Back to Archived Journals » International Journal of Interferon, Cytokine and Mediator Research » Volume 7
TNF-α in the cardiovascular system: from physiology to therapy
Received 30 March 2015
Accepted for publication 21 May 2015
Published 9 July 2015 Volume 2015:7 Pages 9—25
DOI https://doi.org/10.2147/IJICMR.S64894
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Randall Davis
Katharina Urschel,1 Iwona Cicha2
1Laboratory of Molecular Cardiology, Department of Cardiology and Angiology, 2Cardiovascular Nanomedicine Unit, Section of Experimental Oncology and Nanomedicine, ENT Department, University Hospital Erlangen, Germany
Abstract: Signaling pathways induced by the proinflammatory cytokine tumor necrosis factor alpha (TNF-α) play a key role in the cellular responses to inflammation and injury. In the cardiovascular system, TNF-α -activated signal transduction pathways may contribute to vascular dysfunction, development and progression of atherosclerosis, and adverse cardiac remodeling following myocardial infarction and heart failure. This review addresses the role of TNF-α in vascular physiology and disease. Furthermore, the therapeutic benefits of systemic TNF-α antagonism in cardiovascular and autoimmune inflammatory diseases are summarized and critically discussed.
Keywords: TNF-α, vascular inflammation, atherosclerosis, shear stress, heart failure, TNF-α antagonists
TNF-α signaling
Tumor necrosis factor alpha (TNF-α) was described in 1975 as a circulating antitumorigenic cytokine.1 Originally, TNF-α was thought to be produced mainly by immune cells like activated macrophages and lymphocytes,2,3 but further studies reported its expression also in endothelial and epithelial cells,4,5 smooth muscle cells,6 and cardiac myocytes.7 More recent investigations demonstrated that TNF-α is a key proinflammatory cytokine and an important part of the innate immune system which, upon stimulation of pattern recognition receptors, increases the expression of genes required to control tissue inflammation and injury.
The human TNF-α gene, first cloned in 1985,8 is located on chromosome 6p21.3, spans about three kb and contains four exons, whereby more than 80% of the secreted protein is coded for by the last exon.9 TNF-α is expressed as a 26 kDa transmembrane protein arranged in stable homotrimers that are biologically active.10 Upon proteolytic cleavage by the metalloprotease TNF-α-converting enzyme (TACE),11 the soluble cytokine is released into the extracellular space. This secreted 17 kDa protein forms triangular pyramid-shaped trimers and activates two distinct TNF-α receptors,12 ie, TNFR1 (p55) and TNFR2 (p75).13,14 TNFR1 is expressed in nearly all cells, and can be activated by both the membrane-bound and soluble trimeric forms of TNF-α, whereas TNFR2 is found mainly in cells of the immune system and the heart, and responds to the membrane-bound form of TNF-α. Both TNF receptors lack intrinsic enzyme activity and require the recruitment of adaptor molecules to initiate signaling.13 Upon binding TNF-α, TNFR1 forms trimers and the resulting conformational change leads to dissociation of the inhibitory protein SODD (silencer of death domains) from the intracellular death domain. This dissociation enables the adaptor protein TRADD (TNFR1-associated death domain) to bind to the death domain and to initiate signaling by nuclear factor kappa B (NF-κB), mitogen-activated protein kinases (MAPKs) or caspases (Figure 1).15,16
 | Figure 1 TNF-α-initiated signal transduction. TNF-α (soluble or membrane-bound) activates two different TNF-receptors, ie, TNFR1 and TNFR2. After binding of TNF-α, TNFR1 recruits distinct adaptor molecules (TRAF2, TRADD) at the intracellular death domain, thereby activating three major signaling pathways: NF-κB-signaling, MAPK/C-Jun-signaling, and caspase/apoptotic signaling. TNFR2 activation by membrane-bound TNF-α leads to activation of PI3K/Akt and proangiogenic pathways (eg, VEGF/VEGFR2), and is furthermore able to interfere with NF-κB signaling. |
During activation of NF-κB, the adaptor protein TRADD recruits TRAF2 (TNFR-associated factor 2) and the serine-threonine kinase RIP (receptor-interacting protein kinase). Subsequently, IκB kinase is recruited by TRAF2 and activated by RIP, and phosphorylates IκBα (inhibitor of κB), an inhibitory protein that binds to NF-κB and inhibits its translocation. Upon phosphorylation IκBα is degraded, releasing the transcription factor NF-κB, which translocates to the nucleus to mediate the transcription of proteins involved in cell activation, proliferation, and survival.
Among the three major MAPK cascades, TNF-α induces the strongest activation of the c-Jun N-terminal kinases (JNKs) responsive to stress stimuli, whereas the responses evoked in p38-MAPK and extracellular signal-regulated kinase (ERK) activation are moderate to low. The TRAF2/Rac axis phosphorylates the JNK-inducing upstream kinases, which then activate JNK. Upon translocation to the nucleus, JNK activates transcription factors such as c-Jun and activating transcription factor 2. The JNK pathway is involved in apoptosis, neurodegeneration, cell differentiation, and proliferation, but also in more specific regulation of cell functions, eg, chemokine production mediated by AP-1 (activator protein-1), including RANTES (regulated on activation, normal T-cell expressed and secreted), interleukin-8, and granulocyte macrophage colony-stimulating factor.
Although the death-inducing capability of TNF-α is weak compared with other TNF family members (such as Fas), its binding to TNFR1, which contains an intracellular death domain, can induce cell death signaling in certain conditions.17 In this process, TRADD binds FADD (Fas-associated protein with death domain), which then recruits caspase-8. A high concentration of caspase-8 induces its autoproteolytic activation and subsequent cleaving of effector caspases, thus mediating cell apoptosis. It must be noted, however, that compared with its overwhelming functions in the inflammatory response, TNF-α-induced cell death signaling is usually of minor importance in cell physiology.
TNF-α-mediated effects induced by the pathways downstream of TNFR1 are often conflicting, indicating an extensive cross-talk and context dependency. As an example, NF-κB enhances the transcription of proteins that interfere with cell death signaling. In contrast, activated caspases modulate NF-κB signaling by cleaving several components of the NF-κB pathway. Furthermore, the balance between proapoptotic and antiapoptotic signaling can be shifted by activation of TNFR2, which uses Etk (endothelial/epithelial tyrosine kinase, a non-receptor tyrosine kinase family member), to promote cell adhesion, migration, proliferation, survival, and angiogenesis. Activation of TNFR2 by TNF-α may lead to transactivation of vascular endothelial growth factor receptor 2 and Etk. Subsequently, downstream signaling by PI3K/Akt (phosphatidylinositol 3-kinase/Akt)v is initiated, which stimulates endothelial migration and tube formation.18 Other factors, such as increased production of reactive oxygen species (ROS), concurrent cytokine/growth factor expression, or cell type can additionally modulate the cellular effects of TNF-α. Thanks to such complicated regulation of TNF-α signaling, various cells with vastly diverse functions can respond appropriately to inflammation and injury.13
Role of TNF-α in the cardiovascular system
The cardiovascular effects of TNF-α include its effect on endothelial function and interactions with inflammatory cells, as well as effects on vasodilation, smooth muscle cell (SMC) physiology, cardiac myocyte function, and glucose homeostasis. Direct evidence of TNF-α-stimulated vascular dysfunction was provided by a study of intra-arterial TNF-α administration in humans. In healthy volunteers, an acute local vascular inflammation was observed upon intra-arterial infusion of high-dose TNF-α (80 or 240 ng/min) for 30 minutes. In parallel, impaired endothelium-dependent vasomotion and a sustained increase in endothelial tissue plasminogen activator release were detected.19 Administration of a lower TNF-α dose (17 ng/min) for 60 minutes induced an increase in basal vascular resistance in healthy subjects, which was blocked by pretreatment with a nonselective cyclooxygenase inhibitor or a nitric oxide (NO) synthase (NOS) inhibitor.20 The authors concluded that the observed effects of TNF-α were likely mediated not only by the reduced bioavailability of NO, but also by increased cyclooxygenase-dependent production of vasoconstrictors, as also reported in more recent studies.21,22
Role of TNF-α in atherogenesis
Vascular injury associated with atherosclerotic disease is characterized by formation and release of inflammatory cytokines, activation of ROS production, and reduced availability of NO, leading to endothelial dysfunction. Among the proinflammatory cytokines, TNF-α is a key player maintaining low-level systemic inflammation. Several mechanisms were described that contribute to the proatherogenic effects of TNF-α on the endothelium, including its role in ROS production, reducing the bioavailability of NO, and increasing the endothelial permeability to circulating blood components and cells. These proinflammatory atherogenic mechanisms are briefly highlighted below.
TNF-α-induced endothelial barrier dysfunction
TNF-α regulates vascular permeability in order to control inflammation, since the increased permeability of microvessels allows blood macromolecules and inflammatory cells to enter the injured tissues. However, increased permeability also contributes to formation of atherosclerotic plaques, which are initiated by the subendothelial accumulation of blood lipids and inflammatory cells. TNF-α contributes to disruption of the endothelial barrier by several mechanisms. Activation of TNFR1 has been reported to induce protein kinase C-dependent rearrangement of the actin cytoskeleton,23 to increase the monomeric to filament actin pool ratio,24 and to destabilize microtubules in vitro.25 Destabilization of the endothelial cytoskeleton by TNF-α induces formation of intercellular gaps26 and leads to a dose-dependent and time-dependent increase in paracellular movement of macromolecules across endothelial monolayers.26
In parallel with rearrangement of the actin cytoskeleton, TNF-α increases tyrosine phosphorylation of vascular endothelial cadherin, the key molecule in endothelial cell-cell contacts, which allows paracellular passage of blood macromolecules.27 Another mechanism contributing to increased endothelial activation and permeability in response to TNF-α is the degradation of glycocalyx.28 Moreover, TNF-α was recently shown to increase the transcytosis of low-density lipoprotein across human endothelial cells and to contribute to early atherosclerosis by enhancing subendothelial retention of low-density lipoprotein in the vascular walls of apolipoprotein E-deficient mice.29 These effects of TNF-α were substantially blocked not only by transcytosis inhibitors, but also by NF-κB inhibitors and peroxisome proliferator-activated receptor-γ inhibitors, indicating a cross-talk between these pathways.
