")
Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Tissue inhibitor of matrix metalloproteinases-1 loaded poly(lactic-co-glycolic acid) nanoparticles for delivery across the blood–brain barrier
Authors Chaturvedi M, Molino Y, Sreedhar B, Khrestchatisky M, Kaczmarek L
Received 20 September 2013
Accepted for publication 12 October 2013
Published 20 January 2014 Volume 2014:9(1) Pages 575—588
DOI https://doi.org/10.2147/IJN.S54750
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Mayank Chaturvedi,1 Yves Molino,2 Bojja Sreedhar,3 Michel Khrestchatisky,4 Leszek Kaczmarek1
1Laboratory of Neurobiology, Nencki Institute, Warsaw, Poland; 2Vect-Horus, Marseille, France; 3Indian Institute of Chemical Technology, Hyderabad, India; 4Aix-Marseille Université, CNRS, NICN, UMR7259, Marseille, France
Aim: The aim of this study was to develop poly(lactic-co-glycolic acid) (PLGA) nanoparticles (NPs) for delivery of a protein – tissue inhibitor of matrix metalloproteinases 1 (TIMP-1) – across the blood–brain barrier (BBB) to inhibit deleterious matrix metalloproteinases (MMPs).
Materials and methods: The NPs were formulated by multiple-emulsion solvent-evaporation, and for enhancing BBB penetration, they were coated with polysorbate 80 (Ps80). We compared Ps80-coated and uncoated NPs for their toxicity, binding, and BBB penetration on primary rat brain capillary endothelial cell cultures and the rat brain endothelial 4 cell line. These studies were followed by in vivo studies for brain delivery of these NPs.
Results: Results showed that neither Ps80-coated nor uncoated NPs caused significant opening of the BBB, and essentially they were nontoxic. NPs without Ps80 coating had more binding to endothelial cells compared to Ps80-coated NPs. Penetration studies showed that TIMP-1 NPs + Ps80 had 11.21%±1.35% penetration, whereas TIMP-1 alone and TIMP-1 NPs without Ps80 coating did not cross the endothelial monolayer. In vivo studies indicated BBB penetration of intravenously injected TIMP-1 NPs + Ps80.
Conclusion: The study demonstrated that Ps80 coating of NPs does not cause significant toxic effects to endothelial cells and that it can be used to enhance the delivery of protein across endothelial cell barriers, both in vitro and in vivo.
Keywords: PLGA nanoparticles, drug delivery, protein delivery, sustained release, brain delivery, BBB penetration, RBCEC culture
Introduction
Many newly developed therapeutic molecules are biopharmaceuticals, such as recombinant proteins, small interfering ribonucleic acids, monoclonal antibodies, and nonviral gene medicines. However, application of these potential therapeutics for central nervous system (CNS) disorders has been a challenging task, as most of them do not cross the blood–brain barrier (BBB). This is due to the fact that the BBB is a physical barrier formed by the tight junctions between endothelial cells, the presence of astrocytic end-feet, and a physiological barrier endowed with efficient efflux pumps, which prevent most molecules and notably high-molecular-weight molecules from passing through.
During recent years, different strategies have been developed to deliver molecules across the BBB.1 Particularly promising are nanotechnology-based delivery systems, such as liposomes, dendrimers, and nanoparticles (NPs) made of various polymers, eg, poly(lactic-co-glycolic acid) (PLGA) and poly(butyl cyanoacrylate) (PBCA). PLGA-based nano/microparticles have been widely explored as carriers for controlled delivery of macromolecular therapeutics.2 PLGA-based NPs are advantageous, due to their biodegradable and biocompatible nature; moreover, they have been approved by the US Food and Drug Administration for human use.3 Furthermore, NP delivery across the BBB can be facilitated by coating with surfactants, such as polysorbate 80 (Ps80; Tween 80).4
The objective of this study was to develop PLGA NPs loaded with a 28 kDa protein: tissue inhibitor of matrix metalloproteinases 1 (TIMP-1). TIMP-1 is an endogenous inhibitor of matrix metalloproteinases (MMPs), which are enzymes capable of cleaving extracellular matrix, as well as membrane and secreted proteins, under physiological and pathological conditions. For instance, it has been shown that MMP-9 is elevated in various CNS disorders, including Alzheimer’s disease,5 multiple sclerosis,6 brain tumors,7 Guillain–Barré syndrome,8 spinal cord injury,9 ischemic stroke,10 epilepsy,11 and excitotoxic/neuroinflammatory processes.12 Moreover, MMP-9 inhibition by its chemical inhibitors has displayed beneficial effects.13 Therefore, it is suggested that MMP-9 inhibition can be a potential therapeutic target.14,15 At present, available MMP inhibitors are poorly specific and have a wide range of targets, and development of specific inhibitors is always a challenging task. Moreover, exogenous inhibitors may have unanticipated side effects. Therefore, TIMP-1, the endogenous inhibitor of MMPs with particularly strong affinity for MMP-9, holds great therapeutic hope. However, like most proteins, it does not cross the BBB. Furthermore, TIMP-1 in its native form has a short half-life and low bioavailability.16 Recently, there have been attempts to increase the bioavailability of TIMP-1 by polyethylene glycolation (PEGylation).17 Hence, we decided to formulate TIMP-1 in a PLGA-based nanoparticulate formulation to increase its bioavailability and enhance its brain delivery.
Herein, we show formulation optimization of TIMP-1-loaded PLGA NPs and coating with a surfactant (Ps80) to enhance their BBB penetration. These NPs were analyzed using various approaches, such as scanning electron microscopy (SEM), transmission electron microscopy (TEM), dynamic light scattering (DLS), polydispersity index (PDI), zeta potential, protein loading, and drug release. For BBB-penetration studies of NPs, we used in vitro BBB models based on monolayers of an endothelial cell line (rat brain endothelial 4 [RBE4]) and of primary rat brain capillary endothelial cells (RBCEC) to study TIMP-1 NP binding/uptake, toxicity, and passage across the BBB. Finally, we tested them in vivo, by injecting them intravenously via the tail vein in mice.
Materials and methods
Materials
For formulation of polymeric NPs, we used PLGA (molecular weight 45,000–75,000), copolymer ratio 50:50, polyvinyl alcohol (PVA; average molecular weight 30,000–70,000), bovine serum albumin (BSA), Coumarin 6 dye, dimethyl tartaric acid (DMT), dichloromethane (DCM), and Ps80 (Tween 80), all purchased from Sigma-Aldrich (St Louis, MO, USA).
TIMP-1 expression and characterization
We expressed recombinant mouse TIMP-1 as described in our earlier study.18 In brief, the TIMP1 gene was cloned along with Histag and expressed in the human embryonic kidney 293 T-cell line. Purification was done using Talon affinity chromatography (BD, Franklin Lakes, NJ, USA) and to remove imidazole from isolated protein, dialysis was performed at 4°C against 10 mM phosphate-buffered saline (PBS), pH 7.5. The expressed protein was characterized by Western blot, reverse zymography, and gelatinase assay. Purified TIMP-1 was formulated in PLGA NPs.
Formulation
We started by optimizing PLGA NPs loaded with the candidate protein (TIMP-1). For this purpose, different formulations were prepared considering PLGA concentration as a variable, and characterized for various physical parameters. Based on encapsulation efficiency, in vitro release, mean diameter, PDI, and zeta potential, the formulation was chosen for further in vitro studies.
