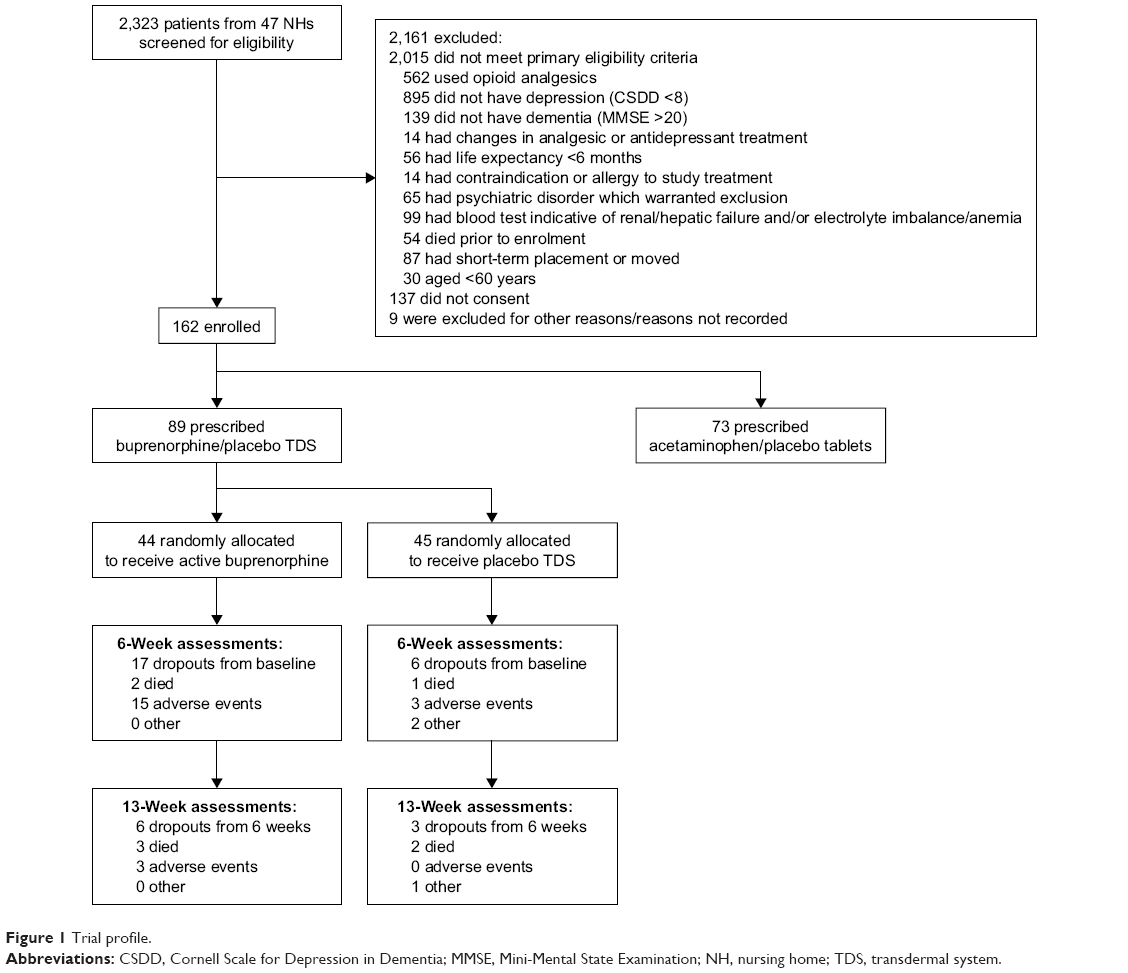 | Figure 1 Trial profile. |
 | Table 2 Background characteristics of included patients at baseline |
Frequency and types of adverse events
All adverse events recorded are presented in Table 3. Because each patient may have had more than one adverse event of each type, the number of adverse events may not correspond to the number of patients affected unless specified. Psychiatric adverse events were reported most frequently, with 17 separate adverse effects recorded in the active treatment group and none in the placebo group (p=0.003). Of psychiatric symptoms, personality changes (ie, changed emotional lability or other behavioral changes described as such) were the most frequent, reported in eight patients (18.2%), followed by confusion reported in five patients (11.4%). Neurological adverse events were the second most commonly reported, with 11 adverse effects recorded in the active treatment group and 2 in the placebo group (p=0.039). The most frequent neurological adverse event and the single most frequent adverse symptom was sedation/somnolence, which was reported in nine patients (20.5%) receiving active treatment and two patients receiving placebo (4.4%, p=0.022).
