Back to Journals » Open Access Rheumatology: Research and Reviews » Volume 14
Tofacitinib Inhibits STAT Phosphorylation and Matrix Metalloproteinase-3, -9 and -13 Production by C28/I2 Human Juvenile Chondrocytes
Authors Thorpe JR, Wilson RA, Mesiano S, Malemud CJ
Received 2 March 2022
Accepted for publication 6 September 2022
Published 4 October 2022 Volume 2022:14 Pages 195—209
DOI https://doi.org/10.2147/OARRR.S363736
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Chuan-Ju Liu
Jessica R Thorpe,1 Rachel A Wilson,2 Sam Mesiano,2 Charles J Malemud1
1Department of Medicine, Division of Rheumatic Diseases, Case Western Reserve University School of Medicine, Cleveland, OH, 44106, USA; 2Department of Reproductive Biology, Case Western Reserve University School of Medicine, University Hospitals Cleveland, Cleveland, OH, 44106, USA
Correspondence: Charles J Malemud, Department of Medicine, Division of Rheumatic Diseases, Foley Medical Building, 2061 Cornell Road, Room 207, Cleveland, OH, 44106-5076, USA, Tel +1 216 844-7846 ; +1 216 536-1945, Fax +1 216 844-2288, Email [email protected]; [email protected]
Purpose: This in vitro study was designed to determine the effect of the pan-Janus kinase inhibitor, Tofacitinib, on basal and interleukin-6 (IL-6)-induced signal transducers and activators of transcription (STAT) phosphorylation and matrix metalloproteinase (MMP) gene expression and MMP production by C28/I2 human chondrocytes.
Methods: C28/I2 chondrocytes were grown to a confluent high-density and treated either with recombinant human IL-6 (rhIL-6; 10– 20ng/mL) or maintained in the basal state for up to 60 min. MMP gene expression was determined using RT-PCR and MMP production by semi-quantitative immunohistochemistry. The effect of IL-6 with or without Tofacitinib on activation of STAT proteins was determined from quantitative Western blots.
Results: C28/I2 chondrocytes produced STAT1, STAT3 and STAT5AB which were phosphorylated (p) following treatment with rhIL-6 for 30 min. Tofacitinib (2.5nM– 100nM) decreased rhIL-6-induced activation of STAT1, STAT3, and STAT5AB as well as decreasing the expression of MMP3 and MMP13 but not MMP9, MMP1 or MMP2. In addition, Tofacitinib (50nM) reduced the number of rhIL-6-induced MMP3-, and MMP13- antibody-positive C28/I2 chondrocytes. However, Tofacitinib did decrease the number of MMP9-antibody-positive C28/I2 chondrocytes.
Conclusion: Taken together, these data showed that Tofacitinib, a pan-JAK small molecule inhibitor employed for the medical therapy of rheumatoid arthritis was a potent inhibitor of rhIL-6-induced STAT phosphorylation that appeared to be coupled to the inhibition of MMP-3, -9 and -13 production by C28/I2 chondrocytes.
Keywords: arthritis, cytokines, Janus kinase, signal transducers and activators of transcription
Introduction
Matrix metalloproteinases (MMPs) play a crucial role in the pathogenesis and progression of rheumatoid arthritis (RA).1 At the cellular level, RA is characterized by the migration of immune and non-immune cells from the peripheral circulation to the synovium and synovial joint fluid where activated synovial fibroblasts, T-cells, B-cells, dendritic cells, macrophages and neutrophils2,3 contribute to an elevated level of pro-inflammatory cytokines, several of which [eg interleukin-1β (IL-1β), IL-6, IL-17, tumor necrosis factor-α (TNF-α)] are intimately involved in upregulating matrix metalloproteinase (MMP) gene expression. In effect, the upregulation of MMP gene expression occurs mainly through the capacity of these pro-inflammatory cytokines to activate the stress-activated/mitogen-activated protein kinase (SAPK/MAPK) and/or the Janus Kinase/Signal Transducers and Activators of Transcription (JAK-STAT) signaling pathways by activated immune cells, synovial tissue fibroblasts and articular chondrocytes.4–6
In RA, the biological activity of the MMP family is principally involved in the degradation of cartilage extracellular matrix (ECM) proteins, typically, Type II collagen, the sulfated proteoglycan, aggrecan, the “minor” proteoglycans, including decorin, and biglycan, all of which are synthesized by chondrocytes.7 These ECM proteins provide the biochemical/biomechanical mechanism crucial for the maintenance of cartilage homeostasis and therefore, synovial joint integrity.7 As such, their loss from articular cartilage results in cartilage degradation in RA, as well as in osteoarthritis (OA) pathology.
The MMP messenger RNAs (mRNAs) produced by cells involved in the RA process are translated and produced as latent pro-enzymes. The pro-MMPs must be activated to be fully functional and MMPs have been implicated in this process as well.8 In addition, in RA a faulty endogenous MMP inhibitor system has been reported. Thus, under homeostatic conditions the biological activity of MMPs are regulated by a protein family referred to as tissue inhibitors of metalloproteinases (TIMPs).8
Importantly, the pro-MMPs that were previously shown to be produced by chondrocytes in vitro in response to stimulation by various pro-inflammatory cytokines, were pro-MMP-1 (collagenase-1), pro-MMP-2 (72kDa gelatinase), pro-MMP-3 (stromelysin-1), pro-MMP-9 (92kDa gelatinase) and pro-MMP-13 (collagenase-3)8–12 as well as A disintegrin and metalloproteinase/A disintegrin and A disintegrin metalloproteinase with thrombospondin motif (ADAM/ADAMTS) families of metalloproteinases.8,9
Several previous “pre-clinical” studies were designed to address the progression of cartilage ECM degradation in RA using MMP small molecule inhibitors (SMIs) (eg p38 MAPK inhibitors).13 This strategy was successfully employed in vitro.13–16 Furthermore, the development of SAPK/MAPK SMIs was then moved ahead into RA clinical trials, after they met several pre-clinical in vitro and in vivo primary endpoints as evidenced by the inhibition of MMP activity and amelioration of the severity of arthritis in a well-validated mouse model of inflammatory arthritis, respectively.17 However, a p38 MAPK SMI then failed to provide clinical efficacy in human RA clinical trials and elicited untoward adverse events such that they were not further developed for clinical use reviewed in.18 Then, more recently, Tocilizumab, a fully humanized monoclonal antibody that blocks the binding of IL-6 to its receptor, IL-6Rα as well as the interaction of IL-6 with the soluble IL-6R19, both pathways requiring gp130 to facilitate signal transduction and, Tofacitinib, a pan-JAK SMI was tested in vitro and in RA animal models.20 At present, both drugs have been successfully employed as drug therapies for human RA.21–23
Based on these findings as well as other experimental evidence, we first determined the extent to which recombinant human IL-6 (rhIL-6) activated STAT proteins, respectively, in the C28/I2 line of immortalized human chondrocytes. In that regard, we had previously shown that blocking IL-6/IL-Rα or IL6/sIL-6R with Tocilizumab inhibited STAT protein activation as well as the production of MMP-9 by C28/I2 chondrocytes.24 Thus, the present study of MMP gene expression was stimulated by these previous experimental outcomes, and here we addressed the hypothesis that Tofacitinib would inhibit human chondrocyte MMP gene expression by blocking STAT protein activation in vitro.
Materials and Methods
C28/I2 Human Juvenile Chondrocytes
The C28/I2 human chondrocyte line has been previously employed as a cell culture model for human chondrocytes in the evaluation of chondrocyte-mediated changes associated with arthritis.25,26 This human chondrocyte line was donated courtesy of Professor Mary B. Goldring; Hospital for Special Surgery/Weill Medical College of Cornell University, New York, New York, USA. The short tandem repeat (STR) profile for C28/I2 is published by Millipore-Sigma that sells the C28/I2 line to research laboratories.
