Back to Journals » International Journal of Nanomedicine » Volume 15
TiO2 Nanoparticles Caused DNA Damage in Lung and Extra-Pulmonary Organs Through ROS-Activated FOXO3a Signaling Pathway After Intratracheal Administration in Rats
Authors Han B, Pei Z, Shi L, Wang Q, Li C, Zhang B, Su X, Zhang N, Zhou L, Zhao B, Niu Y ![]() , Zhang R
, Zhang R
Received 30 March 2020
Accepted for publication 17 July 2020
Published 21 August 2020 Volume 2020:15 Pages 6279—6294
DOI https://doi.org/10.2147/IJN.S254969
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Mian Wang
Bin Han1 ,* Zijie Pei2 ,* Lei Shi,3 Qian Wang,4 Chen Li,1 Boyuan Zhang,1 Xuan Su,1 Ning Zhang,1 Lixiao Zhou,1 Bo Zhao,5 Yujie Niu,3,6 Rong Zhang1,6
1Department of Toxicology, Hebei Medical University, Shijiazhuang 050017, Hebei, People’s Republic of China; 2Department of Pathology, Medical School, China Three Gorge University, Yichang 443002, People’s Republic of China; 3Occupational Health and Environmental Health, Hebei Medical University, Shijiazhuang 050017, Hebei, People’s Republic of China; 4Experimental Center, Hebei Medical University, Shijiazhuang 050017, Hebei, People’s Republic of China; 5Department of Laboratory Diagnosis, Hebei Medical University, Shijiazhuang 050017, Hebei, People’s Republic of China; 6Hebei Key Laboratory of Environment and Human Health, Hebei Medical University, Shijiazhuang 050017, Hebei, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Rong Zhang
Department of Toxicology, Hebei Medical University, 361 Zhongshan East Road, Shijiazhuang, Hebei 050017, People’s Republic of China
Fax +86-311-86265605
Email [email protected]
Introduction: Because of the increased production and application of manufactured Nano-TiO2 in the past several years, it is important to investigate its potential hazards. TiO2 is classified by IARC as a possible human carcinogen; however, the potential mechanism of carcinogenesis has not been studied clearly. The present study aimed to investigate the mechanism of DNA damage in rat lung and extra-pulmonary organs caused by TiO2nanoparticles.
Methods: In the present study, SD rats were exposed to Nano-TiO2 by intratracheal injection at a dose of 0, 0.2, or 1 g/kg body weight. The titanium levels in tissues were detected by ICP-MS. Western blot was used to detect the protein expression levels. The DNA damage and oxidative stress were detected by comet assay and ROS, MDA, SOD, and GSH-Px levels, respectively.
Results: The titanium levels of the 1 g/kg group on day-3 and day-7 were significantly increased in liver and kidney as well as significantly decreased in lung compared to day-1. ROS and MDA levels were statistically increased, whereas SOD and GSH-Px levels were statistically decreased in tissues of rats in dose-dependent manners after Nano-TiO2 treatment. PI3K, p-AKT/AKT, and p-FOXO3a/FOXO3a in lung, liver, and kidney activated in dose-dependent manners. The levels of DNA damage in liver, kidney, and lung in each Nano-TiO2 treatment group were significantly increased and could not recover within 7 days. GADD45α, ChK2, and XRCC1 in liver, kidney, and lung of rats exposed to Nano-TiO2 statistically increased, which triggered DNA repair.
Conclusion: This work demonstrated that Ti could deposit in lung and enter extra-pulmonary organs of rats and cause oxidative stress, then trigger DNA damage through activating the PI3K-AKT-FOXO3a pathway and then promoting GADD45α, ChK2, and XRCC1 to process the DNA repair.