 | Table 3 Adverse events that may be related to study treatment |
Rates and causes of discontinuation
Buprenorphine TDS active treatment was discontinued in 23 patients (52.3%) due to adverse events, compared with 6 patients (13.3%) in the placebo group (p<0.001). Mean time to discontinuation was 61 days (SD 36) in the active treatment group and 82 days (SD 24) in the placebo group. Within the first 14 days, nine patients (20.5%) discontinued active treatment, and two patients (4.4%) discontinued placebo. Nearly half of patients who did not tolerate active treatment reported several types of adverse events (Table 4). Psychiatric adverse events were the most frequent cause of discontinuation reported in 12 of 23 patients (52%). Neurological adverse events were the second most frequent cause of discontinuation reported in nine patients (39%), five of whom also had psychiatric symptoms.
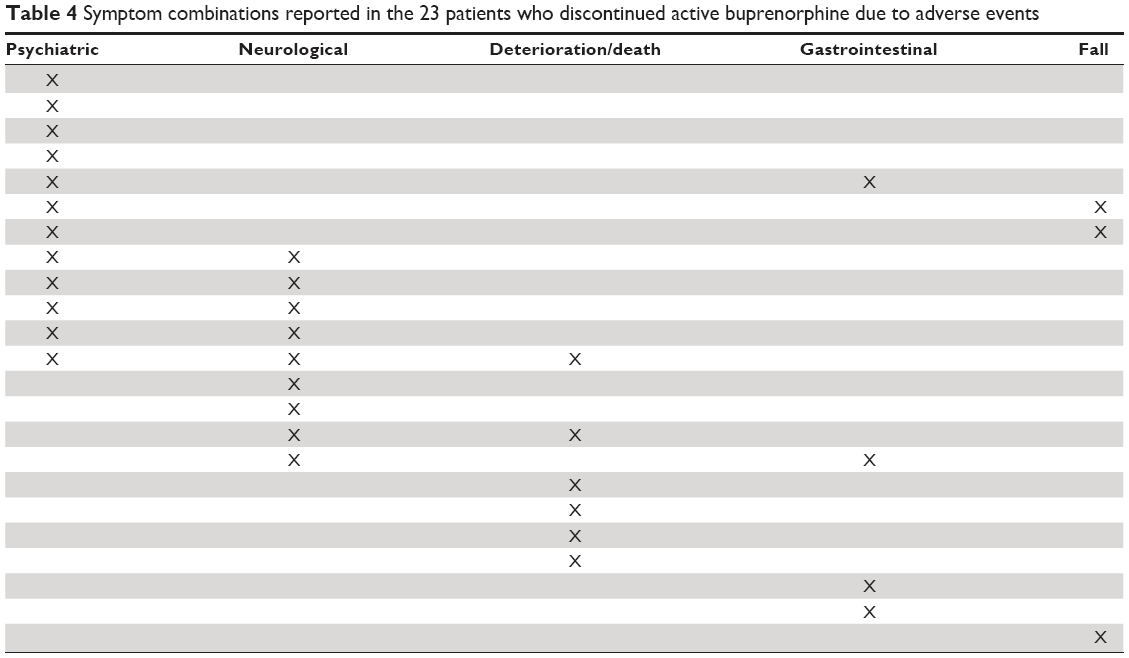 | Table 4 Symptom combinations reported in the 23 patients who discontinued active buprenorphine due to adverse events |
Kaplan–Meier estimates of time to discontinuation are shown in Figure 2. Throughout the study, patients who received active treatment had 4.7 times higher risk of discontinuation compared with those who received placebo (Table 5; Cox proportional hazards model, unadjusted HR, 95% CI: 1.66–13.3, p=0.004). Adjusted for age, sex, MOBID-2, CSDD, and MMSE at baseline, active treatment was associated with 24.0 times higher risk of discontinuation (95% CI: 2.45–235, p=0.006). In this model, age, sex, depressive symptoms, and pain were not significantly associated with discontinuation (age: HR 1.0, 95% CI: 0.99–1.11, p=0.133; sex: HR 0.32, 95% CI: 0.10–1.58, p=0.149; CSDD: HR 1.1, 95% CI: 0.90–1.34, p=0.343; MOBID-2: HR 1.1, 95% CI: 0.91–1.45, p=0.246). Lower MMSE scores were associated with increased risk of discontinuation (HR 0.82, 95% CI: 0.71–0.94, p=0.005), but interaction effects of MMSE score were tested in a new model and were not significant, that is, patients who received active treatment were not at increased risk of discontinuation if they had lower MMSE scores (HR 1.04, 95% CI: 0.82–1.31, p=0.767).
 | Figure 2 Kaplan–Meier survival plot: duration of study treatment. |
 | Table 5 Estimated treatment effect of buprenorphine versus placebo on discontinuation (Cox regression) |
Drug use and rates of discontinuation