Normal Human Chondrocytes
Normal Human Chondrocytes (NHC) were obtained from the commercial vendor ScienCell.
Cell Culture Protocol
In vitro studies with rhIL-6 or rhIL-1β (R&D Systems) activation of the JAK-STAT or SAPK/MAPK pathways were conducted either with the C28/I2 chondrocyte line or normal human chondrocytes (NHC) obtained from a commercial source (ScienCell). C28/I2 chondrocytes were maintained at 37°C in a 5% CO2 humidified incubator in Dulbecco’s Modified Eagle Medium (DMEM)/Ham’s F-12 (1:1) supplemented with 10% fetal bovine serum (FBS; 10% v/v), 1% penicillin/streptomycin, and 2mM L-glutamine (all purchased from Thermo Fisher Scientific). Previously published studies26 indicated that C28/I2 chondrocytes were a suitable surrogate for human chondrocytes. NHC were maintained in “chondrocyte medium” (ScienCell) consisting of basal medium containing Vitamin C, 5% FBS, 1% chondrocyte growth supplement, 1% penicillin/streptomycin solution and grown to a confluent cell density. In this regard, NHC were employed to test the extent to which the activation of STAT proteins by rhIL-6 was similar or different to C28/I2.
Incubation with rhIL-6 and Tofacitinib
C28/I2 chondrocytes or NHC were treated with Tofacitinib using a standard pre-incubation protocol in which chondrocytes were pre-incubated in medium supplemented with 0.1% FBS and Tofacitinib at varying concentrations (100nM, 50nM, 20nM, or 2.5nM; n=4 for each concentration) for 30 min. Following this “pre-incubation”, chondrocytes were washed with Dulbecco’s phosphate-buffered saline (DPBS) (Thermo Fisher Scientific) and then treated again with Tofacitinib (100nM, 50nM, 20nM, or 2.5nM) together with IL-6 (10ng/mL, 20ng/mL or 50ng/mL) in medium supplemented with 0.1% FBS for 30 min. The nanomolar concentrations of Tofacitinib employed in these studies were similar to those found to be at the IC50 level for JAK3 (1nM), JAK2 (20nM) and JAK3 (112nM).20 For the control groups, C28/I2 chondrocytes or NHC were treated with DMEM/Ham’s F-12 supplemented with 0.1% FBS and 10 ng/mL or 20ng/mL IL-6 (n=4) or “chondrocyte medium” for NHC supplemented with 0.1% FBS (n=4) for 30 min at 37°C. Following each treatment, chondrocytes were washed with DPBS and lysed with RIPA buffer containing protease and phosphatase inhibitors. The total cell lysate was then collected, and centrifuged at 16,000 × g for 10 min at 4°C yielding a cell pellet and a supernatant containing the protein lysate.
STAT Protein Analysis by Western Blot
Following these various treatments, the abundance of specific STAT proteins produced by C28/I2 chondrocytes or NHC was then analyzed by a quantitative Western blotting protocol. Following the collection of final protein lysates, the protein content in these supernatants was determined with the bicinchoninic acid protein assay (Thermo Fisher Scientific). Equal amounts of protein were then diluted in gel loading buffer containing 375mM Tris-HCL, 6% sodium dodecyl sulfate (SDS), 48% glycerol, 9% β-mercaptoethanol and 0.03% bromophenol blue, pH 6.8. The samples were heated at 100°C for 5 min and then loaded onto pre-cast 4–20% Tris-glycine polyacrylamide gels for denaturing sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) using the Novex electrophoresis system (Thermo Fisher Scientific). Protein was then electro-transferred from the pre-cast gel to a polyvinylidene difluoride membrane (PVDF) (Bio-Rad Laboratories). Immunodetection was achieved by first incubating PVDF membranes in a blocking buffer consisting of 5% nonfat milk in Tris-buffered saline (TBS) containing 0.1% Tween-20 (TBST-20) at room temperature for 1 hr and then washed 3 times for 5 min each in TBST. The membrane was incubated overnight at 4°C with primary antibodies (Table 1) diluted in 5% w/v BSA, 1X TBST-20. The following day, the PVDF membrane was washed 3 times for 5 min each in TBST-20 and incubated with horseradish peroxidase-conjugated secondary anti-mouse or anti-rabbit antibody diluted in 5% nonfat milk in TBST-20 for 1 hr at room temperature. Finally, the PVDF membrane was washed 3 times in the TBST-20 and imaged using chemiluminescence Radiance Q (Azure Biosystems). The immunoblots were quantified with ImageJ software [developed by National Institutes of Health (NIH) and LOCI, University of Wisconsin].
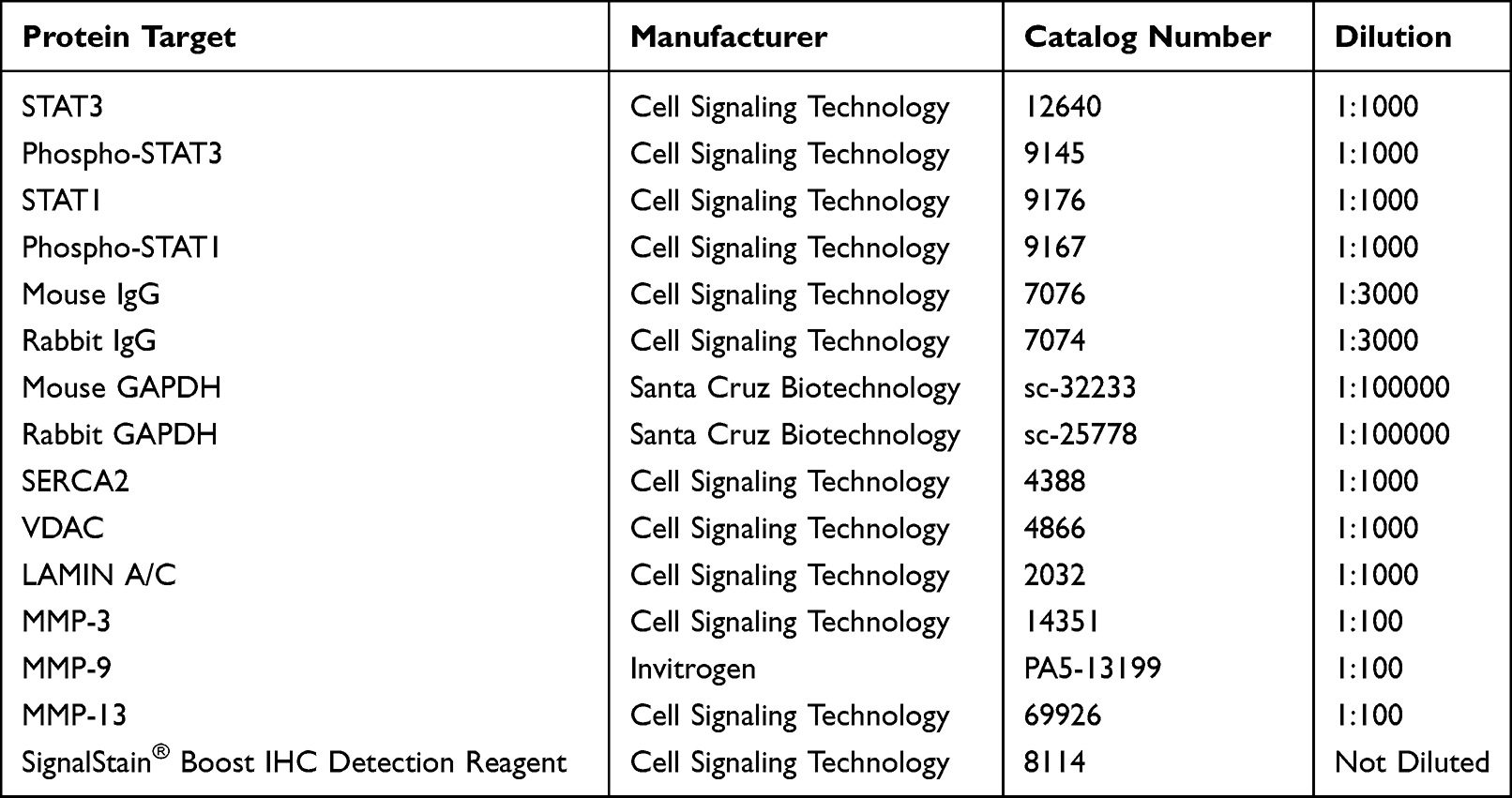 |
Table 1 Antibodies Employed for Western Blotting or Immunohistochemistry |
Cellular Compartment Fractionation
C28/I2 chondrocytes were incubated with 20ng/mL rhIL-6 in medium with 0.1% FBS for either 0 min (treatment placed on the cells then immediately removed), 1 min, 5 min, 30 min or 60 min (n=4 for each time point) at 37°C in a 5% CO2 humidified incubator to determine the extent to which un-phosphorylated and/or phosphorylated STAT proteins were translocated from the cytoplasm to the nucleus. Additionally, cells were treated with medium which was supplemented with 0.1% FBS (n=4) or medium containing 0.1% FBS, 20ng/mL rhIL-6, and 50nM Tofacitinib (n=4) for 30 min at 37°C. Following these procedures, C28/I2 chondrocytes were subjected to a subcellular fractionation protocol as previously described by Baghirova et al.27
Isolation of Subcellular Proteins