Keywords: Nano-TiO2, DNA damage, PI3K/AKT/FOXO3a signaling pathway, DNA repair, GADD45α/ChK2/XRCC1 signaling pathway
Introduction
Nanoparticle titanium dioxide (Nano-TiO2) is a common nanomaterial with a size less than 100 nm. Because of its photocatalytic properties, chemical resistance, and thermal stability, Nano-TiO2 has been widely used in medicine, wastewater treatment, coatings, sterilization, cosmetics, food additives, biomedical ceramics, etc. TiO2 is generally considered to be a low-toxic chemical, but TiO2 is classified by IARC as a group 2B, possible human carcinogen.1 TiO2 nanoparticles could generate amounts of free radicals, which induced indirect genotoxicity mainly by DNA-adduct formation.2 The rutile-based Nano-TiO2, as a result of photocatalysis, have been shown to produce reactive oxygen species (ROS), then causing DNA damage in vitro.3,4 Nano-TiO2 could exist in the air, and enter the body through the respiratory tract, and caused hyperplasia and inflammation in mice.5 National Institute for Occupational Safety and Health (NIOSH) lists recommend airborne exposure limits of 0.3 mg/m3 for ultrafine TiO2 as time-weighted average concentrations in the workplace.6 Furthermore, intratracheal instillation of Nano-TiO2 has been shown to translocate to organs such as the liver7,8 and induce renal fibrosis in mice,9 but the toxicological mechanism in distant organs is still unclear.
Nano-TiO2 exposure by intranasal administration for 90 days could induce ROS accumulation, lead to oxidation of lipids, proteins, and DNA, then trigger brain injury in mice.10 ROS possess the ability to damage the DNA nucleobases.11 Shukla et al12 found that Nano-TiO2 induced ROS and oxidative stress in human epidermal cells (A431), leading to DNA oxidative damage and micronuclei formation. Previous studies have reported that Fork head O (FOXO) factors are involved in oxidative stress responses,13 which increases cellular antioxidant activity.14,15 Furthermore, FOXO3a, a member of the FOXO subfamily, could provide protection against ROS-induced damage13 and protects alveolar epithelial cells from oxidative stress.16 The FOXO subfamily of transcription factors has been implicated in diverse physiologic processes, including cell cycle arrest, cell death, stress resistance, and apoptosis.17 With ROS-induced DNA damage, Forkheads could contribute secondarily to defense through regulating the expression of proteins involved in the repair process.18 FOXO3 has important downstream targets of the phosphatidylinositol 3-kinase (PI3K)-protein kinase B (AKT) pathway.19 ROS triggers PI3K kinase activation, which subsequently regulates AKT activation, leading to cell survival, proliferation, protein synthesis, and transcription.20,21 In addition, the oxidative stress response is regulated in part by FOXO induction of MnSOD, catalase, and growth arrest and DNA damage α (GADD45α).13,14,18 The function of GADD is to protect cells and ensure survival by inducing cell cycle arrest, DNA repair, and promoting apoptosis. Furthermore, decreased GADD45α expression is also considered a survival mechanism, which is essential for escape from programmed cell death.22 Checkpoint kinase (ChK2) is involved in the ataxia telangiectasia mutation (ATM) signaling pathway, which is activated as a downstream effector associated with DNA damage under oxidative stress conditions.23,24 Many studies have shown that X-ray repair cross complementing gene 1(XRCC1) plays a key role in DNA damage caused by oxidative stress.25,26 Previous studies have found that XRCC1 gene deletion exacerbates γ-ray-induced DNA damage and cell cycle arrest in liver cancer cells.27 Therefore, it is hypothesized that the PI3K/AKT/FOXO3a signaling pathway was activated in lung, liver, and kidney when ROS increased after intratracheal instillation of Nano-TiO2. Then the DNA repair was triggered to defense of the DNA damages.
In this study, a Nano-TiO2 exposure rat model through tracheal perfusion was established to evaluate the effects of Nano-TiO2 on oxidative stress and DNA damage in the lung, liver, and kidney of rats. We found FOXO3a did trigger the DNA-repair mechanism after Nano-TiO2 exposure. Our results might provide a theoretical basis for the future research and find the potentially therapeutic target for DNA damages induced by Nano-TiO2.
Materials and Methods
Characterization of Nano-TiO2
The anatase-phase Titanium (IV) oxide (Nano-TiO2, 99.7% purity) was purchased from Sigma Chemical Co. (St. Louis, MO, USA). The size and morphology of Nano-TiO2 were evaluated by Tecnai G220 transmission electron microscope (TEM, FEI, USA) and Sirion 200 scanning electron microscope (SEM, FEI, Holland). Before SEM analysis, the nanoparticles were sputter-coated with gold and then dispersed in buffer and cast onto a carbon-coated copper grid sample holder followed by evaporation at room temperature. The specific surface area of the Nano-TiO2 particles was calculated using the Brunauer-Emmett-Teller (BET) adsorption isotherm.
