Back to Journals » International Journal of Nanomedicine » Volume 14
Titania nanotube-based protein delivery system to inhibit cranial bone regeneration in Crouzon model of craniosynostosis
Authors Bariana M, Kaidonis JA ![]() , Losic D
, Losic D ![]() , Ranjitkar S
, Ranjitkar S ![]() , Anderson PJ
, Anderson PJ
Received 18 January 2019
Accepted for publication 27 June 2019
Published 6 August 2019 Volume 2019:14 Pages 6313—6324
DOI https://doi.org/10.2147/IJN.S202090
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Manpreet Bariana,1 John A Kaidonis,1 Dusan Losic,2 Sarbin Ranjitkar,1,* Peter J Anderson1,3,*
1Adelaide Dental School, The University of Adelaide, Adelaide, SA 5005, Australia; 2School of Chemical Engineering, The University of Adelaide, Adelaide, SA 5005, Australia; 3Australian Craniofacial Unit, Adelaide, SA 5006, Australia
*These authors contributed equally to this work
Background: Craniosynostosis is a developmental disorder characterized by the premature fusion of skull sutures, necessitating repetitive, high-risk neurosurgical interventions throughout infancy. This study used protein-releasing Titania nanotubular implant (TNT/Ti) loaded with glypican 3 (GPC3) in the cranial critical-sized defects (CSDs) in Crouzon murine model (Fgfr2c342y/+ knock-in mutation) to address a key challenge of delaying post-operative bone regeneration in craniosynostosis.
Materials and methods: A 3 mm wide circular CSD was created in two murine models of Crouzon syndrome: (i) surgical control (CSDs without TNT/Ti or any protein, n=6) and (ii) experimental groups with TNT/Ti loaded with GPC3, further subdivided into the presence or absence of chitosan coating (on nanotubes) (n=12 in each group). The bone volume percentage in CSDs was assessed 90 days post-implantation using micro-computed tomography (micro-CT) and histological analysis.
Results: Nano-implants retrieved after 90 days post-operatively depicted well-adhered, hexagonally arranged, and densely packed nanotubes with average diameter of 120±10 nm. The nanotubular architecture was generally well-preserved. Compared with the control bone volume percentage data (without GPC3), GPC3-loaded TNT/Ti without chitosan coating displayed a significantly lower volume percent in cranial CSDs (P<0.001). Histological assessment showed relatively less bone regeneration (healing) in GPC3-loaded CSDs than control CSDs.
Conclusion: The finding of inhibition of cranial bone regeneration by GPC3-loaded TNT/Ti in vivo is an important advance in the novel field of minimally-invasive craniosynostosis therapy and holds the prospect of altering the whole paradigm of treatment for affected children. Future animal studies on a larger sample are indicated to refine the dosage and duration of drug delivery across different ages and both sexes with the view to undertake human clinical trials.
Keywords: craniosynostosis, protein delivery, glypican, titania nanotube, murine
Introduction
Craniosynostosis is a developmental disorder characterized by premature fusion of one or more cranial sutures, affecting approximately 1 in 2,500 children.1–3 Craniosynostosis can result in severe complications, including increased intracranial pressure, impaired cerebral blood flow, airway obstruction, restricted brain growth, impaired vision and hearing, cognitive disability, seizure, and adverse psychological effects associated with deformed craniofacial facial features.4–6 Management of craniosynostosis usually requires complex cranial vault reconstruction to release the synostosed suture, and to correct craniofacial deformities, but rapid post-operative bone re-growth at the craniectomy site often necessitates repetitive high-risk neurosurgical procedures as the dysmorphic growth continues.7
Recent advancements in cranial bone biology and genetic micro-array techniques have unraveled the pathophysiology of craniosynostosis by identifying biochemical pathways involved in both normal and pathological suture morphogenesis.8,9 The osteogenically potent bone morphogenetic protein 2 (BMP2) signaling pathway plays a key role in regulating cellular functions and bone formation around sutures, whereas defects in this pathway have been linked to craniosynostosis.1,2 Molecules that inhibit bone formation (such as noggin and glypicans (GPC1 and GPC3)) downregulate the BMP2-mediated osteogenic activity and are expressed in reduced amounts in prematurely-fusing sutures.10 While noggin has been shown to inhibit bone formation in animal models, its effect is only short-term and is restricted to the initial phase of suture resynostosis.11,12 Furthermore, noggin therapy does not rescue premature suture fusion in delayed-onset craniosynostosis.13 In comparison, glypicans have a longer-term potential in controlling post-operative bone growth, with GPC3 being shown to be more effective than GPC1.14
Murine models of human craniosynostosis have elucidated the phenotypic changes associated with specific genetic mutations, eg, in Crouzon murine model linked to the Fgfr2c342y/+ mutation.15 For craniofacial application, a critical-sized defect (CSD) is created surgically in the mouse cranium that provides a reproducible and non-load bearing orthotopic site for assessing cranial bone regeneration. CSD resembles an operative site in a human skull where a surgical cut is made into the parietal bone to release synostosed (fused) sutures. As bone formation is a continuous process, it is essential for the localized drug delivery system to provide a slow, sustained (extended) release of glypicans. Moreover, the protein concentration is critical as the heterogeneous suture cells can respond paradoxically to the same biomolecule at different concentrations.8 Localized drug delivery of GPC has been recently demonstrated by our team for the first time using Titanium (Ti) implants with a layer of Titania nanotubes (TNTs) fabricated by electrochemical oxidation.16,17 This drug delivery system has the potential to address a key clinical challenge for craniosynostosis management in terms of developing a viable localized drug delivery system with desirable pharmacokinetics and release pattern. Numerous in vitro studies (including cell studies) have demonstrated sustained release of various drugs from TNTs for potential application in neurodegenerative diseases (eg, neurotransmitters),18 orthopedics (eg, antibiotics, anti-inflammatory, and antiresorptive agents),19,20 and cancer treatment (doxorubicin (DOX) and apoptosis-inducing ligand).21 In addition, in vivo animal studies have confirmed the biocompatibility and osseointegration properties of these implants.22,23
Cytokines and therapeutic proteins have been administered locally into the craniofacial regions either directly using a carrier (polymer-based microspheres, absorbable collagen sponges, hydrogels, and implants, lipid nanoparticles, ceramics, titanium fibre-mesh or porous glass) or indirectly using gene based therapies (both viral and non-viral vectors).8,24,25 Nonetheless, they lack therapeutic efficacy due to undesired pharmacokinetics and uncontrolled release patterns, and/or are very complex to formulate. TNTs are easy to fabricate and provide superior control of release rate and duration over other drug delivery methods.25,26 Recently, we have demonstrated the use of Titania nanotubular implant (TNT/Ti) for sustained delivery of GPC3 over a period of 2 weeks in vitro, with improved (prolonged) release for chitosan coated TNT/Ti.16 These in vitro findings provide a strong foundation for further testing of sustained-drug releasing capabilities of TNT/Ti in vivo, which is an important step towards clinical translation.