The NPs were synthesized by multiple emulsion and solvent evaporation, modified from Reddy and Labhasetwar.19 In brief, five formulations with 1%–5% PLGA (50:50), ie, 10, 20, 30, 40, and 50 mg/mL (PLGA1, PLGA2, PLGA3, PLGA4, and PLGA5, respectively), were dissolved in 5 mL of DCM along with 4 mg of DMT. Separately, 500 μg of TIMP-1 and 1 mg of BSA in 500 μL of water were dissolved. The protein was emulsified using a microtip probe sonicator for 2 minutes in an ice bath at 55 W of energy output by dissolving DCM containing PLGA to make a primary emulsion, which was further emulsified in 20 mL of 1% PVA solution in water. In the formulation, BSA was used to stabilize the encapsulated TIMP-1 from interfacial inactivation and DMT was used to facilitate the release of TIMP-1 from NPs. Also, it has been shown that DMT might exert a stabilizing effect by steric inhibition of the interactions between adjacent NPs. In the second aqueous phase we used PVA, although it has been shown that it is difficult to remove PVA after the purification procedures, which eventually affect the physical properties and cellular uptake of NPs, as discussed by Panyam et al.20 As mentioned earlier, we adapted the formulation procedure from Reddy and Labhasetwar,19 who showed high entrapment efficiency and sustained release (up to 60 days) of a 32 kDa protein superoxide dismutase, and thus we followed their study, instead of using any other surfactant. This multiple emulsion was stirred overnight to evaporate DCM, and NPs were collected by centrifugation at 10,000 × g for 20 minutes at 4°C. The NPs were washed thrice using water, and supernatant was collected for protein-loading analysis.
We formulated control PLGA NPs carrying BSA as model protein and also Coumarin 6 dye-loaded NPs (which were used for in vitro BBB-penetration studies). The control NPs were made without TIMP-1 with the same procedure including BSA, and dye-loaded NPs were formulated using 50 μg of Coumarin 6 dye in 5 mL DCM. The particles were washed three times to remove PVA and then lyophilized (VirTis; SP Scientific, Warminster, PA, USA) for 48 hours to obtain a dry pellet. The NPs were analyzed by using SEM, TEM, DLS, PDI, zeta potential, protein loading, and drug release.
Characterization of nanoparticles
Scanning electron microscopy
For studying NP size and surface morphology, an S520 SEM (Hitachi, Tokyo, Japan) was used. A drop of concentrated aqueous suspension (20 mg freeze-dried TIMP-1 PLGA NPs in 10 mL double-distilled water) was spread over a slab and dried under vacuum. The sample was shadowed in a cathodic evaporator with a 20 nm-thick gold layer. The diameter and surface morphology of NPs in each field was observed.
Transmission electron microscopy
A JEM 1400 (JEOL, Tokyo, Japan) equipped with a high-resolution digital camera (charge-coupled device Morada; Olympus, Tokyo, Japan) was used for particle-size evaluation. A drop of the sample solution was placed onto a 400-mesh copper grid coated with carbon. About 1 minute after the deposit, the grid was tapped with filter paper to remove the surface water. The samples were air-dried before measurement.
Size distribution, PDI, and surface charge
Mean particle size, size distribution, PDI, and zeta potential of NPs were determined by photon correlation spectroscopy using a Zetasizer 3000 (Malvern Instruments, Malvern, UK) at a fixed angle of 90° and a temperature of 27°C by measuring electrophoretic mobility of NPs in a U-type tube. Before measurements, the TIMP-1 PLGA NPs were uniformly dispersed in double-distilled water. A suitably diluted aqueous dispersion of NPs was mounted in the instrument, and mean particle size distribution, PDI, and zeta potential were calculated. Each reported value is the average of six measurements.
Encapsulation, yield and in vitro protein-release kinetics
For encapsulation studies, we used an indirect method, as described previously.19 To determine percentage encapsulation efficiency of protein, NP yield, and actual protein loading, the following formulas were used:

For both studies, ie, encapsulation studies and in vitro release assay, we used the Bradford method for protein-concentration determination. In brief, the NPs were suspended in PBS (pH 7.4). Vials containing 10 mg of loaded PLGA NPs dispersed in 5 mL of PBS were incubated at 37°C on a constant shaking mixer. One vial was withdrawn at each time point (days 1, 2, 3, 5, and 7); the contents of the vial were centrifuged at 8,000 rpm for 10 minutes, and the supernatant containing released BSA/TIMP-1 was quantitated by the Bradford method. The Bradford reagent was obtained from Sigma-Aldrich; the procedure was followed according to the manufacturer’s instructions. For protein-stability studies, the same supernatant was used.
Protein stability
For determining protein stability inside NPs, sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot were performed. Based on single bands for TIMP-1, it was assumed that the released protein was stable after release from NPs. For this, the supernatant collected at different time points was run in 12% SDS-PAGE and transferred to polyvinylidene difluoride membrane. The membrane was blocked overnight at 4°C in 20 mM Tris-HCl, pH 7.6, 150 mM NaCl, and 0.02% Ps20 (TBST) containing 10% milk followed by washing three times in TBST for 10 minutes each. For detection, the membrane was incubated overnight in 1:200 dilution of mouse anti-TIMP-1 antibody (AF980; R&D Systems, Minneapolis, MN, USA), and rabbit polyclonal 6× Histag antibody (ab9108; Abcam, Cambridge, UK) in TBST at 4°C. The membrane was washed three times with TBST and incubated with horseradish peroxidase-conjugated secondary antibody, and the bands were visualized using ECL Plus reagent (GE Healthcare Bio-Sciences, Uppsala, Sweden) substrate solution. Recombinant mouse TIMP-1 with a 6× Histag (WBC022; R&D Systems) was used as a standard for both Western blots. The blots shown in this study are representative replicates selected from at least three independent experiments.
Evaluation of nanoparticles
The optimized formulation (PLGA3) was chosen based on its encapsulation efficiency and release kinetics for the rest of the in vitro studies, ie, for toxicity, binding/uptake, and penetration in RBE4 and primary RBCEC in vitro BBB models (schematic representation in Figure 1), and for in vivo studies following NP tail-vein injection.
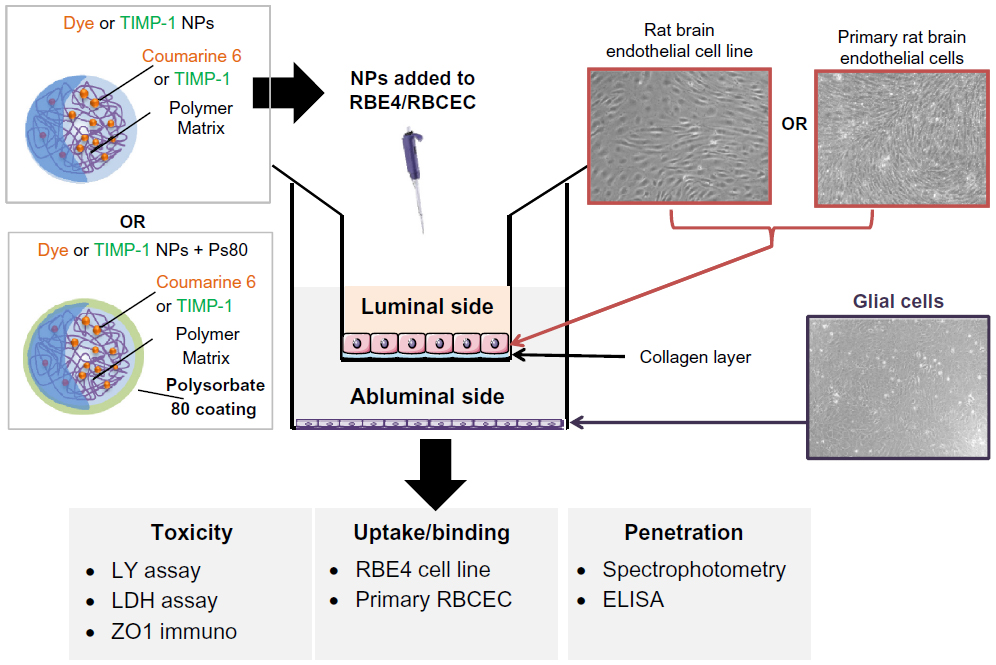 | Figure 1 Schematic illustration of experimental plan for in vitro evaluation of NPs: The Coumarin 6 dye-loaded NPs (Dye NPs) and TIMP-1 loaded NPs (TIMP-1 NPs) without Ps80 coating and with Ps80 coating (Dye NPs + Ps80 and TIMP-1 NPs + Ps80) were evaluated in vitro. The NPs were evaluated using a rat brain endothelial cell line (RBE4) and primary rat brain capillary endothelial cells (RBCEC) co-cultured with glial cells. The NPs were evaluated for toxicity, uptake/binding and penetration. For toxicity studies, LDH assay, LY assay, and ZO1 (tight junction marker) immunocytochemistry were performed. The uptake and binding studies were done on RBCEC. For penetration studies, fluorescence spectrophotometry was used for dye-loaded NPs and ELISA for TIMP-1 NPs. |
Polysorbate 80 coating of nanoparticles
Before all in vitro and in vivo experiments, the optimized NPs were coated with Ps80 to improve their BBB delivery. We evaluated BBB penetration of uncoated NPs (ie, TIMP-1 NPs or Dye NPs) with surfactant-coated NPs (TIMP-1 NPs + Ps80 or Dye NPs + Ps80). For Ps80 coating, the NPs were incubated with 1% (w/v) solution of Ps80 for 45 minutes immediately before in vitro or in vivo experiments, as described by Gelperina et al.4 For initial studies, we used Coumarin 6 dye-loaded NPs, as the dye is easier to detect, and for advanced studies we used TIMP-1 NPs.
