Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 12
Tiotropium bromide exerts anti-inflammatory effects during resistive breathing, an experimental model of severe airway obstruction
Authors Toumpanakis D, Loverdos K, Tzouda V, Vassilakopoulou V ![]() , Litsiou E, Magkou C, Karavana V
, Litsiou E, Magkou C, Karavana V ![]() , Pieper M, Vassilakopoulos T
, Pieper M, Vassilakopoulos T
Received 29 March 2017
Accepted for publication 19 June 2017
Published 28 July 2017 Volume 2017:12 Pages 2207—2220
DOI https://doi.org/10.2147/COPD.S137587
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Richard Russell
Dimitrios Toumpanakis,1,2 Konstantinos Loverdos,1,2 Vassiliki Tzouda,1,2 Vyronia Vassilakopoulou,1,2 Eleni Litsiou,1,2 Christina Magkou,3 Vassiliki Karavana,1,2 Michael Pieper,4 Theodoros Vassilakopoulos1,2
1First Critical Care Department, Pulmonary Unit, National and Kapodistrian University of Athens Medical School, Evangelismos General Hospital, 2George P. Livanos and Marianthi Simou Laboratories, Thorax Foundation, 3Department of Pathology, Evangelismos General Hospital, Athens, Greece; 4Boehringer Ingelheim Pharma GmbH & Co. KG Div. Research Germany, Biberach, Germany
Introduction: Resistive breathing (RB), a hallmark of obstructive airway diseases, is characterized by strenuous contractions of the inspiratory muscles that impose increased mechanical stress on the lung. RB is shown to induce pulmonary inflammation in previous healthy animals. Tiotropium bromide, an anticholinergic bronchodilator, is also shown to exert anti-inflammatory effects. The effect of tiotropium on RB-induced pulmonary inflammation is unknown.
Methods: Adult rats were anesthetized, tracheostomized and breathed spontaneously through a two-way non-rebreathing valve. Resistances were connected to the inspiratory and/or expiratory port, to produce inspiratory resistive breathing (IRB) of 40% or 50% Pi/Pi,max (40% and 50% IRB), expiratory resistive breathing (ERB) of 60% Pe/Pe,max (60% ERB) or combined resistive breathing (CRB) of both 40% Pi/Pi,max and 60% Pe/Pe,max (40%/60% CRB). Tiotropium aerosol was inhaled prior to RB. After 6 h of RB, mechanical parameters of the respiratory system were measured and bronchoalveolar lavage (BAL) was performed. IL-1β and IL-6 protein levels were measured in lung tissue. Lung injury was estimated histologically.
Results: In all, 40% and 50% IRB increased macrophage and neutrophil counts in BAL and raised IL-1β and IL-6 lung levels, tissue elasticity, BAL total protein levels and lung injury score. Tiotropium attenuated BAL neutrophil number, IL-1β, IL-6 levels and lung injury score increase at both 40% and 50% IRB. The increase in macrophage count and protein in BAL was only reversed at 40% IRB, while tissue elasticity was not affected. In all, 60% ERB raised BAL neutrophil count and total protein and reduced macrophage count. IL-1β and IL-6 levels and lung injury score were increased. Tiotropium attenuated these alterations, except for the decrease in macrophage count and the increase in total protein level. In all, 40%/60% CRB increased macrophage and neutrophil count in BAL, IL-1β and IL-6 levels, tissue elasticity, total protein in BAL and histological injury score. Tiotropium attenuated the aforementioned alterations.
Conclusion: Tiotropium inhalation attenuates RB-induced pulmonary inflammation.
Keywords: resistive breathing, inflammation, tiotropium bromide
Introduction
Diseases of airway obstruction, such as asthma and COPD, are characterized by significant alterations in the lung mechanics, mainly increased airway resistance and hyperinflation. During severe COPD exacerbations, resistive breathing (RB) becomes the major pathophysiologic mechanism leading to increased work of breathing and hyperinflation. During RB, strenuous contractions of the inspiratory muscles to overcome the increased load of breathing create increased negative intrathoracic pressures that lead to significant mechanical stress on the lung.1 Following mechanical stress on the lung, there is an increase in alveolar-capillary membrane permeability and activation of inflammatory processes.2 Our group has shown that various forms of RB, pose an injurious insult to the lung, inducing acute lung injury, inflammation and respiratory failure to previously healthy rats3,4 and mice.5 The response to mechanical stress is not solely due to mechanical disruption of the integrity of the alveolar-capillary membrane (stress membrane failure model)6 but can also be mediated by the activation of cellular pathways (cellular mechanotransduction).7
Anticholinergics are currently the major inhaled bronchodilators (together with beta2-agonists) used for the treatment of diseases of airway obstruction, such as COPD and asthma. Tiotropium bromide, a once-daily inhaled muscarinic receptor antagonist, improves lung function in COPD patients and reduces exacerbation risk,8,9 although the mechanism of exacerbation risk reduction is not established. The effect(s) of treatment with tiotropium during severe COPD exacerbations has not been adequately studied.
Cumulative evidence in recent years has shown a potential interaction of the cholinergic system with inflammation in diseases of airway obstruction.10,11 Inflammatory cells express muscarinic receptors,12 and tiotropium exhibits anti-inflammatory properties. Human alveolar macrophages release chemotactic mediators in response to acetylcholine, a phenomenon attenuated by tiotropium.13 Tiotropium exerted anti-inflammatory action in an experimental murine model of asthma14 and prevented allergen-induced airway remodeling.15 Moreover, tiotropium reduced lung inflammation in a gastroesophageal reflux-induced lung injury murine model16 and cigarette-induced lung inflammation in mice.17 However, in a human study with COPD patients, tiotropium failed to reduce markers of airway or systemic inflammation.18 The potential anti-inflammatory effects of tiotropium during severe COPD exacerbations have not been studied.
Thus, more studies are needed to investigate the potential anti-inflammatory action of tiotropium bromide. We hypothesized that tiotropium bromide exerts anti-inflammatory effects in RB-induced pulmonary inflammation. To investigate our hypothesis, we used an in vivo model of acute RB in previous healthy rats, including both inspiratory resistive breathing (IRB) or expiratory resistive breathing (ERB) and their combination, to model (in previous healthy lungs) the mechanical consequences of increased airway resistance, as the airway resistance observed in severe COPD exacerbations.3,4
Methods
Subjects
Adult female rats (age 8–12 weeks, weight 200–250 g) were used in the study. All procedures were in accordance with the European Union Directive for care and use of laboratory animals and were approved by the Greek Veterinary Administration and the ethics committee of Evangelismos Hospital. Animal number per group is noted in figure legends.
Acute RB model in rats
An animal model of acute RB was set, as previously described.1,3 Briefly, rats were anesthetized with an intraperitoneal (ip) injection of a mixture of ketamine (75 mg/kg) and xylazine (5 mg/kg) and tracheostomized (tracheal cannula, 14G). After a short stabilization period (~20 min), the tracheal cannula was connected to a two-sided non-rebreathing valve (Hans-Rudolph, Shawnee, KS, USA). With the use of a pressure transducer, the tracheal pressure was monitored, and the maximum inspiratory pressure (Pi,max) was measured during spontaneous breathing efforts through a totally occluded inspiratory port for 10 s (DirecWin; Raytech Instruments Inc., Vancouver, BC, Canada). The maximum expiratory pressure (Pe,max) was also measured during spontaneous breathing efforts through a totally occluded expiratory port for 10 s.
Three models of RB were used, and a 6-h time frame was used based on previous findings of RB-induced acute lung injury.3
Inspiratory resistive breathing
The inspiratory port of the valve was connected to a gauge (resistance), the internal diameter of which was chosen to provide a peak tidal inspiratory pressure (Pi) of 40% or 50% of maximum (Pi,max).
ERB
The expiratory port of the valve was connected to a gauge (resistance), the internal diameter of which was chosen to provide a peak tidal expiratory pressure (Pe) of 60% of maximum (Pe,max).
Combined (expiratory and inspiratory) resistive breathing (CRB)
Resistances (gages of adequate diameter) were connected to both the inspiratory and expiratory ports to provide a peak tidal inspiratory pressure at 40% of maximum and a peak tidal expiratory pressure at 60% of maximum.