Our aim was to assess the effect of GPC3 released from TNT/Ti (with or without chitosan coating) in inhibiting cranial bone regeneration in a Crouzon murine model involving skeletally-matured mice (3.0–3.5 months old) at 90-day post-implantation period. The experimental design of in vivo protein release via TNT/Ti within the cranial critical-sized defects is represented in Figure 1. We hypothesized that localized release of GPC3 would inhibit bone formation in Crouzon murine model of CSD during that period. To achieve this and provide critical therapeutic concentration of GPC3 with sustained release, we selected the anodization process to make TNTs with large diameters and with higher drug loading capacity. Slow and sustained release of loaded GPC3 inside TNTs for several weeks was tested by coating of layers of biodegradable chitosan on the top of TNTs surface. To prove the proposed hypothesis of inhibiting cranial bone regeneration using GPC3 loaded TNTs implants, we performed a Crouzon murine model with 90-day post-implantation period combined with histological and morphometric studies.
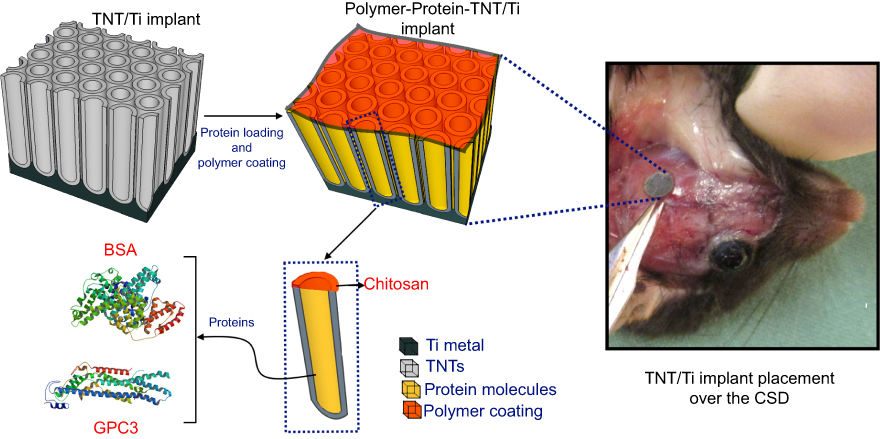 |
Figure 1 An electrochemically anodized Titania nanotubular implant (TNT/Ti) for sustained delivery of a bone antagonizing protein (glypican 3, GPC3) and a control protein (bone serum albumin; BSA) in surgically created critical-sized defect (CSD) as part of craniosynostosis therapy.Abbreviations: Ti, titanium; TNT, Titania nanotube. |
Materials and methods
Sample
Skeletally-matured male mice with a mixed C57BL/6 genetic background were divided into two cohorts (Figure 2): mice with Fgfr2c342y/+ knock-in mutation (representative of human Crouzon syndrome) (n=30) and wildtype littermates (Fgfr2c342+/+) (n=16). They were obtained from an ongoing breeding colony of craniosynostotic miceh at the Women’s and Children’s Hospital, Adelaide. The mice were managed using the existing protocol in a light- and temperature-controlled, pathogen-free environment with unrestricted access to water and food. Welfare assessment was carried out weekly by visual examination. The animal experimentation protocol was approved by the Women’s and Children’s Hospital, North Adelaide (AE977/6/2014) and the Animal Ethics Committees of the University of Adelaide, Adelaide (M-2014–138). The animals were cared for in the animal facility using the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes (7th Edition, 2004). For drug-delivery, nanotubes were anodized on TNT/Ti (substrate) and then loaded with either a non-functional protein (bovine serum albumin (BSA) or an inhibitor of bone production, GPC3).
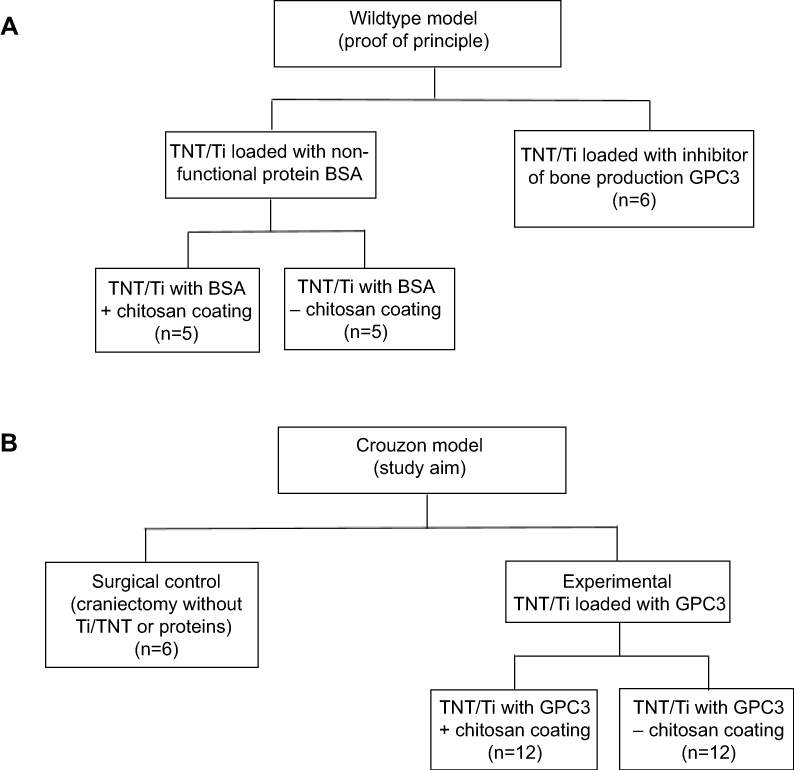 |
Figure 2 Flowchart showing the experimental layout (study design) for different TNT/Ti treatment groups at two different stages: (A) first stage of implant testing in Wildtype mice (proof of principle) to refine the delivery method, and (B) second stage of translating the refined drug delivery system into the Crouzon murine model (study aim).Abbreviations: TNT/Ti, Titania nanotubular implant; BSA, bovine serum albumin; GPC3, glypican 3 protein. |
Study design
This study was conducted in two stages, one relating to each cohort of mice (Figure 2). In the first stage, a wildtype “proof of principle” study was carried out for feasibility testing of the TNT/Ti drug delivery system (Figure 2A). This study was necessary to confirm no foreign body reaction was triggered by the TNT/Ti (eg, bone resorption), and to show GPC3 would inhibit bone growth more effectively than BSA. We included two wildtype groups: (i) TNT/Ti loaded with (BSA), which was subdivided into two groups (presence or absence of chitosan coating on nanotubes; n=5 in each group), and (ii) TNT/Ti loaded with inhibitor of bone production (GPC3) without chitosan coating (n=6). This study also enabled chitosan coating on BSA release to be assessed, and some experimental parameters to be refined for the Crouzon study. In the second stage involving the Crouzon murine model, we included two groups: (i) Surgical control (CSDs without TNT/Ti or any protein, n=6) and (ii) Experimental groups with TNT/Ti loaded with GPC3, subdivided into presence or absence of chitosan coating (on nanotubes) (n=12 in each group). Chitosan coating was included as a variable in the Crouzon (in vivo) murine model because of our previous in vitro finding of its potential to extend drug release from nanotubes.16 All mice were divided into various groups randomly. We only studied males in order to eliminate any potential confounding effect of estrogen that affects bone density and healing in females.