Cell-culture preparation
For preliminary experiments, we used RBE4 cell line monoculture, and for advanced experiments we used RBCEC co-cultured with astrocytes.
Rat brain endothelial cell line monoculture
RBE4 cells are an immortalized cell line derived from RBE cells.21 RBE4 cells were seeded on rat tail collagen type I (3 μg/cm2; BD) on Millipore filters (polyethylene twelve-well, pore size 1.0 μm; Millipore, Billerica, MA, USA) at a density of 9 × 104 cells/cm2 and maintained in Eagle’s minimum essential medium -alpha with GlutaMax™ (Life Technologies, Carlsbad, CA, USA) and Ham’s F10 with GlutaMax (Life Technologies) both at 45% v/v supplemented with 10% fetal calf serum (CliniSciences, Nanterre, France), basic fibroblast growth factor (bFGF; 1 ng/mL; Life Technologies) and Geneticin® (300 μg/mL; Life Technologies). Under these conditions, confluent monolayers were established within 3 days.
Primary rat brain endothelial cells co-cultured with astrocytes
Primary cultures of astrocytes were prepared from newborn rat cerebral cortex. After removal of the meninges, the brain tissue was forced gently through a 70 μm nylon sieve. Dissociated glial cells were seeded into cell culture flasks and the culture media (Dulbecco’s Modified Eagle’s Medium [DMEM] supplemented with 10% fetal bovine serum, 100 units/mL antibiotic penicillin, and 100 μg/mL streptomycin) was replaced twice a week. After 1 week of proliferation, the glial cells were gently shaken for 24 hours to remove the microglial cells. Three weeks after seeding, astrocytes were passaged by treatment with trypsin and frozen in liquid nitrogen. Vials of astrocytes were defrosted 5 days before the establishment of the RBCEC co-culture at a density of 80 × 103 cells per well of a twelve-well plate.
RBCEC culture was adapted from previously described techniques.22–24 Briefly, the endothelial cells were seeded on type IV collagen and fibronectin (both 0.5 μg/cm2; BD) on Millipore filters (polyethylene twelve-well, pore size 1.0 μm) at a density of 1.6 × 105 cells/cm2 and maintained in DMEM/Ham’s F12 supplemented with 20% bovine platelet-poor plasma-derived serum named Endothelial Cell Media (ECM; CliniSciences, Nanterre, France), composed of bFGF, (2 ng/mL), heparin (100 μg/mL; Life Technologies), gentamicin (50 μg/mL; Life Technologies) and HEPES (4-[2-hydroxyethyl]-1-piperazineethanesulfonic acid; 2.5 mM; Life Technologies). Then, the filters were transferred into the wells containing the astrocytes and the culture medium was replaced with ECM supplemented with hydrocortisone at 500 nM for differentiation and expression of junction-related proteins. Under these conditions, in vitro models were established within 3 days.
In vitro investigations
The NPs with and without Ps80 were assessed for three in vitro evaluation parameters: first, toxicity by Lucifer yellow (LY) assay, lactate dehydrogenase (LDH) assay, and immunocytochemistry for ZO1 tight junction markers; second, uptake/binding studies by confocal microscopy of RBCEC; and third, BBB-penetration studies using RBE4 and RBCEC by determining the passage of NPs with and without Ps80 coating across the cell monolayers using spectrophotometry for Dye NPs and TIMP-1 enzyme-linked immunosorbent assay (ELISA) for TIMP-1 NPs.
Toxicity studies
To determine the toxicity of NPs on the BBB, we performed LY and LDH assays.
Lucifer yellow assay: LY (LY CH lithium salt; Sigma-Aldrich) is a small molecule known not to pass the BBB. LY was used as an integrity control of the barrier to ensure preservation of the barrier during the experiments and absence of toxicity of the compound that is co-incubated with LY. The inserts containing the RBCEC monolayer were gently washed and transferred to clean twelve-well plates. Both the upper and lower chambers were washed with prewarmed DMEM/F12 without phenol red. LY was incubated in the upper chamber (apical compartment) of the culture system in contact with endothelial cells for 60 minutes at 37°C. After this time, the medium of the lower chamber was collected, and fluorescence was quantified by fluorometric analysis with a Beckman DTX 800 Multimode Detector (Beckman Coulter, Inc., Pasadena, CA, USA) with excitation at 430/485 nm and emission at 535 nm. During each 60-minute experiment, the average cleared volume was plotted versus time, and the slope estimated by linear regression analysis. The slope of the clearance curve for the collagen IV fibronectin precoated control filter was denoted PSf, and the slope of the clearance curve for the culture was denoted PSt. The PS value for the endothelial monolayer (PSe) was calculated from:
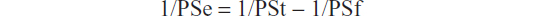
The PSe values were divided by the area of the porous membrane (1.1 cm2 for plates with 12-well Millipore inserts) to establish the permeability coefficient (Pe; 10–3 cm/minute). The results are presented in percentages compared to the control cells alone, expressed as 100%.
Lactate dehydrogenase assay: Cytotoxicity was assessed using the LDH Cytotoxicity Assay Kit II (Abcam). LDH is an enzyme that converts pyruvic acid to lactic acid, and it can be readily detected when cell membranes are no longer intact. For this assay, RBE4 cells were treated with different concentrations of Dye NPs (20, 2, and 0.2 μg/mL) and conditioned culture media was collected from the upper compartment at different time points (30 minutes and 120 minutes). For RBCEC, the highest concentration of NPs (20 μg/mL) was used, and medium was collected after 60 minutes. The activity of LDH in the conditioned cell-culture medium was measured in triplicate with a colorimetric assay at 450 nm wavelength.
Immunocytochemistry for ZO1: To visualize tight junction morphology, immunocytochemistry was performed using ZO1. The NPs were incubated with RBCEC in 20 μg/mL concentration, with or without Ps80 coating for 60 minutes. Cells were fixed by removing the medium from the cultures, washing the cells twice with PBS, and further incubating in 4% paraformaldehyde for 10 minutes and washing three times with PBS followed by mounting. The cells were permeabilized for 10 minutes with TBST followed by washing with PBS twice. Blocking was done using 3% BSA for 30 minutes followed by washing once with PBS again. The cells were then incubated with ZO1 and TIMP-1 (1:100) antibodies in a solution of 1% BSA for 1 hour at room temperature. After two washes, cells were incubated with secondary antibodies – Alexa 488, 594 (1:500) and Hoechst (1:1000) – for 30 minutes at room temperature, followed by three washes with PBS. Finally, cells were mounted with Vectashield® mounting media (Vector Laboratories, Burlingame, CA, USA) and visualized under confocal microscopy (LSM 780; Carl Zeiss, Jena, Germany) equipped with a 63× oil-immersion objective.