Effects of TNF-α on NO formation and bioavailability
NO is the major mediator of endothelium-dependent vasorelaxation. In vivo studies demonstrated that endothelium-dependent vasodilation is impaired by TNF-α in a NO-dependent manner, in various vessels of the arterial tree.30–33 TNF-α decreases the bioavailability of NO both by reducing its production31,34,35 and by enhancing its removal.30 Decreased NO generation results from TNF-α-mediated inhibition of endothelial NO synthase (eNOS) expression36 and activity. TNF-α signaling suppresses gene promoter activity and destabilizes eNOS mRNA, thus reducing eNOS protein expression.37,38 These processes are mediated by TNFR1 signaling, whereas TNFR2 has little effect on eNOS expression.39 Moreover, TNF-α decreases NO bioavailability by accumulation of the endogenous eNOS inhibitor ADMA (asymmetric dimethylarginine)40 and by enhanced removal of NO, for example via its reaction with superoxide, in which peroxynitrite is generated.41
Role of TNF-α in ROS generation
TNF-α stimulates vascular superoxide production by increasing the activity of NADPH-dependent oxidases (NOX) in endothelial cells,30,31,42 smooth muscle cells,43 and neutrophils.44 NOX catalyze the transfer of electrons from NADPH to molecular oxygen, resulting in generation of superoxide. Other potential sources of TNF-α-induced vascular superoxide production include ceramide-activated protein kinase, xanthine oxidase,45 lipoxygenase, mitochondrial oxidase, and uncoupled eNOS.46
Expression of different NOX subunits (NOX4 [Nox 4A and 4B],47 NOX2, NOX548), as well as the regulatory proteins associated with NOX, namely p22phox, p47phox, and p67phox, has been described in human arterial endothelial cells. Upon exposure to TNF-α, expression of several of these proteins (NOX4A, p22phox, p47phox, and p67phox) is upregulated, and the activity of NOX is increased by nearly threefold.47 NOX activation by TNF-α is furthermore Rac1-dependent, as shown in endothelial cells in vitro49 and in cardiac-specific Rac1-deficient mice.50
Furthermore, studies in human microvascular endothelial cells demonstrated that TNF-α induces rapid phosphorylation of p47phox, which then binds to TRAF4 and translocates to the membrane, resulting in increased p47phox-p22phox complex formation and NOX activation. Binding of phosphorylated p47phox by TRAF4 occurs in a protein kinase C-dependent manner,51,52 which may link superoxide generation to the pathways involved in endothelial barrier disruption by TNF-α.53 This is further underscored by the fact that TNF-α-induced vascular permeability is prevented by an antiperoxynitrite agent.54
Apart from increasing monolayer permeability and decreasing NO bioavailability, the generation of superoxide upon TNF-α exposure has further implications for induction of proatherogenic pathways in endothelium, as activation of NOX has been shown to contribute to TNF-α-induced NF-κB activation.53
TNFα-induced endothelial- leukocyte interactions
Vascular endothelial cells are the primary cellular target for the actions of circulating proinflammatory cytokines. TNF-α binding to TNFR1 initiates the expression of many proinflammatory, proatherogenic proteins55,56 via activation of the transcription factors NF-κB and c-Jun/ATF-2,57–60 and thus contributes to increased recruitment of leukocytes by the endothelium.61
Proinflammatory TNF-α signaling induces rapid endothelial expression of cell adhesion molecules, which mediate the recruitment and transmigration of circulating leukocytes into the vascular wall.62 Adhesion molecules, including E-selectin, vascular cell adhesion molecule 1 (VCAM-1), and intercellular cell adhesion molecule 1 (ICAM-1) are synthesized de novo within 30–120 minutes of TNF-α stimulation63 and transported to the endothelial surface.
Chandrasekharan et al62 investigated in detail the involvement of the two TNF-α receptors in endothelial adhesion molecule expression. Studies utilizing cultured endothelial cells derived from wild-type, TNFR2-deficient, or TNFR1-deficient mice showed that TNFR2 was required for TNF-α-induced E-selectin, VCAM-1, and ICAM-1 expression. Furthermore, TNF-α-stimulated leukocyte rolling, firm adhesion to the endothelium, and transmigration were dramatically reduced in TNFR2-deficient mice, indicating that endothelial TNFR2 is also essential for TNF-α-induced leukocyte-endothelial cell interaction.62
Role of shear stress in endothelial responses to TNF-α
TNF-α plays an important role in regulating the endothelial response to inflammation and injury by promoting migration of immune cells into the diseased or injured tissues in order to protect the tissues from harmful pathogens, damaged cells or irritants. However, the chronic presence of increased systemic levels of TNF-α in the context of cardiovascular or inflammatory diseases may be deleterious to the endothelium in medium and large arteries, where constant low-level stimulation of endothelial activation and vascular permeability contributes to atherogenesis. There are several endothelial protection mechanisms that prevent the inflammatory response in arteries exposed to TNF-α, the most important of them being the signaling pathways activated by laminar shear stress (Figure 2).
 | Figure 2 Shear stress dependency of TNF-α signaling in endothelial cells. (A) Laminar shear stress prevents proinflammatory activation of endothelial cells by TNF-α by inhibiting NF-κB signaling (eg, preventing expression of adhesion molecules and inducing production of NO) and therefore results in a quiescent endothelial phenotype. (B) Proatherogenic non-uniform shear stress synergistically enhances proinflammatory TNF-α signaling by activating NF-κB, AP-1, or c-Jun, leading to endothelial dysfunction characterized by increased permeability, enhanced oxidative stress, lack of cell alignment with flow and adhesion molecule expression, followed by adhesion of leukocytes to the activated endothelium. |
Laminar shear stress exerts a profound antiapoptotic effect on endothelial cells treated with TNF-α via upregulation of NO synthesis64,65 and post-transcriptional activation of eNOS.66 Immediately after the onset of shear stress, eNOS is activated by phosphorylation at serine 1177 through PI3K/Akt, which increases NO formation within seconds. Moreover, laminar flow mediates prosurvival signaling by inhibiting TNF-α-induced caspase-3 and JNK activity in vitro and ex vivo.67 Downstream of TNFR1, laminar shear stress furthermore reduces c-Jun and NF-κB transcriptional activation in vitro.68 Ex vivo studies on perfused rabbit aortas69 showed that pre-exposure to laminar shear stress (12 dyne/cm2) inhibited TNF-α-induced activation of JNK, p38-MAPK, and ERK, and prevented VCAM-1 expression in endothelial cells. Furthermore, laminar shear stress was shown to prevent the association of TNFR1 with TRAF-269 and to suppress the capacity of TNF-α to induce proinflammatory gene expression in endothelial cells by enhancing induction of NF-κB-dependent cytoprotective transcripts.70
In vivo studies showed that endothelial VCAM-1 expression depends on NF-κB activation in response to disturbed shear stress.71 Hajra et al72 detected high levels of NF-κB proteins in endothelial cells at sites of low shear in the aortic arch and suggested that these regions may be primed for enhanced NF-κB activation in response to proinflammatory agonists. In accordance with this, significantly stronger VCAM-1 expression was detected at the atherosclerosis-prone region of the murine aorta (lesser curvature of the aortic arch), as compared with the greater curvature region with a low probability of atherosclerosis.73 This corresponds to the observation that the pattern of shear stress determines the localization of atherosclerotic plaques at the arterial wall regions typically characterized by non-uniform blood flow and low shear stress, ie, at bifurcations and curvatures. Our in vitro studies demonstrated that prolonged preconditioning of endothelial cells with either laminar or non-uniform shear stress strongly affects their responsiveness to circulating TNF-α.74 Endothelial cells exposed to laminar shear stress were essentially protected against TNF-α-induced upregulation of adhesion molecules and recruitment of monocytic cells. In contrast, in cells exposed to non-uniform shear stress, TNF-α induced a dramatic increase in the numbers of adhering monocytic cells, and in the expression of VCAM-1 and E-selectin, which was sensitive to NF-κB inhibition. These findings indicated that the TNF-α-activated signaling pathways are largely convergent with non-uniform shear stress-triggered signaling involving NF-κB, c-Jun, or AP-1. Additionally, upon exposure to non-uniform shear stress, endothelial glycocalyx becomes stiffer and loses its buffering function,75,76 thus further potentiating the glycocalyx-degrading effect of TNF-α.28 Taken together, chronic exposure to laminar shear stress strengthens endothelial cell resistance to TNF-α, whereas non-uniform shear stress increases endothelial susceptibility to TNF-α stimulus (Figure 2).74 In endothelial cells constantly exposed to blood flow, shear stress-activated mechanisms are thus major modulators of the biologic responses to cytokines.
Role of TNF-α in apoptosis of endothelial cells
TNF-α normally acts as a potent activator of endothelial cells (Figure 3) and does not induce endothelial apoptosis.57 However, when combined with agents that release ceramide (eg, ultraviolet irradiation or cytotoxic drugs) or with ceramide mimetics such as lipopolysaccharide, TNF-α can mediate cell death in endothelial cells.57 According to studies by Csiszar et al32,77 in the carotid arteries of young rats, recombinant TNF-α elicits endothelial dysfunction, oxidative stress, and proinflammatory gene expression. In aged coronary arteries, TNF-α expression is increased and contributes to apoptosis of endothelial cells by caspase-3 activation. Furthermore, elevated levels of plasma TNF-α during septic shock can induce both microvascular dysfunction and NF-κB-dependent endothelial apoptosis,78 which is associated with the severity and outcome of sepsis. The balance between activation and injury of endothelial cells induced by TNF-α and other cytokines may thus govern local vascular responses to these mediators in disease conditions in vivo.
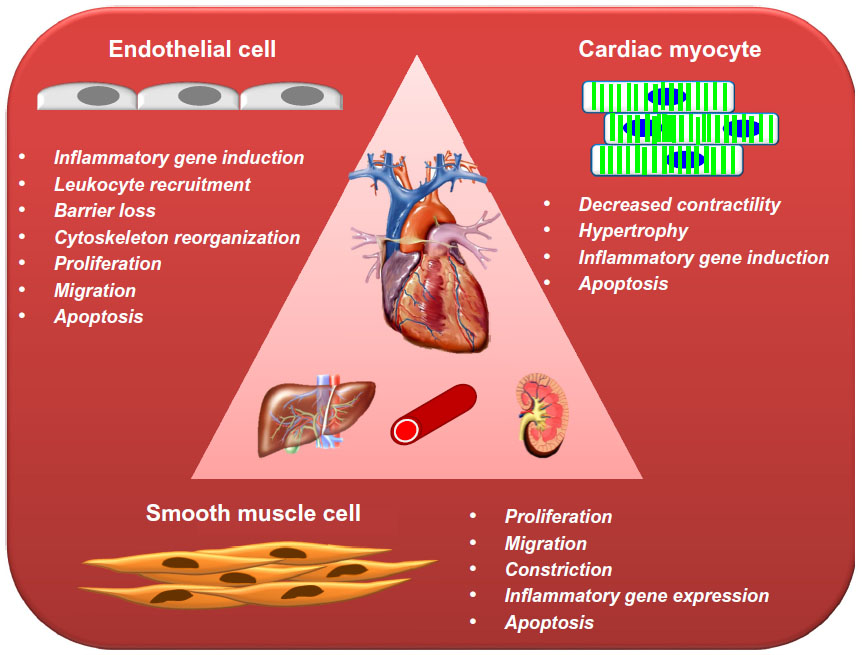 | Figure 3 Schematic presentation of the most important effects elicited by TNF-α in the cells of cardiovascular system. |
Role in vascular insulin resistance
Chronic inflammation contributes to vascular insulin resistance. Upon binding to its receptor on endothelial cells, insulin enhances microvascular perfusion of skeletal muscle by activating PI3K/Akt, which mediates an antiapoptotic effect and also increases gene expression and activation of eNOS.79 TNF-α interferes with endothelial insulin signaling and induces insulin resistance both in vivo80 and in vitro.81 In bovine aortic endothelial cells, TNF-α increases p38-MAPK phosphorylation and potently stimulates serine phosphorylation of the insulin receptor, thus inactivating signaling. In parallel, insulin-stimulated receptor tyrosine phosphorylation, Akt phosphorylation, and eNOS activity,81 as well as insulin uptake,82 are strongly inhibited by TNF-α in a p38-MAPK-dependent manner. TNF-α-mediated vascular insulin resistance may thus contribute to generalized endothelial dysfunction, eg, in diabetes and the cardiometabolic syndrome.
Effects on vascular smooth muscle cells
Phenotypic transition of vascular SMCs from a differentiated to a proliferative state during vascular injury and inflammation plays an important role in early atherosclerosis. TNF-α signaling induces SMC migration and proliferation mediated by TRAIL, ERK, and p38MAPK activation downstream of TNFR1.23,83,84 Moreover, TNF-α stimulates NOX and superoxide dismutase in SMCs, thus enhancing the formation of ROS and H2O2, which further induce activation of ERK and p38MAPK.85 During the development of atherosclerotic lesions, matrix metalloproteinase (MMP)-9 is upregulated and is required for migration of SMCs. TNF-α signaling upregulates nuclear FoxO4, which in turn activates transcription of the MMP-9 gene, inducing the ability of SMCs to migrate in vitro and in vivo.86
SMCs are normally resistant to Fas or cytokine-induced apoptosis, and even prolonged stimulation with a cytokine cocktail (interferon-γ, TNF-α, and interleukin-1β) or sFas ligand did not induce any significant change in SMC morphology or viability in vitro.87 However, in the context of atherosclerosis, SMCs isolated from the neointima of injured rat aorta were characterized by inhibited DNA synthesis and a decrease in cell numbers in response to TNF-α, as compared with medial SMCs.88 TNF-α also induced terminal dUTP nick end-labeling (TUNEL) positivity and caspase-3 expression in intimal SMCs, indicating increased cell death by apoptosis, whereas medial SMCs were markedly less sensitive to the TNF-α stimulus. Apoptosis in intimal SMCs was effectively inhibited by treatment with blocking antibodies against TNF-α receptors, suggesting that endogenous activation of TNF-α receptors may represent a way to limit hyperplasia in injured arteries.88 However, this mechanism may also be involved in intimal SMC apoptosis in advanced atherosclerotic plaques. Taken together, TNF-α affects SMC functions in multiple ways (Figure 3), consequently inducing the proatherogenic SMC phenotype.