During RB, 100% of oxygen was supplemented to prevent hypoxemia. Quietly breathing animals (breathing 100% of oxygen against no load for an equal time frame) served as controls (ctrs). During experiment, additional doses of anesthesia (30 mg/kg ketamine ip) were supplemented, as needed.
Tiotropium administration
Prior to resistive or quietly breathing (30 min), rats received a nebulized dose of an aqueous solution of tiotropium bromide (1 mg/mL for 3 min) using a jet nebulizer (PARI TurboBOY, Starnberg, Germany), which produced a mean particle diameter of 3.5 μm. Animals that did not receive tiotropium were exposed to vehicle alone. The selection of dose was based on consultation with Boehringer Ingelheim and a pilot study (data not shown), where this dose was efficient in blunting airway reactivity to methacholine challenge in our model.
Evaluation of lung injury and pulmonary inflammation
Respiratory system mechanics
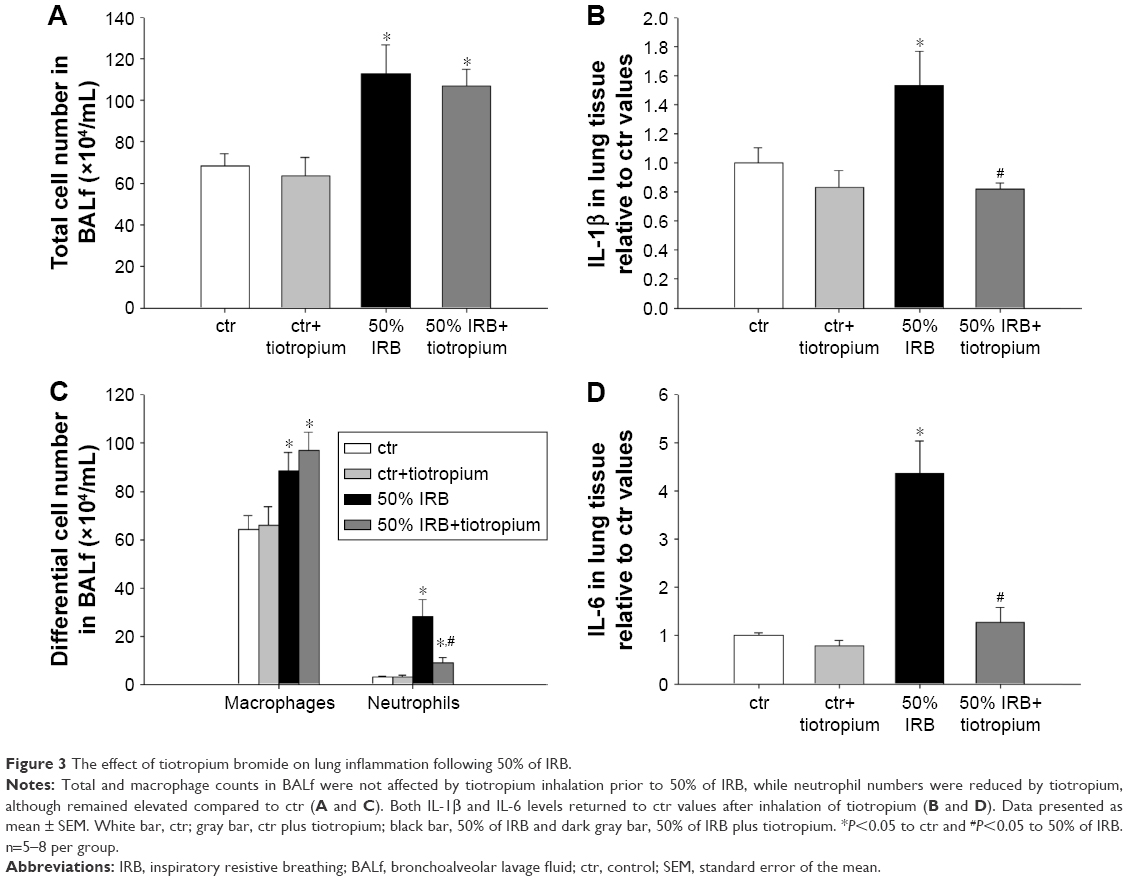
The mechanical properties of the respiratory system were estimated with the use of the forced oscillation technique and static pressure–volume (P–V) curves. Following the end of RB session or quietly breathing for ctr animals, the animals received an additional dose of anesthesia (ketamine 75 mg/kg, xylazine 10 mg/kg ip), the resistances and the two-way valve were removed and the tracheal cannula was connected to a small animal ventilator (SCIREQ, Montreal, QC, Canada). After 3 min of ventilation (tidal volume: 10 mL/kg, respiratory rate: 90 breaths·min−1, positive end-expiratory pressure [PEEP]: 3 cm H2O, FiO2: 0.21), a dose of succinylcholine (8 mg/kg ip) was administered to cease spontaneous breathing. Prior to measurements, a deep inflation to 30 cm H2O was performed to standardize lung volume history, and during measurements, the heart rate was monitored to ensure adequate depth of anesthesia. Measurements of respiratory impedance were performed with the low-frequency forced oscillation technique, as previously described.3 Impedance (Z) was then fitted to constant phase model:19
|
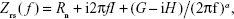
where Rn is the Newtonian resistance of the airways, i is the imaginary unit, f is the frequency, I is the inertance of the gas in the airways, G represents tissue viscance (viscous dissipation of energy), H represents tissue elasticity and alpha can be calculated through the equation α=(2/π)arctan(H/G).
A static P–V curve was also constructed, and the static compliance of the respiratory system (Cst) was derived from the mid linear segment of the expiratory part of the P–V curve. Hysteresis was also calculated.
Following the measurement of respiratory system mechanics, the animals were sacrificed by exsanguination (vena cava dissection) under anesthesia and the thoracic cavity was exposed. The left lung was temporarily ligated, and the right lung was lavaged by gentle instillation of three aliquots of 2.5 mL normal saline. Approximately 7 mL of bronchoalveolar lavage fluid (BALf) was withdrawn and was immediately centrifuged (300× g, 10 min, 4°C). The supernatant was stored at −80°C. The cell pellet was resuspended to 1 mL of normal saline. Then, the right lung was removed and stored at −80°C for further analysis. The left lung was fixed with 4% formaldehyde under 20 cm H2O pressure, was removed and after 24 h was embedded in paraffin using conventional techniques.
BALf analysis
Total cell count in resuspended BALf and differential cell count on Giemsa-stained cytospins were performed, and percentages of macrophages, neutrophils, eosinophils, basophils and lymphocytes were determined. Total protein concentration of BALf was also estimated, as an index of alveolar-capillary protein leakage, using a colorimetric protein assay according to the manufacturer (Bio-Rad Laboratories Inc., Hercules, CA, USA).
Protein levels of inflammatory cytokines
Interleukin (IL)-6 and IL-1β were measured with ELISA from lung tissue protein extracts, with the standard protocol supplied by the manufacturer (DuoSet; R&D Systems, Inc., Minneapolis, MN, USA). Protein extraction was performed, as previously described.3
Histology
A total lung injury score was estimated in hematoxylin and eosin (H&E)-stained lung tissue sections based on the following histological features: 1) septal thickening; 2) capillary congestion; 3) intra-alveolar hemorrhage; 4) interstitial neutrophil infiltration and 5) intra-alveolar neutrophil infiltration. Each feature was scored from 0 to 3 based on its absence (0) or presence to a mild (1), moderate (2) or severe (3) degree.3
Statistical analysis
Results are presented as mean ± standard error of the mean (SEM). Statistical analysis was performed with analysis of variance (ANOVA) and Fisher’s least significant difference (LSD) post hoc test (Statistica software; StatSoft Inc., Tulsa, OK, USA). Data from histology were ordinal and were analyzed with the non-parametric Kruskal–Wallis ANOVA and Mann–Whitney U-test for post hoc analysis. A P-value <0.05 was chosen as statistically significant.