The size and zeta potential of Nano-TiO2 were performed on DelsaTM Nano particle analysis instrument (Beckman Coulter, CA, USA). Additionally, the materials were examined for endotoxin content using a Toxin SensorTM Chromogenic LAL Endotoxin Assay Kit (Genscript, USA). No endotoxins were detected in Nano-TiO2.
Animals and Treatment
Forty-five male laboratory-bred SD rats weighing 200±20 g were purchased from the Experimental Animal Center of Hebei Medical University (Shijiazhuang, China). Before experiment, the animals were fed in the experimental room for 2–3 days to habituate them to the environment of the laboratory. An ambient temperature of 25±2°C, relative humidity of 50±2%, and photoperiod of 12 hours were maintained throughout the study. All animals were fed a diet and water ad libitum in stainless cages, and they received humane treatment in compliance with the Principles of Laboratory Animal Care formulated by the National Society for Medical Research. The present protocol was approved by the Committee of the Ethics Animal Experiments of Hebei Medical University (IACUC-Hebmu-20,160,048). The food was purchased from the Experimental Animal Center of Hebei Medical University.
The rats were randomly divided into three groups (15 rats/group): the control group, and two TiO2 treatment groups (0.2 and 1.0 g/kg body weight). The suspensions of Nano-TiO2 were sonicated for 20 minutes and administrated to rats via one intratracheal injection at a dose of 0.6 mL/200 g body weight, respectively.28 Rats in the control group were instilled with 0.6 mL PBS per 200 g body-weight. Before injection, the rats were anesthetized by ether. Food and water were provided 2 hours later. On the 1st, 3rd, and 7th days after exposure, five rats were sacrificed in each dose group. After anesthetizing by pentobarbital sodium, rats were sacrificed. Lung, liver, and kidney were separated to further detect at days 1, 3, and 7 after instillation. After weighting the body and tissues, the coefficients of lung, liver, and kidney were calculated as the ratio of tissues (wet weight, mg) to body weight (g).
Titanium Burden Analysis
About 0.1–0.3 g of lung, liver, and kidney tissue was digested and analyzed for titanium contents, respectively. Tissues were digested in nitric acid (ultrapure grade, Beijing Fine Chemical Ltd., Beijing, China) overnight. After adding 0.5 mL H2O2, the mixed solution was completely digested by a microwave digestion system (MDS, Sineo Microwave Chemistry Technology Co., Ltd., China). Then, the solution was heated at 120°C to remove the remaining nitric acid until it was colorless and clear. The remaining solutions were diluted to 3 mL with 2% nitric acid. Then, the concentration in digested samples was detected by inductively coupled plasma-mass spectrometry (ICP-MS, ELAN DRC-e, PerkinElmer Co., USA). Then 0.5 μg/L Rhodium was chosen as an internal standard element. Data were expressed as ppb (nanograms per gram fresh tissue, ng/g).
The deposited alveolar fraction was predicted using the “multiple path particle dosimetry (MPPD) model” version 2.11, which is based on the aerosol characteristics.29 The initial baseline sets of MPPD inputs based on data from the ICRP report (ICRP 1994) are listed in Supplementary Table 1. The maximum reported Nano-TiO2 concentration (0.3 mg/m3) was referred to as an input parameter for the MPPD model to estimate the retained dose in the rat lung.
Histopathology
The lung, liver, and kidney tissues were separated and fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned into 5 μm-thick slices. The resulting slides were stained with hematoxylin and eosin (HE) for histological examination according to the standard techniques. After staining, the histopathological changes were observed by optical microscope (Olympus, Japan).
Oxidative Stress Detection
The activities of anti-oxidase glutathione peroxidase (GSH-Px) and superoxide dismutase (SOD), and the levels of the lipid peroxidation product malonaldehyde (MDA) in the lung, liver, and kidney of rats were determined according to the methods described in our previous study using commercial kits (Nanjing JianchengBioeng Inst., China).30 The data is expressed as units of activity or nanomoles per milligram of protein.