Intra- and post-operative digital photographs of both wildtype and Crouzon skulls were obtained for visual analysis of CSDs. Scanning electron microscopy (SEM) was used to assess the implant morphology before and after the implantation period. High-resolution micro-CT imaging and 3D image reconstruction yielded quantitative bone volume percentage data in the non-mineralized medullary space of CSDs at day 90 (ie, 3 months) post-operatively. Qualitative histological analysis of the craniectomy sites was carried out using Haemotoxylin and Eosin (H&E) and Movat Pentachrome staining to assess the quality of newly regenerated bone and fibrous tissues between different groups.
Fabrication of TNT/Ti
High purity titanium foil (99.6% of Ti 0.20 mm thickness) was supplied by Nilaco (Japan). All standard electrolytes, polymers, and solvents for TNT implant fabrication were obtained from Sigma-Aldrich (Australia). Titanium foils were annealed in air for 2 hours at 450°C, mechanically polished, cut into 3 mm circular Ti discs (via ultrasonic milling using DMG Ultrasonic 20 linear) and cleaned/degreased by successive sonication in acetone, ethanol, and isopropanol. Then, the discs were rinsed with deionized water and dried with nitrogen gas. TNT arrays on Ti discs were fabricated by using a modified two-step electrochemical anodization process in lactic acid containing organic electrolyte (comprising of ethylene glycol, ammonium fluoride, and DL-Lactic acid (Sigma-Aldrich), as described previously.16,17 Special temperature-controlled electrochemical apparatus was set up using anodization conditions at 60°C and 120 V for 5 minutes. Subsequently, the TNT layer was removed by sonication in methanol, and the second anodization was carried out under similar conditions to prepare well-defined TNT/Ti. We chose the dimensions of nanoimplants based on our previous studies that have also undertaken detailed characterization using various methods, including scanning electron microscopy, X-ray power diffraction, and contact angle measurement.16,17
Protein loading and polymer coating
GPC3 and BSA were loaded into the nanotubes using a vacuum drying technique (5 µg/sample). The amount of protein was determined from our observation of the amount required to show optimal regulatory effect of glypicans on the BMP pathway within therapeutic limit.2,16 Nanotube arrays were surface coated with 2% chitosan solution (spin-coated at 1500 rpm for 15 seconds) for five wildtype mice and 12 Fgfr2c342y/+ knock-in mice (Figure 2). We used a field-emission scanning electron microscope for morphological characterization of implants (FEI Quanta 450, Eidenhoven, the Netherlands). Prior to animal experiments, the protein-loaded implants were sterilized using low-temperature hydrogen peroxide gas plasma (STERRAD® 100 NXTM System, Advanced sterilization Products, Irvine, CA, USA).
Surgical procedure
The mice were anesthetized by injecting a mixture of 100 mg/kg ketamine hydrochloride and 10 mg/kg xylazine via intraperitoneal route, followed by subcutaneous injection of 0.05 mg/kg of the analgesic, buprenorphine. Once unresponsive to paw-pinching, the eyes were protected by lacrilube to prevent corneal damage. The hair on the scalp was disinfected using 70% ethanol, and a C-shaped incision was made on the skin over the parietal bone. The skin flap was gently lifted to expose the cranium and a 3 mm circular CSD was created using a fine biopsy puncture lateral to the right coronal suture with continuous saline buffer irrigation under well-lit condition. The bone disc (from the CSD) was removed gently, ensuring that the underlying dura remained intact. The sterilized protein-loaded TNT/Ti were carefully inserted into the CSD. After craniectomy and implantation, the surgical sites were sutured and the mice were allowed to recover on a heat pad/incubator or held in the palm of hand to establish rhythmic breathing before being transferred into individual cages. The mice were assessed for any skin reaction over the implant, weight loss, or distress twice daily for 3 months. Histological analysis in a separate sample showed that there was no adverse reaction, including necrosis, lipoma formation, subdural hematoma, or chronic inflammation (eg, influx of macrophages, giant cells, or neutrophils) on the skin or bone over the implantation site (unpublished data).
Assessment of bone regeneration
At 90 days post-operatively (post-implantation period), the mice were euthanized by asphyxiation with carbon dioxide (at a flow rate of 0.5%). The skulls were dissected from the soft tissues and the implant discs removed carefully so that the surgical procedure did not cause any damage to the underlying bone and other tissues. Then, the skulls were subjected to micro-CT imaging (Skyscan 1076 small animal micro-CT, Bruker, Kontich, Belgium) using these parameters: 50 kV, 110 µA with a rotation step of 0.6, 0.5 mm Al filter, scanning width of 35 mm, imaging time of 48 minutes, and image resolution of 8.7 µm. We used Skyscan NRecon software for image reconstruction as BMP files (ring artefact correction of 15, beam hardening correction of 30%, smoothing at 1 pixel, misalignment correction of <10, and thresholding limits of 0.00–0.11). We used Data viewer, CTan, and Aviso 9.0 software for realignment, segmentation, 3D image reconstruction, and morphometric analysis. We calculated the volume of newly formed bone in each CSD by creating a 3 mm wide cylindrical volume of interest at the centre of each reconstructed CSD axially over the entire bone thickness. The ratio of the bone volume to the total tissue volume in the CSD provided the percentage bone volume data. We did not use medication such as tetracycline antibiotic that could have affected bone metabolism.