A fractionation protocol with minor modifications was employed27 in order to enrich for proteins in a specific cellular compartment. This protocol is a method for separating total cellular proteins into three cellular compartments, namely, cytosolic, membrane-bound organelles, and nuclear compartments. Following the treatment of C28/I2 cells as described above, the culture media was decanted. Chondrocytes were washed with DPBS, and then 160μL of ice cold lysis buffer A [NaCl (150mM), HEPES, pH 7.4 (50mM), digitonin (25μg/mL), hexylene glycol, (1M)] was added to each dish. The cell layer was then scraped off of the dish and the lysis buffer and chondrocytes were collected in a 1.7 mL centrifuge tube. Cells were incubated using an end-over-end rotator for 10 min at 4℃ and then centrifuged at 2000 × g for 10 min at 4℃. The resulting supernatant contained cytosolic proteins which were collected leaving a residual cell pellet. Next, 110ul of lysis buffer B [NaCl 150mM), HEPES pH 7.4 (50mM), IGEPAL CA-640 (1% v/v), hexylene glycol, (1M)] was added. The cell pellet was re-suspended with a vortex mixer. Samples were incubated on ice for 30 min and then centrifuged at 7000 × g for 10 min at 4℃. The resulting supernatant contained proteins from membrane-bound organelles except for those proteins in the nuclear compartment. The supernatant was collected and separated from the cell pellet. Lastly, 110uL of lysis buffer C [NaCl (150mM), HEPES, pH7.4 (50mM), sodium deoxycholate (0.5% v/v), SDS (0.1%), hexylene glycol (1M)] was added and the cell pellet was re-suspended by vortex mixing. Samples were incubated by end-over-end rotation for 30 min at 4℃ and then centrifuged at 7800 × g for 10 min at 4℃. The resulting supernatant containing proteins from the nuclear compartment was collected and saved at −80℃.
Proteins from each subcellular compartment were separated by SDS-PAGE as described above. Immune blotting for glyceraldehyde-3-phosphate dehydrogenase (GAPDH: marker for cytosolic and nuclear protein28) sarco endoplasmic reticulum calcium-ATPase 2 (SERCA2), voltage-dependent anion channel 1 (VDAC) were used as markers for membrane-bound proteins, and lamin A/C (LMNA) was used as a maker for nuclear proteins to confirm the relative purity of the subcellular fractions. The PDVF membrane was incubated overnight at 4°C with primary antibodies reactive with these biomarkers (Table 1) diluted in 5% w/v BSA, 1X TBST-20. The following day the PDVF membrane was washed 3 times for 5 min each in TBST-20 and incubated with horseradish peroxidase-conjugated secondary anti-mouse or anti-rabbit antibody diluted in 5% nonfat milk in TBST-20 for 1 hr at room temperature. Finally, the PDVF membrane was washed 3 times in TBST-20 containing chemiluminescence Radiance Q and quantified using ImageJ.
MMP Gene Expression
The repertoire of MMP gene expression by chondrocytes was previously reported.29,30 In that regard, IL-1β a known activator of MMP gene expression for cultured human chondrocytes9 regulated, in part, via activation of SAPK/MAPK signaling was employed to determine the extent to which the MMP1, 2, 3, 9 and 13 were expressed by C28/I2 chondrocytes in response to the addition of IL-1β (Cell Signaling Technologies).
RNA Isolation
After C28/I2 chondrocytes had reached a confluent state, they were sub-cultured into 24-well dishes at high density (5x105cells/mL; 106cells/cm2). After ~3 days, the sub-cultured chondrocytes had again reached confluency. The medium was decanted and C28/I2 chondrocytes were rinsed with PBS, then pre-incubated for 30 min in DMEM/Ham’s F-12 supplemented with 0.1% FBS and Tofacitinib (50nM). Following that, C28/I2 chondrocytes were briefly washed with PBS and then incubated with Tofacitinib (50nM) or Tofacitinib (50nM) together with rhIL-6 at either 10ng/mL, 20ng/mL or 50ng/mL (n=4 for each concentration of Tofacitinib) for 1 hr. In addition, C28/I2 chondrocytes were treated with medium alone as the primary control (n=4) or supplemented with Tofacitinib (50nM) (n=4). C28/I2 chondrocytes were then washed with PBS prior to RNA isolation.
Analysis of mRNA by Quantitative Reverse Transcriptase Polymerase Chain Reaction (qRT-PCR)
A commercial kit (Total RNA Isolation/NucleoSpin® RNA; Macherey-Nagel) was used to isolate total RNA from C28/I2 chondrocytes. The yield of RNA was analyzed by absorbance at 260nm. The SuperScript™ II Reverse Transcriptase kit for RT-PCR (Thermo Fisher Scientific) with random hexamers was employed to reverse transcribe 387ng of total RNA. Following that, the cDNA was used as a template for qRT-PCR by employing MMP gene primer sets that were designed with the PrimerExpress application; (Thermo Fisher Scientific) (Table 2).
 |
Table 2 Primer Pair Sequences, Melting Temperatures, and Resultant Amplicon Size (Bp) for MMP/GAPDH Analysis by qRT-PCR |
The StepOnePlus real-time PCR system (Thermo Fisher Scientific) was employed for qRT-PCR. Melting curves estimated the relative specificity of the amplified products where SYBR Green (Thermo Fisher Scientific) was the fluorescent detector for the measurements of amplicon abundance in real-time. In that regard, the abundance of specific mRNA relative to GAPDH mRNA was calculated using the ∆CT method [relative mRNA abundance = 2−(CT target gene of interest – CT GAPDH)].31 The relative gene expression assays were conducted and validated for primer pair specificity and each primer pair was optimized for equivalent qRT-PCR efficiency as previously described.31
Immunohistochemical (IHC) Analysis of MMP3, MMP9 and MMP13 Production in Response to rhIL-6 or rhIL-6 Plus Tofacitinib (50nM)
Preparation of C28/I2 Chondrocytes for IHC
Confluent C28/I2 chondrocytes were sub-cultured into 8-well Labtek dishes (10,000 cells/well; 14,300 cells/cm2). After ~3 days, the sub-cultured chondrocytes had again reached a confluent state at which time the medium was decanted and C28/I2 chondrocytes rinsed with PBS, then pre-incubated for 30 min in medium (DMEM/Ham’s F-12 supplemented with 0.1% FBS) containing Tofacitinib (50nM). C28/I2 chondrocytes were then briefly washed with PBS followed by incubation with Tofacitinib (50nM) or Tofacitinib (50nM) plus rhIL-6 (10ng/mL) for 1 hr. In addition, C28/I2 chondrocytes were treated with medium alone (primary control; n=4) or supplemented with Tofacitinib alone (50nM) (n=4). C28/I2 chondrocytes were then washed with PBS prior to being fixed in 4% paraformaldehyde (PF) for 15 min at room temperature.