Total number of prescribed drugs, ACB score, total number of psychotropic drugs, and use of any individual psychotropic drug (N06A, N06D, N05A, N05B, N05C, or N03A; dichotomized) were all not independently associated with discontinuation of the study treatment (active or placebo). However, patients who received active treatment and used antidepressants had 21.6 times increased risk of discontinuation compared with patients who used antidepressants and received placebo (95% CI: 2.75–170, p=0.003; Table 5). The interaction between active buprenorphine and antidepressant use remained unchanged when adjusting for age, sex, and MMSE (HR 23.2, 95% CI: 2.95–182, p=0.003). In this model, active buprenorphine was not significantly associated with increased risk of discontinuation in patients who did not use antidepressants (HR 2.95, 95% CI: 0.53–16.6, p=0.218), as shown in Figure 3 (Table 5). Interaction effects were calculated separately for each variable for drug use, and none except antidepressant use had significant interactions with active buprenorphine.
 | Figure 3 Cox proportional hazard plot: discontinuation risk stratified on treatment allocation and antidepressant use. |
Although patients who used antidepressants and received active treatment had significantly increased risk of discontinuation, we were not able to detect any significant difference in the total number of adverse events and treatment discontinuations compared with those who did not use antidepressants. Nine of the 14 patients (64.3%) who received active treatment and used antidepressants reported adverse events and discontinued treatment. Of the 30 patients who received active treatment and did not use antidepressants, 16 (53.3%) reported adverse events and 14 (46.7%) discontinued treatment. Using χ2-tests, the rates of adverse events and discontinuation in patients who received active treatment and used antidepressants were compared to those who did not use antidepressants (groups defined by the number of prescriptions for antidepressants at baseline), but no significant differences were found (p=0.599 and 0.419, respectively). We did not find that patients who used antidepressants reported any single type of adverse event more frequently, except confusion which was reported in three patients who used antidepressants (21.4%) and two patients who did not use antidepressants (6.7%, p=0.013, χ2-test).