Histological analysis was carried out by decalcifying the cranium with 10% EDTA in 0.1 M Tris Buffer (pH 7.4) and 7% sucrose for up to 4 weeks at room temperature, followed by embedding in paraffin wax. The sections were then cut into 7 µm thick serial sagittal slices using a rotary microtome (Leica Microsystems, Wetzlar, Germany). Sagittal sections were made around each CSD and then stained with H&E and Movat Pentachrome stains using standard protocols. The slides were imaged using a bright field microscope (Carl Zeiss, Jena, Germany), equipped with a DFC480 digital camera (Leica Microsystems).
Statistical analysis
Statistical analysis was conducted using the IBM SPSS Statistics software (version 26) (IBM Corporation, Armonk, NY, USA). The Shapiro-Wilk test confirmed normal distribution of the bone volume percentage data. One-way ANOVA and Bonferroni’s post-hoc tests were carried out separately for the wildtype and Crouzon models to assess whether bone volume percentage (outcome) varied significantly between treatment groups (variables). Statistical significance was set at the 0.05 probability level. Statistical interpretation was made by taking into account both the P-values and effect size (Cohen’s f). Effect size was calculated by using the formula: f=√(ƞ2/(1–ƞ2)), where ƞ2 (partial ita-squared) was obtained as a MANOVA output. The effect size (f) of 0.10 is mild, 0.25 is moderate, and 0.40 is high.
Results
Characterization of fabricated TNT/Ti
The typical structure of fabricated TNTs/Ti implant is presented in SEM images in Figure 3 (the whole implant disc in A and TNT structure at different magnifications in B and C). These images showed well-adhered, hexagonally arranged, and densely packed Titania nanotubes with average diameter of 120±10 nm even after being implanted in CSDs for 90 days, indicating that the nanotubular architecture was well-preserved with ordered arrays of open-pored structure. However, the nanotube wall was rough and flaky at the top, probably because of implant interaction with fibrous tissue/protein from the extracellular matrix within the CSD. We only observed minor superficial wound infection at the incision site (away from the implant) in two mice that were treated with a short course of oral tetracycline for 3 days. Although tetracycline has been shown to accelerate bone healing, a short-term treatment is unlikely to have caused major issues in our study. There was no adverse reaction on the soft tissues or the bone during the study period, indicating that the TNT/Ti, proteins, and chitosan coating were biocompatible.
 |
Figure 3 SEM images showing the surface topography of representative TNT/Ti delivery platforms after day 90 of the in vivo study showing (A) the whole fabricated implant disc with TNT structures, and (B–C) only the TNT structures (top view) at different magnifications.Abbreviations: TNT/Ti, Titania nanotubular implant; TNT, Titania nanotube. |
Protein release in the wildtype model
The 3D micro-CT reconstructions and 2D sagittal sections of CSDs in wildtype skulls at day 90 post-operatively are shown in Figures 4A and B. The BSA-loaded CSDs, either with or without chitosan-coating, displayed some reossification. In contrast, GPC3 treated CSDs were larger, with less bone formation over the dura mater. One-way ANOVA showed a significant effect of the treatment on bone volume percentage (P<0.01), and Bonferroni post-hoc tests revealed a significantly lower bone volume percentage for the GPC3-treated CSD (TNT-GPC3) than either of the two BSA-treated CSD with or without chitosan coating (TNT-BSA-CH or TNT-BSA) (P<0.01 for each comparison; effect size=1.1 (large) and 1.2 (large), respectively) (Figure 4C). These findings provided proof of principle validation that GPC3-loaded TNT/Ti can therapeutically downregulate the BMP2 activity and inhibit bone regeneration in vivo.
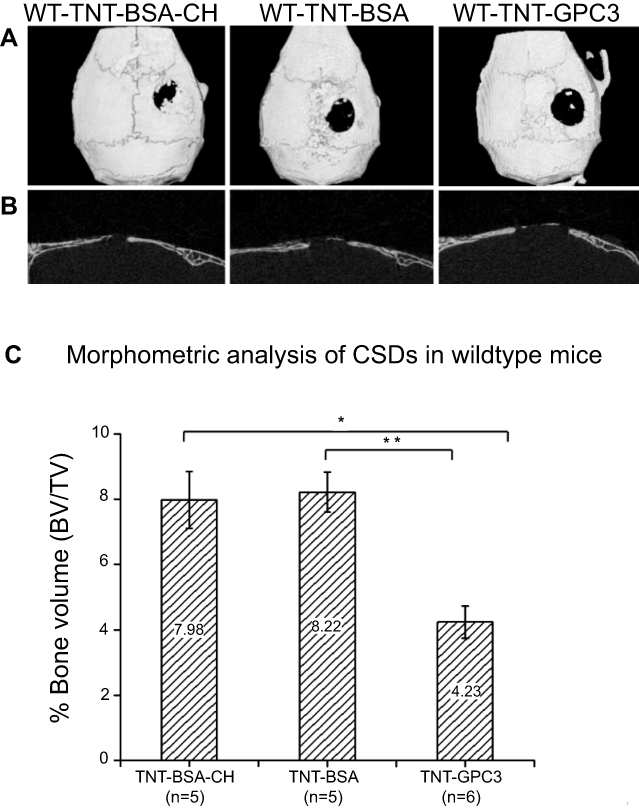 |
Figure 4 Micro-CT images and bone volume percentage data for newly formed bone in wildtype mice treated with non-functional BSA protein (either with chitosan coating [TNT-BSA-CH] or no coating [TNT-BSA]), and GPC3 protein (TNT-GPC3) at day 90 post-operatively; (A) Top view of the 3D micro-CT reconstructions showing surgically created critical-sized defects (CSDs), (B) Sagittal sections through the middle of the CSDs , and (C) bone volume percentage within a 3 mm wide cylindrical volume of interest around the CSDs. * and ** indicate significant differences at P<0.01 between the TNT-GPC3 group and the TNT-BSA-CH group, and the TNT-GPC3 group and the TNT-BSA group, respectively. There was no significant difference in bone volume percentage between the TNT-BSA-CH and TNT-BSA groups. Effect size: TNT-GPC3 vs TNT-BSA-CH=1.13 (large); TNT-GPC3 vs TNT-BSA=1.20 (large).Abbreviations: BSA, bovine serum albumin; BV, bone volume; CH, chitosan; GPC3, glypican 3 protein; Ti, titanium; TNT, Titania nanotube; TV, total volume; WT, wildtype. |
The micro-CT data was consistent with histological analysis (Figures 4 and 5). The H&E staining for the BSA groups showed prominent bone reossification and defect healing (with thick acellular fibrous tissue) in both uncoated and chitosan-coated TNT/Ti (implantation sites), with the latter displaying isolated bony islands interpolating the fibrous tissue inside the defect. GPC3-treated CSDs showed larger, unhealed, and less ossified (fibrous) defect sites compared with the BSA-treated groups, confirming inhibition of bone regeneration by GPC3-loaded TNT/Ti.