Antigen retrieval was initiated on PF-fixed C28/I2 chondrocytes. The slides were placed in cold 10mM sodium citrate containing 0.05% Tween20, pH 6.0, then brought to 125°C for 20 min in a “decloaking” chamber. Following antigen retrieval, the slides were washed with PBS. The chondrocytes were then treated with 0.1% Triton-X100 in PBS for 5 min at room temperature, and rinsed with PBS. Chondrocytes were then treated with “blocking serum” (Vectastain® ABC Kit; Vector Laboratories) for 30 min at room temperature. Following that, 1:100 dilutions of the MMP-3, MMP-9, or MMP-13 antibodies (Table 1) were added and the chondrocytes incubated at room temperature for 30 min, then rinsed with PBS. Secondary antibody was added and incubated at room temperature for 30 min before being rinsed with PBS. The Cell Signaling SignalStain(R) DAB Substrate Kit (Cell Signaling Technologies) was used to bind the second antibody. Brown staining was monitored by microscopic inspection. A blank was produced with DAB and hematoxylin.
Slides were rinsed with deionized water then incubated for 30 sec with hematoxylin at room temperature, and then rinsed again with deionized water. The samples were dehydrated using increasing concentrations of ethanol and then mounted on a coverslip with xylene and Permount (Fisher Scientific).
Imaging and Semi-Quantitative Analysis of IHC
The slides were imaged at 40X magnification using a Keyence BZ-X800 microscope. Representative images of each of the slides were collected. ImageJ Fiji was employed to semi-quantify the presence of an MMP-positive antibody protein signal.32,33
Statistical Analyses
Experiments on C28/I2 chondrocytes were conducted in quadruplicate for each condition or for each of the Tofacitinib concentrations used to determine the effect on responses to rhIL-6. Replicates were analyzed for outliers using Dixon’s Q test where critical Q-values corresponded to a 95% confidence level. Normally distributed data were compared for significance between various groups using the Analysis of Variance (ANOVA) One-Way ANOVA Calculator followed by Tukey’s Honest Significant Difference test. Non-normally distributed data between groups was compared using the Tukey–Kramer Method. A p value <0.05 implied a significant difference between a control group and various experimental groups.
Results
MMP Gene Expression in Response to rhIL-6 and Effect of Tofacitinib
In a preliminary experiment, we tested the effect of Tofacitinib (2.5nM–100nM) on rhIL-6-treated (20ng/mL) C28/I2 chondrocytes. The results indicated no effect on the expression of any of the MMPs employing that experimental design. Therefore, in a follow-up analysis, we first found that Tofacitinib (50nM) by itself did not alter MMP expression (Figure 1). However, Tofacitinib (50nM) inhibited rhIL-6-induced expression of MMP3 (∆%= −55.6) and MMP13 (∆%= −48.6) only when the concentration of rhIL-6 was maintained below 20ng/mL (Figure 1). There was also a trend towards a reduction in MMP2 and MMP9. This result did not reach statistical significance (Figure 1).
 |
Figure 1 Effect of Tofacitinib on rhIL6-induced relative changes in MMP gene expression. C28/I2 chondrocytes were grown to a confluent state and then treated under control conditions, or with Tofacitinib (50nM) for 30 min. The medium was decanted and chondrocytes re-treated under control conditions or with Tofacitinib (50nM) in presence or absence of rhIL-6 at either 10ng/mL, 20ng/mL, or 50ng/mL for 1 hr. Total RNA was then extracted and used to measure the relative abundance of MMP mRNA by qRT-PCR. Average ΔCT data is representative of Mean ± Standard Deviation, N=4; *p<0.05. |
Effect of Tofacitinb on MMP Gene Expression by C28/I2 Chondrocytes Incubated with rhIL-1β
Cross-talk between a cytokine-activated SAPK/MAPK pathway and the JAK-STAT pathway has been a source of conjecture.5 In that regard, we evaluated the possibility that Tofacitinib could inhibit rhIL-1β-mediated MMP expression if activation of SAPK/MAPK signaling via rhIL-1β resulted in further activation of JAK-STAT signaling. In that case, we found that rhIL-1β significantly increased the relative expression of MMP3 and MMP13 (Figure 2). However, Tofacitinib did not inhibit rhIL-1ß-induced expression of MMP3 or MMP13 (Figure 2). Of note, inhibition of JNK activation with the JNKi, SP600125, significantly decreased rhIL-1ß-induced expression of MMP13 (Figure 2).
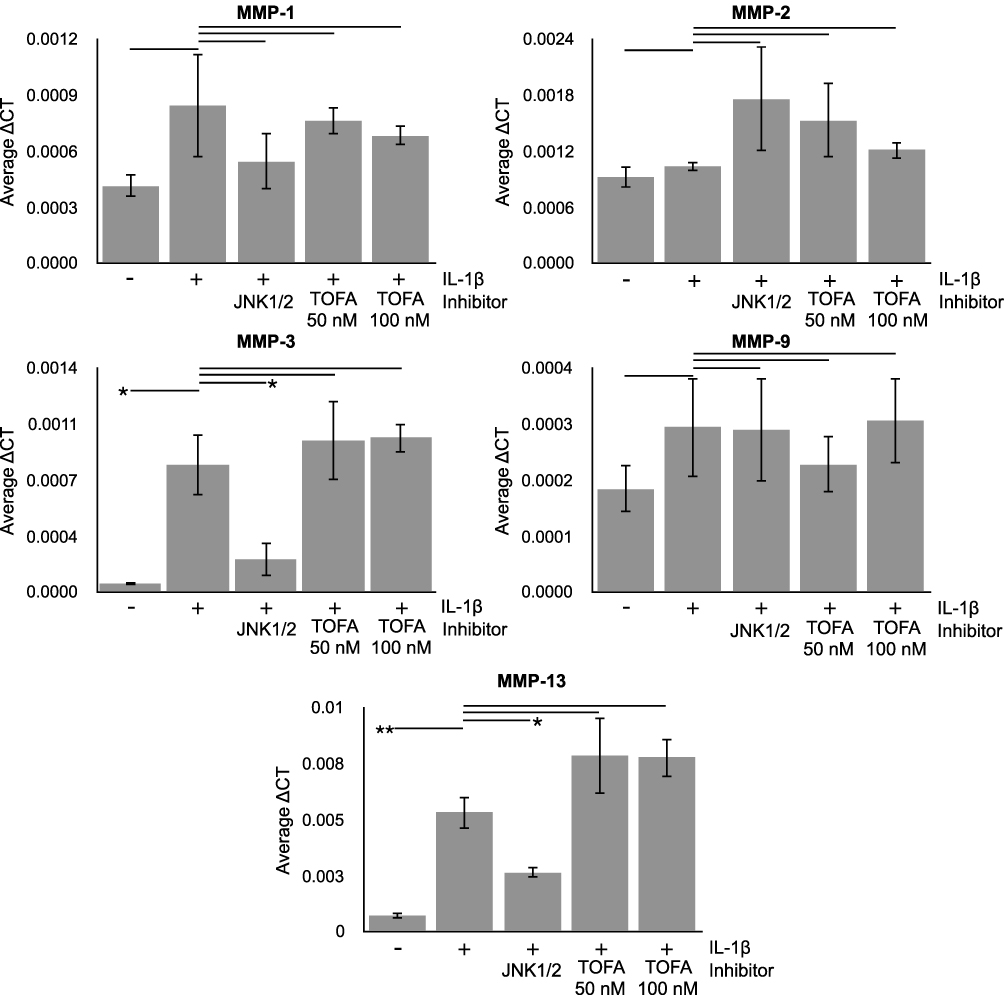 |
Figure 2 Effect of Tofacitinib (TOFA) on rhIL-1β-Induced MMP Relative Gene Expression. C28/I2 chondrocytes were grown to confluency and then treated under basal conditions (control) or with Tofacitinib (TOFA; 50nM or 100nM) or SP600125 (JNK inhibitor (JNK1/2); 100μM) for 1 hr. The medium was decanted and C28/I2 chondrocytes were treated either under basal conditions, with Tofacitinib (as shown) or with the JNKi, SP600125 (100μM) in the presence or absence of rhIL-1β (10ng/mL) for 1 hr. Total RNA was extracted and used to measure the relative abundance of MMP mRNA by qRT-PCR. Average ΔCT data is Mean ± Standard Deviation; N=4; *p<0.05; **p<0.02. |
Tofacitinib Reduced the Number of C28/I2 Chondrocytes with Immunoreactive MMP3, MMP9 and MMP13
Incubation of C28/I2 chondrocytes with rhIL-6 (10ng/mL) increased the number of MMP3, MMP9- and MMP13-positive chondrocytes compared to the control (basal) conditions and these values were decreased by Tofacitinib (Figure 3; Table 3). These results were derived from a semi-quantitative immunohistochemistry (IHC) analysis that determined the extent to which Tofacitinib (50nM) altered the number of MMP3, MMP9 or MMP13 antibody-positive chondrocytes in response to IL-6. In this analysis using the protocol of Crowe et al,32,33 the higher value in each group represents the number of chondrocytes that were antibody-positive for a particular MMP whereas the lower value is the number of chondrocytes that were MMP-antibody-negative for a particular MMP (Table 3).