The intracellular ROS was measured using 2ʹ,7ʹ-dichlorofluorescein diacetate (DCFH-DA) as a probe.31 About 50 mg of lung, liver, and kidney tissue were cut into pieces on the ice, and then incubated with 0.25% trypsin for 30 minutes at 37°C. After adding 250 μL FBS in order to stop digestion, the suspension was filtered through a nylon filter to yield individual cells. The cell suspension was centrifuged at 800 r/min for 10 minutes, and the supernatant was discarded. Thereafter, 1 mL of PBS containing 1 μL DCFH-DA dye was added to each tube. All the tubes were incubated for 30 minutes at 37°C and then DCFH-DA was discarded. After washing three times with PBS, 1 mL of PBS was added to each tube and fluorescence intensity was measured in flow cytometry (BD Biosciences, San Jose, CA, USA) using software at excitation and emission wavelengths of 488 nm and 525 nm, respectively.
DNA Damage Detection
DNA damages induced by Nano-TiO2 were detected by Comet assay. Then 100 mg lung, liver, and kidney samples from each group were cut into pieces on ice, and then incubated with 0.25% trypsin for 30 minutes at 37°C. After adding 250 μL FBS to stop digestion, the suspension was filtered at nylon filter to yield individual cells. Then, cells suspended in 0.5% low melting agarose were spread on the normal melting agarose-coated (1%) slides. After lysing at 4°C overnight, the slides were soaked in electrophoretic buffer solution for 30 minutes and electrophoresed for 25 minutes at 25 V (300 mA) at 4°C. The cells were stained with 10 mg/mL ethidium bromide. Two hundred randomly chosen cells were scored visually using a fluorescence microscope (Olympus, Japan) coupled with a 515–560 nm wavelength excitation filter and 590 nm wavelength emission filter. The olive tail moment (OTM) of each comet was calculated using CASP analyzed software.
Western Blot Analysis
Tissue lysates were homogenized with modified RIPA buffer containing 1% phenylmethylsulfonyl fluoride (PMSF) using a high throughput tissue grinder (Scientz, Ningbo, China). Total protein extracts were determined by using a Pierce®BCA protein assay kit (Thermo Fisher Scientific, USA). An equal amount of lysate proteins was loaded onto 10 or 12% SDS-polyacrylamide gels and electrophoretically transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Danvers, MA) using a wet transfer system (Liuyi, Beijing, China). After blocking with 5% nonfat milk in Tris-buffered saline containing 0.05% Tween-20 (TBST) for 3 hours at room temperature, membranes were incubated with primary antibody overnight at 4°C: including anti-FOXO3a antibody (Cell Signaling Technologies, Danvers, MA, USA) at 1:1000, rabbit anti-GADD45α antibody (Proteintech, Wuhan, China) at 1:200, rabbit anti-AKT antibody (Proteintech) at 1:2000, rabbit anti-XRCC1 antibody (Bioworld, Nanjing, China) at 1:500, rabbit anti-ChK2 antibody (Proteintech) at 1:600, rabbit anti-PI3K antibody (Abcam, Cambridge, UK) at 1:5000, rabbit anti-phosphor-AKT antibody (Abcam) at 1:5000, rabbit anti-phosphor-FOXO3a antibody (AffinityBiosciences, USA) at 1:1000, and monoclonal rabbit anti-GAPDH antibody (Proteintech) was used at 1:20,000. GAPDH served as an internal standard for Western blot analysis. After washing three times with TBST, all membranes were incubated with a horseradish peroxidase-conjugated anti-rabbit IgG secondary antibody (Cell Signaling Technologies) for 1 hour at 37°C. After being washed three times with TBST, the antibody-bound proteins were detected using enhanced chemiluminescence (TIANGEN, Beijing, China). Analysis of the Western blot was performed using Chemiluminescent Imaging System (Tanon, China) and quantified by Gel-Pro Analyzer 3.0.
Statistical Analysis
Results were expressed as mean ± SE and data were analyzed using one-way analysis of variance (ANOVA) with Dunnett’s multiple comparison tests to determine significance. Homogeneity of variance between all groups was ascertained. In all cases, P-values <0.05 were considered statistically significant.