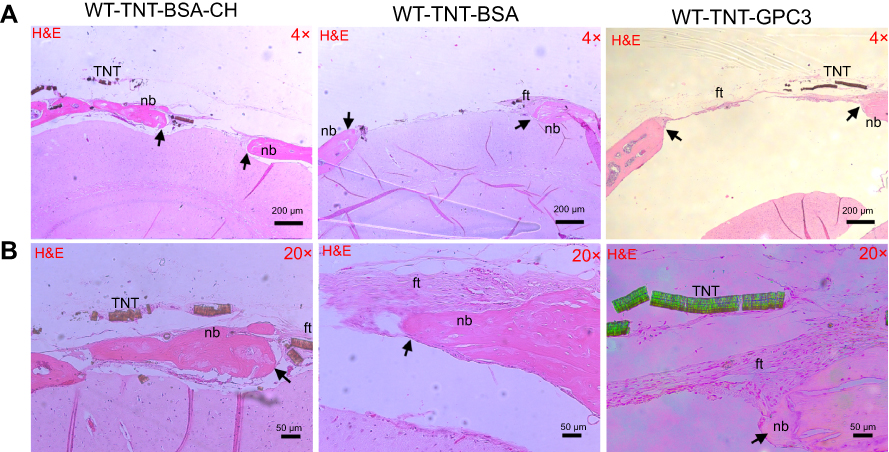 |
Figure 5 Histological (H&E) images showing bone regeneration in the critical-sized defects (CSDs) of the three wildtype groups at day 90 post-operatively at (A) a low magnification (×4) and (B) a high magnification (×20). Images A and B were prepared from different slices of the same CSD (for each group) that displayed the features most clearly at each magnification. Black arrows mark the new bone edge.Notes: WT-TNT-BSA-CH, wildtype mice in which Titania nanotubes were loaded with bovine serum albumin and also coated with chitosan; WT-TNT-BSA, Wildtype mice in which Titania nanotubes were loaded with bovine serum albumin but not coated with chitosan; WT-TNT-GPC3, Wildtype mice in which Titania nanotubes were loaded with glypican 3 protein. Abbreviations: nb, new bone; ft, fibrous tissue; TNT, Titania nanotube (delaminated); WT, wildtype; BSA, bovine serum albumin; TNT, Titania nanotube; CH, chitosan; GPC3, glypican 3 protein. |
Protein release in the Crouzon murine model
The 3D micro-CT reconstructions and 2D sagittal sections of CSDs are shown in Figures 6A and B. The micro-CT images showed near obliteration of CSDs in the control group due to accelerated bone morphogenesis associated with the Fgfr2c342y/+ knock-in mutation. One-way ANOVA showed a significant effect of the GCP3 treatment (either with or without chitosan coating) on bone volume percentage (Figure 6C). Bonferroni’s post-hoc tests revealed a significantly lower bone volume percentage for the GPC-treated CSD without chitosan (TNT-GPC3) compared with the craniectomy control (P<0.001, effect size=0.86 (large)), and for the GPC-treated CSD without chitosan (TNT-GPC3) compared with the GPC-treated CSD with chitosan (TNT-GPC3-CH) (P<0.05, effect size=0.57 (large)).
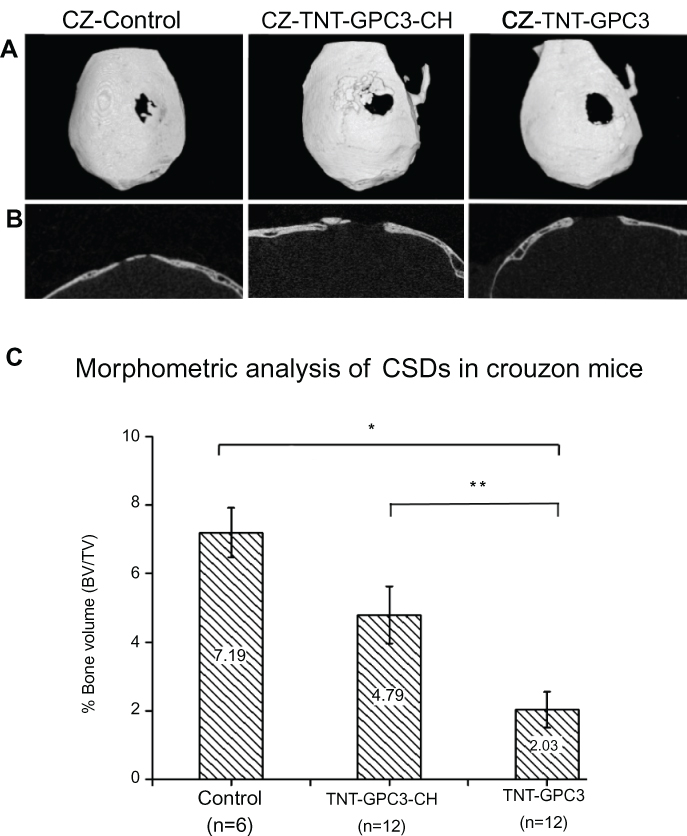 |
Figure 6 Micro-CT images and bone volume percentage data for newly formed bone in three groups of the Crouzon murine model (with Fgfr2c342y/+ knock-in mutation) including the surgical control (craniectomy only) and the two experimental groups, including Titania nanotubes loaded with glypican 3 protein and then either coated with chitosan (TNT-GPC3-CH) or not (TNT-GPC3), at day 90 post-operatively; (A) Top view of the 3D reconstructed skulls from micro-CT scans showing surgically created critical-sized defects (CSDs), (B) Sagittal sections through the middle of the CDSs, and (C) bone volume percentage within a 3 mm wide cylindrical volume of interest around the CSDs. *Significant difference between the TNT-GPC3 group and the control group at P<0.001, and **Significant difference between the TNT-GPC3 group and the TNT-GPC3-CH group at P<0.05. There was no significant difference in bone volume percentage between the control and TNT-GPC3-CH groups. Effect size: TNT-GPC3 vs control=0.86 (large); TNT-GPC3 vs TNT-GPC3-CH=0.57 (large). CZ represents Crouzon murine model in image A.Abbreviations: BV, bone volume; CH, chitosan; GPC3, glypican 3 protein; TNT, Titania nanotube; TV, total volume. |
Histological findings were consistent with the micro-CT data (Figure 7). The H&E and Movat Pentachrome staining of the control CSDs mainly exhibited mature lamellar bone and a thick band of fibrous tissue bridging the gaps at a low magnification (×4). The high magnification images (×20) displayed newly regenerated bony edges with ectopic bony islands interweaved within the collagen fibres. Chitosan coating in GPC3-loaded nanotubes resulted in incomplete healing with ectopic bone formation, with a trend of relatively greater bone regeneration in coated than uncoated nanotubes.