Role in cardiac myocytes
In healthy individuals, cardiac TNF-α concentrations are low and do not affect contractile function.89–91 The presence of TNF-α protein and transcripts is usually restricted to microvessels in the normal heart.92 However, human cardiac myocytes express functional TNFR1 and respond to TNF-α.93 In vivo administration of exogenous TNF-α exerts cardiodepressant effects in rat94,95 and dog96,97 hearts. TNF-α dose-dependently reduces sarcoplasmic reticulum Ca2+ uptake and myofilament Ca2+ sensitivity,98–100 and this effect is mediated by p38 MAPK, a negative regulator of cardiac contractility. Intracellular calcium handling is suppressed by TNF-α via inhibition of sarcoplasmic reticulum calcium adenosine triphosphatase expression and activity.101 Furthermore, leakage of Ca2+ from the sarcoplasmic reticulum, which contributes to the depressed Ca2+ transient and reduced contractility is also increased by TNF-α.102 This effect is mediated by increased activation of cardiac caspase-8, leading to myocardial NO and mitochondrial ROS production, which results in S-nitrosylation of the ryanodine receptor and Ca2+ leak from the sarcoplasmic reticulum.103 TNF-α exposure is also associated with another NO-independent, but calcium-dependent, early depressant mechanism that is manifested by reduced contractile function and cyclic adenosine monophosphate response to beta-adrenergic stimulation.104
Apart from reducing contractility in cardiac myocytes (Figure 3), TNF-α induces cell hypertrophy manifested by cell enlargement, increased global protein synthesis, induction of sarcomere organization, and expression of embryonic genes, such as ANF (atrial natriuretic factor, a marker of hypertrophy).105 These phenomena are mediated by activation of p38MAPK and NF-κB106 in a ROS-dependent manner.105,107–109 In line with these findings, in vivo studies demonstrated a hypertrophic response in cardiac myocytes, leading to left ventricular hypertrophy and dilatation upon cardiac-specific overexpression or chronic infusion of high doses of TNF-α.94,105,110
Cardiac myocytes have further been shown to undergo apoptosis after stimulation with TNF-α in vitro.111 Al-Lamki et al92 investigated in detail the independent regulation and differential functions of TNFRs in myocardium. In biopsies from normal human hearts, TNFR1 but not TNFR2, was predominantly and strongly expressed by cardiac myocytes and rarely seen in fibroblasts. In organ cultures of the wild-type mouse heart, TNF-α stimulation upregulated TNFR2, activated signaling downstream of both receptors, and caused apoptosis and entry into the cell cycle. In TNFR1-deficient mouse hearts treated with TNF-α, activation of TNFR2, negligible apoptosis, and increased entry into the cell cycle were observed. In contrast, TNFR2-deficient organ cultures showed increased TUNEL positivity and lower levels of cell cycle entry.92 These findings are thus consistent with TNFR1-mediated cardiac cell death and TNFR2-mediated repair.
TNF-α in myocardial infarction
Occlusion of coronary arteries rapidly induces expression of proinflammatory cytokines, including TNF-α.112 However, conflicting findings were reported regarding the role of TNF-α in animal models of myocardial infarction (MI). TNF-α deficiency was shown to either decrease113 or have no effect on infarct size as compared with wild-type mice.114–117 Similarly, TNFR1-deficiency either reduced115 or did not affect infarct size.117–119 In a mouse model of coronary ligation, TNFR2-deficiency did not influence infarct size.118 Interestingly, knockout of both TNFRs in mice was shown to increase infarct size after 1 day, and this increase was associated with enhanced apoptosis of cardiac myocytes.120 These data are consistent with the finding that whereas an early increase in TNF-α post-MI contributes to stabilization of left ventricular function, prolonged stimulation with TNF-α induces left ventricular dysfunction in the later phase post-MI.121 In a recent study by Zhang et al, knockouts of TNFR1 or TNFR2 were shown to have opposite effects on cardiac function post-MI, whereby deletion of TNFR1 improved the cardiac dysfunction and the absence of TNFR2 increased the cardiac injury.122 Thus, both TNFR appear to play an important role post-MI, explaining at least in part the conflicting findings from in vivo knockout studies. Ideally, upregulation of TNF-α post-MI should be rapid and transient, in order to activate expression of TNFR2 and cardioprotective signaling. However, chronic exposure to increased TNF-α levels leads to TNFR1-mediated left ventricular dysfunction and increased MMP-2 activity, resulting in degradation of the matrix, and finally increased apoptosis of cardiomyocytes.117,123
TNF-α in heart failure
Heart failure is characterized by a debilitating decline in cardiac function. TNFR1 signaling is detrimental in murine models of heart failure, whereas TNFR2 mediates cardioprotection.124 Contractile dysfunction downstream of TNFR1 involves ß-adrenergic receptor uncoupling,125 as well as increased oxidative stress due to ROS formation and enhanced inducible NOS synthesis leading to production of NO and peroxynitrite.126 Furthermore, TNF-α downregulates the expression of contractile proteins, such as α-myosin heavy chain and cardiac α-actin.127 Apart from reducing contractility, TNF-α enhances the transcription of hypertrophic genes in the failing heart.105 Together with increased cardiac myocyte apoptosis128 and cardiac fibrosis,129 this hypertrophic effect contributes to structural alterations and heart failure. Consequently, long-lasting neutralization of TNF-α was shown to attenuate cardiac myocyte hypertrophy and adverse myocardial remodeling in a rat model of heart failure.130
Contribution of TNF-α signaling to hyper/hypotension
In sepsis, massive overproduction of TNF-α provokes life-threatening refractory hypotension, and according to clinical trials, this hypotensive effect constitutes a limiting factor for the use of TNF-α as a systemic antitumor agent. Upon administration in dogs, TNF-α (at 60 mg/kg body weight) induces cardiovascular collapse leading to the lethal shock syndrome.131 This hypotensive effect of TNF-α is mediated by excessive production of NO and ROS and activation of caspase.132
In contrast, a constant low-level increase in TNF-α production in the vascular system activates multiple pathways that contribute to vasoconstriction and can induce hypertension. Serum TNF-α is significantly and independently associated with blood pressure in apparently healthy subjects.133,134 In a study by Bautista et al,134 mean plasma levels of TNF-α were four times higher in hypertensive subjects than in non-hypertensive controls. The prevalence of hypertension increased progressively from 30% in the lowest TNF-α quartile to nearly 53% in the highest quartile, corresponding to an almost twofold significant increase in the prevalence.
In physiological conditions, a complex cross-regulation between the renin-angiotensin system and TNF-α signaling takes place. TNF-α was shown to inhibit renin gene expression in adrenal cells135 and renal juxtaglomerular cells,136 although TNF-α deficiency did not affect physiological regulation of renin expression by salt.136 Furthermore, a reduction of angiotensinogen production in renal proximal tubular cells by TNF-α was reported.137 However, this reduction may be counterbalanced by increased angiotensinogen expression in hepatocytes,138 which is an important contributor to circulating angiotensinogen levels. In the heart, no effects of cardiac-specific overexpression of TNF-α on renin or angiotensin production were observed, but the activation of myocardial renin-angiotensin system was increased, leading to enhanced left ventricular remodeling, increased collagen content, and apoptosis of cardiac myocytes.139
In multiple animal studies, TNF-α was found to promote hypertension, both in the presence and absence of renal injury.140,141 According to a recent report, kidney-derived TNF-α plays a decisive role in promoting the angiotensin II-induced increase in blood pressure.142 In a mouse model of chronic hypertensive kidney disease, TNF-α deficiency resulted in a blunted hypertensive response and reduced organ damage, mediated by enhanced eNOS expression and NO bioavailability. The same effect, along with reduced cardiac hypertrophy, was observed in wild-type animals transplanted with a TNF-α-knockout kidney, confirming that TNF-α produced by renal parenchymal cells potentiates the hypertensive response by suppressing bioavailability of NO within the kidney.
In the vasculature, TNF-α is known to participate in vascular contraction, acting either at the level of the endothelium or the SMCs. The major pathways involved in this effect include those for induction of vasoconstrictive preproendothelin-1 mRNA expression and endothelin-1 protein secretion in endothelial cells.143,144 Activation of the NFkB pathway is furthermore responsible for TNFα-induced upregulation of expression of the endothelin B2 receptor in SMCs.145 The endothelin B2 receptor mediates vasoconstriction146,147 and is not expressed in SMCs under normal conditions, but is upregulated in patients with cardiovascular disease (eg, peripheral artery disease,148 pulmonary hypertension,149 ischemic heart disease,150 and cerebral ischemic stroke146), suggesting possible involvement of TNF-α signaling. Moreover, TNF-α was reported to induce production of thromboxane A2 in endothelial cells21 and SMCs22 and to abolish insulin-mediated vasodilation in a JNK-dependent manner.151 Taken together, the existing studies indicate that TNF-α is an important regulator of blood pressure, both at the level of the kidney and at the level of the vasculature and myocardium.
Inactivation of TNF-α in atherosclerosis, heart failure, and inflammatory diseases
TNF-α promotes the inflammatory response, thus contributing to the clinical problems related to cardiovascular diseases and autoimmune disorders, which are strongly associated with cardiovascular comorbidity. The currently available TNF-α inhibitors include two types of biologic therapeutics, whereby blockade of TNF-α is achieved using either monoclonal antibodies (eg, infliximab, adalimumab, certolizumab, golimumab) or a soluble recombinant fusion protein consisting of the extracellular ligand-binding domain of TNFR2 and the Fc fragment of human IgG1 (etanercept).
TNF-α blockade in animal models
Experimental data show that genetic inactivation of TNF-α diminishes the size of atherosclerotic lesions, both in wild-type mice deficient in TNF-α and fed an atherogenic diet and in apolipoprotein E/TNF-α double-deficient mice, irrespective of the type of diet.152–155
The effect of anti-TNF-α antibodies on neointima formation following balloon injury in rabbits was investigated in detail by Zhou et al.156 Balloon angioplasty increased tissue expression of TNF-α by 100,000-fold over baseline, and this increase persisted over 6 days following arterial injury. Anti-TNF-α treatment neutralized tissue TNF-α activity by 60%–75% and suppressed macrophage infiltration, but did not inhibit neointima formation.156
Regarding the vascular dysfunction associated with increased insulin resistance, inhibition of TNF-α was shown to result in a marked increase in insulin sensitivity in obese rats. Moreover, neutralization of TNF-α by etanercept significantly reduced TNF-α levels in β2-adrenergic receptor-deficient mice, leading to increased insulin receptor tyrosine phosphorylation157 and alleviation of insulin resistance.
TNF-α antagonism was also shown to confer cardioprotection. A single administration of soluble TNFR-Fc fusion protein at the time of reperfusion after MI was able to limit infarct size and to reduce early left ventricular diastolic dysfunction in rats subjected to 60 minutes of coronary occlusion.158 Moreover, systemic TNF-α inhibition was cardioprotective in rats subjected to a continuous TNF-α infusion,94 in mice with cardiac-restricted overexpression of TNF-α,159 and in animal models of heart failure.126,160
Collectively, these preclinical observations suggest that blockade of TNF-α might inhibit progression of atherosclerotic lesions, prevent insulin resistance and accelerated atherosclerosis in the metabolic syndrome, and improve clinical outcomes in patients with heart failure.