Results
Characterization of Nano-TiO2
SEM and TEM analysis clearly showed a three-dimensional nanostructure of Nano-TiO2, which consisted of globular shaped particles (Figure 1A and B). The size reported by SEM and TEM ranged from 10–25 nm. The larger particles showed many spherical protrusions on the surface, which indicates that they were formed by fusing many smaller particles during the spray drying process. The BET results showed that the surface area of the particles was about 50 m2/g, and the diameter of Nano-TiO2 in PBS was 392.7±99.4 nm. The zeta potential of Nano-TiO2 was −26.49 mV, which indicated that the Nano-TiO2 were scattered in PBS.
 |
Figure 1 Characterization of Ti and the contents of Ti in organs. (A and B) Scanning electron microscopy and transmission electron microscopy images of Nano-TiO2. The powder was deposited on 200-mesh copper grids. The particle primary structure was 10–30 nm and globular. (C) On the 7th day after instillation, the contents of Ti in tissues of rats in the 0, 0.2, and 1 g/kg Nano-TiO2 treatment groups. N=5, *P<0.05, in comparison to the control group. (D) The contents of Ti in tissues of rats after 1 g/kg Nano-TiO2 treatment for 1, 3, and 7 days. N=5, *P<0.05, in comparison to the 1-day group. |
Body Weight Coefficients of Liver, Kidney, and Lung
After treatment for 1, 3, and 7 days, rats were sacrificed and the body weight and tissues/organs weight were determined. The weight of rats is shown in Supplementary Figure 1. No obvious differences were found in the body weight coefficient between the control and treatment groups (Supplementary Table 2).
Titanium Burden After Nano-TiO2 Treatment
We measured the content of Ti in tissues in different exposure groups after 7 days of exposure. The contents of titanium in tissues are shown in Figure 1C. After Nano-TiO2 post-treatment for 7 days, the titanium levels in liver and kidney were significantly increased in the 1 g/kg Nano-TiO2 group (P<0.05), respectively, whereas no significantly changes in the 0.2 g/kg Nano-TiO2 group compared with the control.
In order to detect the time-response and the clearance in different tissues after Nano-TiO2 treatment, we measured contents of Ti in tissues of rats after 1 g/kg Nano-TiO2 treatment for 1, 3, and 7 days (Figure 1D). Compared with post-treatment for 1 day, the titanium levels in lung were significantly decreased in post-treatment for 3 and 7 days, respectively (P<0.05). Moreover, the titanium levels in liver and kidney were significantly increased in post-treatment for 7 days compared with 1 day (P<0.05). The titanium standard curve and response analysis of ICP-MS are shown in Supplementary Figure 2.
For the MPPD model, a steady-state retention dose would take 26.74 days or 4.3 months to achieve for 0.2 g/kg (about 40 mg) or 1 g/kg (about 200 mg) based on inhalation of an aerosol concentration of 0.3 mg/m3 (Time-weighted average concentrations in workplace) for 8 hours/day, 5 days/week.
Histopathological Changes of Liver, Kidney, and Lung Tissue in Rats After Nano-TiO2 Exposure
After Nano-TiO2 exposure, there appeared to be histopathologic changes in the liver, kidney, and lung of rats.
Lung: In the control group, we can observe the neat bronchial epithelium and regular lumen, intact alveolar ducts and alveolar sacs, and normal elastic fibers and reticular fibers of alveolar septa with the capillary network normal dyed (Figure 2A1). In the 0.2 g/kg group, inflammatory cell infiltration in the bronchus and irregular bronchial lumen could be found (Figure 2A2). In the 1 g/kg group, aggravating irregular bronchial lumen, thickening of the alveolar wall, and alveolar septal were observed (Figure 2A3).