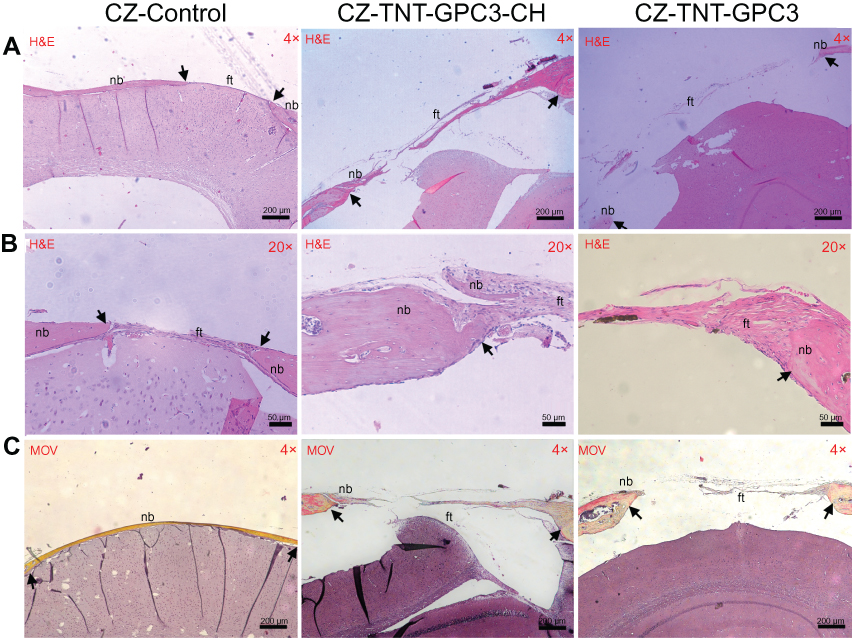 |
Figure 7 Histological images showing bone regeneration in the critical-sized defects (CSDs) of three groups of Crouzon murine model (with Fgfr2c342y/+ knock-in mutation) at day 90 post-operatively for (A) H&E staining at a low magnification (×4), (B) H&E staining at a high magnification (×20), and (C) Movat Pentachrome (MOV) staining at a low magnification (×4). Images A and B were prepared from different slices of the same CSD (for each group) that displayed the features most clearly at each magnification. Black arrows mark the new bone in the CSDs.Notes: CZ-Control, Crouzon murine model without Titania nanoimplants or proteins (i.e. surgical control); CZ-TNT-GPC3-CH, Crouzon murine model with Titania nanotubes loaded with glypican 3 protein and also coated with chitosan; CZ-TNT-GPC3, Crouzon murine model with Titania nanotubes loaded with glypican 3 protein (but no chitosan coating). Abbreviations: nb, new bone; ft, fibrous tissue; CH, chitosan; GPC3, glypican 3 protein; TNT, Titania nanotube. |
Discussion
Our results prove our hypothesis of inhibition of cranial bone regeneration in Fgfr2c342y/+ knock-in mice by GPC3-loaded TNT/Ti. Given that existing in vitro, ex vivo, and in vivo studies on these implants (for dental and orthopedic applications) are at an early stage of research and development, our pre-clinical study is a major advance in the novel field of minimally-invasive craniosynostosis therapy and holds the prospect of clinical translation to reduce the morbidity and mortality in affected children. This nanoimplant system also has additional potential craniofacial applications, including orthopedic correction of malocclusion (eg, maxillary expansion by releasing fused mid-palatal suture) and correction of various skeletal defects (eg, in hemifacial macrosomia). Such drug delivery systems will require precise control over the release of various drugs that promote and inhibit bone regeneration. This approach could also benefit prolonged, sustained delivery of antimicrobial and remineralizing agents intraorally, such as dental caries and erosion.
Various nano-drug delivery systems, including those based on natural or synthetic polymeric, metallic, and organic nanoparticles, have been explored for clinical application.27–31 In craniofacial clinical practice, the TNT-based implant system is likely to have an advantage over its zero-dimensional counterparts (quantum dots, nanoclusters, etc) as they do not penetrate or mediate transport across the blood–brain barrier (due to relatively high aspect ratio).27 Traditionally, TNTs have been believed to be mechanically robust, non-toxic, and chemically stable/inert and stable in vivo.32 After drug delivery is complete, the TNT/Ti may not require surgical removal, just as titanium plates and screws used for surgical repair of fractured bones are left in place in the long-term. However, there is some emerging evidence to suggest potential issues with peri-implant and orthopedic complications associated with metal release.33 Long-term immune response to nano-drug delivery systems remains a largely unexplored area,34 and further research is needed to investigate this.
We designed TNT/Ti specially for easy insertion into the 3 mm wide CSD, without damaging the nanotube arrays. While our previous in vitro study showed glypican release from TNT/Ti for around 2 weeks in buffer,16 the release duration is likely to be prolonged in our current study (evidenced by decrease in bone regeneration), probably because of a limited diffusion gradient in vivo. In uncoated TNT/Ti, proteins are released into the CSD and the perisutural region by passive diffusive transport in the cranial environment (extracellular matrix and interstitial fluids).35 In comparison, the release kinetics of the proteins in chitosan-coated TNT/Ti (polymer thickness ~1 µm) is modified by polymer degradation and transformation.16,17 Chitosan undergoes slow enzymatic hydrolysis in the presence of several proteases, mainly lysozymes (present in body fluids), which contributes to prolonged drug release,36 and partially open pores (with patches of undissolved polymer) have been shown to be responsible for sustained drug release.37 There was some trend of less bone regeneration in the TNT-GPC3-CH group compared with the control group in the Crouzon murine model, but it did not reach statistical significance. Further study is indicated using a larger sample size to investigate this.