Nanoparticle uptake/binding
For NP-uptake/binding studies, we first tested dye-loaded NPs on the RBE4 cells. For this, NPs without Ps80 coating (Dye NPs) and with Ps80 coating (Dye NPs + Ps80) were incubated in different concentrations of 40, 4 and 0.4 μg/well. The cells were incubated at 37°C for 120 minutes followed by cutting of the insert membranes, fixing the cells, and mounting on slides.
Fluorescence microscopy and intensity analysis: Fluorescence photomicrographs were taken using a DMI 6000 B microscope (Leica Microsystems, Wetzlar, Germany) using Leica Application Suite Advanced Fluorescence software. Pictures were taken with a 2.5× optical zoom lens objective using an L5 blue excitation filter (450±64 nm) with a pixel resolution of 1,392×1,024.
Penetration of nanoparticles across cell monolayers
To evaluate NP penetration across the RBE4 or RBCEC cell monolayers, the Dye NPs and Dye NPs + Ps80 were incubated for 30/120 minutes at 37°C in different concentrations (20, 2, and 0.2 μg/well). To estimate BBB penetration we determined the amount of NPs in the lower compartment by fluorescence spectrophotometry. For TIMP-1 NPs and TIMP-1 NPs + Ps80, a TIMP-1 ELISA was used.
Fluorescence spectrophotometric assay: In the group incubated with Dye NPs or Dye NPs + Ps80, fluorescence spectrophotometry was done. For assessing the Coumarin 6 amount in the lower compartment, the medium was collected from the lower compartment from each group, and fluorescence was quantified by a microplate reader (444/505 nm). The assay was done using the SpectraMax® M5e (Molecular Devices, Sunnyvale, CA, USA). Each experiment was repeated at least three times.
TIMP-1 ELISA: For determining the penetration of NPs across cell monolayers, we determined the amount of TIMP-1 in the lower compartment. For this, we used a TIMP-1 Mouse ELISA Kit (Abcam). All reagents and samples were prepared as instructed by the supplier. The medium from the lower compartment of the cultures was collected after 30 minutes for the RBE4 cell line and 60 minutes for RBCEC. The NPs were further incubated overnight at 37°C to release the protein. The next day, protein release was determined by a TIMP-1 ELISA. The method and dilution of samples were followed as per the manufacturer’s instruction, and the plate was read at 450 nm in an ELISA 96-well plate reader.
In vivo evaluation of nanoparticles for BBB penetration
For in vivo evaluation of BBB penetration, TIMP-1 NPs + Ps80 were intravenously injected in mice, and immunocytochemistry for Histag was done to detect BBB penetration of TIMP-1 NPs + Ps80.
Animals
All the experiments with animals were carried out according to guidelines established by the Animal Ethical Committee of the Nencki Institute of Experimental Biology, based on national and EU laws. Special care was taken to minimize suffering and the number of animals used. For in vivo studies, we used twelve mice. The animals were kept in the laboratory animal facility, with free access to food and water, with a 12-hour light/dark cycle.
Intravenous injection and perfusion
For intravenous administration, TIMP-1 NPs + Ps80 were suspended in 500 μL of PBS (pH 7.4) to obtain the required concentration of TIMP-1. The intravenous injections were done using Bioseb® MTI Mouse Tail Illuminator (Bioseb, Vitrolles, France). Each animal was given intravenously a preparation containing 100 μg of TIMP-1 or PBS (pH 7.4) as vehicle. TIMP-1 loaded NPs were injected through the tail vein of mice (n=4). Three hours after injection, mice were injected with pentobarbital (200 mg/kg, intraperitoneally) and perfused transcardially, first with PBS and then with 4% paraformaldehyde. Brains were isolated, and tissue sectioning was done by cryoembedding the brain tissue in tissue freezing medium, (Tissue-Tek® Sacura, the Netherlands) and 300 μm-thick slices were cut.
Immunohistochemistry
For immunohistochemistry, slices were mounted on slides and blocked for 2 hours in 5% normal donkey serum in PBST. After 2 hours, slices were incubated overnight at 4°C with a rabbit polyclonal anti-6× Histag antibody (1:500) (Abcam). Subsequently, slices were washed with PBST for 15 minutes at room temperature. After washing, samples were incubated with a fluorescent Alexa 555 secondary antibody (1:500; Life Technologies) for 2 hours at room temperature and washed again with PBST 4 times for 10 min. Finally, slices were mounted with Vectashield mounting media, counterstained with DAPI (4′,6-diamidino-2-phenylindole) and visualized under a confocal microscope (TCS SP5; Leica Microsystems) equipped with a 40× oil-immersion objective using the 555 nm Argon laser (for excitation of Alexa 555) at a pixel resolution of 1,024×1,024 and 4.5 optical zoom. The Z-stacks of optical slices were acquired in 0.4 μm steps. The compiled Z-stack was analyzed using ImageJ software version 1.46r (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
The values for evaluation of physicochemical parameters in Tables 1 and 2 are means ± standard deviation, where n=3. The rest of the data in graphs are represented as means ± standard error of mean, and statistical comparisons were carried out between the groups using two sample t-test assuming unequal variances. A significance value of P<0.05 was accepted and considered as relevant.
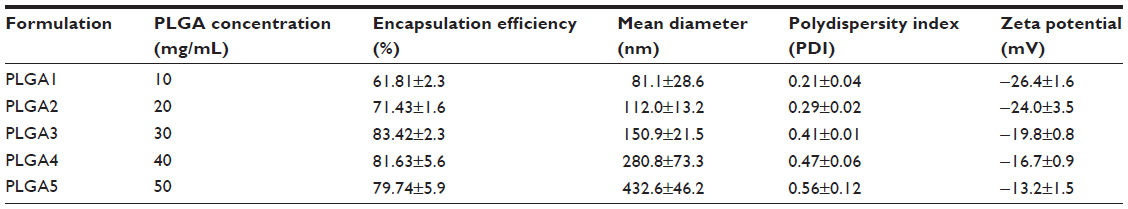 | Table 1 Physicochemical parameters of the TIMP-1-loaded PLGA nanoparticles with different polymer concentrations |
 | Table 2 Physicochemical parameters of PLGA3 formulation |
Results
Formulation optimization and characterization
We tested five different concentrations of 50:50 PLGA as a process variable (1% w/v to 5% w/v), keeping the remaining parameters constant. The formulations were characterized for various physical parameters: encapsulation efficiency, mean diameter, PDI, and zeta potential (Figure 2). On the basis of these parameters, the optimal formulation was chosen for the rest of the in vitro and in vivo experiments.
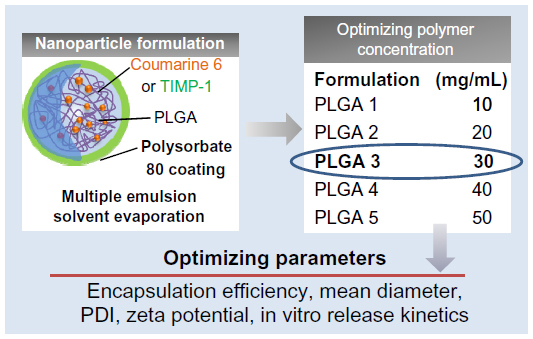 | Figure 2 Formulation optimization. |
Particle morphology, size, size distribution, PDI, and zeta potential
The particles were mostly spherical in shape, and mean size of TIMP-1 NPs was 81.1±28.6 nm for 1% w/v (for PLGA1), which increased to 432.6±46.2 nm for 5% w/v (for PLGA5) (see Table 1). The representative SEM and TEM for PLGA3 are shown in Figure 3A and B, respectively. The mean hydrodynamic diameter of NPs was much higher when measured by DLS, eg, PLGA3 NPs had a mean hydrodynamic diameter of 330 nm with a PDI of 0.159 (Figure 3C). This discrepancy in size between the SEM and DLS measurements may be due to hydration of the multilayered PVA that remained associated with the NPs at the interface despite repeated washing.19,25 The PDI and zeta potential were found to increase with the increase in PLGA concentration (Table 1). The results are in consensus with results published by Dey et al.26 The zeta potential of TIMP-1-NP PLGA3 formulation was −19.8±0.8 mV.
 | Figure 3 (A–C) Characterization results of PLGA3 NPs loaded with TIMP-1. (A) Representative scanning electron micrograph of TIMP-1-loaded PLGA NPs. (B) Representative transmission electron micrograph of TIMP-1-loaded PLGA NPs; scale bar 500 nm. (C) Particle-size analysis by dynamic light scattering. |
Encapsulation efficiency, nanoparticle yield, and actual protein loading
Based on the results of various formulations, increasing the amount of polymer (1%–3% w/v) increased the encapsulation efficiency, but further increasing the polymer concentration did not further increase encapsulation (Table 1).