Results
IRB – load 40%
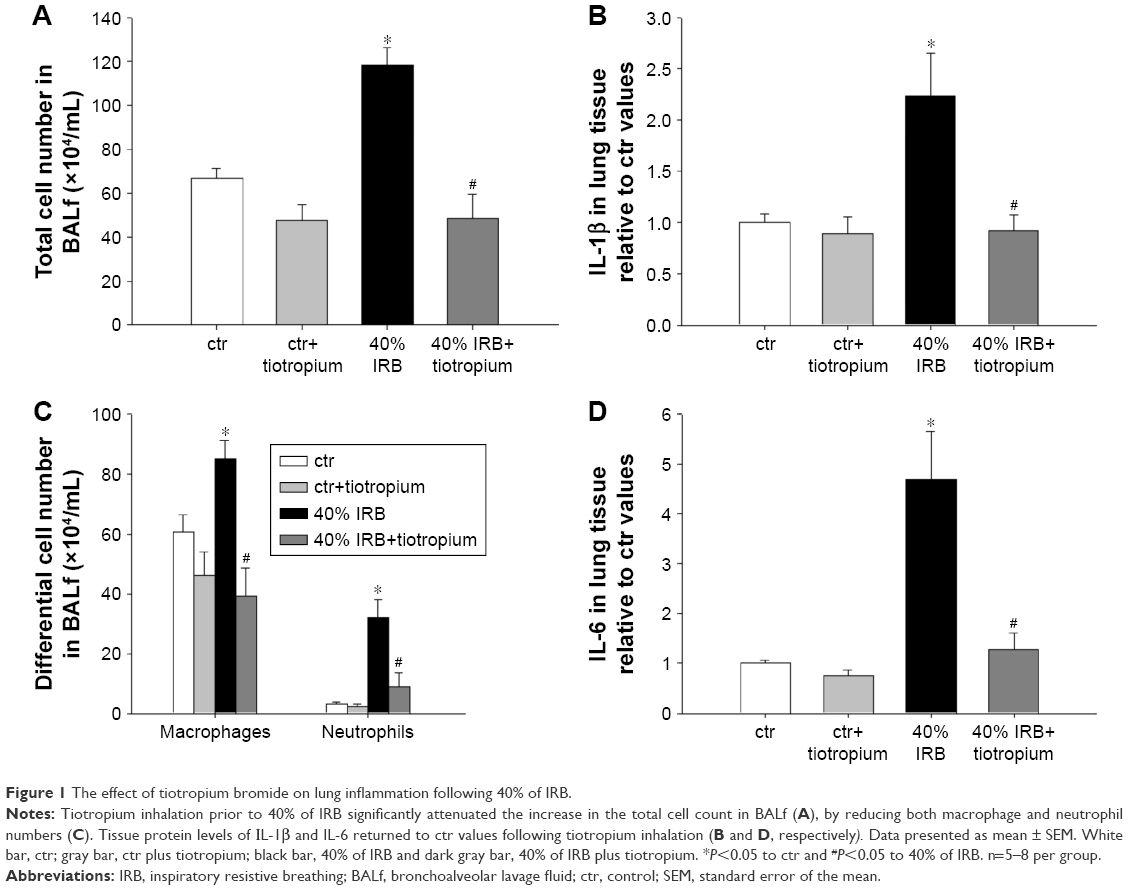
IRB at 40% of Pi,max was associated with induction of pulmonary inflammation. In detail, after 6 h of 40% IRB, increased total cell number was found in the BALf compared to ctr (P<0.001; Figure 1A) due to elevation of both macrophages and neutrophils (P=0.02 and P=0.001, respectively; Figure 1C). Moreover, 6 h of 40% of IRB resulted in increased protein levels of IL-1β and IL-6 in the lung tissue (P=0.002 and P<0.001, respectively; Figure 1B and D). Total protein in BALf was significantly increased after 40% of IRB, compared to ctr (P<0.001; Figure 2A). Regarding respiratory system mechanics, 6 h of 40% IRB was followed by an increase in tissue elasticity (P=0.04) (Figure 2B) and a decrease in static compliance (P<0.001). Airway resistance, tissue viscance and P–V curve hysteresis were not affected by 40% IRB (Table 1). In all, 6 h of 40% of IRB increased total lung injury score in H&E lung tissue section compared to ctr (P=0.002; Figure 2C). Regarding the different histological features, the increase in total lung injury score was mainly due to an increase in capillary congestion, intra-alveolar hemorrhage and interstitial neutrophil infiltration.
 | Figure 1 The effect of tiotropium bromide on lung inflammation following 40% of IRB. |
 | Figure 2 The effect of tiotropium bromide on lung permeability and injury following 40% of IRB. |
 | Table 1 Effect of tiotropium on respiratory system mechanics after 6 h of 40% IRB |
Effects of tiotropium on 40% IRB
Tiotropium bromide inhalation prior to 40% IRB significantly attenuated IRB-induced pulmonary inflammation. Total cell, macrophages and neutrophils counts were reduced to ctr levels after tiotropium inhalation compared to 40% IRB without tiotropium (P<0.001, P<0.001 and P=0.004, respectively; Figure 1A and C). Tiotropium inhalation also reduced IL-1β and IL-6 protein levels in lung tissue after 40% IRB (P=0.001 and P<0.001, respectively; Figure 1B and D). In addition, tiotropium attenuated the increase in bronchoalveolar lavage (BAL) protein following 40% of IRB (P=0.02; Figure 2A). In contrast, tiotropium inhalation did not influence the increase in tissue elasticity after 40% of IRB (Figure 2B and Table 1). Following tiotropium inhalation, no difference in total lung injury score, compared to ctr values, was observed. Indeed, after tiotropium inhalation, there was a tendency (P=0.07) for a reduction in total lung injury score, compared with 6 h of 40% IRB (Figure 2C).
IRB – load 50%
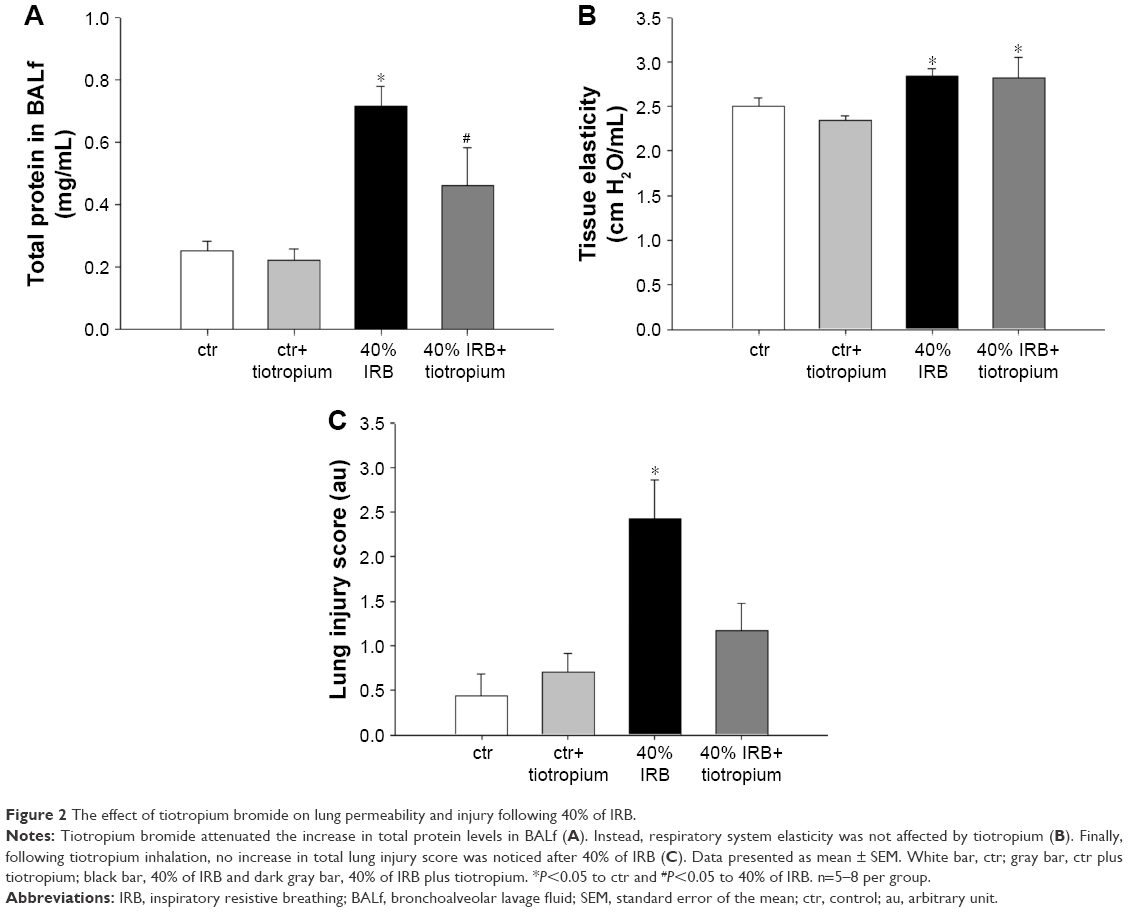
In all, 6 h of 50% IRB resulted in a significant increase in total cell number in BALf, compared to ctr values (P=0.004; Figure 3A), due to an increase in both macrophage and neutrophil counts (P=0.02 and P<0.001, respectively; Figure 3C). Moreover, lung tissue levels of the inflammatory cytokines IL-1β and IL-6 raised after 6 h of 50% of IRB (P<0.001 for both; Figure 3B and D). In all, 50% IRB was also associated with increased lung permeability, as seen by the increased total protein levels in BALf, compared to ctr (Figure 4A). Moreover, 50% of IRB led to a significant deterioration of the mechanical parameters of the respiratory system by increasing tissue viscance and tissue elasticity (P=0.002 and P<0.001, respectively). In accordance, static compliance was also decreased after 50% IRB (P<0.001), and the hysteresis of P–V curve of the respiratory system was increased, compared to ctr (P=0.01; Table 2). In all, 50% of IRB resulted in an increased total lung injury score, compared to ctr values (P=0.02; Figure 4C), mainly due to an increase in septal thickening, intra-alveolar hemorrhage and interstitial neutrophil infiltration and capillary congestion to a lesser degree.