Clinical studies in patients with heart failure
Circulating levels of TNF-α and soluble TNFRs are independent predictors of mortality in patients with heart failure.161 Although preclinical data suggested that TNF-α blockade in heart failure would be beneficial, randomized trials of anti-TNF-α therapy (etanercept in the RENEWAL trial162 and infliximab in the ATTACH trial)163 in human heart failure unexpectedly demonstrated a time-related and dose-related increase in mortality and heart failure-related hospitalization. In further studies, the mechanisms of new and/or worsening heart failure in etanercept-treated patients were investigated,164 demonstrating a significant 70-fold increase in plasma levels of immunoreactive TNF-α. Moreover, etanercept enhanced TNF-α bioactivity and increased the immunoreactive mass of homotrimeric TNF-α in heart failure patients, and this was suggested as a potential mechanism of the detrimental effects of this therapy in some patients.164 In light of the unique and divergent inflammatory responses evoked specifically by each TNF-α receptor in the failing heart, global TNF-α inhibition can therefore dysregulate both adverse and protective signaling, leading to dramatic worsening of cardiac function.
TNF-α antagonism in autoimmune diseases
Anti-TNF-α therapy is approved for use in autoimmune inflammatory conditions, such as rheumatoid arthritis, ankylosing spondylitis, inflammatory bowel disease, and psoriasis.165 These disorders are often associated with an increased risk of cardiovascular comorbidities, and the benefits of TNF-α inhibition, apart from anti-inflammatory efficacy and improved survival,166 are thought to include cardiovascular protection. Consequently, many studies have been conducted to investigate the vascular effects of TNF-α inhibition in these patients, and they have produced an abundance of controversial results.
In patients with rheumatoid arthritis, use of anti-TNF-α therapy (etanercept or infliximab) was shown to reduce the incidence of first cardiovascular events.167 Moreover, in rheumatoid arthritis patients with pre-existing cardiovascular disease, the risk of heart failure was significantly lower in anti-TNF-α-treated patients than in the remaining patients.168 Other studies reported an increased risk of developing heart failure, after adjustment for traditional cardiovascular risk factors, in patients who had a higher disease activity score at follow-up,169 and a slightly increased relative risk of heart failure in a large population of relatively young rheumatoid arthritis patients.170
Increased aortic stiffness, an independent predictor of cardiovascular mortality, is a common feature of inflammatory diseases. Several studies have demonstrated improved aortic stiffness in patients with inflammatory arthropathies treated by TNF-α antagonists,171–174 whereas carotid intima-media thickness was either reduced174 or remained stable.175 In contrast, arterial stiffness, lipid profiles, and other traditional cardiovascular risk factors were not improved after 6 and 12 months of anti-TNF-α therapy in patients with ankylosing spondylitis, although the treatment decreased their disease activity.176
Regarding endothelial function, flow-mediated dilatation in patients with inflammatory arthropathies is strongly impaired when compared with healthy controls.177,178 Anti-TNF-α treatment was shown to significantly improve endothelium-dependent vasodilation in patients with rheumatoid arthritis.175,179,180 Other studies in rheumatoid arthritis patients showed a significant but transient effect of infliximab infusion on endothelium-dependent vasodilatation, which returned to baseline within 2–4 weeks post-infusion.177,181 Beneficial effects on capillary recruitment and endothelium-dependent vasodilatation were also reported in ankylosing spondylitis patients treated either with etanercept,178 or with infliximab, which was furthermore shown to reduce nitrite concentrations.182 In contrast, Irace et al reported transient vasoconstriction and increased wall shear stress in the common carotid and brachial arteries following treatment with infliximab in patients with rheumatoid arthritis, with no effect on flow-mediated dilatation.183 Progression of atherosclerosis in psoriatic arthritis patients despite TNF-α blockade was also observed; these patients showed no recovery of flow-mediated dilatation after 2 years of anti-TNF-α treatment, and their serum TNF-α levels were increased at 2 years when compared with baseline.184 There was a significant progression in both mean intima-media thickness and mean maximum intima-media thickness, despite improvement in the disease score and the lipid profile.
The effects of systemic blockade of TNF-α on insulin resistance remain controversial. While some studies reported improved insulin sensitivity in infliximab-treated patients with rheumatoid arthritis,185,186 other authors found that insulin resistance was not alleviated by antagonism of TNF-α.187 Gonzalez-Gay et al188 suggested that the effects may depend on the degree and severity of insulin resistance before the start of therapy, whereby the beneficial effect is observed in patients with the most severe disease.
In obese insulin-resistant patients, no improvement in insulin sensitivity was observed following a single intravenous administration of soluble TNFR-Fc fusion protein.189 Similarly, no effects of systemic TNF-α blockade on vascular or metabolic insulin sensitivity were detected in either healthy obese patients190 or in obese patients with type 2 diabetes mellitus.191
The effects of direct TNF-α inhibition on lipid metabolism in patients with rheumatoid arthritis are similarly controversial: In two independent studies,183,192 adverse effects were observed, including elevated triglycerides and reduced high-density lipoprotein cholesterol levels. One further study in infliximab-treated patients with rheumatoid arthritis and ankylosing spondylitis showed no influence on the overall lipid profile.193 In contrast, several studies demonstrated increases in serum concentrations of high-density lipoprotein cholesterol upon treatment with infliximab.194–196 Similar beneficial effects of etanercept treatment on the lipid profile were observed in patients with ankylosing spondylitis.197 The detailed interplay of the proinflammatory activity of TNF-α with insulin resistance and lipid metabolism in the context of atherosclerosis and cardiometabolic disorders thus warrants further investigation.
Toward the next generation of anti-TNF-α therapies
The currently available TNF-α blockers neutralize the activity of soluble TNF-α and prevent its binding to both TNFR1 and TNFR2. Although these compounds are effective in the treatment of inflammatory arthritis, ankylosing spondylitis, and psoriasis, long-term use of TNF-α blockers can have adverse effects due to inhibition of the pleiotropic functions of TNF-α. Safety considerations here include an increased incidence of pulmonary and skin infections, new-onset autoimmune diseases, malignancies, and congestive heart failure, resulting from inhibition of the immunomodulatory effects of TNF-α.198 Since many of the pathological and protective signals diverge either at the ligand level (soluble versus membrane-bound TNF-α) or at the receptor level (TNFR1 or TNFR2), selective blockade of soluble TNF-α or of TNFR1 signaling has been postulated to be preferable to complete TNF-α blockade in the treatment of chronic inflammatory diseases.199,200
Based on the results obtained in animal models of myocardial infarction122 and rheumatoid arthritis,201 therapeutic strategies that target TNFR1 while sparing TNFR2 are expected to both inhibit inflammation and promote the activity of regulatory T-cells, and might be superior to systemic TNF-α blockade. Consequently, there is a growing interest in use of specific receptor-targeting compounds as anti-TNF-α therapeutic agents. These compounds are still in the research and development phase, but many recent studies in animal models of inflammatory disease indicate that increased therapeutic benefit and a reduced risk of side effects can be achieved by specific inhibition of soluble TNF or TNFR1, while leaving TNFR2 signaling fully functional. A variety of approaches to the pharmacological blockade of TNFR1 have been proposed thus far, among them monoclonal antibodies (atrosab)202 and antagonistic TNF-α mutants specific for TNFR1.203,204 Therapeutic administration of soluble TNFR1 PLAD (preligand-binding assembly domain) protein potently inhibited inflammatory arthritis in animal models.205 In addition to these inhibitory strategies, a short hairpin RNA directed against TNFR1 was successfully used to downregulate TNFR1 expression and to reduce experimental arthritis in vivo.206
Furthermore, several reports from the field of targeted drug delivery and nanomedicine appear to open promising possibilities for local or tissue-targeted TNF-α gene silencing. Two different topical delivery systems (cationic amphiphilic lipid particles207 and capsaicin-loaded cationic lipid-polymer hybrid nanoparticles)208 have recently demonstrated efficacy in local transport of anti-TNF-α small interfering (si)RNA and in the treatment of experimental psoriasis. Moreover, the problem of insufficient endothelial siRNA transfection efficiency in vivo has recently been overcome by Dahlman et al,209 who developed a polymeric nanoconstruct (7C1) capable of delivering siRNAs specifically to endothelial cells, which allowed concurrent silencing of multiple endothelial genes. This approach was successfully utilized for the purpose of RNA-based therapies, as demonstrated in animal models of various diseases involving endothelial dysfunction.209 It seems plausible that the combination of specific siRNAs with local delivery systems that allow blocking of TNF-α/TNFR1 signaling in vascular cells without affecting gene expression in other tissues or immune cells may prove a safe and efficient strategy to control cardiovascular inflammation in the future.
Conclusion
TNF-α-induced signaling plays an important role in the cellular response to inflammation and injury, being central to the development and homeostasis of the immune system. The protective mechanisms induced by TNF-α include the host defense against infections and its inhibitory role in tumorigenesis. However, persistent overproduction of TNF-α has deleterious effects on tissues by inducing a chronic inflammatory response. In the cardiovascular system, TNF-α-activated signal transduction pathways contribute to vascular dysfunction, atherogenesis, hypertension, and adverse cardiac remodeling after myocardial infarction. The existing clinical studies seem to support the notion that treatment with anti-TNF-α may have a favorable effect on endothelial dysfunction and atherosclerotic processes in patients with inflammatory arthropathies. However, an increased risk of heart failure, as a consequence of systemic TNF-α blockade, remains a major concern in patients with cardiovascular disease. Selective inhibition of TNFR1 with specific antibodies, antagonists, or siRNAs provides a promising opportunity to neutralize the proinflammatory, proatherogenic effects of TNF-α, while maintaining the cardioprotective immunomodulatory responses mediated by TNFR2. Further studies on TNF-α inhibition in atherosclerosis and cardiometabolic disease are therefore urgently needed in order to assess the safety and efficacy of novel therapeutic strategies focused on blocking the responses specific to TNFR1, as compared with global TNF-α inhibition.