Changes in activity during the first week of treatment
Day-to-day activity counts in the first week of treatment, measured by actigraphy, are shown in Figure 4 with the mean activity counts during the week before treatment started as the baseline activity score. Patients who received active treatment had significantly reduced daytime activity on day 2 of treatment compared with placebo (mixed model; Total AC: −16,967, p=0.005). This corresponds to a 21.4% decrease in total daytime activity in those who received active treatment. Comparing the mean daytime activity in the first week of treatment with baseline activity, we found that active treatment was associated with a 12.9% decrease in mean Total AC, but this effect was not statistically significant (mixed model; p=0.053).
 | Figure 4 Daytime activity during the first week of study treatment. |
Discussion
To our knowledge, this is the first placebo-controlled study investigating the tolerability and observed adverse events of buprenorphine TDS in nursing home patients with moderate to severe dementia. Patients who used antidepressants and received active treatment had the highest risk of discontinuation; this suggests a clinically relevant interaction between antidepressants and buprenorphine in people with dementia. Buprenorphine significantly reduced daytime activity as measured by actigraphy on the second day of treatment compared with placebo, supporting reports from nursing home staff of increased sedation/somnolence as the most frequent adverse effect. The poor tolerability of buprenorphine TDS due to the high risk of neurological and psychiatric adverse events should be considered carefully by clinicians before prescribing to people with dementia, and particularly to patients who are also using antidepressants, which may further reduce tolerability. This study does not assess the efficacy of buprenorphine TDS for treating different types of pain in dementia, which should be addressed in future research.
In the active treatment group, 57% had reported adverse events. A recent meta-analysis of six randomized controlled studies (five were placebo controlled) found that 82% of elderly patients (≥65) had adverse events of buprenorphine TDS.27 The lower rate of reported adverse events in our study can most likely be attributed to our reliance on proxy observations of adverse events. Although self-report of symptoms is considered the gold standard, people with advanced dementia often have impaired ability to reliably report their symptom burden. For example, less than half of patients with MMSE ≤6 are able to comprehend any assessment scale used to self-report painful symptoms.28 In our study, mean MMSE was 7 at baseline; therefore, many could not self-report adverse effects of buprenorphine TDS. Mild adverse effects of buprenorphine are subjective, they may not be easily observable, and subtle changes such as reduced appetite, confusion, or agitation could be misinterpreted as neuropsychiatric symptoms in dementia which may not be attributed to study treatment by the proxy raters. Because people with communication difficulty due to advanced dementia cannot be expected to reliably self-report mild adverse events, the true prevalence of adverse events is likely to have been underestimated in our study due to observer bias. Therefore, this should be interpreted as a tolerability study presenting adverse events associated with discontinuation of treatment, rather than the absolute frequency of adverse events in people with dementia.
Although very frail patients with short life expectancy were not included in the trial, sudden clinical deterioration is difficult to predict and must be expected to occur during an extended follow-up period in nursing home patients with advanced dementia, regardless of exposure to a clinical intervention. The number of patients who were withdrawn from the study because of severe clinical deterioration with short life expectancy did not differ significantly between the active treatment and placebo groups, and our sample size and follow-up period were not designed to investigate whether buprenorphine use may be associated with increased mortality. Adverse events were registered on suspicion, based on detailed reports of clinical changes from nursing home staff. Even though we did not assess the likelihood of causality between the study treatment and each reported adverse event, we conclude that the difference in the total number of adverse events between active treatment and placebo can likely be attributed to adverse effects of buprenorphine.
Previous studies indicate that buprenorphine TDS is well tolerated in elderly patients, with studies reporting similar or lower rates of adverse events in healthy elderly patients compared with younger controls.29–31 In elderly patients without dementia, the most common adverse events associated with discontinuation of buprenorphine treatment are gastrointestinal – nausea (8.2%), vomiting (3.9%), and constipation (2.0%) – followed by neurological symptoms – dizziness (5.1%), somnolence (2.0%), and headache (2.0%).27 In our study, psychiatric and neurological adverse effects were frequent, reported in 16 of patients who discontinued treatment (36.4%). Four patients (9.1%) who discontinued treatment had gastrointestinal symptoms. This indicates that psychiatric adverse events of buprenorphine may occur more frequently in people with dementia compared with cognitively intact elderly patients. As buprenorphine has similar pharmacokinetic properties in elderly patients including those with renal impairment,29,32 this reduced tolerability is most likely explained by pharmacodynamic changes in people with dementia.33,34
Buprenorphine TDS is absorbed slowly, and it reaches active concentration after ~24 hours and steady state during the first 72 hours in young healthy patients.35 Although pharmacokinetics have not been investigated in the very old and frail, buprenorphine TDS appears to be absorbed at a similar rate in people aged ≥75 years compared to a younger control group.29 The largest drop in daytime activity observed in our study (recorded ~24–36 hours after administration) may therefore correspond to the first systemic exposure to buprenorphine. While the reduction in daytime activity during the first week of active treatment was not statistically significant, this was probably due to low sample size.