 | Figure 3 The effect of tiotropium bromide on lung inflammation following 50% of IRB. |
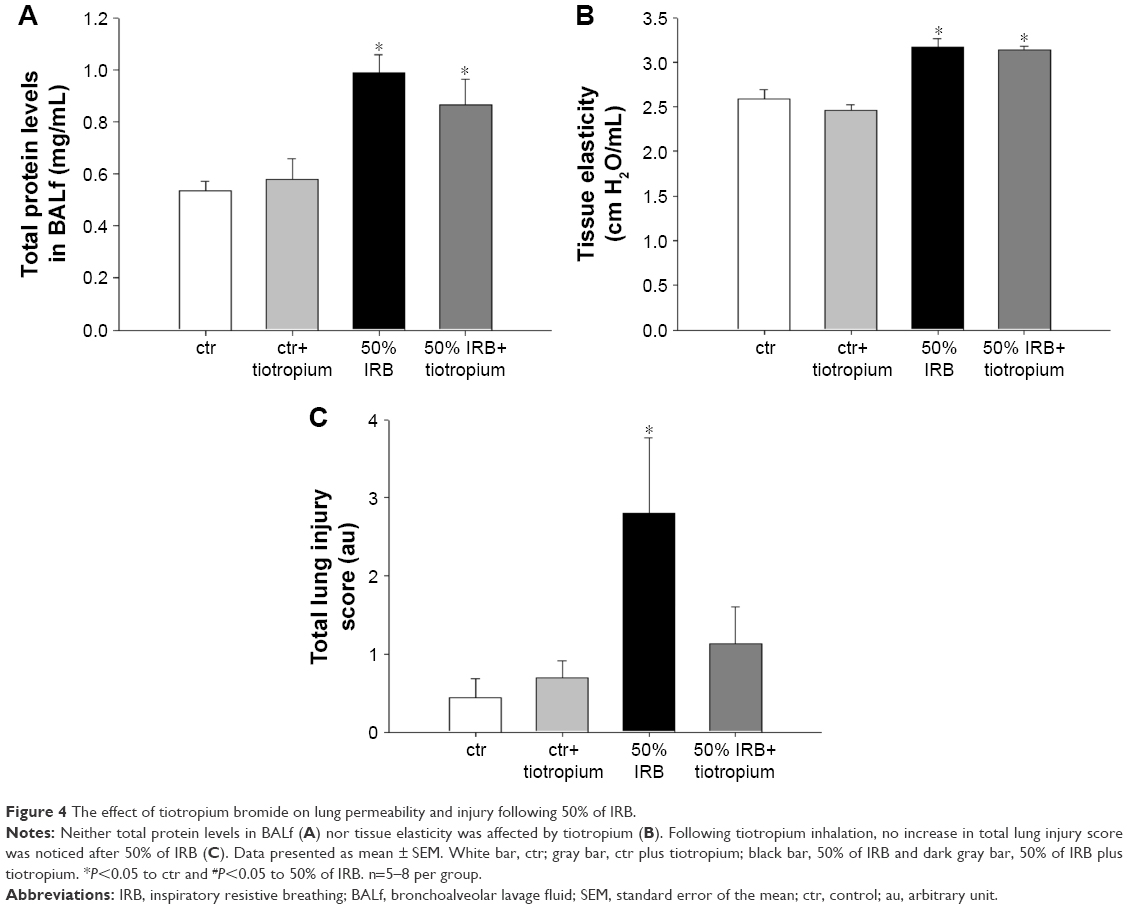 | Figure 4 The effect of tiotropium bromide on lung permeability and injury following 50% of IRB. |
 | Table 2 Effect of tiotropium on respiratory system mechanics after 6 h of 50% IRB |
Effects of tiotropium on 50% IRB
Tiotropium bromide inhalation prior to 50% of IRB did not affect total cell and macrophage number in BALf. Neutrophil count, although remaining significantly increased compared to ctr (P=0.006), was also reduced compared to 50% of IRB (P<0.001; Figure 3A). Tiotropium also attenuated the increase of IL-1β and IL-6 following 50% IRB (P=0.001 and P<0.001; Figure 3B and C). Following tiotropium, no alteration was noticed in protein levels in BALf or most of the mechanical parameters of the respiratory system compared to 50% IRB without tiotropium inhalation (Figure 4B and D and Table 2). However, it should be noted that tiotropium significantly reduced P–V curve hysteresis (P=0.01). Following tiotropium, the elevation in the lung injury score after 50% of IRB was attenuated (Figure 4C).
ERB – load 60%
In all, 6 h of ERB at 60% of Pe,max (60% ERB) did not alter total cell number in BALf compared to ctr (Figure 5A). However, 60% of ERB significantly altered differential cell count by decreasing the number of macrophages (P=0.002) and increasing the number of neutrophils in the BALf (P<0.001), compared to ctr (Figure 5C). Compared to ctr, 6 h of 60% ERB was also associated with an increase in the protein levels of IL-1β and IL-6 (P=0.01 and P<0.001, respectively; Figure 5B and D). After 6 h of 60% ERB, no difference in tissue elasticity or static lung compliance was noticed compared to ctr values. However, an unexpected rise in P–V curve hysteresis was found after 60% of ERB compared to ctr (P=0.01; Table 3). Moreover, total protein levels were increased after 6 h of 60% of ERB compared to ctr values (Figure 6A). In all, 6 h of 60% ERB resulted in an increased total lung injury score, mainly due to an observed rise in interstitial neutrophil infiltration and capillary congestion, compared to ctr values (P=0.001; Figure 6C).