 |
Table 3 3,3′-Diaminobenzidine (DAB) Staining Intensity Normalized to the Number of Positively Brown-Stained C28/I2 Chondrocytes |
 |
Figure 3 Immunohistochemistry of MMP3, MMP9 and MMP13 Antibody-Positive C28/I2 Chondrocytes in Response to IL-6 and the Effect of Tofacitinib (50nM) on MMP Antibody-Positive Chondrocytes. The scale is 50μm (A–J). Confluent C28/I2 chondrocytes were pre-treated with Tofacitinib (50nM) for 30 min followed by incubation with rhIL-6 (10ng/mL) or rhIL-6 plus Tofacitinib (50nM) for 1 hr. The “no-additions” group served as the control for rhIL-6-treated chondrocytes. The arrows in each panel indicate an MMP3, MMP9 or MMP13-positive C28/I2 chondrocyte. Panel J is a “blank” which shows the appearance of individual chondrocytes stained with hematoxylin. MMP3 (A–C); MMP9 (D–F); MMP13 (G–I); Appearance of confluent C28/I2 chondrocytes stained with DAB and hematoxylin (J). |
When primary MMP antibodies were omitted the chondrocytes looked identical in appearance to Figure 3J with a dull brown stain similar to those chondrocytes that were read-out as MMP negative. Thus, no immunoreactive signal was detected when primary antibody was replaced with non-immune IgG of the same isotype.
The results showed that rhIL-6 increased the proportion of cells staining positive for MMP3 (+80.4%), MMP9 (+67.6%) and MMP13 (+55.4%). Those values were decreased by Tofacitinib for each MMP. Thus, rhIL-6 effectively increased MMP3, MMP9 and MMP13 production by C28/I2 chondrocytes whereas Tofacitinib (50nM) decreased MMP3, MMP3 and MMP13 production by C28/I2 chondrocytes in response to rhIL-6.
Tofacitinib Inhibited STAT Protein Phosphorylation in Response to rhIL-6
C28/I2 chondrocytes and NHC were maintained in a basal state or induced with 20ng/mL rhIL-6 (R&D Systems) to activate the JAK/STAT pathway. Analyses were conducted to determine the extent to which Tofacitinib affects the activation of STAT proteins by C28/I2 chondrocytes or NHC. Both non-phosphorylated (ie total STAT) as well as p-STAT1 and p-STAT3 were detected by immunoblotting. For C28/I2 chondrocytes, total STAT3 and STAT1 were detected in the “no additions” control and in the rhIL-6 (20ng/mL)-treated groups (Figure 4A and B). By contrast, p-STAT3 and p-STAT1 were detected only at low levels in the “no additions” control group (Figure 4A and B). However, p-STAT3 and p-STAT1 were increased when C28/I2 chondrocytes were incubated with rhIL-6 (Figure 4C). We also determined the STAT activation profile by NHC. In that case, rhIL-6 increased total and p-STAT3 and p-STAT1 (Figure 4D and E). However, STAT5AB was not detected in NHC [data not shown] although STAT5AB was detected in C28/I2 chondrocytes as previously reported35 and rhIL-6 activated STAT5AB [data not shown]. The percent increase in total STAT proteins and p-STAT proteins in response to rhIL-6 is shown as a function of GAPDH provided in Table 4.
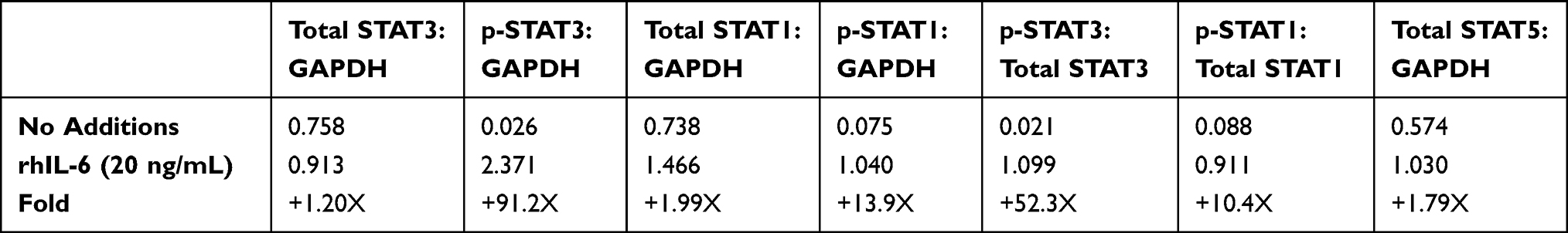 |
Table 4 Percent Change in STAT Proteins in Response to IL-6 |
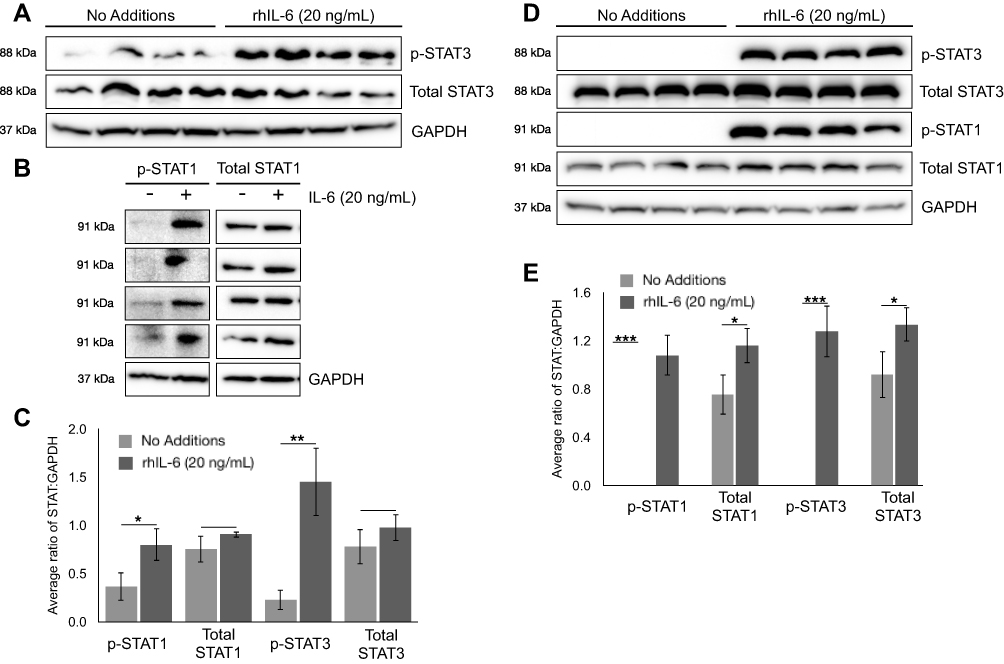 |
Figure 4 Cytokine rhIL-6 induced phosphorylation of STAT1 and STAT3 in C28/I2 chondrocytes and NHC. (A) Immunoblot analysis of p-STAT3, total STAT3, and GAPDH in whole cell lysate from C28/I2 cells. (B) Immunoblot analysis of p-STAT1, total STAT1, and GAPDH in whole cell lysate from C28/I2 cells. (C) Relative abundance of p-STAT3, total STAT3, p-STAT1, and total STAT1 normalized to GAPDH in C28/I2 cells treated as described. (D) Immunoblot analysis of p-STAT3, total STAT3, p-STAT1, total STAT1, and GAPDH in whole cell lysate from NHC. (E) Relative abundance of p-STAT3, total STAT3, p-STAT1, and total STAT1 normalized to GAPDH in NHC cells treated as described. Immunoblot data is representative of Mean ± Standard Deviation; N=4; *p<0.05, **p<0.01, ***p<0.003. |
The effect of Tofacitinib on STAT activation in C28/I2 chondrocyte and NHC was determined. Tofacitinib inhibited total STAT1, and STAT3 content in both cell types and decreased rhIL-6-induced p-STAT1 and p-STAT3 in a dose-dependent manner (Figure 5).