The PLGA3 formulation (3% w/v) had maximum encapsulation efficiency, 83.42%±2.3%, ie, 83.42% of the added protein was entrapped into NPs. Percentage NP yield and percentage actual protein loading were calculated as described earlier by the formulas, and were found to be 65.43%±2.3% and 1.07%±0.5% respectively, for the PLGA3 formulation (Table 2). As the results show, the PLGA3 formulation with 30 mg/mL polymer concentration had the highest encapsulation efficiency.
In vitro release profile and protein stability
The in vitro release profiles of different formulations were assessed (Table 3) to identify the best release kinetics in order to select an optimal formulation. The release kinetics of all formulations demonstrated a similar sustained release, showing an initial burst followed by diffusion. The release of TIMP-1 from NPs was sustained; in particular, PLGA3 NPs had 21.0%±0.8% of release in 24 hours, and 72.0%±2.8% in 7 days (Figure 4). SDS-PAGE and Western blots were performed for protein stability, and we observed a single band for TIMP-1, which suggested that the released protein was stable after release from NPs. The representative blot for PLGA3 NPs is shown in the inset in Figure 4. The control formulations, using BSA as model protein, demonstrated almost identical physical properties as TIMP-1 NPs. Hence, here we show the characterization data only from TIMP-1-loaded NPs.
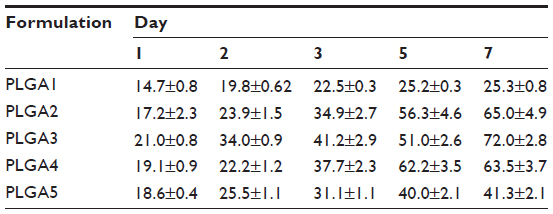 | Table 3 In vitro release profile of TIMP-1 loaded PLGA nanoparticles with different polymer concentrations |
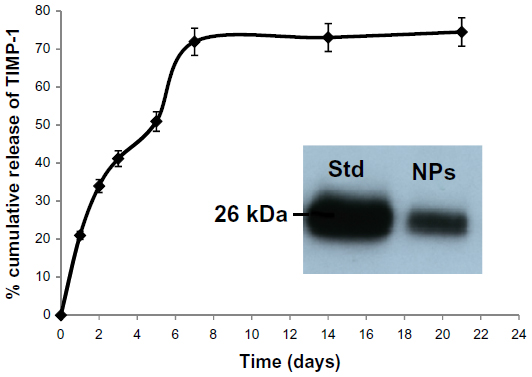 | Figure 4 TIMP-1 release and stability: release of TIMP-1 from optimal PLGA3 NPs under in vitro conditions. Inset is representative Western blot of the protein released from the NPs, suggesting that release of TIMP-1 is stable after 48 hours. Data are means ± standard error of mean; n=3. |
From the characterization data obtained, the PLGA3 formulation with 3% w/v polymer (Figure 2) was selected as optimal for the in vitro and in vivo experiments, considering encapsulation efficiency and in vitro release profile.
In vitro evaluation
In vitro studies were done on the RBE4 cell line and RBCEC for toxicity, uptake/binding and, BBB-penetration studies.
Toxicity studies
For studying toxicity of NPs on endothelial cells, we used an LY assay and an LDH assay. The LY assay on RBCEC showed that in neither of the cell inserts treated with Dye NPs/TIMP-1 NPs or Dye NPs + Ps80/TIMP-1 NPs + Ps80 was there significant increase of endothelial permeability due to toxicity (Figure 5A). Moreover, the LDH assay also showed that none of these NPs were toxic to the RBCEC (Figure 5B). As a complementary approach, we assessed the effects of NPs on the expression and distribution of the tight junction marker ZO1. The immunocytochemistry from RBCEC incubated with TIMP-1 NPs + Ps80 and TIMP-1 NPs showed that the NPs caused no significant change in ZO1 distribution (Figure 5C).
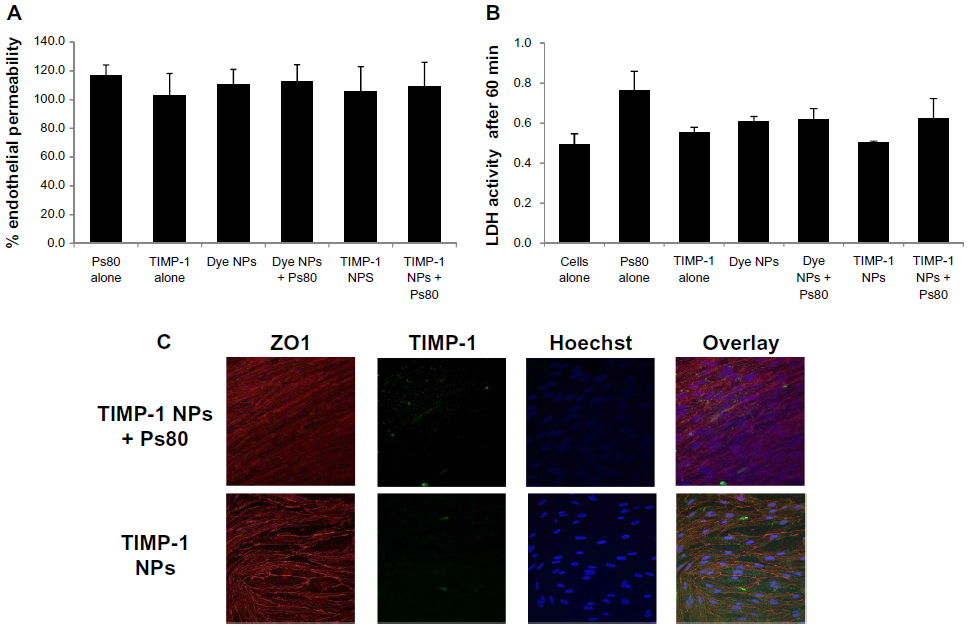 | Figure 5 (A–C) Toxicity studies. (A) LY assay on RBCEC, showing no significant impact of NPs on endothelial cell permeability for LY compared to control (no NPs), indicating that NPs caused no significant opening of the endothelial cell monolayer. Ps80 alone and TIMP-1 alone were used as controls. (B) LDH assay, also showing that NPs were not toxic to the RBCEC compared to control. Ps80 alone and TIMP-1 alone were used as controls. (C) Representative fluorescence photomicrographs of ZO1 immunocytochemistry on RBCEC treated with TIMP-1 NPs and TIMP-1 NPs + Ps80, indicating no significant difference between the groups. |
Uptake/binding of nanoparticles
In the NP-uptake/binding studies, we tested Dye NPs and Dye NPs + Ps80. The results showed that NPs that were coated with Ps80 had less binding to NPs compared to uncoated NPs on the RBE4 cell line (Figure 6). The binding of NPs was concentration-dependent, ie, both NP types at higher concentration bound more to the cells, and this value decreased as the concentration decreased.