Acknowledgments
This work was supported by the German Research Foundation (DFG CI 162/2-1), ELAN-Fonds of University Hospital Erlangen (research grant to KU), and by the EU “NanoAthero” project (FP7-NMP-2012-LARGE-6-309820). The authors would like to thank Margarete Goppelt-Struebe for critical reading and editing of the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci U S A. 1975;72(9):3666–3670. | |
Rich EA, Panuska JR, Wallis RS, Wolf CB, Leonard ML, Ellner JJ. Dyscoordinate expression of tumor necrosis factor-alpha by human blood monocytes and alveolar macrophages. Am Rev Respir Dis. 1989; 139(4):1010–1016. | |
Williamson BD, Carswell EA, Rubin BY, Prendergast JS, Old LJ. Human tumor necrosis factor produced by human B-cell lines: synergistic cytotoxic interaction with human interferon. Proc Natl Acad Sci U S A. 1983;80(17):5397–5401. | |
Neuhaus T, Totzke G, Gruenewald E, et al. Tumour necrosis factor-alpha gene expression and production in human umbilical arterial endothelial cells. Clin Sci. 2000;98(4):461–470. | |
von Asmuth EJ, Dentener MA, Ceska M, Buurman WA. IL-6, IL-8 and TNF production by cytokine and lipopolysaccharide-stimulated human renal cortical epithelial cells in vitro. Eur Cytokine Netw. 1994;5(3):301–310. | |
Warner SJ, Libby P. Human vascular smooth muscle cells. Target for and source of tumor necrosis factor. J Immunol. 1989;142(1):100–109. | |
Doyama K, Fujiwara H, Fukumoto M, et al. Tumour necrosis factor is expressed in cardiac tissues of patients with heart failure. Int J Cardiol. 1996;54(3):217–225. | |
Old LJ. Tumor necrosis factor (TNF). Science. 1985;230(4726):630–632. | |
Nedwin GE, Naylor SL, Sakaguchi AY, et al. Human lymphotoxin and tumor necrosis factor genes: structure, homology and chromosomal localization. Nucleic Acids Res. 1985;13(17):6361–6373. | |
Canault M, Peiretti F, Mueller C, et al. Proinflammatory properties of murine aortic endothelial cells exclusively expressing a non cleavable form of TNFalpha. Effect on tumor necrosis factor alpha receptor type 2. Thromb Haemost. 2004;92(6):1428–1437. | |
Black RA, Rauch CT, Kozlosky CJ, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997;385(6618):729–733. | |
Tartaglia LA, Weber RF, Figari IS, Reynolds C, Palladino MA Jr, Goeddel DV. The two different receptors for tumor necrosis factor mediate distinct cellular responses. Proc Natl Acad Sci U S A. 1991;88(20):929–9296. | |
Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104(4):487–501. | |
Theiss AL, Simmons JG, Jobin C, Lund PK. Tumor necrosis factor (TNF) alpha increases collagen accumulation and proliferation in intestinal myofibroblasts via TNF receptor 2. J Biol Chem. 2005;280(43):36099–36109. | |
Chen G, Goeddel DV. TNF-R1 signaling: a beautiful pathway. Science. 2002;296(5573):1634–1635. | |
Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10(1):45–65. | |
Gaur U, Aggarwal BB. Regulation of proliferation, survival and apoptosis by members of the TNF superfamily. Biochem Pharmacol. 2003;66(8):1403–1408. | |
Zhang R, Xu Y, Ekman N, et al. Etk/Bmx transactivates vascular endothelial growth factor 2 and recruits phosphatidylinositol 3-kinase to mediate the tumor necrosis factor-induced angiogenic pathway. J Biol Chem. 2003;278(51):51267–51276. | |
Chia S, Qadan M, Newton R, Ludlam CA, Fox KA, Newby DE. Intra-arterial tumor necrosis factor-alpha impairs endothelium-dependent vasodilatation and stimulates local tissue plasminogen activator release in humans. Arterioscler Thromb Vasc Biol. 2003;23(4):695–701. | |
Nakamura M, Yoshida H, Arakawa N, Saitoh S, Satoh M, Hiramori K. Effects of tumor necrosis factor-alpha on basal and stimulated endothelium-dependent vasomotion in human resistance vessel. J Cardiovasc Pharmacol. 2000;36(4):487–492. | |
Fang WR, Wei J, Han D, et al. MC-002 exhibits positive effects against platelets aggregation and endothelial dysfunction through thromboxane A(2) inhibition. Thromb Res. 2014;133(4):610–615. | |
Muzaffar S, Shukla N, Massey Y, Angelini GD, Jeremy JY. NADPH oxidase 1 mediates upregulation of thromboxane A(2) synthase in human vascular smooth muscle cells: inhibition with iloprost. Eur J Pharmacol. 2011;658(2–3):187–192. | |
Goetze S, Kintscher U, Kaneshiro K, et al. TNF alpha induces expression of transcription factors c-FOS, EGR-1, and ETS-1 in vascular lesions through extracellular signal-regulated kinases 1/2. Atherosclerosis. 2001;159(1):93–101. | |
Goldblum SE, Ding X, Campbell-Washington J. TNF-alpha induces endothelial cell F-actin depolymerization, new actin synthesis, and barrier dysfunction. Am J Physiol. 1993;264(4 Pt 1):C894–C905. | |
Petrache I, Birukova A, Ramirez SI, Garcia JG, Verin AD. The role of the microtubules in tumor necrosis factor-alpha-induced endothelial cell permeability. Am J Respir Cell Mol Biol. 2003;28(5):574–581. | |
Goldblum SE, Sun WL. Tumor necrosis factor-alpha augments pulmonary arterial transendothelial albumin flux in vitro. Am J Physiol. 1990;258(2 Pt 1):L57–L67. | |
Angelini DJ, Hyun SW, Grigoryev DN, et al. TNF-alpha increases tyrosine phosphorylation of vascular endothelial cadherin and opens the paracellular pathway through fyn activation in human lung endothelia. Am J Physiol Lung Cell Mol Physiol. 2006;291(6):L1232–L1245. | |
Chappell D, Hofmann-Kiefer K, Jacob M, et al. TNF-alpha induced shedding of the endothelial glycocalyx is prevented by hydrocortisone and antithrombin. Basic Res Cardiol. 2009;104(1):78–89. | |
Zhang Y, Yang X, Bian F, et al. TNF-alpha promotes early atherosclerosis by increasing transcytosis of LDL across endothelial cells: crosstalk between NF-kappaB and PPAR-gamma. J Mol Cell Cardiol. 2014;72:85–94. | |
Gao X, Belmadani S, Picchi A, et al. Tumor necrosis factor-alpha induces endothelial dysfunction in Lepr(db) mice. Circulation. 2007; 115(2):245–254. | |
Picchi A, Gao X, Belmadani S, et al. Tumor necrosis factor-alpha induces endothelial dysfunction in the prediabetic metabolic syndrome. Circ Res. 2006;99(1):69–77. | |
Csiszar A, Labinskyy N, Smith K, Rivera A, Orosz Z, Ungvari Z. Vasculoprotective effects of anti-tumor necrosis factor-alpha treatment in aging. Am J Pathol. 2007;170(1):388–398. | |
Wimalasundera R, Fexby S, Regan L, Thom SA, Hughes AD. Effect of tumour necrosis factor-alpha and interleukin 1beta on endothelium-dependent relaxation in rat mesenteric resistance arteries in vitro. Br J Pharmacol. 2003;138(7):1285–1294. | |
Goodwin BL, Pendleton LC, Levy MM, Solomonson LP, Eichler DC. Tumor necrosis factor-alpha reduces argininosuccinate synthase expression and nitric oxide production in aortic endothelial cells. Am J Physiol Heart Circ Physiol. 2007;293(2):H1115–H1121. | |
Greenberg S, Xie J, Wang Y, et al. Tumor necrosis factor-alpha inhibits endothelium-dependent relaxation. J Appl Physiol. 1993;74(5):2394–2403. | |
Agnoletti L, Curello S, Bachetti T, et al. Serum from patients with severe heart failure downregulates eNOS and is proapoptotic: role of tumor necrosis factor-alpha. Circulation. 1999;100(19):1983–1991. | |
Neumann P, Gertzberg N, Johnson A. TNF-alpha induces a decrease in eNOS promoter activity. Am J Physiol Lung Cell Mol Physiol. 2004;286(2):L452–L459. | |
Yoshizumi M, Perrella MA, Burnett JC Jr, Lee ME. Tumor necrosis factor downregulates an endothelial nitric oxide synthase mRNA by shortening its half-life. Circ Res. 1993;73(1):205–209. | |
Valerio A, Cardile A, Cozzi V, et al. TNF-alpha downregulates eNOS expression and mitochondrial biogenesis in fat and muscle of obese rodents. J Clin Invest. 2006;116(10):2791–2798. | |
Ito A, Tsao PS, Adimoolam S, Kimoto M, Ogawa T, Cooke JP. Novel mechanism for endothelial dysfunction: dysregulation of dimethylarginine dimethylaminohydrolase. Circulation. 1999;99(24):3092–3095. | |
Didion SP, Ryan MJ, Didion LA, Fegan PE, Sigmund CD, Faraci FM. Increased superoxide and vascular dysfunction in CuZnSOD-deficient mice. Circ Res. 2002;91(10):938–944. | |
Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res. 2000;86(5):494–501. | |
De Keulenaer GW, Alexander RW, Ushio-Fukai M, Ishizaka N, Griendling KK. Tumour necrosis factor alpha activates a p22phox-based NADH oxidase in vascular smooth muscle. Biochem J. 1998;329 Pt 3:653–657. | |
Mazor R, Itzhaki O, Sela S, et al. Tumor necrosis factor-alpha: a possible priming agent for the polymorphonuclear leukocyte-reduced nicotinamide-adenine dinucleotide phosphate oxidase in hypertension. Hypertension. 2010;55(2):353–362. | |
White CR, Darley-Usmar V, Berrington WR, et al. Circulating plasma xanthine oxidase contributes to vascular dysfunction in hypercholesterolemic rabbits. Proc Natl Acad Sci U S A. 1996;93(16):8745–8749. | |
Vasquez-Vivar J, Kalyanaraman B, Martasek P, et al. Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci U S A. 1998;95(16):9220–9225. | |
Yoshida LS, Tsunawaki S. Expression of NADPH oxidases and enhanced H(2)O(2)-generating activity in human coronary artery endothelial cells upon induction with tumor necrosis factor-alpha. Int Immunopharmacol. 2008;8(10):1377–1385. | |
Pandey D, Patel A, Patel V, et al. Expression and functional significance of NADPH oxidase 5 (Nox5) and its splice variants in human blood vessels. Am J Physiol Heart Circ Physiol. 2012;302(10):H1919–H1928. | |
Chen XL, Zhang Q, Zhao R, Medford RM. Superoxide, H2O2, and iron are required for TNF-alpha-induced MCP-1 gene expression in endothelial cells: role of Rac1 and NADPH oxidase. Am J Physiol Heart Circ Physiol. 2004;286(3):H1001–H1007. | |
Zhu H, Shan L, Peng T. Rac1 mediates sex difference in cardiac tumor necrosis factor-alpha expression via NADPH oxidase-ERK1/2/p38 MAPK pathway in endotoxemia. J Mol Cell Cardiol. 2009;47(2):264–274. | |
Li JM, Fan LM, Christie MR, Shah AM. Acute tumor necrosis factor alpha signaling via NADPH oxidase in microvascular endothelial cells: role of p47phox phosphorylation and binding to TRAF4. Mol Cell Biol. 2005;25(6):2320–2330. | |
Xu YC, Wu RF, Gu Y, et al. Involvement of TRAF4 in oxidative activation of c-Jun N-terminal kinase. J Biol Chem. 2002;277(31):28051–28057. | |
Gertzberg N, Neumann P, Rizzo V, Johnson A. NAD(P)H oxidase mediates the endothelial barrier dysfunction induced by TNF-alpha. Am J Physiol Lung Cell Mol Physiol. 2004;286(1):L37–L48. | |
Neumann P, Gertzberg N, Vaughan E, et al. Peroxynitrite mediates TNF-alpha-induced endothelial barrier dysfunction and nitration of actin. Am J Physiol Lung Cell Mol Physiol. 2006;290(4):L674–L684. | |
Bergh N, Ulfhammer E, Glise K, Jern S, Karlsson L. Influence of TNF-alpha and biomechanical stress on endothelial anti- and prothrombotic genes. Biochem Biophys Res Commun. 2009;385(3):314–318. | |