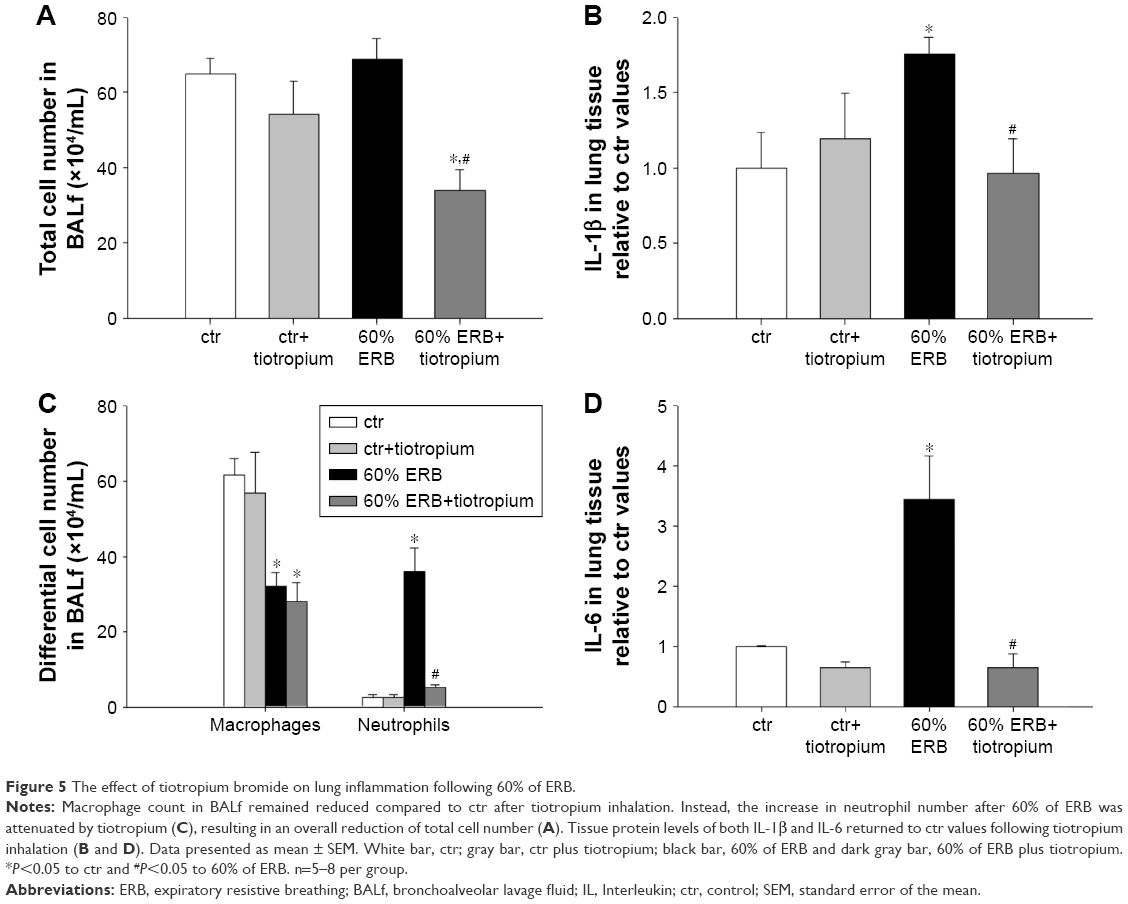 | Figure 5 The effect of tiotropium bromide on lung inflammation following 60% of ERB. |
 | Table 3 Effect of tiotropium on respiratory system mechanics after 6 h of 60% ERB |
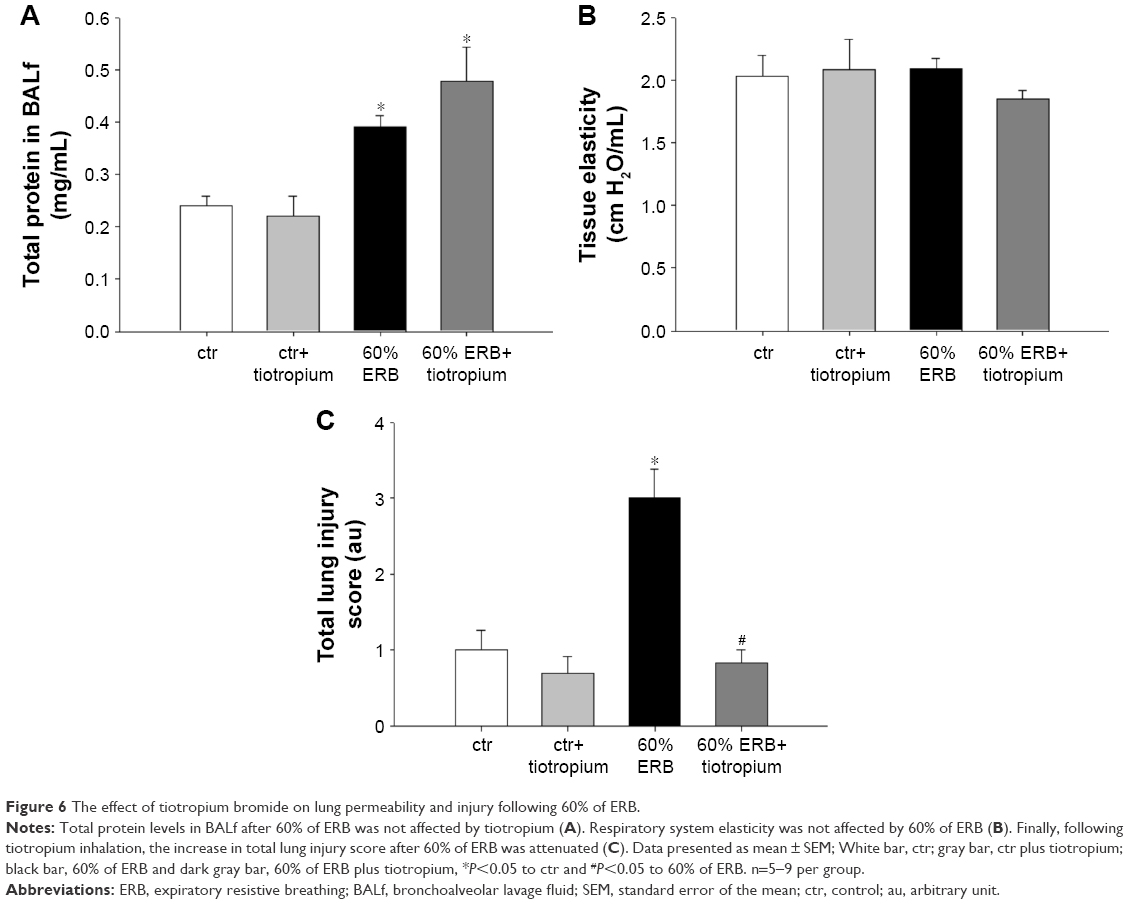 | Figure 6 The effect of tiotropium bromide on lung permeability and injury following 60% of ERB. |
Effect of tiotropium on 60% ERB
Tiotropium bromide inhalation prior to 60% of ERB resulted in a significant attenuation of the effects of ERB to the lung. More explicitly, tiotropium bromide inhibited the increase in the neutrophil number secondary to 60% of ERB (P<0.001) but did not alter the reduction in the macrophage count (Figure 5C). As a consequence, the total cell number after tiotropium inhalation was also decreased compared to both ctr and 6 h of 60% ERB (P=0.005 and P=0.002, respectively; Figure 5A). Regarding cytokine production following 6 h of 60% ERB, tiotropium bromide attenuated the increase in both IL-1β and IL-6 (P=0.01 and P<0.001 compared to 60% of ERB, respectively; Figure 5B and D). Tiotropium bromide inhalation had no effect on tissue elasticity (Figure 6B), static compliance (Table 3) or total protein levels in BALf (Figure 6A). However, the increase in hysteresis of the P–V curve was prevented after tiotropium inhalation (P=0.003 compared to 60% of ERB; Table 3). Following tiotropium inhalation, a significant reduction in total lung injury score was measured, compared to 6 h of 60% ERB (P<0.001; Figure 6C).