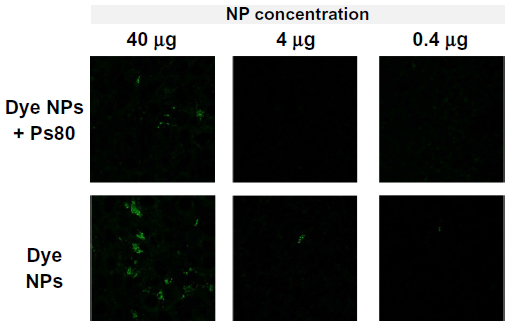 | Figure 6 Uptake/binding studies using Coumarin 6 dye-loaded NPs (Dye NPs): representative fluorescence photomicrographs showing that the Dye NPs (without Ps80 coating) have more binding/uptake to RBE4 compared to Dye NPs + Ps80 (with Ps80 coating) and hence we do not see any signal. With decrease in NP concentration, the binding is also decreased. |
In vitro BBB penetration
For penetration studies, the Dye NPs/Dye NPs + Ps80 were incubated in different concentrations with the RBE4 cell line, and after 60 minutes the amount of dye in the lower compartment was determined and compared to the initial amount of dye incubated. The results indicated (Figure 7A) that the Dye NPs (without Ps80 coating) did not penetrate across the monolayer. On the other hand, Dye NPs + Ps80 crossed the cell monolayer, and, unexpectedly the Dye NPs + Ps80 at lower concentration (4.0 μg/mL) had higher penetration, 23.99%±2.3%, than at the higher concentration (40.0 μg/mL), which was characterized by 3.34%±0.7% penetration. When we further lowered the Dye NPs + Ps80 concentration (down to 0.4 μg/mL), we observed no penetration. When the same experiment was done on the RBCEC with Dye NPs and Dye NPs + Ps80 with the 4 μg/mL concentration, Dye NPs + Ps80 showed 14.32%±2.31% penetration, while Dye NPs showed no penetration.
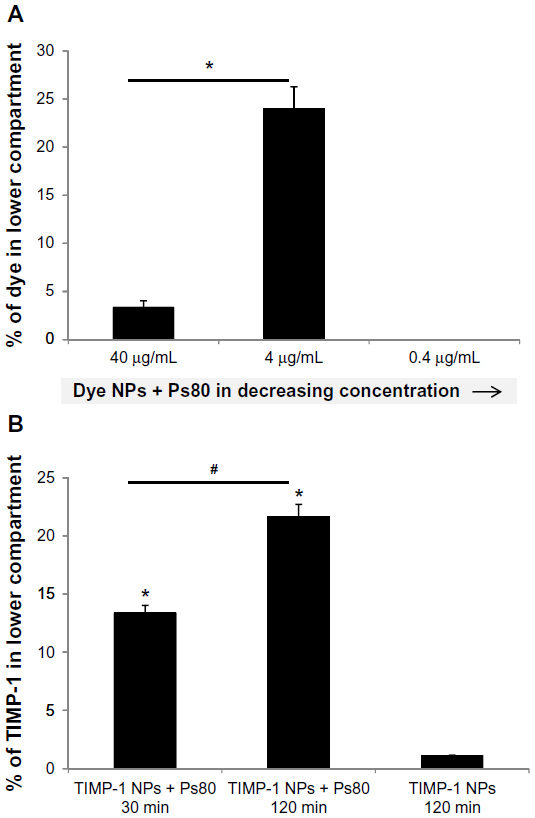 | Figure 7 In vitro blood–brain barrier penetration. |
For assaying penetration of TIMP-1 NPs and TIMP-1 NPs + Ps80, we used a TIMP-1 ELISA. NPs (4 μg/mL) were incubated on the RBE4 cell line monolayer, and samples of the lower-compartment media were taken at 30 minutes and 120 minutes. TIMP-1 content was compared to amounts of TIMP-1 in NPs based on actual protein loading in NPs. On the RBE4 cell line (Figure 7B), TIMP-1 NPs + Ps80 penetration across the cell monolayer was 13.3%±1.1% and 21.6%±1.7% after 30 and 120 minutes, respectively. Uncoated TIMP-1 NP penetration was only 1.10%±0.3%. On RBCEC, after 60 minutes, 11.21%±1.35% of TIMP-1 was detected in the lower compartment in cell inserts treated with TIMP-1 NPs + Ps80. TIMP-1 NPs and TIMP-1 (native protein alone) showed no permeability across the endothelial cell monolayer. The in vitro experiments provide strong evidence that with the help of the Ps80-coated NPs, protein could successfully traverse the BBB.
In vivo BBB penetration
For in vivo evaluation of NP passage across the BBB and/or accumulation in the nervous tissue, we injected the TIMP-1 NPs + Ps80 into the tail vein and cardiacally perfused the mice 3 hours later. The immunohistochemistry was done using a 6× Histag antibody, to recognize solely the recombinant TIMP-1 protein. The results showed that TIMP-1 NPs + Ps80 penetrated into the brain, as we found Histag signal in the brain sections (Figure 8). As a control, we used PBS as vehicle, and no signal was detected in this group.
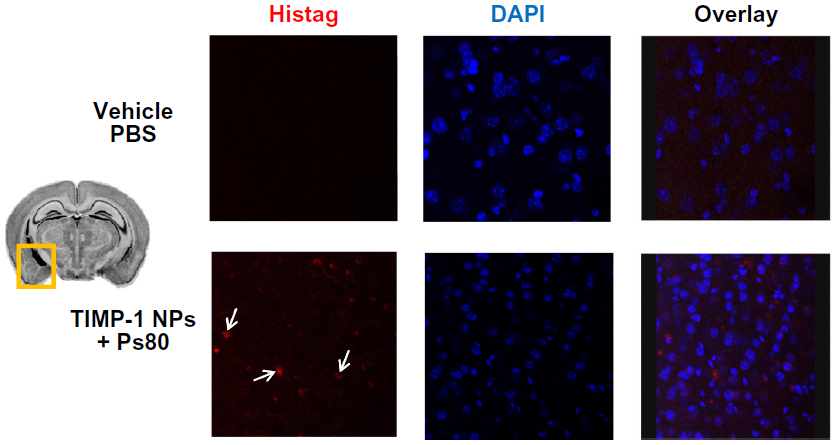 | Figure 8 In vivo blood–brain barrier (BBB) penetration. |
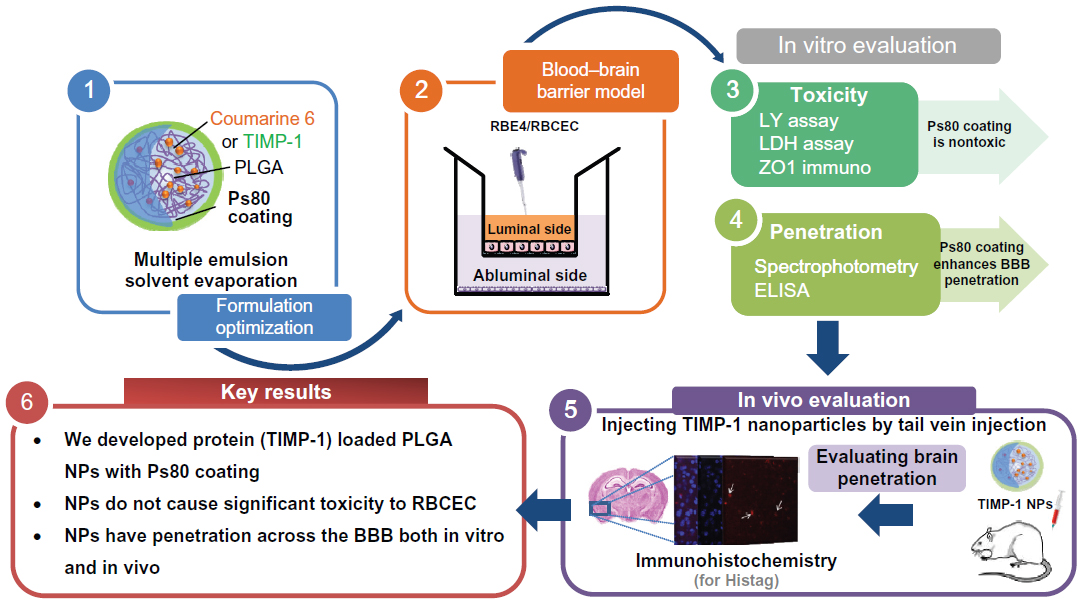 | Figure 9 Graphical Summary. |
Discussion
Proteins are the most rapidly growing class of pharmaceuticals for which controlled or targeted release is used to increase specificity, to lower toxicity, and to decrease the risk associated with treatment.27 The objective of this study was to develop protein-loaded PLGA NPs for brain delivery of a protein. The protein of interest in our study was TIMP-1, an inhibitor of MMP-9, a potential therapeutic target for various disorders (see earlier). However, native TIMP-1 has low bioavailability and does not cross the BBB. We assessed its delivery using PLGA NPs coated with Ps80, and we found that such entrapment of TIMP-1 allows for the sustained – over the course of days – protein release and the efficient penetration of TIMP-1 through the BBB, both in vitro and in vivo.