Our observation of near obliteration of the CSD in Crouzon murine model is consistent with accelerated bone regeneration after craniectomy in a patient with coronal synostosis (without any bone graft).38 In contrast, GPC3 releasing implants (chitosan-coated and uncoated TNTs) reduced re-ossification at the craniectomy site, with the chitosan-coated implants, demonstrating a greater number of bony islands over the dura than uncoated implants (Figures 6 and 7). Our observation of reduced bone formation in chitosan-coated GPC3 nanoimplants can be attributed to non-uniform degradation of chitosan by lysozymes in the cranial environment (failing to provide continuous protein release).39 Alternatively, bone growth might have occurred centripetally towards the chitosan surfaces. Even with a low degree of acetylation, the positively charged coating could have stimulated osteoblast adhesion and differentiation that are evident in the form of bony islands.40,41 However, it appears to have been counteracted by inhibition of bone development by GPC3 (Figures 4 and 5).
Future animal studies on a larger sample are indicated to refine the dosage and duration of the TNT/Ti drug delivery system (at various time periods) on various age groups and both sexes, which will provide a sound foundation to conduct human clinical trials. Clinically, TNTs can be fabricated easily on metal/3D printed fixation devices used in surgical procedures (eg, bone plates and screws). Ultimately, successful clinical translation could alter the whole paradigm of treatment for children affected by craniosynostosis.
Conclusion
Ease of fabrication, physiologically relevant nano-architecture, and the capability for localized protein delivery make TNT/Ti an attractive option for craniofacial drug delivery. To our knowledge, this is the first report that demonstrates the effectiveness of TNT/Ti delivery systems in inhibiting bone regeneration in a Crouzon murine model of craniosynostosis. This minimally invasive non-surgical adjuvant holds the prospect of clinical translation in treating affected children, improving the likelihood of reducing the morbidity and mortality associated with repeated cranial vault reconstruction.
Acknowledgments
The authors gratefully acknowledge the financial support provided by the Australian Dental Research Foundation (ADRF 18/2013), the Australian Research Council (FT110100711 and IH150100003), and the Australian Craniomaxillofacial Foundation. Ms Ruth Williams from Adelaide Microscopy provided assistance with micro-CT imaging and histological work. Titania nanotubular implants were fabricated at the School of Chemical Engineering, University of Adelaide and at the OptoFab node of the Australian National Fabrication Facility utilizing Commonwealth and South Australia State Government funding.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Coussens AK, Wilkinson CR, Hughes IP, et al. Unravelling the molecular control of calvarial suture fusion in children with craniosynostosis. BMC Genomics. 2007;8(1):458–482. doi:10.1186/1471-2164-8-458
2. Dwivedi P, Grose R, Hii C, Filmus C, Anderson P, Powell B. Regulation of bone morphogenetic protein signalling and osteogenesis by glypicans in human cranial suture cells. Bone. 2013;48(2):243–254. doi:10.1016/j.bone.2011.03.588
3. Wilkie AO. Craniosynostosis: genes and mechanisms. Hum Mol Gen. 1997;6:1647–1656. doi:10.1093/hmg/6.10.1647
4. Gosain AK, McCarthy JG, Wisoff JH. Morbidity associated with increased intracranial pressure in Apert and Pfeiffer syndromes: the need for long-term evaluation. Plast Reconstr Surg. 1996;97(2):292–301.
5. Gonsalez S, Hayward R, Jones B, Lane R. Upper airway obstruction and raised intracranial pressure in children with craniosynostosis. Eur Respir J. 1997;10(2):367–375.
6. Panchal J, Uttchin V. Management of craniosynostosis. Plast Reconstr Surg. 2003;111(5):2032–2048. doi:10.1097/01.PRS.0000056839.94034.47
7. Hermann CD, Hyzy SL, Olivares-Navarrete R, et al. Craniosynostosis and resynostosis: models, imaging, and dental implications. J Dent Res. 2016;95(8):846–852. doi:10.1177/0022034516643315
8. Mooney MP, Moursi AM, Opperman LA, Siegel MI. Cytokine therapy for craniosynostosis. Expert Opin Biol Ther. 2004;4(3):279–299. doi:10.1517/14712598.4.3.279
9. Derderian C, Seaward J. Syndromic craniosynostosis. Semin Plast Surg. 2012;26(2):64–75. doi:10.1055/s-0032-1320064
10. Dwivedi P, Lam N, Powell B. Boning up on glypicans - opportunities for new insights into bone biology. Cell Biochem Funct. 2013;31(2):91–114. doi:10.1002/cbf.2939
11. Cooper GM, Curry C, Barbano TE, et al. Noggin inhibits postoperative resynostosis in craniosynostotic rabbits. J Bone Min Res. 2007;22(7):1046–1054. doi:10.1359/jbmr.070410
12. Cooper GM, Usas A, Olshanski A, Mooney MP, Losee JE, Huard J. Ex vivo Noggin gene therapy inhibits bone formation in a mouse model of postoperative resynostosis. Plast Reconstr Surg. 2009;123(2 Suppl):S94–S103. doi:10.1097/PRS.0b013e318191c05b
13. Cray J, Burrows AM, Vecchione L, et al. Blocking bone morphogenetic protein function using in vivo noggin therapy does not rescue premature suture fusion in rabbits with delayed-onset craniosynostosis. Plast Reconstr Surg. 2011;127(3):1163–1172. doi:10.1097/PRS.0b013e318205f23b
14. Coussens AK, Hughes IP, Wilkinson CR, et al. Identification of genes differentially expressed by prematurely fused human sutures using a novel in vivo – in vitro approach. Differentiation. 2008;76(5):531–545. doi:10.1111/j.1432-0436.2007.00244.x
15. Khominsky A, Yong R, Ranjitkar S, Townsend GC, Anderson P. Extensive phenotyping of orofacial features in Crouzon syndrome. Arch Oral Biol. 2018;86:123–130. doi:10.1016/j.archoralbio.2017.10.022
16. Bariana M, Dwivedi P, Ranjitkar S, Kaidonis JA, Losic D, Anderson PJ. Glypican-based drug releasing titania implants to regulate BMP2 bioactivity as a potential approach for craniosynostosis therapy. Nanomedicine. 2018;14:2365–2374. doi: 10.1016/j.nano.2017.06.005
17. Bariana M, Dwivedi P, Ranjitkar S, Kaidonis JA, Losic D, Anderson PJ. Biological response of human suture mesenchymal cells to Titania nanotube-based implants for advanced craniosynostosis therapy. Colloids Surf B. 2017;150:59–67. doi:10.1016/j.colsurfb.2016.11.019
18. Gulati K, Aw M, Losic D. Nanoengineered drug-releasing Ti wires as an alternative for local delivery of chemotherapeutics in the brain. Int J Nanomed. 2012;7:2069–2076.