CRB – load 40%/60%
CRB at a load of 40% of Pi,max and 60% of Pe,max resulted in induction of pulmonary inflammation, as indicated by increased BALf cellularity (P<0.001; Figure 7A), due to infiltration by both macrophages and neutrophils (P=0.01 and P<0.001, respectively; Figure 7C). Moreover, increases in protein levels of IL-1β and IL-6 (P<0.001 and P=0.008, respectively; Figure 7B and D) in lung tissue were noticed. Total protein in BALf was also increased (P=0.009; Figure 8A), and respiratory system elasticity raised, as measured by both the forced oscillation technique and static P–V curves (P=0.02 and P=0.01, respectively; Table 4). CRB was also associated with an increased total lung injury score compared to ctr values (P<0.001; Figure 8C), which was mainly due to an increase in septal thickening, capillary congestion and interstitial neutrophil infiltration.
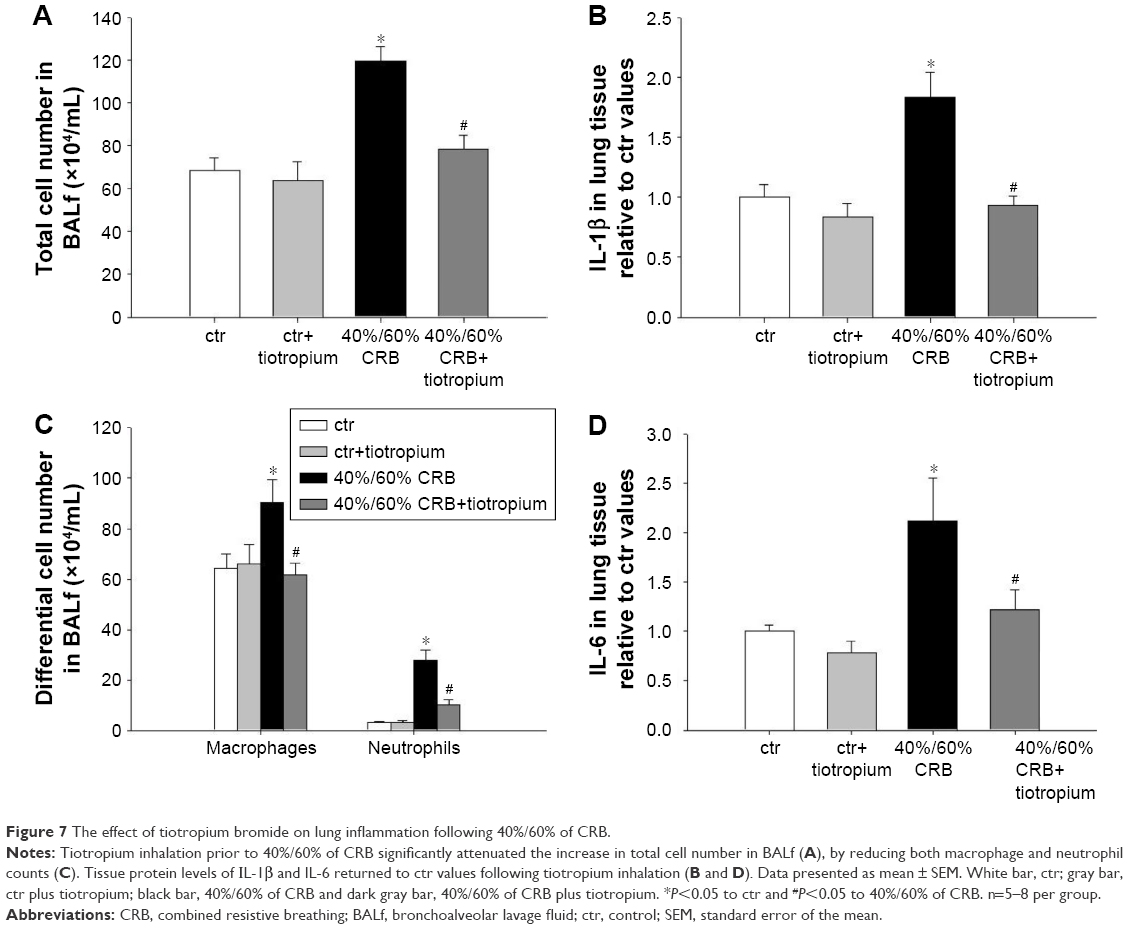 | Figure 7 The effect of tiotropium bromide on lung inflammation following 40%/60% of CRB. |
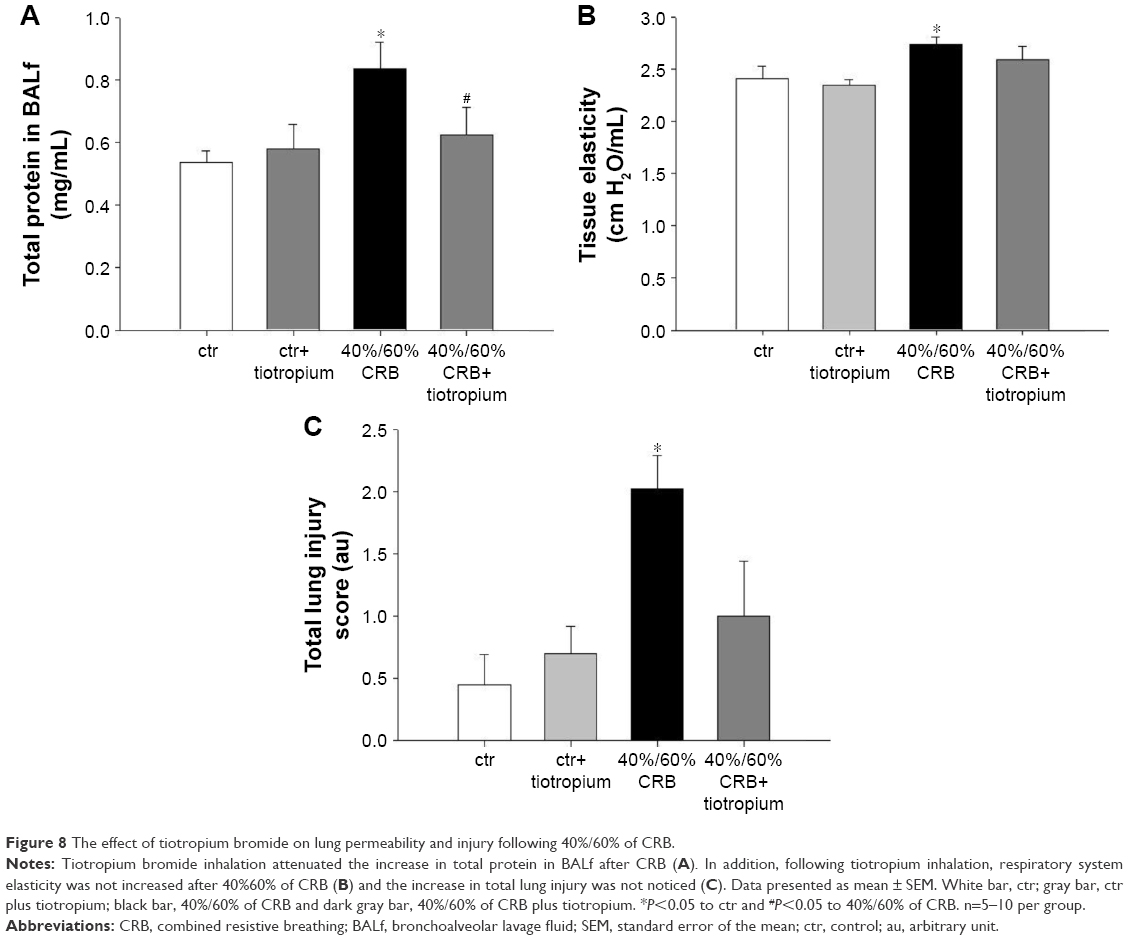 | Figure 8 The effect of tiotropium bromide on lung permeability and injury following 40%/60% of CRB. |
 | Table 4 Effect of tiotropium on respiratory system mechanics after 6 h of 40%/60% CRB |
Effects of tiotropium on 40%/60% CRB
Tiotropium bromide inhalation significantly prevented the increase in BAL cellularity (P<0.001 to 6 h of 40%/60% CRB) by reducing both macrophage and neutrophil counts (P=0.01 and P<0.001 to 6 h of 40%/60% CRB, respectively; Figure 7C). Indeed, after tiotropium inhalation, there was only a tendency (P=0.06) for increased neutrophil count compared to ctr. Moreover, tiotropium attenuated the increase in IL-1β and IL-6 after 6 h of CRB (P<0.001 and P=0.03 to 40%/60% CRB; Figure 7B and D, respectively). Tiotropium bromide reduced total protein level in BALf after 6 h of 40%/60% CRB (P=0.04; Figure 8A). Finally, regarding the mechanical parameters of the respiratory system, a moderate effect of tiotropium bromide inhalation was noticed, since tissue elasticity and static compliance were not statistically different compared to either ctr or 6 h of 40%/60% of CRB after tiotropium inhalation (Figure 8B and Table 4). Following tiotropium inhalation, no significant change in total lung injury score over ctr values was observed (Figure 8C).