Depression is associated with the use of antidepressants in nursing home patients with dementia.17 Because depression was an inclusion criterion, we may have selected patients who used more antidepressants relative to other psychotropic drugs. This may have enabled us to find a significant interaction between antidepressant use and buprenorphine discontinuation, while potential interactions between buprenorphine and other psychotropic drugs may have gone unnoticed. However, patients in the active treatment group generally used less psychotropic drugs compared with the placebo group and had a lower prevalence of antidepressant use (31.8%) than that expected from recent reports in people with dementia (~40%).36,37 Thus, the observed interaction between antidepressants and buprenorphine is likely to be of clinical relevance. While we did not find significant interaction effects between other psychotropic drugs and active buprenorphine, this may be due to insufficient power rather than the absence of such effects.
As shown in two recent studies, the use of opioid analgesics in the oldest nursing home patients with dementia is increasing.7,9 Age is associated with increased pain, frailty, and dementia. In patients with severe pain, or very frail patients, it may be difficult to achieve full analgesic effect as the type or dose of analgesics required may not be tolerated by the patient. Because patients with dementia have not been included in safety studies, the evidence base to ensure appropriate prescribing is lacking. In the DEP.PAIN.DEM trial, neither did we find a significant change in pain intensity in either of the buprenorphine or placebo TDS groups during follow-up, nor did we find a significant treatment effect on pain between these groups.39 However, this may be due to insufficient sample size as the DEP.PAIN.DEM trial was not powered to assess the effect of buprenorphine on pain. Further studies should investigate the efficacy and tolerability of buprenorphine and other opioid analgesics for pain in nursing home patients with dementia and painful symptoms. Use of opioids in people with dementia should be based on a careful risk–benefit evaluation, including regular assessments of pain and potential adverse effects, in combination with nonpharmacological strategies as appropriate.34
This study has limitations. The included patients had clinically significant depressive symptoms at baseline, but not all had pain. Therefore, our results may not be generalizable to patients who receive buprenorphine for pain. Prescribers may have had a lower threshold for discontinuation upon adverse events in this study, for instance, the risk–benefit consideration may have been shifted toward a greater awareness of adverse events as the treatment was prescribed off-label. The patients’ level of vulnerability to adverse events, and the relative prevalence of different types of adverse events, may also be different in people with advanced dementia and severe pain as opposed to the current sample which consisted of people with advanced dementia and depression without severe pain. Similarly, the adverse effects of buprenorphine TDS may differ between subgroups of pain patients (neuropathic/nociceptive; acute/chronic pain). Since the focus of the DEP.PAIN.DEM trial was to investigate the efficacy of pain treatment on depression, we did not diagnose the type and duration of pain. Therefore, future studies should investigate the safety and efficacy of buprenorphine TDS in people with dementia and different types of pain. Patients who were prescribed buprenorphine rather than acetaminophen used more regular analgesics and/or had difficulty swallowing tablets. This means that we may have selected more frail/multimorbid patients to receive buprenorphine/placebo as opposed to acetaminophen/placebo in the DEP.PAIN.DEM trial. However, this prescribing strategy mirrors clinical practice with a stepwise increase from non-opioid to opioid analgesics and the choice of transdermal formulation for patients who cannot swallow tablets; therefore, our sample should be similar to nursing home patients with dementia who receive buprenorphine TDS. We included a mixture of opioid-naïve patients, patients who had previously discontinued or received sporadic as-needed treatment with an opioid, and patients who received ongoing buprenorphine treatment; this is likely to have affected the observed pattern of adverse events which is not representative of an opioid-naïve population. Despite randomization, we