Xanthoulea S, Thelen M, Pottgens C, Gijbels MJ, Lutgens E, de Winther MP. Absence of p55 TNF receptor reduces atherosclerosis, but has no major effect on angiotensin II induced aneurysms in LDL receptor deficient mice. PLoS One. 2009;4(7):e6113. | |
Pober JS. Activation and injury of endothelial cells by cytokines. Pathol Biol (Paris). 1998;46(3):159–163. | |
Deisher TA, Haddix TL, Montgomery KF, Pohlman TH, Kaushansky K, Harlan JM. The role of protein kinase C in the induction of VCAM-1 expression on human umbilical vein endothelial cells. FEBS Lett. 1993;331(3):285–290. | |
Rajan S, Ye J, Bai S, Huang F, Guo YL. NF-kappaB, but not p38 MAP kinase, is required for TNF-alpha-induced expression of cell adhesion molecules in endothelial cells. J Cell Biochem. 2008;105(2):477–486. | |
Zhou Z, Connell MC, MacEwan DJ. TNFR1-induced NF-kappaB, but not ERK, p38MAPK or JNK activation, mediates TNF-induced ICAM-1 and VCAM-1 expression on endothelial cells. Cell Signal. 2007;19(6):1238–1248. | |
Cavender D, Saegusa Y, Ziff M. Stimulation of endothelial cell binding of lymphocytes by tumor necrosis factor. J Immunol. 1987;139(6):1855–1860. | |
Chandrasekharan UM, Siemionow M, Unsal M, et al. Tumor necrosis factor alpha (TNF-alpha) receptor-II is required for TNF-alpha-induced leukocyte-endothelial interaction in vivo. Blood. 2007;109(5):1938–1944. | |
Urschel K, Worner A, Daniel WG, Garlichs CD, Cicha I. Role of shear stress patterns in the TNF-alpha-induced atherogenic protein expression and monocytic cell adhesion to endothelium. Clin Hemorheol Microcirc. 2010;46(2–3):203–210. | |
Dimmeler S, Haendeler J, Rippmann V, Nehls M, Zeiher AM. Shear stress inhibits apoptosis of human endothelial cells. FEBS Lett. 1996; 399(1–2):71–74. | |
Hoffmann J, Haendeler J, Aicher A, et al. Aging enhances the sensitivity of endothelial cells toward apoptotic stimuli: important role of nitric oxide. Circ Res. 2001;89(8):709–715. | |
Dimmeler S, Hermann C, Galle J, Zeiher AM. Upregulation of superoxide dismutase and nitric oxide synthase mediates the apoptosis-suppressive effects of shear stress on endothelial cells. Arterioscler Thromb Vasc Biol. 1999;19(3):656–664. | |
Garin G, Abe J, Mohan A, et al. Flow antagonizes TNF-alpha signaling in endothelial cells by inhibiting caspase-dependent PKC zeta processing. Circ Res. 2007;101(1):97–105. | |
Lerner-Marmarosh N, Yoshizumi M, Che W, et al. Inhibition of tumor necrosis factor-[alpha]-induced SHP-2 phosphatase activity by shear stress: a mechanism to reduce endothelial inflammation. Arterioscler Thromb Vasc Biol. 2003;23(10):1775–1781. | |
Yamawaki H, Lehoux S, Berk BC. Chronic physiological shear stress inhibits tumor necrosis factor-induced proinflammatory responses in rabbit aorta perfused ex vivo. Circulation. 2003;108(13):1619–1625. | |
Partridge J, Carlsen H, Enesa K, et al. Laminar shear stress acts as a switch to regulate divergent functions of NF-kappaB in endothelial cells. FASEB J. 2007;21(13):3553–3561. | |
Harry BL, Sanders JM, Feaver RE, et al. Endothelial cell PECAM-1 promotes atherosclerotic lesions in areas of disturbed flow in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 2008;28(11):2003–2008. | |
Hajra L, Evans AI, Chen M, Hyduk SJ, Collins T, Cybulsky MI. The NF-kappa B signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc Natl Acad Sci U S A. 2000;97(16):9052–9057. | |
Zakkar M, Chaudhury H, Sandvik G, et al. Increased endothelial mitogen-activated protein kinase phosphatase-1 expression suppresses proinflammatory activation at sites that are resistant to atherosclerosis. Circ Res. 2008;103(7):726–732. | |
Cicha I, Beronov K, Ramirez EL, et al. Shear stress preconditioning modulates endothelial susceptibility to circulating TNF-alpha and monocytic cell recruitment in a simplified model of arterial bifurcations. Atherosclerosis. 2009;207(1):93–102. | |
Haldenby KA, Chappell DC, Winlove CP, Parker KH, Firth JA. Focal and regional variations in the composition of the glycocalyx of large vessel endothelium. J Vasc Res. 1994;31(1):2–9. | |
van den Berg BM, Nieuwdorp M, Stroes ES, Vink H. Glycocalyx and endothelial (dys) function: from mice to men. Pharmacol Rep. 2006; 58 Suppl:75–80. | |
Csiszar A, Ungvari Z, Koller A, Edwards JG, Kaley G. Proinflammatory phenotype of coronary arteries promotes endothelial apoptosis in aging. Physiol Genomics. 2004;17(1):21–30. | |
Liang YJ, Li X, Zhang XJ, et al. Elevated levels of plasma TNF-alpha are associated with microvascular endothelial dysfunction in patients with sepsis through activating the NF-B-k and p38 mitogen-activated protein kinase in endothelial cells. Shock. 2014;41(4):275–281. | |
Vincent MA, Barrett EJ, Lindner JR, Clark MG, Rattigan S. Inhibiting NOS blocks microvascular recruitment and blunts muscle glucose uptake in response to insulin. Am J Physiol Endocrinol Metab. 2003; 285(1):E123–E129. | |
Zhang L, Wheatley CM, Richards SM, Barrett EJ, Clark MG, Rattigan S. TNF-alpha acutely inhibits vascular effects of physiological but not high insulin or contraction. Am J Physiol Endocrinol Metab. 2003; 285(3):E654–E660. | |
Li G, Barrett EJ, Barrett MO, Cao W, Liu Z. Tumor necrosis factor-alpha induces insulin resistance in endothelial cells via a p38 mitogen-activated protein kinase-dependent pathway. Endocrinology. 2007;148(7):3356–3363. | |
Wang H, Wang AX, Liu Z, Barrett EJ. Insulin signaling stimulates insulin transport by bovine aortic endothelial cells. Diabetes. 2008;57(3):540–547. | |
Goetze S, Xi XP, Kawano Y, et al. TNF-alpha-induced migration of vascular smooth muscle cells is MAPK dependent. Hypertension. 1999;33(1 Pt 2):183–189. | |
Secchiero P, Zerbinati C, Rimondi E, et al. TRAIL promotes the survival, migration and proliferation of vascular smooth muscle cells. Cell Mol Life Sci. 2004;61(15):1965–1974. | |
Xu S, He Y, Vokurkova M, Touyz RM. Endothelial cells negatively modulate reactive oxygen species generation in vascular smooth muscle cells: role of thioredoxin. Hypertension. 2009;54(2):427–433. | |
Li H, Liang J, Castrillon DH, DePinho RA, Olson EN, Liu ZP. FoxO4 regulates tumor necrosis factor alpha-directed smooth muscle cell migration by activating matrix metalloproteinase 9 gene transcription. Mol Cell Biol. 2007;27(7):2676–2686. | |
Knapp AC, Huang J, Starling G, Kiener PA. Inhibitors of HMG-CoA reductase sensitize human smooth muscle cells to Fas-ligand and cytokine-induced cell death. Atherosclerosis. 2000;152(1):217–227. | |
Niemann-Jonsson A, Ares MP, Yan ZQ, et al. Increased rate of apoptosis in intimal arterial smooth muscle cells through endogenous activation of TNF receptors. Arterioscler Thromb Vasc Biol. 2001;21(12):1909–1914. | |
Aker S, Belosjorow S, Konietzka I, et al. Serum but not myocardial TNF-alpha concentration is increased in pacing-induced heart failure in rabbits. Am J Physiol Regul Integr Comp Physiol. 2003;285(2):R463–R469. | |
Arras M, Hoche A, Bohle R, Eckert P, Riedel W, Schaper J. Tumor necrosis factor-alpha in macrophages of heart, liver, kidney, and in the pituitary gland. Cell Tissue Res. 1996;285(1):39–49. | |
Frangogiannis NG, Lindsey ML, Michael LH, et al. Resident cardiac mast cells degranulate and release preformed TNF-alpha, initiating the cytokine cascade in experimental canine myocardial ischemia/reperfusion. Circulation. 1998;98(7):699–710. | |
Al-Lamki RS, Brookes AP, Wang J, et al. TNF receptors differentially signal and are differentially expressed and regulated in the human heart. Am J Transplant. 2009;9(12):2679–2696. | |
Yokoyama T, Vaca L, Rossen RD, Durante W, Hazarika P, Mann DL. Cellular basis for the negative inotropic effects of tumor necrosis factor-alpha in the adult mammalian heart. J Clin Invest. 1993;92(5):2303–2312. | |
Bozkurt B, Kribbs SB, Clubb FJ Jr, et al. Pathophysiologically relevant concentrations of tumor necrosis factor-alpha promote progressive left ventricular dysfunction and remodeling in rats. Circulation. 1998; 97(14):1382–1391. | |
Grandel U, Fink L, Blum A, et al. Endotoxin-induced myocardial tumor necrosis factor-alpha synthesis depresses contractility of isolated rat hearts: evidence for a role of sphingosine and cyclooxygenase-2-derived thromboxane production. Circulation. 2000;102(22):2758–2764. | |
Murray DR, Freeman GL. Tumor necrosis factor-alpha induces a biphasic effect on myocardial contractility in conscious dogs. Circ Res. 1996;78(1):154–160. | |
Dorge H, Schulz R, Belosjorow S, et al. Coronary microembolization: the role of TNF-alpha in contractile dysfunction. J Mol Cell Cardiol. 2002;34(1):51–62. | |
Friedrichs GS, Swillo RE, Jow B, et al. Sphingosine modulates myocyte electrophysiology, induces negative inotropy, and decreases survival after myocardial ischemia. J Cardiovasc Pharmacol. 2002;39(1):18–28. | |
Thielmann M, Dorge H, Martin C, et al. Myocardial dysfunction with coronary microembolization: signal transduction through a sequence of nitric oxide, tumor necrosis factor-alpha, and sphingosine. Circ Res. 2002;90(7):807–813. | |
Skyschally A, Gres P, van Caster P, et al. Reduced calcium responsiveness characterizes contractile dysfunction following coronary microembolization. Basic Res Cardiol. 2008;103(6):552–559. | |
Tsai CT, Wu CK, Lee JK, et al. TNF-alpha down-regulates sarcoplasmic reticulum Ca2+ ATPase expression and leads to left ventricular diastolic dysfunction through binding of NF-kappaB to promoter response element. Cardiovasc Res. 2015;105(3):318–329. | |
Duncan DJ, Yang Z, Hopkins PM, Steele DS, Harrison SM. TNF-alpha and IL-1beta increase Ca2+ leak from the sarcoplasmic reticulum and susceptibility to arrhythmia in rat ventricular myocytes. Cell Calcium. 2010;47(4):378–386. | |
Fauconnier J, Meli AC, Thireau J, et al. Ryanodine receptor leak mediated by caspase-8 activation leads to left ventricular injury after myocardial ischemia-reperfusion. Proc Natl Acad Sci U S A. 2011; 108(32):13258–13263. | |
Kumar A, Paladugu B, Mensing J, Kumar A, Parrillo JE. Nitric oxide-dependent and -independent mechanisms are involved in TNF-alpha-induced depression of cardiac myocyte contractility. Am J Physiol Regul Integr Comp Physiol. 2007;292(5):R1900–R1906. | |
Higuchi Y, Otsu K, Nishida K, et al. Involvement of reactive oxygen species-mediated NF-kappa B activation in TNF-alpha-induced cardiomyocyte hypertrophy. J Mol Cell Cardiol. 2002;34(2):233–240. | |
Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336(15):1066–1071. | |
Nakamura K, Fushimi K, Kouchi H, et al. Inhibitory effects of antioxidants on neonatal rat cardiac myocyte hypertrophy induced by tumor necrosis factor-alpha and angiotensin II. Circulation. 1998;98(8):794–799. | |