Discussion
The major findings of our study are as follows: 1) RB in its various forms induces pulmonary inflammation and 2) tiotropium bromide inhalation prior to RB prevents various aspects of the RB-induced pulmonary inflammation.
RB is the hallmark of diseases of airway obstruction, especially during exacerbations. RB produces large negative intrathoracic pressures during inspiration, due to the strenuous contractions of the inspiratory muscles, which may provoke significant mechanical stress to lung-resident cells. Indeed, our research group has previously shown in an experimental model that moderate-to-severe inspiratory resistive loading at 50% of maximum can induce acute pulmonary inflammation in previously healthy rats.3
However, obstructive lung diseases, such as COPD and asthma, are also characterized by expiratory flow limitation and subsequent hyperinflation. Intrinsic PEEP (PEEPi) induction during expiration, as a result of expiratory loading addition to inspiratory, could oppose to the large negative intrathoracic pressures producing pulmonary edema during inspiration,20 thus preventing the occurrence of lung injury observed after IRB alone. On the other hand, a potential proinflammatory role for dynamic hyperinflation per se has been proposed, mainly attributable to cellular stretch.21 Our data suggest that the combination of ERB and IRB exerts similar proinflammatory and injurious action, while ERB alone at the load we used, although able to induce BAL neutrophilia and cytokine upregulation in the BAL, did not induce other indices of lung injury. This is in accordance with a recently published study by our group.4 Moreover, the injurious effect of CRB has been also shown in a murine model of RB through tracheal banding for 24 h.5
The anti-inflammatory effect of tiotropium in our experimental models suggests that an activation of the cholinergic signaling may exist during RB under our experimental settings. The source of this activation may be dual. First, resistive loading has been shown to activate vagal sensory pathways.22 Second, cumulative evidence has also suggested the presence of a non-neuronic cholinergic system, since both lung epithelial and inflammatory cells have been shown to release acetylcholine and to express functional muscarinic and nicotinic acetylcholine receptors (AchR).23 In vitro, inflammatory and apoptotic signaling after mechanical stretch of human bronchial epithelial cells was affected by the activity of a7 nicotinic AchR, suggesting that cholinergic pathways may be activated in lung-resident cells by mechanical stress per se.24
Interestingly, reduction in neutrophil infiltration following various forms of acute RB was a consistent effect of tiotropium inhalation, noticed even after the most severe inspiratory obstruction of 50% of Pi,max. Acetylcholine has been found to induce neutrophilic chemotactic activity,25 and M3 receptors are also expressed in neutrophils of sputum samples of COPD patients.25 Tiotropium bromide attenuated the release of chemotactic mediators in vitro, a finding in accordance with the reduced influx of neutrophils seen in our model of RB following tiotropium inhalation.13 Moreover, in an in vivo model of cigarette smoke exposure in mice, tiotropium bromide exerted an anti-neutrophilic effect,17 mediated by inhibition of muscarinic M3 receptors on structural lung cells.26 Blockage of M3 AchR has been also shown to attenuate BAL neutrophilia in a mouse model of lipopolysaccharide (LPS)-induced pulmonary inflammation.27
The reduction in the protein levels of the inflammatory cytokines, IL-1β and IL-6, was also a consistent protective effect of tiotropium inhalation prior to RB. Tiotropium has also been reported to reduce IL-6 levels in the BALf of cigarette smoke-exposed mice.17 In contrast, tiotropium failed to reduce IL-6 protein levels in the sputum of COPD patients, although this may be attributed to the reduction in sputum production by tiotropium, causing decreased sputum volume and increased concentration of solutes.18
Some indices of the preventive action of tiotropium against RB-induced lung injury were also dependent on the severity and type of RB. Regarding pure IRB, tiotropium was relatively more effective in reversing lung injury after 40% IRB compared to 50% IRB. Total protein in BALf, an index of lung permeability, was reversed at 40% of IRB, while respiratory system mechanics remained deranged in both groups after tiotropium. It is reasonable to assume that a therapeutic intervention at a constant dose might produce relatively less effects when a higher insult is used. On the other hand, more severe mechanical stress may also cause injury through stress failure6 and not through signaling-mediated mechanisms (mechanotransduction),2 the first expected to be hardly reversible by therapeutic interventions. In accordance, macrophage count increase was only affected by tiotropium at 40% of IRB but not at 50% of IRB, and the reduction in the macrophage count at pure ERB was not affected by tiotropium, suggesting an acetylcholine-independent mechanism for this latter effect. On the other hand, tiotropium was also effective in preventing lung permeability and respiratory system mechanics derangement at the CRB model, which is of course the most clinically relevant model, because during COPD exacerbations both the inspiratory and the expiratory resistance are increased, the latter more than the former.
The role of the presumed increased parasympathetic activity and/or non-neuronic acetylcholine release following RB to the induction of pulmonary inflammation can be dual since acetylcholine may not only bind to the muscarinic receptors but also to the nicotinic receptors that have been shown to exert anti-inflammatory action.28 Nicotinic receptors are detected in airway epithelium and inflammatory cells23 and have been found to exert a protective effect to mechanical stress-induced stimuli in the lung. Activation of the a7nAchRc inhibited the proinflammatory and proapoptotic signaling in airway epithelial cells (BEAS-2B) exposed to cyclic stretch in vitro24 and in vivo protected against ventilator-induced lung injury.29 Indeed, an anti-inflammatory vagal reflex has been well described to be mediated in part by acetylcholine nicotinic receptors.30 Thus, it is possible that blockage of muscarinic receptor in our model may favor the activation of nicotinic pathway and produce an anti-inflammatory balance.
A limitation of our study is that tiotropium was only given in a preventive manner, ie, prior to initiation of RB. The administration of tiotropium in a therapeutic protocol, ie, after the initiation of the RB and the induction of pulmonary inflammation, would offer significant additional information. However, in clinical practice, tiotropium bromide is mainly used as a maintenance therapy and not as a reliever; therefore, this strategy is in accordance with our experimental protocol. Thus, although patients may continue to use maintenance therapy during an exacerbation, initial establishment of tiotropium therapy usually precedes the onset of an exacerbation, except possibly in treatment-naïve patients.
In addition, we studied previously healthy animals to investigate the injurious effect of RB (as a mechanical stimulus) per se. Performing RB in animals with preexisting airway inflammation, as seen in severe COPD and/or during an infectious COPD exacerbation, although it would have added significant information, would not have allowed us to discriminate the protective effect of tiotropium on the effects of the mechanical stimulus of RB per se. Thus, it would stand beyond the scope of our study and could be the subject of future studies.
The magnitude of resistive loading in our experiment has been chosen, so as to mimic severe obstructive disease and especially disease exacerbations.3,4,31 Thus, the efficacy of tiotropium inhalation to attenuate the inflammatory response to RB (neutrophil infiltration, cytokine upregulation) is clinically significant relevant to both COPD and severe asthma, which are both characterized by increased sputum neutrophil number.32–34 Our data support previously reported studies on the anti-inflammatory action of tiotropium,11 irrespective of its bronchodilator action, since in our experiment, obstruction is fixed, artificially produced and not induced by bronchoconstriction.