found that patients who received active treatment used significantly less psychotropic and anticholinergic drugs, and fewer used antidepressants. This could potentially influence the results, as these drugs are associated with adverse outcomes in people with dementia.38 However, because these drugs were more prevalent in the control group, the high occurrence of adverse events in the active treatment group is likely caused by buprenorphine. We have not controlled for changes in concomitant drug use during study treatment. Physicians were instructed to avoid changes, particularly to psychotropic and analgesic drugs, but drug changes were not assessed in the 17 patients who discontinued treatment before week 6 assessment. The DEP.PAIN.DEM trial was designed with 90% power to detect a 2-point difference in depression (CSDD) from baseline to 13-week follow-up between active treatment (acetaminophen or buprenorphine) and placebo.39 For the secondary outcomes reported in the present study, no a priori power analyses have been conducted. This is an important limitation, and the findings from the current analyses should therefore be interpreted with caution, in particular for the subgroup analyses with lower sample sizes. Because the estimated effect sizes have very wide CIs, the exact magnitude of increased risk remains uncertain. However, we have identified significant between-group differences in reported adverse events and discontinuation risk. Although adverse events were assessed by proxy, and are therefore likely to be affected by observer bias, the placebo-controlled design provides strong evidence that the difference in adverse events is caused by the active drug rather than observer bias. Therefore, we find it important to share the presented results. Further studies are needed to provide evidence of the safety and efficacy of transdermal buprenorphine for different types of pain in people with dementia.
Conclusion
Buprenorphine appears to be poorly tolerated in people with dementia, with a higher prevalence of psychiatric adverse events compared with previous studies in cognitively intact elderly patients. Initiation of buprenorphine therapy is associated with reduced daytime activity. Although no dose adjustment is recommended for buprenorphine in elderly patients, our data suggest that people with dementia are susceptible to adverse events even at the lowest initial dose. When buprenorphine is administered to people with dementia, the patients’ general condition pre- and posttreatment should therefore be monitored carefully, including assessments of intended and adverse treatment effects, particularly in patients using antidepressants.
Acknowledgments
We wish to thank the patients, their relatives, and the nursing home staff for their willingness and motivation that made this work possible. The DEP.PAIN.DEM trial and AE and EF received grants from the Research Council of Norway. BSH would like to thank the G.C. Rieber Foundation and the Norwegian Government for supporting our work at the Centre for Elderly and Nursing Home Medicine, University of Bergen, Norway. DA is a Royal Society Wolfson Research Merit Award Holder and would like to thank the Wolfson Foundation and the Royal Society for their support. Parts of the work by DDS were carried out at the Biostatistics and Data analysis core facility (BIOS), University of Bergen. This work was funded by the Research Council of Norway (sponsor’s protocol code 221951) and has received a grant from the University of Bergen. The funders played no role in the design of the study; the collection, analysis, and interpretation of the data; writing of the report; and the decision to submit the article for publication.
This paper represents independent research [part] funded by the National Institute for Health Research (NIHR) Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health and Social Care.
Author contributions
BSH, DA, and CB contributed in the conception of the study and in obtaining funding. All authors contributed to the study design, in carrying out the study, and in writing the manuscript. DDS, AE, EF, and BSH contributed to the statistical analysis. BSH and AE are guarantors for the study. All authors have approved the final article.
Disclosure
Although Mundipharma Research Limited has supplied study medication, the company has had no influence on study design, data collection, analyses and interpretation of data, or final publications. The authors report no conflicts of interest in this work.