 |
Figure 2 Effects of Nano-TiO2 exposure on the histopathological changes in the lung, liver, and kidney at 7 days after treatment. (A) The representative images of lung tissue of rats stained with HE. (B) The representative images of liver tissue of rats stained with HE. (C) The representative images of kidney tissue of rats stained with HE. Scale bar represents 50 μm. The arrow in A2 indicated irregular luminal inflammatory cell infiltration; The arrow in A3indicated the alveolar wall thickening. The arrow in B2 indicated cell disorder. The arrow in B3 indicated the spotty necrosis. The arrow in C2 indicated the gap increases. The arrow in C3 indicated necrosis. 1: Control group, 2: 0.2 g/kg group, 3: 1 g/kg group. |
Liver: In the control group, the hepatocytes were arranged regularly and neatly around the central vein. In the 0.2 g/kg group, the cells were disordered, and distal cells of the central vein were stained more darkly than in the control group (Figure 2B1 and B2). In the 1 g/kg group, the vacuoles were observed in the cytoplasm and punctate necrosis appeared in the inflammatory cells (Figure 2B3).
Kidney: In the control group, the cells were arranged regularly, and the glomeruli and tubules were closely arranged. In the 0.2 g/kg group, the renal tubular gap increased compared with the control group (Figure 2C1 and C2). In the 1 g/kg group, the renal tubular showed edema accompanied with partial necrosis (Figure 2C3).
Changes of Oxidative Stress Levels Induced by Nano-TiO2Rats
Lung: Compared with the control group, SOD, GSH-Px activities were significantly decreased (P<0.05) and MDA content was significantly increased (P<0.05) in a concentration dependent manner. Compared with that on day 1 after Nano-TiO2 exposure, SOD activity was significantly decreased in the 0.2 and 1.0 g/kg groups at 1, 3, and 7 days post-exposure (P<0.05). There was no significant change in GSH-Px activity in each dose group at 3 days and 7 days post-exposure compared to 1 day. MDA content was significantly increased in the 0.2 g/kg group at 7 days post-exposure (P<0.05), but no significant changes were found in the 1 g/kg group at 3 days and 7 days after Nano-TiO2 exposure compared to 1 day (Figure 3A–C).
 |
Figure 3 The oxidative stress levels in the lung, liver, and kidney of rats after Nano-TiO2 treatment. (A–C) The SOD, GSH-Px activities, and MDA content in lung of rats. (D–F) The SOD, GSH-Px activities, and MDA content in liver of rats. (G–I) The SOD, GSH-Px activities, and MDA content in kidney of rats. (J–L) ROS levels induced by Nano-TiO2 in organs of rats. N=5, *P<0.05, in comparison to the control group; #P<0.05, in comparison to the 0.2 g/kg group.Abbreviations: SOD, superoxide dismutase; GSH-Px, glutathione peroxidase; MDA, malonaldehyde. |
Liver: SOD, GSH-Px activities were significantly decreased (P<0.05) and MDA content was significantly increased (P<0.05) in a dose dependent manner compared with the control group. Compared with that on day 1 after Nano-TiO2 exposure, SOD activity was significantly increased (P<0.05) in the 0.2 g/kg group at 3 days and 7 days after Nano-TiO2 exposure; SOD activity was significantly increased in the 1 g/kg group at 7 days after Nano-TiO2 exposure (P<0.05); GSH-Px activity was significantly increased in each group at 3 days and 7 days after Nano-TiO2 exposure, (P<0.05); MDA content was not significantly changed in each group at 3 days after Nano-TiO2 exposure compared to 1 day, whereas MDA content was significantly decreased in each group at 7 days after Nano-TiO2 exposure (P<0.05) (Figure 3D–F).
Kidney: Compared with the control group, SOD, GSH-Px activities were significantly decreased (P<0.05), MDA content was significantly increased (P<0.05). Compared with that on day 1 after Nano-TiO2 exposure, SOD activity was significantly increased in the 0.2 g/kg and 1 g/kg groups at 3 days and 7 days after Nano-TiO2 exposure (P<0.05); GSH-Px activity was significantly increased in each dose at 3 days and 7 days after Nano-TiO2 exposure (P<0.05); MDA content showed no significant change in each dose at 3 days after Nano-TiO2 exposure compared to 1 day; MDA content was significantly decreased in the 0.2 g/kg and 1 g/kg groups at 7 days after Nano-TiO2 exposure (P<0.05) (Figure 3G–I).