These results have certain clinical implications. During severe COPD exacerbations, especially those leading to hospitalization, patients face RB of similar magnitude to our model due to airway narrowing caused by bronchoconstriction and airway inflammation and edema.31 This airway narrowing per se causes inflammation augmenting the preexisting inflammation.31 Our results suggest that tiotropium would attenuate the inflammation imposed by RB. This would reduce the total inflammatory burden associated with the severe COPD exacerbation and would thus be expected to lead to more rapid resolution of the inflammation and thus hastened recovery. In fact, anti-inflammatory treatments such as systemic steroids hasten recovery and reduce length of hospitalization and rate of relapse of COPD exacerbations.35 More importantly, tiotropium treatment has been shown to reduce exacerbation days, antibiotic days and hospital days in COPD patients.36 It stands to speculate that the reduction in the duration of exacerbations and hospitalization with tiotropium was in part due to a hastened resolution/decline of lung inflammation related to the COPD exacerbation-resistive breathing, as our results with the anti-inflammatory action of tiotropium in our RB model suggest.
Conclusion
We have shown that tiotropium inhalation attenuates the pulmonary inflammation induced by RB.
Acknowledgment
This study was funded by Boehringer Ingelheim.
Disclosure
The author Michael Pieper is an employee of Boehringer Ingelheim Pharma GmbH & Co. KG. The other authors report no conflict of interest in this work.
References
Vassilakopoulos T, Divangahi M, Rallis G, et al. Differential cytokine gene expression in the diaphragm in response to strenuous resistive breathing. Am J Respir Crit Care Med. 2004;170(2):154–161. | ||
Vlahakis NE, Hubmayr RD. Cellular stress failure in ventilator-injured lungs. Am J Respir Crit Care Med. 2005;171(12):1328–1342. | ||
Toumpanakis D, Kastis GA, Zacharatos P, et al. Inspiratory resistive breathing induces acute lung injury. Am J Respir Crit Care Med. 2010;182(9):1129–1136. | ||
Loverdos K, Toumpanakis D, Litsiou E, et al. The differential effects of inspiratory, expiratory, and combined resistive breathing on healthy lung. Int J Chron Obstruct Pulmon Dis. 2016;11:1623–1638. | ||
Glynos C, Toumpanakis D, Loverdos K, et al. Guanylyl cyclase activation reverses resistive breathing-induced lung injury and inflammation. Am J Respir Cell Mol Biol. 2015;52(6):762–771. | ||
West JB. Invited review: pulmonary capillary stress failure. J Appl Physiol (1985). 2000;89(6):2483–2489. | ||
Jaalouk DE, Lammerding J. Mechanotransduction gone awry. Nat Rev Mol Cell Biol. 2009;10(1):63–73. | ||
Tashkin DP, Celli B, Senn S, et al. A 4-year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med. 2008;359(15):1543–1554. | ||
Karner C, Chong J, Poole P. Tiotropium versus placebo for chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2014;7:CD009285. | ||
Alagha K, Palot A, Sofalvi T, et al. Long-acting muscarinic receptor antagonists for the treatment of chronic airway diseases. Ther Adv Chronic Dis. 2014;5(2):85–98. | ||
Kistemaker LE, Gosens R. Acetylcholine beyond bronchoconstriction: roles in inflammation and remodeling. Trends Pharmacol Sci. 2015;36(3):164–171. | ||
Gosens R, Zaagsma J, Meurs H, Halayko AJ. Muscarinic receptor signaling in the pathophysiology of asthma and COPD. Respir Res. 2006;7:73. | ||
Buhling F, Lieder N, Kuhlmann UC, Waldburg N, Welte T. Tiotropium suppresses acetylcholine-induced release of chemotactic mediators in vitro. Respir Med. 2007;101(11):2386–2394. | ||
Ohta S, Oda N, Yokoe T, et al. Effect of tiotropium bromide on airway inflammation and remodelling in a mouse model of asthma. Clin Exp Allergy. 2010;40(8):1266–1275. | ||
Bos IS, Gosens R, Zuidhof AB, et al. Inhibition of allergen-induced airway remodelling by tiotropium and budesonide: a comparison. Eur Respir J. 2007;30(4):653–661. | ||
Cui Y, Devillier P, Kuang X, et al. Tiotropium reduction of lung inflammation in a model of chronic gastro-oesophageal reflux. Eur Respir J. 2010;35(6):1370–1376. | ||
Wollin L, Pieper MP. Tiotropium bromide exerts anti-inflammatory activity in a cigarette smoke mouse model of COPD. Pulm Pharmacol Ther. 2010;23(4):345–354. | ||
Powrie DJ, Wilkinson TM, Donaldson GC, et al. Effect of tiotropium on sputum and serum inflammatory markers and exacerbations in COPD. Eur Respir J. 2007;30(3):472–478. | ||
Hantos Z, Daroczy B, Suki B, Nagy S, Fredberg JJ. Input impedance and peripheral inhomogeneity of dog lungs. J Appl Physiol (1985). 1992;72(1):168–178. | ||
Willms D, Shure D. Pulmonary edema due to upper airway obstruction in adults. Chest. 1988;94(5):1090–1092. | ||
Agusti A, Soriano JB. Dynamic hyperinflation and pulmonary inflammation: a potentially relevant relationship? Eur Respir Rev. 2006;15:68–71. | ||
Mathiot MJ, Jammes Y, Grimaud C. Role of vagal and spinal sensory pathways in diaphragmatic response to resistive loading. Neurosci Lett. 1987;73(2):131–136. | ||
Gwilt CR, Donnelly LE, Rogers DF. The non-neuronal cholinergic system in the airways: an unappreciated regulatory role in pulmonary inflammation? Pharmacol Ther. 2007;115(2):208–222. | ||
dos Santos CC, Shan Y, Akram A, Slutsky AS, Haitsma JJ. Neuroimmune regulation of ventilator-induced lung injury. Am J Respir Crit Care Med. 2011;183(4):471–482. | ||
Profita M, Giorgi RD, Sala A, et al. Muscarinic receptors, leukotriene B4 production and neutrophilic inflammation in COPD patients. Allergy. 2005;60(11):1361–1369. | ||
Kistemaker LE, van Os RP, Dethmers-Ausema A, et al. Muscarinic M3 receptors on structural cells regulate cigarette smoke-induced neutrophilic airway inflammation in mice. Am J Physiol Lung Cell Mol Physiol. 2015;308(1):L96–L103. | ||
Xu ZP, Yang K, Xu GN, et al. Role of M3 mAChR in in vivo and in vitro models of LPS-induced inflammatory response. Int Immunopharmacol. 2012;14(3):320–327. | ||
Kolahian S, Gosens R. Cholinergic regulation of airway inflammation and remodelling. J Allergy (Cairo). 2012;2012:681258. | ||
Bregeon F, Xeridat F, Andreotti N, et al. Activation of nicotinic cholinergic receptors prevents ventilator-induced lung injury in rats. PLoS One. 2011;6(8):e22386. | ||
Andersson U, Tracey KJ. Neural reflexes in inflammation and immunity. J Exp Med. 2012;209(6):1057–1068. | ||
Vassilakopoulos T, Toumpanakis D. Can resistive breathing injure the lung? Implications for COPD exacerbations. Int J Chron Obstruct Pulmon Dis. 2016;11:2377–2384. | ||
Hoenderdos K, Condliffe A. The neutrophil in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2013;48(5):531–539. | ||
Moore WC, Meyers DA, Wenzel SE, et al. Identification of asthma phenotypes using cluster analysis in the Severe Asthma Research Program. Am J Respir Crit Care Med. 2010;181(4):315–323. | ||
Moore WC, Hastie AT, Li X, et al. Sputum neutrophil counts are associated with more severe asthma phenotypes using cluster analysis. J Allergy Clin Immunol. 2014;133(6):1557–1563. | ||
Woods JA, Wheeler JS, Finch CK, Pinner NA. Corticosteroids in the treatment of acute exacerbations of chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2014;9:421–430. | ||
Rice KL, Leimer I, Kesten S, Niewoehner DE. Responses to tiotropium in African-American and Caucasian patients with chronic obstructive pulmonary disease. Transl Res. 2008;152(2):88–94. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.