Funakoshi H, Kubota T, Machida Y, et al. Involvement of inducible nitric oxide synthase in cardiac dysfunction with tumor necrosis factor-alpha. Am J Physiol Heart Circ Physiol. 2002;282(6):H2159–H2166. | |
Moe K, Khairunnisa K, Yin N, Chin-Dusting J, Wong P, Wong M. Tumor necrosis factor-α-induced nuclear factor-kappaB activation in human cardiomyocytes is mediated by NADPH oxidase. J Physiol Biochem. 2014;70(3):769–779. | |
Funakoshi H, Zacharia LC, Tang Z, et al. A1 adenosine receptor upregulation accompanies decreasing myocardial adenosine levels in mice with left ventricular dysfunction. Circulation. 2007;115(17):2307–2315. | |
Krown KA, Page MT, Nguyen C, et al. Tumor necrosis factor alpha-induced apoptosis in cardiac myocytes. Involvement of the sphingolipid signaling cascade in cardiac cell death. J Clin Invest. 1996;98(12):2854–2865. | |
Herskowitz A, Choi S, Ansari AA, Wesselingh S. Cytokine mRNA expression in postischemic/reperfused myocardium. Am J Pathol. 1995;146(2):419–428. | |
Maekawa N, Wada H, Kanda T, et al. Improved myocardial ischemia/reperfusion injury in mice lacking tumor necrosis factor-alpha. J Am Coll Cardiol. 2002;39(7):1229–1235. | |
Dawn B, Guo Y, Rezazadeh A, et al. Tumor necrosis factor-alpha does not modulate ischemia/reperfusion injury in naive myocardium but is essential for the development of late preconditioning. J Mol Cell Cardiol. 2004;37(1):51–61. | |
Flaherty MP, Guo Y, Tiwari S, et al. The role of TNF-alpha receptors p55 and p75 in acute myocardial ischemia/reperfusion injury and late preconditioning. J Mol Cell Cardiol. 2008;45(6):735–741. | |
Labruto F, Yang J, Vaage J, Valen G. Role of tumor necrosis factor alpha and its receptor I in preconditioning by hyperoxia. Basic Res Cardiol. 2005;100(3):198–207. | |
Sun M, Dawood F, Wen WH, et al. Excessive tumor necrosis factor activation after infarction contributes to susceptibility of myocardial rupture and left ventricular dysfunction. Circulation. 2004;110(20):3221–3228. | |
Monden Y, Kubota T, Inoue T, et al. Tumor necrosis factor-alpha is toxic via receptor 1 and protective via receptor 2 in a murine model of myocardial infarction. Am J Physiol Heart Circ Physiol. 2007;293(1):H743–H753. | |
Ramani R, Mathier M, Wang P, et al. Inhibition of tumor necrosis factor receptor-1-mediated pathways has beneficial effects in a murine model of postischemic remodeling. Am J Physiol Heart Circ Physiol. 2004;287(3):H1369–H1377. | |
Kurrelmeyer KM, Michael LH, Baumgarten G, et al. Endogenous tumor necrosis factor protects the adult cardiac myocyte against ischemic-induced apoptosis in a murine model of acute myocardial infarction. Proc Natl Acad Sci U S A. 2000;97(10):5456–5461. | |
Berthonneche C, Sulpice T, Boucher F, et al. New insights into the pathological role of TNF-alpha in early cardiac dysfunction and subsequent heart failure after infarction in rats. Am J Physiol Heart Circ Physiol. 2004;287(1):H340–H350. | |
Zhang YQ, Zhao JL, Lau WB, et al. Tumor necrosis factor-alpha and lymphotoxin-alpha mediate myocardial ischemic injury via TNF receptor 1, but are cardioprotective when activating TNF receptor 2. PLoS One. 2013;8(5). | |
Hayashidani S, Tsutsui H, Ikeuchi M, et al. Targeted deletion of MMP-2 attenuates early LV rupture and late remodeling after experimental myocardial infarction. Am J Physiol Heart Circ Physiol. 2003;285(3):H1229–H1235. | |
Hamid T, Gu Y, Ortines RV, et al. Divergent tumor necrosis factor receptor-related remodeling responses in heart failure: role of nuclear factor-kappaB and inflammatory activation. Circulation. 2009; 119(10):1386–1397. | |
Gulick T, Chung MK, Pieper SJ, Lange LG, Schreiner GF. Interleukin 1 and tumor necrosis factor inhibit cardiac myocyte beta-adrenergic responsiveness. Proc Natl Acad Sci U S A. 1989; 86(17):6753–6757. | |
Moe GW, Marin-Garcia J, Konig A, Goldenthal M, Lu X, Feng Q. In vivo TNF-alpha inhibition ameliorates cardiac mitochondrial dysfunction, oxidative stress, and apoptosis in experimental heart failure. Am J Physiol Heart Circ Physiol. 2004;287(4):H1813–H1820. | |
Patten M, Kramer E, Bunemann J, et al. Endotoxin and cytokines alter contractile protein expression in cardiac myocytes in vivo. Pflugers Arch. 2001;442(6):920–927. | |
Zhu J, Liu M, Kennedy RH, Liu SJ. TNF-alpha-induced impairment of mitochondrial integrity and apoptosis mediated by caspase-8 in adult ventricular myocytes. Cytokine. 2006;34(1–2):96–105. | |
Awad AE, Kandalam V, Chakrabarti S, et al. Tumor necrosis factor induces matrix metalloproteinases in cardiomyocytes and cardiofibroblasts differentially via superoxide production in a PI3Kgamma-dependent manner. Am J Physiol Cell Physiol. 2010;298(3):C679–C692. | |
Jobe LJ, Melendez GC, Levick SP, Du Y, Brower GL, Janicki JS. TNF-alpha inhibition attenuates adverse myocardial remodeling in a rat model of volume overload. Am J Physiol Heart Circ Physiol. 2009;297(4):H1462–H1468. | |
Eichacker PQ, Hoffman WD, Farese A, et al. TNF but not IL-1 in dogs causes lethal lung injury and multiple organ dysfunction similar to human sepsis. J Appl Physiol. 1991;71(5):1979–1989. | |
Cauwels A, Brouckaert P. Survival of TNF toxicity: Dependence on caspases and NO. Arch Biochem Biophys. 2007;462(2):132–139. | |
Ito H, Ohshima A, Tsuzuki M, et al. Association of serum tumour necrosis factor-alpha with serum low-density lipoprotein-cholesterol and blood pressure in apparently healthy Japanese women. Clin Exp Pharmacol Physiol. 2001;28(3):188–192. | |
Bautista LE, Vera LM, Arenas IA, Gamarra G. Independent association between inflammatory markers (C-reactive protein, interleukin-6, and TNF-alpha) and essential hypertension. J Hum Hypertens. 2005;19(2):149–154. | |
Chen LS, Cuddy MP, LaVallette LA. Regulation of human renin gene promoter activity: a new negative regulatory region determines the responsiveness to TNF alpha. Kidney Int. 1998;54(6):2045–2055. | |
Todorov V, Muller M, Schweda F, Kurtz A. Tumor necrosis factor-alpha inhibits renin gene expression. Am J Physiol Regul Integr Comp Physiol. 2002;283(5):R1046–R1051. | |
Satou R, Miyata K, Katsurada A, Navar LG, Kobori H. Tumor necrosis factor-alpha suppresses angiotensinogen expression through formation of a p50/p50 homodimer in human renal proximal tubular cells. Am J Physiol Cell Physiol. 2010;299(4):C750–C759. | |
Brasier AR, Li JY, Wimbish KA. Tumor necrosis factor activates angiotensinogen gene expression by the Rel A transactivator. Hypertension. 1996;27(4):1009–1017. | |
Flesch M, Hoper A, Dell’Italia L, et al. Activation and functional significance of the renin- angiotensin system in mice with cardiac restricted overexpression of tumor necrosis factor. Circulation. 2003; 108(5):598–604. | |
Sriramula S, Haque M, Majid DS, Francis J. Involvement of tumor necrosis factor-alpha in angiotensin II-mediated effects on salt appetite, hypertension, and cardiac hypertrophy. Hypertension. 2008;51(5):1345–1351. | |
Tran LT, MacLeod KM, McNeill JH. Chronic etanercept treatment prevents the development of hypertension in fructose-fed rats. Mol Cell Biochem. 2009;330(1–2):219–228. | |
Zhang JD, Patel MB, Griffiths R, et al. Tumor necrosis factor-alpha produced in the kidney contributes to angiotensin II-dependent hypertension. Hypertension. 2014;64(6):U1275–U1246. | |
Sury MD, Frese-Schaper M, Muhlemann MK, et al. Evidence that N-acetylcysteine inhibits TNF-alpha-induced cerebrovascular endothelin-1 upregulation via inhibition of mitogen- and stress-activated protein kinase. Free Radic Biol Med. 2006;41(9):1372–1383. | |
Ohkita M, Takaoka M, Shiota Y, Nojiri R, Matsumura Y. Nitric oxide inhibits endothelin-1 production through the suppression of nuclear factor kappa B. Clin Sci. 2002;103 Suppl 48:68S–71S. | |
Zhang W, Li XJ, Zeng X, et al. Activation of nuclear factor-kappaB pathway is responsible for tumor necrosis factor-alpha-induced up-regulation of endothelin B2 receptor expression in vascular smooth muscle cells in vitro. Toxicol Lett. 2012;209(2):107–112. | |
Stenman E, Malmsjo M, Uddman E, Gido G, Wieloch T, Edvinsson L. Cerebral ischemia upregulates vascular endothelin ET(B) receptors in rat. Stroke. 2002;33(9):2311–2316. | |
Xu CB, Sun Y, Edvinsson L. Cardiovascular risk factors regulate the expression of vascular endothelin receptors. Pharmacol Ther. 2010; 127(2):148–155. | |
Lind H, Adner M, Erlinge D, Brunkwall J, Edvinsson L. Selective increase of the contractile response to endothelin-1 in subcutaneous arteries from patients with essential hypertension. Blood Press. 1999; 8(1):9–15. | |
Bauer M, Wilkens H, Langer F, Schneider SO, Lausberg H, Schafers HJ. Selective upregulation of endothelin B receptor gene expression in severe pulmonary hypertension. Circulation. 2002;105(9):1034–1036. | |
Dimitrijevic I, Edvinsson ML, Chen Q, Malmsjo M, Kimblad PO, Edvinsson L. Increased expression of vascular endothelin type B and angiotensin type 1 receptors in patients with ischemic heart disease. BMC Cardiovasc Disord. 2009;9:40. | |
Eringa EC, Stehouwer CD, Walburg K, et al. Physiological concentrations of insulin induce endothelin-dependent vasoconstriction of skeletal muscle resistance arteries in the presence of tumor necrosis factor-alpha dependence on c-Jun N-terminal kinase. Arterioscler Thromb Vasc Biol. 2006;26(2):274–280. | |
Canault M, Peiretti F, Mueller C, et al. Exclusive expression of transmembrane TNF-alpha in mice reduces the inflammatory response in early lipid lesions of aortic sinus. Atherosclerosis. 2004;172(2):211–218. | |
Canault M, Peiretti F, Poggi M, et al. Progression of atherosclerosis in ApoE-deficient mice that express distinct molecular forms of TNF-alpha. J Pathol. 2008;214(5):574–583. | |
Branen L, Hovgaard L, Nitulescu M, Bengtsson E, Nilsson J, Jovinge S. Inhibition of tumor necrosis factor-alpha reduces atherosclerosis in apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol. 2004; 24(11):2137–2142. | |
Ohta H, Wada H, Niwa T, et al. Disruption of tumor necrosis factor-alpha gene diminishes the development of atherosclerosis in ApoE-deficient mice. Atherosclerosis. 2005;180(1):11–17. | |
Zhou Z, Lauer MA, Wang K, et al. Effect of anti-tumor necrosis factor-alpha polyclonal antibody on restenosis after balloon angioplasty in a rabbit atherosclerotic model. Atherosclerosis. 2002;161(1):153–159. | |
Jiang Y, Zhang Q, Ye EA, Steinle JJ. Etanercept restores normal insulin signal transduction in beta2-adrenergic receptor knockout mice. J Neuroinflammation. 2014;11:137. | |
Toufektsian MC, Robbez-Masson V, Sanou D, et al. A single intravenous sTNFR-Fc administration at the time of reperfusion limits infarct size – implications in reperfusion strategies in man. Cardiovasc Drugs Ther. 2008;22(6):437–442. | |