19. Gulati K, Prideaux M, Kogawa M, et al. Anodized 3D-printed titanium implants with dual micro- and nano-scale topography promote interaction with human osteoblasts and osteocyte-like cells. J Tissue Eng Regen Med. 2017;11:3313–3325. doi:10.1002/term.2239
20. Maher S, Kaur G, Lima-Marques L, Evdokiou A, Losic D. Engineering of micro- to nanostructured 3D-Printed drug-releasing titanium implants for enhanced osseointegration and localized delivery of anticancer drugs. ACS Appl Mater Interfaces. 2017;9(35):29562–29570. doi:10.1021/acsami.7b09916
21. Popat KC, Leoni L, Grimes CA, Desai TA. Influence of engineered titania nanotubular surfaces on bone cells. Biomateials. 2007;28(21):3188–3197. doi:10.1016/j.biomaterials.2007.03.020
22. von Wilmowsky C, Bauer S, Lutz R, et al. In vivo evaluation of anodic TiO2 nanotubes: an experimental study in the pig. J Biomed Mater Res B. 2009;89(1):165–171. doi:10.1002/jbm.b.31201
23. Bjursten LM, Rasmusson L, Oh S, Smith GC, Brammer KS, Jin S. Titanium dioxide nanotubes enhance bone bonding in vivo. J Biomed Mater Res A. 2010;92(3):1218–1224. doi:10.1002/jbm.a.32463
24. Moioli EK, Clark PA, Xin X, Lal S, Mao JJ. Matrices and scaffolds for drug delivery in dental, oral and craniofacial tissue engineering. Adv Drug Deliv Rev. 2007;59(4–5):308–324. doi:10.1016/j.addr.2007.03.019
25. Springer IN, Warnke PH, Terheyden H, et al. Craniectomy and noggin application in an infant model. J Craniomaxillofac Surg. 2007;35(3):177–184. doi:10.1016/j.jcms.2007.04.003
26. Shen K, Krakora S, Cunningham M, et al. Medical treatment of craniosynostosis: recombinant Noggin inhibits coronal suture closure in the rat craniosynostosis model. Orthod Craniofac Res. 2009;12(3):254–262. doi:10.1111/j.1601-6343.2009.01460.x
27. Suri SS, Fenniri H, Singh B. Nanotechnology-based drug delivery systems. J Occup Med Toxicol. 2007;2:16. doi:10.1186/1745-6673-2-16
28. Garcia LM, Leitune VCB, Kist TL, Takimi A, Samuel SMW, Collares SM. Quantum dots as nonagglomerated nanofillers for adhesive resins. J Dent Res. 2016;95(12):1401–1407. doi:10.1177/0022034516656838
29. Huang J, Zhang X, Yan W, et al. Nanotubular topography enhances the bioactivity of titanium implants. Nanomedicine. 2017;13:1913–1923. doi:10.1016/j.nano.2017.03.017
30. Li G, Zhou T, Lin S, Shi S, Lin Y. Nanomaterials for craniofacial and dental tissue engineering. J Dent Res. 2017;96(7):725–732. doi:10.1177/0022034517706678
31. Lin X, Yang S, Lai K, Yang H, Webster T, Yang L. Orthopedic implant biomaterials with both osteogenic and anti-infection capacities and associated with in vivo evaluation methods. Nanomedicine. 2017;13:123–142. doi:10.1016/j.nano.2016.08.003
32. Santos A, Aw MS, Bariana M, Kumeria T, Wang Y, Losic D. Drug-releasing implants: current progress, challenges and perspectives. J Mater Chem B. 2014;2:6157–6182. doi:10.1039/C4TB00548A
33. Fretwurst T, Nelson K, Tarnow DP, Wang H-L, Giannobile WV. Is metal particle release associated with peri-implant bone destruction? An emerging concept. J Dent Res. 2018;97(3):259–265. doi:10.1177/0022034517740560
34. Halamoda-Kenzaoui B, Holzwarth U, Roebben G, Bogni A. Mapping of the available standards against the regulatory needs for nanomedicines. Nanomed Nanobiotechnol. 2018;e1531. doi:10.1002/wnan.1531
35. Lunt SJ, Kalliomaki TM, Brown A, Yang VX, Milosevic M, Hill RP. Interstitial fluid pressure, vascularity and metastasis in ectopic, orthotopic and spontaneous tumours. BMC Cancer. 2008;8(1):2–15. doi:10.1186/1471-2407-8-2
36. Lim SM, Song DK, Oh SH, Lee-Yoon DS, Bae EH, Lee JH. In vitro and in vivo degradation behavior of acetylated chitosan porous beads. J Biomater Sci Polym Ed. 2008;19(4):453–466. doi:10.1163/156856208783719482
37. Gulati K, Johnson L, Karunagaran R, Findlay D, Losic D. In situ transformation of chitosan films into microtubular structures on the surface of nanoengineered titanium implants. Biomacromolecules. 2016;17(4):1261–1271. doi:10.1021/acs.biomac.5b01037
38. Drake DB, Persing JA, Berman DE, Ogle RC. Calvartal deformity regeneration following subtotal craniectomy for craniosynostosis: a case report and theoretical implications. J Craniofac Surg. 1993;4(2):85–89.
39. Tomihata K, Ikada Y. In vitro and in vivo degradation of films of chitin and its deacetylated derivatives. Biomaterials. 1997;18(7):567–575.
40. Lee JY, Nam SH, Im SY, et al. Enhanced bone formation by controlled growth factor delivery from chitosan-based biomaterials. J Control Release. 2002;78(1–3):187–197.
41. Oktay EO, Demiralp B, Demiralp B, et al. Effects of platelet-rich plasma and chitosan combination on bone regeneration in experimental rabbit cranial defects. J Oral Implantol. 2010;36(3):175–184. doi:10.1563/AAID-JOI-D-09-00023
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.