Nano-TiO2 exposure resulted ina significant increase in ROS, which was evident through increased fluorescence intensity in lung and liver compared to the control group (Figure 3J and K). A significant increase was noted in these groups in a dose dependent manner. In our study, although an increase of 10.39% and 19.08% at 0.2 and 1 g/kg, respectively, was noted at 7 days post-treatment in kidney, only the 1 g/kg group was statistically significant compared with the control group (Figure 3L).
DNA Damage Induced by Nano-TiO2
A significant induction (P<0.05) in DNA damage was observed in the liver, kidney, and lung of rats exposed to Nano-TiO2 at 0.2 and 1 g/kg concentrations compared to control, as evidenced by the Comet assay parameters ie OTM and tail DNA (%). Compared with that on day 1 after Nano-TiO2 exposure, OTM and Tail DNA% in the lung (Figure 4A and B), kidney (Figure 4C and D), and liver (Figure 4E and F) tissue of all dose group showed no significant differences at 3 days and 7 days post-exposure. There was an apparent dose-response relationship and representative pictures of the lung are shown in Figure 4G1–G3.
 |
Figure 4 The DNA damage levels in the lung, liver, and kidney of rats after Nano-TiO2 treatment. (A and B): The OTM value and Tail DNA% value changed in the lung after Nano-TiO2 treatment at different times. (C and D) The OTM value and Tail DNA% value changed in the liver after Nano-TiO2 treatment at different times. (E and F) The OTM value and Tail DNA% value changed in the lung after Nano-TiO2 treatment at different times. (G) Representative images of DNA damage in the lung of rats after Nano-TiO2 treatment for 7 days (400x). (G1: control group; G2: 0.2 g/kg Nano-TiO2 treatment group; G3: 1 g/kg Nano-TiO2 treatment group). The result was the average of at least three independent experiments. N=5, *P<0.05: day 1 in comparison to the control group; #P<0.05: day 3 in comparison to the control group; ○P<0.05: day 7 in comparison to the control group. |
Activation of PI3K-AKT-FOXO3a Signal Pathway in Tissues of Rats After Nano-TiO2 Treatment
The PI3K/AKT pathway is well known to be involved in endothelial cell proliferation and cell survival and also plays an important role in DNA repair. To investigate whether Nano-TiO2 could induce the activation of the PI3K/AKT/FOXO3a signaling pathway, we examined the amount of PI3K, AKT, phosphorylated AKT, FOXO3a, and phosphorylated FOXO3a in the lung, liver, and kidney after single treatment with Nano-TiO2 for 7 days and the control group. Nano-TiO2 treatment had no effect on AKT protein expression, but it increased the expression of the PI3K, phosphorylated AKT, and phosphorylated FOXO3a after Nano-TiO2 treatment (Figure 5). As shown in Figure 5, the ratios of p-AKT/AKT and p-FOXO3a/FOXO3a in the lung (Figure 5C and D), liver (Figure 5G and H), and kidney (Figure 5K and L) were significantly increased in a dose-dependent manner after exposure to Nano-TiO2. These results suggested that DNA damage induced by Nano-TiO2 might relate with the activation of the PI3K-AKT-FOXO3a signaling pathway.
 |
Figure 5 Activation of the PI3K-AKT-FOXO3a signal pathway in the lung, liver, and kidney of rats after Nano-TiO2 treatment for 7 days. (A) The represented protein bands of PI3K, AKT, FOXO3a, p-AKT, and p-FOXO3a in the lung. (B–D) The fold changes of PI3K, p-AKT/AKT, and p-FOXO3a/FOXO3a levels in the lung. (E–H) The represented protein bands and fold changes of PI3K, AKT, FOXO3a, p-AKT, and p-FOXO3a in the liver. (I–L) The represented protein bands and fold changes of PI3K, AKT, FOXO3a, p-AKT, and p-FOXO3a in the kidney. N=5, *P<0.05, in comparison to the control group. #P<0.05, in comparison to the 0.2 g/kg group. Abbreviations: PI3K, phosphatidyl inositol 3-kinase; AKT, protein kinase B; FOXO3a, the Fork head box class O 3a. |
Changes of DNA Repair Signal-Related Proteins in Tissues of Rats Exposed to Nano-TiO2
GADD45α as a downstream signal molecule of FOXO3a can reflect DNA damage repair. Further DNA repair pathways were confirmed through Western blot analysis of various DNA damage repair markers such as XRCC1 and ChK2 (Figure 6A). Significant increases of XRCC1 (Figure 6B), ChK2 (Figure 6C), and GADD45α (Figure 6D) expression were found in lung of rats in a dose dependent manner after Nano-TiO2 treatment.