Li X, Moody MR, Engel D, et al. Cardiac-specific overexpression of tumor necrosis factor-alpha causes oxidative stress and contractile dysfunction in mouse diaphragm. Circulation. 2000;102(14):1690–1696. | |
Iversen PO, Nicolaysen G, Sioud M. DNA enzyme targeting TNF-alpha mRNA improves hemodynamic performance in rats with postinfarction heart failure. Am J Physiol Heart Circ Physiol. 2001;281(5):H2211–H2217. | |
Mann DL, Deswal A, Bozkurt B, Torre-Amione G. New therapeutics for chronic heart failure. Annu Rev Med. 2002;53:59–74. | |
Mann DL, McMurray JJ, Packer M, et al. Targeted anticytokine therapy in patients with chronic heart failure: results of the Randomized Etanercept Worldwide Evaluation (RENEWAL). Circulation. 2004;109(13):1594–1602. | |
Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT; Anti-TNF Therapy Against Congestive Heart Failure Investigators. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation. 2003;107(25):3133–3140. | |
Mann DL, Bozkurt B, Torre-Amione G, Soran OZ, Sivasubramanian N. Effect of the soluble TNF-antagonist etanercept on tumor necrosis factor bioactivity and stability. Clin Transl Sci. 2008;1(2):142–145. | |
Karampetsou MP, Liossis SN, Sfikakis PP. TNF-alpha antagonists beyond approved indications: stories of success and prospects for the future. QJM. 2010;103(12):917–928. | |
Marchesoni A, Zaccara E, Gorla R, et al. TNF-alpha antagonist survival rate in a cohort of rheumatoid arthritis patients observed under conditions of standard clinical practice. Ann N Y Acad Sci. 2009;1173:837–846. | |
Jacobsson LT, Turesson C, Gulfe A, et al. Treatment with tumor necrosis factor blockers is associated with a lower incidence of first cardiovascular events in patients with rheumatoid arthritis. J Rheumatol. 2005;32(7):1213–1218. | |
Wolfe F, Michaud K. Heart failure in rheumatoid arthritis: rates, predictors, and the effect of anti-tumor necrosis factor therapy. Am J Med. 2004;116(5):305–311. | |
Listing J, Strangfeld A, Kekow J, et al. Does tumor necrosis factor alpha inhibition promote or prevent heart failure in patients with rheumatoid arthritis? Arthritis Rheum. 2008;58(3):667–677. | |
Curtis JR, Kramer JM, Martin C, et al. Heart failure among younger rheumatoid arthritis and Crohn’s patients exposed to TNF-alpha antagonists. Rheumatology. 2007;46(11):1688–1693. | |
Maki-Petaja KM, Hall FC, Booth AD, et al. Rheumatoid arthritis is associated with increased aortic pulse-wave velocity, which is reduced by anti-tumor necrosis factor-alpha therapy. Circulation. 2006;114(11):1185–1192. | |
Wong M, Oakley SP, Young L, et al. Infliximab improves vascular stiffness in patients with rheumatoid arthritis. Ann Rheum Dis. 2009;68(8):1277–1284. | |
Angel K, Provan SA, Gulseth HL, Mowinckel P, Kvien TK, Atar D. Tumor necrosis factor-alpha antagonists improve aortic stiffness in patients with inflammatory arthropathies: a controlled study. Hypertension. 2010;55(2):333–338. | |
Angel K, Provan SA, Fagerhol MK, Mowinckel P, Kvien TK, Atar D. Effect of 1-year anti-TNF-alpha therapy on aortic stiffness, carotid atherosclerosis, and calprotectin in inflammatory arthropathies: a controlled study. Am J Hypertens. 2012;25(6):644–650. | |
Sidiropoulos PI, Siakka P, Pagonidis K, et al. Sustained improvement of vascular endothelial function during anti-TNF alpha treatment in rheumatoid arthritis patients. Scand J Rheumatol. 2009;38(1):6–10. | |
Mathieu S, Pereira B, Couderc M, Rabois E, Dubost JJ, Soubrier M. No significant changes in arterial stiffness in patients with ankylosing spondylitis after tumour necrosis factor alpha blockade treatment for 6 and 12 months. Rheumatology. 2013;52(1):204–209. | |
Bosello S, Santoliquido A, Zoli A, et al. TNF-alpha blockade induces a reversible but transient effect on endothelial dysfunction in patients with long-standing severe rheumatoid arthritis. Clin Rheumatol. 2008;27(7):833–839. | |
van Eijk IC, Peters MJ, Serne EH, et al. Microvascular function is impaired in ankylosing spondylitis and improves after tumour necrosis factor alpha blockade. Ann Rheum Dis. 2009;68(3):362–366. | |
Hurlimann D, Forster A, Noll G, et al. Anti-tumor necrosis factor-alpha treatment improves endothelial function in patients with rheumatoid arthritis. Circulation. 2002;106(17):2184–2187. | |
Komai N, Morita Y, Sakuta T, Kuwabara A, Kashihara N. Anti-tumor necrosis factor therapy increases serum adiponectin levels with the improvement of endothelial dysfunction in patients with rheumatoid arthritis. Mod Rheumatol. 2007;17(5):385–390. | |
Gonzalez-Juanatey C, Testa A, Garcia-Castelo A, Garcia-Porrua C, Llorca J, Gonzalez-Gay MA. Active but transient improvement of endothelial function in rheumatoid arthritis patients undergoing long-term treatment with anti-tumor necrosis factor alpha antibody. Arthritis Rheum. 2004;51(3):447–450. | |
Syngle A, Vohra K, Sharma A, Kaur L. Endothelial dysfunction in ankylosing spondylitis improves after tumor necrosis factor-alpha blockade. Clin Rheumatol. 2010;29(7):763–770. | |
Irace C, Mancuso G, Fiaschi E, Madia A, Sesti G, Gnasso A. Effect of anti TNF alpha therapy on arterial diameter and wall shear stress and HDL cholesterol. Atherosclerosis. 2004;177(1):113–118. | |
Ramonda R, Puato M, Punzi L, et al. Atherosclerosis progression in psoriatic arthritis patients despite the treatment with tumor necrosis factor-alpha blockers: a two-year prospective observational study. Joint Bone Spine. 2014;81(5):421–425. | |
Gonzalez-Gay MA, De Matias JM, Gonzalez-Juanatey C, et al. Anti-tumor necrosis factor-alpha blockade improves insulin resistance in patients with rheumatoid arthritis. Clin Exp Rheumatol. 2006;24(1):83–86. | |
Kiortsis DN, Mavridis AK, Vasakos S, Nikas SN, Drosos AA. Effects of infliximab treatment on insulin resistance in patients with rheumatoid arthritis and ankylosing spondylitis. Ann Rheum Dis. 2005;64(5):765–766. | |
Ferraz-Amaro I, Arce-Franco M, Muniz J, et al. Systemic blockade of TNF-alpha does not improve insulin resistance in humans. Horm Metab Res. 2011;43(11):801–808. | |
Gonzalez-Gay MA, Gonzalez-Juanatey C, Miranda-Filloy JA, Llorca J. The potential effect of TNF-alpha antagonist therapy in rheumatoid arthritis may depend on the degree and severity of insulin resistance before the onset of this therapy. Horm Metab Res. 2012;44(7):558–559. | |
Paquot N, Castillo MJ, Lefebvre PJ, Scheen AJ. No increased insulin sensitivity after a single intravenous administration of a recombinant human tumor necrosis factor receptor: Fc fusion protein in obese insulin-resistant patients. J Clin Endocrinol Metab. 2000;85(3):1316–1319. | |
Wascher TC, Lindeman JH, Sourij H, Kooistra T, Pacini G, Roden M. Chronic TNF-alpha neutralization does not improve insulin resistance or endothelial function in “healthy” men with metabolic syndrome. Mol Med. 2011;17(3–4):189–193. | |
Dominguez H, Storgaard H, Rask-Madsen C, et al. Metabolic and vascular effects of tumor necrosis factor-alpha blockade with etanercept in obese patients with type 2 diabetes. J Vasc Res. 2005;42(6):517–525. | |
Cauza E, Cauza K, Hanusch-Enserer U, Etemad M, Dunky A, Kostner K. Intravenous anti TNF-alpha antibody therapy leads to elevated triglyceride and reduced HDL-cholesterol levels in patients with rheumatoid and psoriatic arthritis. Wien Klin Wochenschr. 2002;114(23–24):1004–1007. | |
Kiortsis DN, Mavridis AK, Filippatos TD, Vasakos S, Nikas SN, Drosos AA. Effects of infliximab treatment on lipoprotein profile in patients with rheumatoid arthritis and ankylosing spondylitis. J Rheumatol. 2006;33(5):921–923. | |
Popa C, Barrea P, Netea MG, Stalenhoef AF, van der Meer JW. Anti-TNF therapy and plasma HDL cholesterol concentration. Atherosclerosis. 2005;182(2):375. | |
Spanakis E, Sidiropoulos P, Papadakis J, et al. Modest but sustained increase of serum high density lipoprotein cholesterol levels in patients with inflammatory arthritides treated with infliximab. J Rheumatol. 2006;33(12):2440–2446. | |
Vis M, Nurmohamed MT, Wolbink G, et al. Short term effects of infliximab on the lipid profile in patients with rheumatoid arthritis. J Rheumatol. 2005;32(2):252–255. | |
van Eijk IC, de Vries MK, Levels JH, et al. Improvement of lipid profile is accompanied by atheroprotective alterations in high-density lipoprotein composition upon tumor necrosis factor blockade: a prospective cohort study in ankylosing spondylitis. Arthritis Rheum. 2009;60(5):1324–1330. | |
Lin J, Ziring D, Desai S, et al. TNF alpha blockade in human diseases: an overview of efficacy and safety. Clin Immunol. 2008;126(1):13–30. | |
Sfikakis PP, Tsokos GC. Towards the next generation of anti-TNF drugs. Clin Immunol. 2011;141(3):231–235. | |
Van Hauwermeiren F, Vandenbroucke RE, Libert C. Treatment of TNF mediated diseases by selective inhibition of soluble TNF or TNFR1. Cytokine Growth Factor Rev. 2011;22(5–6):311–319. | |
McCann FE, Perocheau DP, Ruspi G, et al. Selective tumor necrosis factor receptor I blockade is antiinflammatory and reveals immunoregulatory role of tumor necrosis factor receptor II in collagen-induced arthritis. Arthritis Rheumatol. 2014;66(10):2728–2738. | |
Zettlitz KA, Lorenz V, Landauer K, et al. ATROSAB, a humanized antagonistic anti-tumor necrosis factor receptor one-specific antibody. MAbs. 2010;2(6):639–647. | |
Nomura T, Abe Y, Kamada H, et al. Novel protein engineering strategy for creating highly receptor-selective mutant TNFs. Biochem Biophys Res Commun. 2009;388(4):667–671. | |
Shibata H, Yoshioka Y, Abe Y, et al. The treatment of established murine collagen-induced arthritis with a TNFR1-selective antagonistic mutant TNF. Biomaterials. 2009;30(34):6638–6647. | |
Deng GM, Zheng LX, Chan FK, Lenardo M. Amelioration of inflammatory arthritis by targeting the pre-ligand assembly domain of tumor necrosis factor receptors. Nat Med. 2005;11(10):1066–1072. | |
Arntz OJ, Geurts J, Veenbergen S, et al. A crucial role for tumor necrosis factor receptor 1 in synovial lining cells and the reticuloendothelial system in mediating experimental arthritis. Arthritis Res Ther. 2010;12(2). | |
Marepally S, Boakye CH, Patel AR, et al. Topical administration of dual siRNAs using fusogenic lipid nanoparticles for treating psoriatic-like plaques. Nanomedicine (Lond). 2014;9(14):2157–2174. | |
Desai PR, Marepally S, Patel AR, Voshavar C, Chaudhuri A, Singh M. Topical delivery of anti-TNF alpha siRNA and capsaicin via novel lipid-polymer hybrid nanoparticles efficiently inhibits skin inflammation in vivo. J Control Release. 2013;170(1):51–63. | |
Dahlman JE, Barnes C, Khan OF, et al. In vivo endothelial siRNA delivery using polymeric nanoparticles with low molecular weight. Nat Nanotechnol. 2014;9(8):648–655. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.