 |
Figure 5 Effect of Tofacitinib on rhIL-6 induced p-STAT3 and p-STAT1 in NHC and C28/I2. NHC and C28/I2 were pre-incubated with Tofacitinib (2.5nM, 20nM, 50nM, or 100 nM) for 30 min and then treated with rhIL-6 (20ng/mL) and Tofacitinib (2.5nM, 20nM, 50nM, or 100nM) concomitantly for 30 min. (A) Immunoblot analysis of p-STAT3, total STAT3, p-STAT1, total STAT1, and GAPDH in whole cell lysate from NHC or C28/I2 cells. (B) Relative abundance of p-STAT1 or p-STAT3 normalized to GAPDH in NHC or C28/I2 cells treated as described. Immunoblot data is representative of Mean ± Standard Deviation; N=4; *p<0.05, **p<0.01, ***p<0.002. |
Tofacitinib Inhibited p-STAT3 in the Cytoplasmic and Nuclear Compartments
Tofacitinib inhibited rhIL-6-induced p-STAT3 in both the cytoplasmic and nuclear compartments (Figures 6A–C). Thus, Tofacitinib immediately (ie as soon as 0 min) inhibited p-STAT3 in the cytoplasmic and nuclear compartment (Figure 6A–C). Furthermore, Tofacitinib significantly inhibited p-STAT3, in the cytoplasm after 1 min which was sustained after 60 min (Figure 6A). Moreover, in the nuclear compartment, Tofacitinib significantly inhibited p-STAT3 after translocation from the cytoplasmic compartment at 1 min, 5 min (significant according to Tukey’s Honest Significant Difference test), and at 60 min (Figure 6C). Tofacitinib also significantly inhibited STAT3 content in the nuclear compartment after 5 min, 30 min or 60 min (Figure 6D).
 |
Figure 6 Quantification and comparison of rhIL-6 to rhIL-6+Tofacitinib. Western blots were quantified for the relative abundance of total STAT3 or p-STAT3 against GAPDH. (A) Relative abundance of p-STAT3 normalized to GAPDH in the cytoplasmic fraction. (B) Relative abundance of total STAT3 normalized to GAPDH in the cytoplasmic fraction. (C) Relative abundance of p-STAT3 normalized to GAPDH in the nuclear fraction. (D) Relative abundance of total STAT3 normalized to GAPDH in the nuclear fraction. Mean ±Standard Deviation; N=4; *p<0.05; **p<0.02, ***p<0.001. |
Discussion
This study primarily employed the C28/I2 line of immortalized human juvenile chondrocytes to determine the extent to which rhIL-6 modulated MMPs. We employed the pan-JAK SMI, Tofacitinib to examine the expression of MMPs in response to rhIL-6 in order to determine if Tofacitinib affected MMPs by inhibiting STAT protein activation. Furthermore, we compared the results of STAT activation in C28/I2 chondrocytes to STAT activation in NHC.
The development of JAK-STAT inhibitors, including the pan-JAK inhibitor, Tofacitinib, has made its way into the drug armamentarium for the treatment of adult RA. Currently, Tofacitinib is mainly employed in the treatment of RA patients who have shown an inadequate clinical response to conventional synthetic disease modifying anti-rheumatic drugs (csDMARDs) and/or biologic drugs.34–36 In that regard, we examined the extent to which Tofacitinib inhibited both rhIL-1β and rhIL-6-induced MMPs. As expected, Tofacitinib did not inhibit rhIL-1β-induced MMP expression. Conversely, Tofacitinib significantly inhibited rhIL-6-induced expression of MMP3 and MMP13. In this regard, it should also be noted that Tofacitinib was previously shown to effectively reduce RA synovial fibroblast MMP production.37 However, under these conditions, the combination of rhIL-6 and Tofacitinib failed to reduce MMP9. We originally considered the possibility that effect size (n=4) was responsible for this result, but a second analysis where we increased the number of determinations to n=8 confirmed the previous result.
However, using a semi-quantitative analysis of IHC-stained chondrocytes, we found that Tofacitinib (50nM) suppressed MMP3, and MMP13 production in response to rhIL-6. We also found that MMP9 production was also reduced by Tofacitinib in response to rhIL-6 even though MMP9 expression was not significantly reduced by Tofacitinib. Using a similar IHC detection approach, we had previously shown that Tocilizumab which neutralizes rhIL-6-mediated JAK-STAT pathway activation inhibited MMP9 production by C28/I2 chondrocytes.26 Therefore, additional studies will have to be performed to address an apparent lack of association between the effect of Tofacitinib on MMP9 expression and its effect on MMP9 production. At present, we can only speculate as to the possible mechanism(s) underlying this apparent dissociation between the relatively small effect of rhIL-6 plus Tofacitinib on MMP9 expression compared to the effect of rhIL-6 plus Tofacitinib on MMP9 production. The fidelity of MMP9 mRNA translation could be a potential mechanism by which MMP9 mRNA is more efficiently translated to pro-MMP9 protein. This could account for the strength of the rhIL-6-effect on MMP9 protein as shown in Figure 3; Table 4 with only a small decrement in MMP9 mRNA in the rhIL-6 plus Tofacitinib group as depicted in Figure 1.
Based on the results of the C28/I2 chondrocyte compartmental analyses, it is highly likely that the Tofacitinib-mediated reduction in the relative expression of MMP3 and MMP13 in C28/I2 chondrocytes occurred, in part, via suppression of the translocation of activated STAT and non-phosphorylated STAT protein to the nuclear compartment.