References
Helvik AS, Engedal K, Benth JS, Selbaek G. Prevalence and severity of dementia in nursing home residents. Dement Geriatr Cogn Disord. 2015;40:166–177. | ||
Zwakhalen SM, Koopmans RT, Geels PJ, Berger MP, Hamers JP. The prevalence of pain in nursing home residents with dementia measured using an observational pain scale. Eur J Pain. 2009;13:89–93. | ||
Hadjistavropoulos T, Herr K, Prkachin KM, et al. Pain assessment in elderly adults with dementia. Lancet Neurol. 2014;13:1216–1227. | ||
Reynolds KS, Hanson LC, DeVellis RF, Henderson M, Steinhauser KE. Disparities in pain management between cognitively intact and cognitively impaired nursing home residents. J Pain Symptom Manage. 2008;35:388–396. | ||
Achterberg WP, Pieper MJ, van Dalen-Kok AH, et al. Pain management in patients with dementia. Clin Interv Aging. 2013;8:1471–1482. | ||
Tan EC, Jokanovic N, Koponen MP, Thomas D, Hilmer SN, Bell JS. Prevalence of analgesic use and pain in people with and without dementia or cognitive impairment in aged care facilities: a systematic review and meta-analysis. Curr Clin Pharmacol. 2015;10:194–203. | ||
Jensen-Dahm C, Gasse C, Astrup A, Mortensen PB, Waldemar G. Frequent use of opioids in patients with dementia and nursing home residents: a study of the entire elderly population of Denmark. Alzheimers Dement. 2015;11:691–699. | ||
Bauer U, Pitzer S, Schreier MM, Osterbrink J, Alzner R, Iglseder B. Pain treatment for nursing home residents differs according to cognitive state – a cross-sectional study. BMC Geriatr. 2016;16:124. | ||
Sandvik R, Selbaek G, Kirkevold O, Aarsland D, Husebo BS. Analgesic prescribing patterns in Norwegian nursing homes from 2000 to 2011: trend analyses of four data samples. Age Ageing. 2016;45:54–60. | ||
Tan EC, Visvanathan R, Hilmer SN, et al. Analgesic use and pain in residents with and without dementia in aged care facilities: a cross-sectional study. Australas J Ageing. 2016;35:180–187. | ||
Pergolizzi J, Boger RH, Budd K, et al. Opioids and the management of chronic severe pain in the elderly: consensus statement of an International Expert Panel with focus on the six clinically most often used World Health Organization Step III opioids (buprenorphine, fentanyl, hydromorphone, methadone, morphine, oxycodone). Pain Pract. 2008;8:287–313. | ||
Pergolizzi J, Aloisi AM, Dahan A, et al. Current knowledge of buprenorphine and its unique pharmacological profile. Pain Pract. 2010;10:428–450. | ||
Vadivelu N, Hines RL. Management of chronic pain in the elderly: focus on transdermal buprenorphine. Clin Interv Aging. 2008;3:421–430. | ||
Gallagher AM, Leighton-Scott J, van Staa TP. Utilization characteristics and treatment persistence in patients prescribed low-dose buprenorphine patches in primary care in the United Kingdom: a retrospective cohort study. Clin Ther. 2009;31:1707–1715. | ||
Plosker GL. Buprenorphine 5, 10 and 20μg/h transdermal patch: a review of its use in the management of chronic non-malignant pain. Drugs. 2011;71:2491–2509. | ||
van Ojik AL, Jansen PA, Brouwers JR, van Roon EN. Treatment of chronic pain in older people: evidence-based choice of strong-acting opioids. Drugs Aging. 2012;29:615–625. | ||
Erdal A, Flo E, Selbaek G, et al. Associations between pain and depression in nursing home patients at different stages of dementia. J Affect Disord. 2017;218:8–14. | ||
Farina N, Morrell L, Banerjee S. What is the therapeutic value of antidepressants in dementia? A narrative review. Int J Geriatr Psychiatry. 2017;32:32–49. | ||
Boustani M, Campbell N, Munger S, Maidment I, Fox C. Impact of anticholinergics on the aging brain: a review and practical application. Aging Health. 2008;4:311–320. | ||