Since every protein is unique and there is no a priori approach for precise prediction of the entrapment levels and possibly protein stabilization in the NPs, we started with formulation optimization for TIMP-1 NPs based on entrapment efficiency, in vitro release, and other parameters. Hence, we made five different formulations considering PLGA concentration as a variable, while other formulation parameters remained constant. The PLGA3 NPs were chosen as optimal based on various physicochemical parameters. In light of the optimization results presented in this study, modification of PLGA concentration in formulation has significant effects on encapsulation efficiency, particle size, and in vitro release. Initially, increase in polymer concentration increases particle size with increase in encapsulation efficiency, as reported previously with protein-loaded PLGA NPs.28 However, in our study, further increase in polymer concentration did not increase encapsulation efficiency, although the particle size indeed increased.
Release of cargo from NPs takes place through several mechanisms, including surface and bulk erosion, disintegration, diffusion, and desorption. In our study, the five different PLGA formulations with concentrations ranging from 1% to 5% showed sustained release of protein from the PLGA matrix. The initial burst effect was observed with all of the five formulations, which indicated a homogeneous encapsulation of TIMP-1 in the PLGA matrix. During the later phases, the release was mediated through both diffusion of protein and degradation of the polymer matrix itself. PLGA3 formulation with 3% polymer showed maximum cumulative release of protein (almost 72%) from the polymer matrix after 7 days.
For enhancing BBB penetration of NPs, we coated the NPs with Ps80 (Dye NPs + Ps80 and TIMP-1 NPs + Ps80), as Ps80 has been shown to enhance the penetration of NPs across endothelial cells.4 Protein-loaded NPs with Ps80 coating have never been evaluated for delivery of TIMP-1. Therefore, we evaluated NP-based TIMP-1 delivery both in vitro and in vivo. Toxicity studies using LDH assay, LY assay, and ZO1 immunocytochemistry for both Ps80-coated and uncoated NPs suggest that the NPs are nontoxic and do not affect tight junction organization, although one of the various hypothesized mechanisms of BBB penetration of NPs is solubilization of the endothelial cell-membrane lipids by the surfactant used on formulation.29 Our toxicity results can be interpreted in various ways. Firstly, if solubilization of cell-plasma membranes occurs, it does not induce significant toxicity to be detected by the LDH assay. Secondly, Ps80-coated NPs may penetrate the endothelial cells by solubilization of endothelial membrane by the surfactant, which may cause reversible cytotoxicity for a short time, which we could not detect in our results. The reversible-disruption hypothesis was also mentioned earlier by Rempe et al in their experiment using Ps80-coated PBCA NPs.30 Finally, it may be possible that the surface solubilization is not one of the mechanisms for Ps80-coated NPs to penetrate through endothelial cells, as we did not detect cytotoxicity. Instead, there may be receptor-mediated transcytosis or some other mechanism involved in transportation.
Uptake/binding studies from NPs revealed that in in vitro conditions, NPs without Ps80 coating had better binding to endothelial cells as compared to Ps80-coated NPs. This is in contrast with the results generated by the penetration studies of NPs across the cell monolayer, where NPs + Ps80 had a systematic higher penetration rate, while uncoated NPs showed very low or no penetration. This suggests that although uncoated NPs had higher binding on cells, they stayed entrapped and did not penetrate across the cell monolayer. We therefore expect that coating with hydrophilic Ps80 (similar to PEGylation) may extend the circulation time of NPs. Further investigation on uptake mechanisms, drug-uptake kinetics, and retention in the endothelial cells using NPs compared to NPs coated with Ps80 in vivo will be useful to establish the efficacy of NPs in therapeutic applications.
The in vitro BBB-penetration experiments showed strong evidence that the NPs with Ps80 coating have the ability to cross cell monolayers of primary endothelial cells. As shown in the result, due to some unknown reasons, only an optimum concentration of Ps80-coated NPs (4 μg/mL) had peak penetration, while increasing or decreasing the concentration of Ps80-coated NPs decreased the penetration. These results remain unexplained, and further study in this direction may give an insight into the mechanism of transport of these NPs. The transport mechanisms of NPs across the endothelial cell monolayer are yet to be understood;31 there are various hypothesized mechanisms in the literature, such as the surfactants used in the coating of NPs may solubilize the endothelial cell-membrane lipids, thus enhancing NP transport.32,33 Another hypothesized mechanism is through adsorption of the surface of plasma proteins (like apolipoprotein E) on the NP surface, which can lead to NP uptake by endothelial cells via the low-density lipoprotein receptor.29 Likewise, in our case, the Ps80 coating may have promoted adsorption of plasma proteins on the NP surface, leading to BBB uptake and transcytosis via receptors involved in receptor-mediated transcytosis, such as members of the low-density lipoprotein-receptor family. The immunohistochemistry results following in vivo studies showed direct evidence that the NPs have some BBB-penetration properties. Further studies are needed to understand the mechanism of NP transport across the BBB in vivo and to assess the pharmacological potential of TIMP-1 delivery into the brain.
Future perspectives
Therapeutic potential of TIMP-1
As mentioned earlier, there is enhanced expression and activity of MMP-9 in numerous CNS disorders, such as ischemic stroke, epilepsy, and excitotoxic/neuroinflammatory processes, which causes deleterious effects. Therefore, it is suggested that MMP-9 inhibition can be used for neuroprotection. At present, available MMP-9 inhibitors are poorly specific and have a wide range of targets. Development of specific inhibitors is always a challenging task. Moreover, exogenous inhibitors may have unanticipated side effects. Therefore, TIMP-1, being an endogenous inhibitor of MMP-9, has therapeutic potential.15 TIMP-1 has shown neuroprotective effects in various in vitro and in vivo studies using viral vectors. The major obstacle of using TIMP-1 as a therapeutic agent is its short half-life in vivo and brain permeability. There have been attempts to extend its bioavailability, such as by PEGylation,17 so in future it can be developed as a neuroprotective. We believe that Ps80-coated PLGA NPs may significantly improve in bioavailability and will be able to be used as a neuroprotective in future. Previously, we have shown that these TIMP-1-loaded NPs have neuroprotective effects in organotypic hippocampal cultures.18 Furthermore, we have also shown their ability to control MMP-9 activity in the brain, following direct injection.34 In the future, we would like to do further in vivo studies using Ps80-coated TIMP-1 PLGA NPs to evaluate their pharmacological effects.
Brain delivery of NPs
For improving brain delivery of NPs, there are various approaches, such as appending endothelial receptor-specific antibodies, which can improve brain penetration through transcytosis. However, these strategies require chemical conjugation involving complexity in NP formulation. While Ps80 coating of PLGA NPs is a fast and simple method to improve the brain penetration of drugs, including macromolecules as we have shown here, this method should be explored more.
Conclusion
To our knowledge, this is the first study showing delivery of a protein (TIMP-1) using Ps80-coated NPs across the BBB. We developed TIMP-1-loaded PLGA NPs and coated them with Ps80 for enhancing BBB penetration. We performed toxicity studies of the NPs, which suggested that these NPs do not cause significant toxicity to RBCEC. Further, we tested uptake/binding of these NPs on RBE cells, which showed NPs without Ps80 have more binding to cells compared to NPs with Ps80 coating. The study also demonstrated that Ps80 coating of NPs can be used to enhance the delivery of protein across endothelial cell barriers, both in vitro and in vivo. Further studies are required to evaluate their penetration across the BBB.