To summarize, the pan-JAK inhibitor, Tofacitinib was found to be an effective inhibitor of the relative expression of MMP3 and MMP13 genes which are two critical MMPs relevant to promoting the degradation of articular cartilage extracellular matrix proteins in RA synovial joints. Using a semi-quantitative IHC readout method,32,33 we also found that Tofacitinib (50nM) reduced the number of MMP3-, and MMP13 antibody-positive chondrocytes and under the present incubation strategy Tofacitinib also reduced the number of MMP9 antibody-positive chondrocytes. Through these in vitro studies, we also established that Tofacitinib (50nM) likely reduced MMP3, MMP9 and MMP13 production in response to rhIL-6 through the inhibition of STAT3 protein as well as p-STAT3 protein nuclear translocation. It is in the nucleus where phosphorylated as well as un-phosphorylated STAT proteins bind to promoter regions containing STAT protein DNA motifs.38 This interaction is likely responsible for the inhibition of MMP3 MMP9 and MMP13 production. Although this is a proposed mechanism to explain these in vitro results, direct evidence that STAT proteins bind to MMP promoter sequences, such as AP-1 in C28/I2 chondrocytes remains to be determined. In addition, although the relative production of MMP13 was up-regulated by rhIL-6 and MMP13 by rhIL-1β, MMP1 was not. Huang et al39 reported that MMP1 as well as MMP3 and MMP13 were expressed in normal and OA cartilage and also by Chen et al.40 These findings indicated that some variation exists in MMP expression by intact human and OA cartilage compared to cultured C28/I2 human chondrocytes.
Our results also showed that rhIL-1β activated MMP gene expression in C28/I2 chondrocytes. Thus, it is highly unlikely that the mere inhibition of JAK-STAT signaling alone will retard the loss of articular cartilage extracellular matrix proteins in adult RA making it pertinent to continue to strive to discover additional MMP inhibitors perhaps those working through SAPK/MAPK signaling as well.25 Interestingly, recent studies with MAPKAPK2 (MK2) inhibitors41,42 may therefore be instructive in that regard. Thus, MK2 inhibitors in combination with a JAK inhibitor may be required to achieve a more effective inhibition of chondrocyte MMP gene expression in adult RA as well as other forms of inflammatory arthritis, such as OA.
Of course, an important limitation of the present study of MMPs is that the results presented here represent only in vitro responses by the C28/I2 line of immortalized human juvenile chondrocytes. However, it is important to note that there were more similarities than differences in the pattern of STAT protein activation via rhIL-6 by C28/I2 chondrocytes and NHC.
Finally, studies of MMP gene expression and MMP production in response to Tofacitinib in well-validated animal models of RA should also be performed going forward. It is noteworthy that several previously published pre-clinical studies on the effect of Tofacitinib in well-validated animal models of RA showed that the drug (also known as CP-690550) preserved articular cartilage integrity in collagen-induced arthritis and adjuvant-induced arthritis accompanied by a reduction in the influx of inflammatory cells,43 reduced serum levels of IL-6 and IL-8,44 inhibited cytokine receptor signaling mediated by JAK1 heterodimers, but not JAK2 homodimers,45 and reduced the overall inflammatory state measured by 18F-fluorodeoxyglucose positron emission tomography.46
Conclusion
The human juvenile immortalized chondrocyte cell line, C28/I2 expressed MMP1, MMP2, MMP3, MMP9 and MMP13 that were modulated in response to rhIL-1β in a manner similar to treatment with vascular endothelial growth factor-A (VEGF-A).30 The pan-JAK inhibitor, Tofacitinib, significantly inhibited only the relative expression of MMP3 and MMP13 in response to rhIL-6. There was a trend towards the inhibition of the relative expression of MMP9 as well. Using a semi-quantitative IHC analysis, we found that rhIL-6 increased MMP3, MMP9 and MMP13 production whereas Tofacitinib reduced the number of MMP3, MMP9 and MMP13-antibody-positive chondrocytes. As predicted on the basis of previously published JAK studies,20,36 Tofacitinib inhibited the phosphorylation of STAT-1, STAT3 and STAT5AB (data not shown) by C28/I2 chondrocytes in response to rhIL-6. This finding implied that the inhibition of the relative production of MMP3, MMP9 and MMP13 production was functionally related, at least in part, to inhibition of STAT protein activation. Our data also suggested that the inhibition of the relative expression of MMP3 and MMP13 as well as MMP3, MMP9 and MMP13 production by C28/I2 chondrocytes was regulated, in part, by STAT protein trafficking to the nucleus.
Acknowledgments
The authors acknowledge funding, in part, through an investigator-initiated project under contract # WI205444 awarded to CWRU from Pfizer, Incorporated (Principal Investigator, Charles J. Malemud, PhD) and a grant from the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health (R01-HD69819; Sam Mesiano, PhD, Principal Investigator). We also gratefully acknowledge the excellent technical assistance of Nathan Lu, Mihir Panchal and Callista Smith.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agreed to be accountable for all aspects of the work.
Disclosure
Dr. Malemud and Ms. Thorpe received salary support through the Pfizer, Inc. contract. Although the “Tofacitinib Committee” at Pfizer approved the experimental study design for funding, they have played no role in the implementation of this study. The authors report no other conflicts of interest in this work.
References
1. Itoh Y. Metalloproteinases in rheumatoid arthritis. Potential therapeutic targets to improve current therapies. Prog Mol Biol Trans Sci. 2017;148:327–338.
2. Firestein GS, McInnes IB. Immunopathogenesis of rheumatoid arthritis. Immunity. 2017;46:183–196. doi:10.1016/j.immuni.2017.02.006
3. Veale DJ, Orr C, Fearon U. Cellular and molecular perspectives in rheumatoid arthritis. Semin Immunopathol. 2017;39:343–354. doi:10.1007/s00281-017-0633-1
4. Kim EK, Choi EJ. Compromised MAPK signaling in human diseases. An update. Arch Toxicol. 2015;89:867–882. doi:10.1007/s00204-015-1472-2
5. Malemud CJ. The role of the JAK/STAT signal pathway in rheumatoid arthritis. Ther Adv Musculoskel Dis. 2018;10:117–127. doi:10.1177/1759720X18776224
6. Malemud CJ. Defective JAK-STAT pathway signaling contributes to autoimmune diseases. Curr Pharmacol Rep. 2018;4:358–366. doi:10.1007/s40495-018-0151-4
7. Malemud CJ, Stevenson S, Mehraban F, et al. The proteoglycan synthesis repertoire of rabbit chondrocytes maintained in Type II collagen gels. Osteoarthritis Cartilage. 1994;2(1):29–42. doi:10.1016/S1063-4584(05)80004-4
8. Gargiiulo S, Gamba G, Poli G, et al. Metalloproteinases and metalloproteinase inhibitors in age-related diseases. Curr Pharm Des. 2014;20:2993–3018. doi:10.2174/13816128113196660701
9. Malemud CJ. Inhibition of MMPs and ADAM/ADAMTS. Biochem Pharmacol. 2019;165:33–40. doi:10.1016/j.bcp.2019.02.033
10. Troeberg L, Nagase H. Proteases involved in cartilage degradation in osteoarthritis. Biochim Biophys Acta. 2012;1824:133–145. doi:10.1016/j.bbapap.2011.06.020
11. Karouzakis E, Neidhart M, Gay RE, et al. Molecular and cellular basis of rheumatoid arthritis destruction. Immunol Lett. 2006;106:8–13. doi:10.1016/j.imlet.2006.04.011