Salahudeen MS, Duffull SB, Nishtala PS. Anticholinergic burden quantified by anticholinergic risk scales and adverse outcomes in older people: a systematic review. BMC Geriatr. 2015;15:31. | ||
Van Remoortel H, Raste Y, Louvaris Z, et al; PROactive consortium. Validity of six activity monitors in chronic obstructive pulmonary disease: a comparison with indirect calorimetry. PLoS One. 2012;7:e39198. | ||
Lambiase MJ, Gabriel KP, Chang YF, Kuller LH, Matthews KA. Utility of actiwatch sleep monitor to assess waking movement behavior in older women. Med Sci Sports Exerc. 2014;46:2301–2307. | ||
Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198. | ||
Perneczky R, Wagenpfeil S, Komossa K, Grimmer T, Diehl J, Kurz A. Mapping scores onto stages: mini-mental state examination and clinical dementia rating. Am J Geriatr Psychiatry. 2006;14:139–144. | ||
Husebo BS, Ostelo R, Strand LI. The MOBID-2 pain scale: reliability and responsiveness to pain in patients with dementia. Eur J Pain. 2014;18:1419–1430. | ||
Alexopoulos GS, Abrams RC, Young RC, Shamoian CA. Cornell scale for depression in dementia. Biol Psychiatry. 1988;23:271–284. | ||
Pergolizzi JV, Raffa RB, Marcum Z, Colucci S, Ripa SR. Safety of buprenorphine transdermal system in the management of pain in older adults. Postgrad Med. 2017;129:92–101. | ||
Pautex S, Michon A, Guedira M, et al. Pain in severe dementia: self-assessment or observational scales? J Am Geriatr Soc. 2006;54:1040–1045. | ||
Al-Tawil N, Odar-Cederlof I, Berggren AC, Johnson HE, Persson J. Pharmacokinetics of transdermal buprenorphine patch in the elderly. Eur J Clin Pharmacol. 2013;69:143–149. | ||
Likar R, Vadlau EM, Breschan C, Kager I, Korak-Leiter M, Ziervogel G. Comparable analgesic efficacy of transdermal buprenorphine in patients over and under 65 years of age. Clin J Pain. 2008;24:536–543. | ||
Gianni W, Madaio AR, Ceci M, et al. Transdermal buprenorphine for the treatment of chronic noncancer pain in the oldest old. J Pain Symptom Manage. 2011;41:707–714. | ||
Kress HG. Clinical update on the pharmacology, efficacy and safety of transdermal buprenorphine. Eur J Pain. 2009;13:219–230. | ||
Naples JG, Gellad WF, Hanlon JT. Managing pain in older adults: the role of opioid analgesics. Clin Geriatr Med. 2016;32:725–735. | ||
McLachlan AJ, Bath S, Naganathan V, et al. Clinical pharmacology of analgesic medicines in older people: impact of frailty and cognitive impairment. Br J Clin Pharmacol. 2011;71:351–364. | ||
Kapil RP, Cipriano A, Friedman K, et al. Once-weekly transdermal buprenorphine application results in sustained and consistent steady-state plasma levels. J Pain Symptom Manage. 2013;46:65–75. | ||
Janus SI, van Manen JG, IJzerman MJ, Zuidema SU. Psychotropic drug prescriptions in Western European nursing homes. Int Psychogeriatr. 2016;28:1775–1790. | ||
Gulla C, Selbaek G, Flo E, Kjome R, Kirkevold O, Husebo BS. Multi-psychotropic drug prescription and the association to neuropsychiatric symptoms in three Norwegian nursing home cohorts between 2004 and 2011. BMC Geriatr. 2016;16:115. | ||
Johnell K, Bergman GJ, Fastbom J, Danielsson B, Borg N, Salmi P. Psychotropic drugs and the risk of fall injuries, hospitalisations and mortality among older adults. Int J Geriatr Psychiatry. 2017;32:414–420. | ||
Erdal A, Flo E, Aarsland D, et al. Efficacy and safety of analgesic treatment for depression in people with advanced dementia: randomised, multicentre, double-blind, placebo-controlled trial (DEP.PAIN.DEM). Drugs Aging. In Press 2018. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.