Acknowledgments
This study was supported by European regional development funds of the European Union funded through the Foundation of Polish Science (grant MPD4-5071). Also, we would like to thank the Laboratory of Electron Microscopy at Nencki Institute for their help. YM acknowledges the support of the French National Research Agency (ANR; VECtoBrain project). MK acknowledges support from the National Center for Scientific Research (CNRS), Aix-Marseille Université (AMU), and grants from the ANR (TIMPAD, VECtoBrain, and PREVENTAD). Finally, we would like to thank COST Action ECMNet (BM1001) for providing STSM fellowship for MC’s stay in UMR 7259 – CNRS Marseille, France.
Disclosure
Michel Khrestchatisky is director of the UMR7259 National Center for Scientific Research Aix-Marseille Université Laboratory and founder and shareholder in the biotechnology company Vect-Horus. The other authors report no conflicts of interest in this work.
References
Vlieghe P, Khrestchatisky M. Medicinal chemistry based approaches and nanotechnology-based systems to improve CNS drug targeting and delivery. Med Res Rev. 2012;33(3):457–516. | |
Mundargi RC, Babu VR, Rangaswamy V, Patel P, Aminabhavi TM. Nano/micro technologies for delivering macromolecular therapeutics using poly(lactide-co-glycolide) and its derivatives. J Control Release. 2008;125(3):193–209. | |
Panyam J, Labhasetwar V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv Drug Deliv Rev. 2003;55(3):329–347. | |
Gelperina S, Maksimenko O, Khalansky A, et al. Drug delivery to the brain using surfactant-coated poly(lactide-co-glycolide) nanoparticles: influence of the formulation parameters. Eur J Pharm Biopharm. 2010;74(2):157–163. | |
Lorenzl S, Albers DS, Relkin N, et al. Increased plasma levels of matrix metalloproteinase-9 in patients with Alzheimer’s disease. Neurochem Int. 2003;43(3):191–196. | |
Sang QX, Muroski M, Roycik M, et al. Matrix metalloproteinase-9/gelatinase B is a putative therapeutic target of chronic obstructive pulmonary disease and multiple sclerosis. Curr Pharm Biotechnol. 2008;9(1):34–46. | |
Rooprai H, McCormick D. Proteases and their inhibitors in human brain tumours: a review. Anticancer Res. 1997;17(6B):4151–4162. | |
Sharshar T, Durand MC, Lefaucheur JP, et al. MMP-9 correlates with electrophysiologic abnormalities in Guillain-Barré syndrome. Neurology. 2002;59(10):1649–1651. | |
Zhang H, Chang M, Hansen CN, Basso DM, Noble-Haeusslein LJ. Role of matrix metalloproteinases and therapeutic benefits of their inhibition in spinal cord injury. Neurotherapeutics. 2011;8(2):206–220. | |
Clark AW, Krekoski CA, Bou SS, Chapman KR, Edwards DR. Increased gelatinase A (MMP-2) and gelatinase B (MMP-9) activities in human brain after focal ischemia. Neurosci Lett. 1997;238(1–2):53–56. | |
Yin P, Yang L, Zhou HY, Sun RP. Matrix metalloproteinase-9 may be a potential therapeutic target in epilepsy. Med. Hypotheses. 2011;76(2):184–186. | |
Jourquin J, Tremblay E, Decanis N, et al. Neuronal activity-dependent increase of net matrix metalloproteinase activity is associated with MMP-9 neurotoxicity after kainate. Eur J Neurosci. 2003;18(6):1507–1517. | |
Koistinaho M, Malm TM, Kettunen MI, et al. Minocycline protects against permanent cerebral ischemia in wild type but not in matrix metalloprotease-9-deficient mice. J Cereb Blood Flow Metab. 2005;25(4):460–467. | |
Kaczmarek L. MMP-9 inhibitors in the brain: can old bullets shoot new targets? Curr Pharm Des. 2012;19(6):1085–1089. | |
Chaturvedi M, Kaczmarek L. MMP-9 inhibition: a therapeutic strategy in ischemic stroke. Mol Neurobiol. Epub September 12, 2013. | |
Sa Y, Hao J, Samineni D, et al. Brain distribution and elimination of recombinant human TIMP-1 after cerebral ischemia and reperfusion in rats. Neurol Res. 2011;33(4):433–438. | |
Batra J, Robinson J, Mehner C, et al. PEGylation extends circulation half-life while preserving in vitro and in vivo activity of tissue inhibitor of metalloproteinases-1 (TIMP-1). PLoS One. 2012;7(11):e50028. | |
Chaturvedi M, Figiel I, Sreedhar B, Kaczmarek L. Neuroprotection from tissue inhibitor of metalloproteinase-1 and its nanoparticles. Neurochem Int. 2012;61(7):1065–1071. | |
Reddy MK, Labhasetwar V. Nanoparticle-mediated delivery of superoxide dismutase to the brain: an effective strategy to reduce ischemia-reperfusion injury. FASEB J. 2009;23(5):1384–1395. | |
Panyam J, Dali MM, Sahoo SK, et al. Polymer degradation and in vitro release of a model protein from poly(d,l-lactide-co-glycolide) nano- and microparticles. J Control Release. 2003;92(1–2):173–187. | |
Roux F, Durieu-Trautmann O, Chaverot N, et al. Regulation of gamma-glutamyl transpeptidase and alkaline phosphatase activities in immortalized rat brain microvessel endothelial cells. J Cell Physiol. 1994;159(1):101–113. | |
Roux FS, Mokni R, Hughes CC, Clouet PM, Lefauconnier JM, Bourre JM. Lipid synthesis by rat brain microvessel endothelial cells in tissue culture. J Neuropathol Exp Neurol. 1989;48(4):437–447. | |
Roux F, Couraud PO. Rat brain endothelial cell lines for the study of blood-brain barrier permeability and transport functions. Cell Mol Neurobiol. 2005;25(1):41–57. | |
Deli MA, Ábrahám CS, Kataoka Y, Niwa M. Permeability studies on in vitro blood-brain barrier models: physiology, pathology, and pharmacology. Cell Mol Neurobiol. 2005;25(1):59–127. | |
Prabha S, Zhou WZ, Panyam J, Labhasetwar V. Size-dependency of nanoparticle-mediated gene transfection: studies with fractionated nanoparticles. Int J Pharm. 2002;244(1–2):105–115. | |
Dey SK, Mandal B, Bhowmik M, Ghosh LK. Development and in vitro evaluation of Letrozole loaded biodegradable nanoparticles for breast cancer therapy. Braz J Pharm Sci. 2009;45(3):585–591. | |
Makadia HK, Siegel SJ. Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers. 2011;3(3):1377–1397. | |
Bilati U, Allémann E, Doelker E. Poly(D, L-lactide-co-glycolide) protein-loaded nanoparticles prepared by the double emulsion method-processing and formulation issues for enhanced entrapment efficiency. J Microencapsul. 2005;22(2):205–214. | |
Kreuter J. Mechanism of polymeric nanoparticle-based drug transport across the blood-brain barrier (BBB). J Microencapsul. 2013;30(1):49–54. | |
Rempe R, Cramer S, Hüwel S, Galla HJ. Transport of poly(n-butylcyano- acrylate) nanoparticles across the blood-brain barrier in vitro and their influence on barrier integrity. Biochem Biophys Res Commun. 2011;406(1):64–69. | |
Reimold I, Domke D, Bender J, Seyfried CA, Radunz HE, Fricker G. Delivery of nanoparticles to the brain detected by fluorescence microscopy. Eur J Pharm Biopharm. 2008;70(2):627–632. | |
Kreuter J. Nanoparticulate systems for brain delivery of drugs. Adv Drug Deliv Rev. 2001;47(1):65–81. | |
Kreuter J. Influence of the surface properties on nanoparticle-mediated transport of drugs to the brain. J Nanosci Nanotechnol. 2004;4(5):484–488. | |
Knapska E, Lioudyno V, Kiryk A, et al. Reward learning requires activity of matrix metalloproteinase-9 in the central amygdala. J Neurosci. 2013;33(36):14591–14600. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.