12. Matten S, Zafar A, Moin S, et al. Understanding the role of cytokines in the pathogenesis of rheumatoid arthritis. Clin Chim Acta. 2016;455:161–171. doi:10.1016/j.cca.2016.02.010
13. Westra J, Limburg PC. p38 mitogen-activated protein kinase (MAPK) in rheumatoid arthritis. Mini Rev Med Chem. 2006;6(8):867–874. doi:10.2174/138955706777934982
14. Nakai R, Salisbury CM, Rosen H, et al. Ranking the selectivity of PubMed screening hits by activity-based protein profiling: MMP-13 as a case study. Bioorg Med Chem. 2009;17:1101–1108. doi:10.1016/j.bmc.2008.03.018
15. Georgiadis D, Yiotakis A. Specific targeting of metzincin family members with small molecule inhibitors: progress towards a multifarious challenge. Bioorg Med Chem. 2008;16:8781–8794. doi:10.1016/j.bmc.2008.08.058
16. Wada Y, Shimada K, Kimura T, et al. Novel p38 MAP kinase inhibitor R-130823 suppresses IL-6, IL-8 and MMP-13 production in spheroid culture of human synovial sarcoma cell line SW 982. Immunol Lett. 2005;101(1):50–59. doi:10.1016/j.imlet.2005.04.010
17. Medicherla S, Ma JY, Mangadu R, et al. A selective p38 alpha mitogen-activated protein kinase inhibitor reverses cartilage and bone destruction in mice with collagen-induced arthritis. J Pharmacol Exp Ther. 2006;318:132–141. doi:10.1124/jpet.105.098020
18. Hammaker D, Firestein GS. “Go upstream, young man”: lessons learned from the p38 saga. Ann Rheum Dis. 2010;69(Suppl 1):77–82. doi:10.1136/ard.2009.119479
19. Malemud CJ, Pearlman E. Targeting JAK/STAT signaling pathway in inflammatory diseases. Curr Signal Transduct Ther. 2009;4:201–221. doi:10.2174/157436209789057467
20. Yamaoka K. Janus kinase inhibitors for rheumatoid arthritis. Curr Opin Chem Biol. 2016;32:29–33. doi:10.1016/j.cbpa.2016.03.006
21. Lee FB, Fleischmann R, Hall S, et al. Tofacitinib versus methotrexate in rheumatoid arthritis. N Engl J Med. 2014;370:2377–2386. doi:10.1056/NEJMoa1310476
22. Vyas D, O’Dell KM, Bandy JL, et al. Tofacitinib: the first Janus kinase (JAK) inhibitor for the treatment of rheumatoid arthritis. Ann Pharmacother. 2013;47(11):1524–1531. doi:10.1177/1060028013512790
23. Shetty A, Hanson R, Korsten P, et al. Tocilizumab in the treatment of rheumatoid arthritis and beyond. Drug Des Devel Ther. 2014;8:349–364. doi:10.2147/DDDT.S41437
24. Meszaros EC, Dahoud W, Mesiano S, et al. Blockade of recombinant human IL-6 with tocilizumab suppresses matrix metalloproteinase-9 production in the C28/I2 immortalized human chondrocyte cell line. Integr Mol Med. 2015;2:304–310. doi:10.15761/IMM.1000158
25. Goldring MB. Culture of immortalized chondrocytes and their use as models of chondrocyte function. Methods Mol Med. 2004;100:37–52. doi:10.1385/1-59259-810-2:037
26. Hamamura K, Goldring MB, Yokota H. Involvement of p38 MAPK in regulation of MMP13 mRNA in chondrocytes in response to surviving stress to endoplasmic reticulum. Arch Oral Biol. 2009;54:279–286. doi:10.1016/j.archoralbio.2008.11.003
27. Baghirova S, Hughes BG, Hendzel MJ, et al. Sequential fractionation and isolation of subcellular proteins from tissue or cultured cells. MethodsX. 2015;2:440–445. doi:10.1016/j.mex.2015.11.001
28. Ventura M, Mateo F, Serratosa L, et al. Nuclear translocation of glyceraldehyde-3-phosphate dehydrogenase is regulated by acetylation. Int J Biochem Cell Biol. 2010;42:1672–1680. doi:10.1016/j.biocel.2010.06.014
29. Liang Z-J, Zhuang H, Wang G-X, et al. MiRNA-140 is a negative feedback regulator of MMP-13 in IL-1β-stimulated human articular chondrocyte C28/I2 cells. Inflamm Res. 2012;61:503–509. doi:10.1007/s00011-012-0438-6
30. Pufe T, Harde V, Petersen W, et al. Vascular endothelial growth factor (VEGF) induces matrix metalloproteinase expression in immortalized chondrocytes. J Pathol. 2004;202:367–374. doi:10.1002/path.1527
31. Amini P, Wilson R, Wang J, et al. Progesterone and cAMP synergize to inhibit responsiveness of myometrial cells to pro-inflammatory/pro-labor stimuli. Mol Cell Endocrinol. 2019;479:1–11. doi:10.1016/j.mce.2018.08.005
32. Crowe AR, Yue W. Semi-quantitative determination of protein expression using immunohistochemistry staining and analysis. An integrated protocol. Bio Protoc. 2012;9:1–15.
33. Crowe A, Zheng W, Miller J, et al. Characterization of plasma membrane localization and phosphorylation status of organic anion transporting polypeptide (OATP) 1B1 c.521 T>C polymorphism. Pharm Res. 2019;36:101. doi:10.1007/s11095-019-2634-3
34. Kettle JG, Åstrand A, Catley M, et al. Inhibitors of JAK-family kinases: an update on the patent literature 2013–2015, part 1. Expert Opin Ther Pat. 2017;27:127–143. doi:10.1080/13543776.2017.1252753
35. Baker KF, Isaacs JD. Novel therapies for immune-mediated inflammatory diseases: what can we learn from their use in rheumatoid arthritis, spondyloarthritis, systemic lupus erythematosus, psoriasis, Crohn’s disease and ulcerative colitis? Ann Rheum Dis. 2018;75:175–187. doi:10.1136/annrheumdis-2017-211555
36. El Jammal T, Gerfaud-Valentin M, Sève P, et al. Inhibition of JAK/STAT signaling in rheumatologic disorders: the expanding spectrum. Joint Bone Spine. 2012;87:119–129. doi:10.1016/j.jbspin.2019.09.005
37. Pérez-Baos S, Gratal P, Barrasa JL, et al. Inhibition of pSTAT1 by tofacitinib accounts for the early improvement of experimental chronic synovitis. J Inflamm. 2019;16:2. doi:10.1186/s12950-019-0206-2
38. Malemud CJ. Regulation of chondrocyte matrix metalloproteinase gene expression. In: Dhalla NS, Chakraborti S, editors. Role of Proteases in Cellular Dysfunction. UK: Springer Science; 2013:63–77.
39. Huang G, Chubinskya S, Liao W, et al. Wnt5a induces catabolic signaling and matrix metalloproteinase production in human articular chondrocytes. Osteoarthritis Cartilage. 2017;25:1505–1515. doi:10.1016/j.joca.2017.05.018
40. Chen W-P, Xiong Y, Shi Y-X, et al. Astaxanthin reduces matrix metalloproteinase expression in human chondrocytes. Int Immunopharmacol. 2014;19(1):174–177. doi:10.1016/j.intimp.2013.12.007
41. Soni S, Anand P, Padwad YS. MAPKAPK2; the master regulator of RNA-binding proteins modulate transcript stability and tumor progression. J Exp Clin Cancer Res. 2019;38:121. doi:10.1186/s13046-019-1115-1
42. Haller V, Nahidino P, Forster M, et al. An updated patent review of p38 kinase inhibitors (2014–2019). Expert Opin Ther Pat. 2020;30:453–466. doi:10.1080/13543776.2020.1749263
43. Milici AJ, Kudlacz EM, Audoly L, et al. Cartilage preservation by inhibition of Janus kinase 3 in two rodent models of rheumatoid arthritis. Arthritis Res Ther. 2008;10(1):R14. doi:10.1186/ar2365
44. Tanaka Y, Maeshima K, Yamaoka K. In vitro and in vivo analysis of a JAK inhibitor in rheumatoid arthritis. Ann Rheum Dis. 2012;71(Suppl 2):i70–i74. doi:10.1136/annrheumdis-2011-200595
45. Dowty ME, Jesson MJ, Ghosh S, et al. Preclinical to clinical translation of tofacitinib, a Janus kinase inhibitor, in rheumatoid arthritis. J Pharmacol Exp Ther. 2014;348:165–173. doi:10.1124/jpet.113.209304
46. Raychaudhuri S, Abria C, Harmany ZT, et al. Quantitative tracking of inflammatory activity at the peak and trough plasma levels of tofacitinib, a Janus kinase inhibitor, via in vivo 18F-FDG PET. Int J Rheum Dis. 2019;22:2165–2169. doi:10.1111/1756-185X.13732
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.