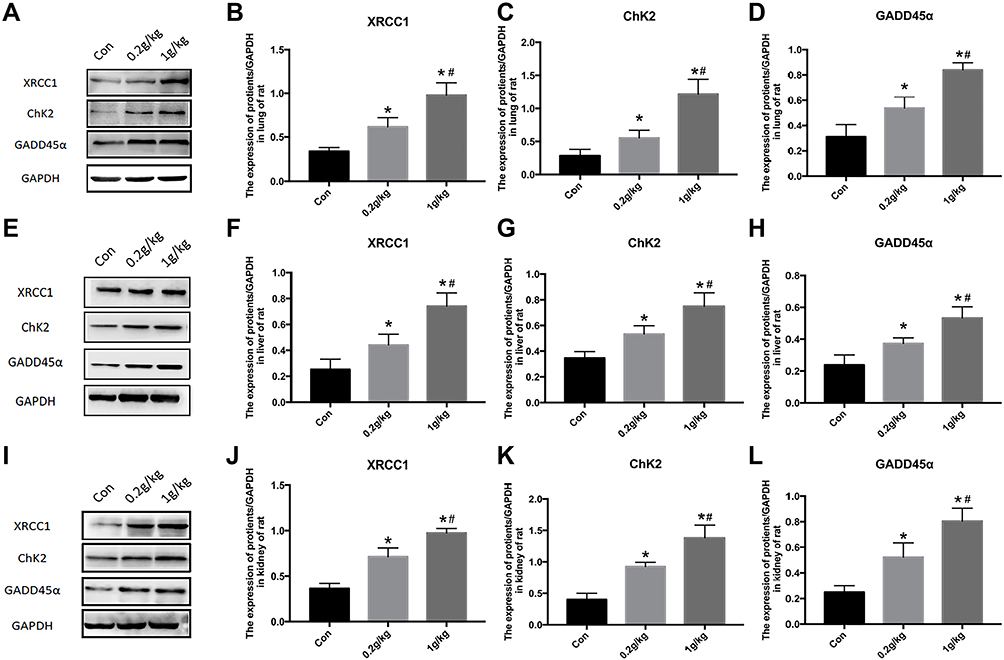 |
Figure 6 Changes of DNA repair proteins in the lung, liver, and kidney of rats exposed to Nano-TiO2 for 7 days. (A–D) The represented protein bands and fold changes of XRCC1, ChK2, and GADD45α in the lung. (E–H) The represented protein bands and fold changes of XRCC1, ChK2, and GADD45α in the liver. (I–L) The represented protein bands and fold changes of XRCC1, ChK2, and GADD45α in the kidney. N=5, *P<0.05, in comparison to the control group. #P<0.05, in comparison to the 0.2 g/kg group. Abbreviations: XRCC1, x-ray repair cross complementing gene 1; ChK2, checkpoint kinase 2; GADD45α, growth arrest and DNA damage α. |
In the liver and kidney of rats, Nano-TiO2 exposure resulted in dose-dependent increases of XRCC1, ChK2, and GADD45α expression (P<0.05) (Figure 6E–L).
Discussion
In recent years, with the rapid development of nanotechnology, Nano-TiO2 has been widely used in coatings, paints, and other fields.32 The National Institute for Occupational Safety and Health (NIOSH) lists Nano-TiO2 as a potential occupational carcinogen6 and recommends airborne exposure limits of 2.4 mg/m3 for fine TiO2 and 0.3 mg/m3 for ultrafine (including engineered nanoscale) TiO2 as time-weighted average concentrations. Although the most reliable studies for exploring the pulmonary toxicity of nanomaterials are inhalation studies, intratracheal instillation studies are useful for examining the dose-dependent manner of nanomaterials.33,37
The pattern of pulmonary inflammation ranking of nanomaterials is the same in inhalation studies and intratracheal instillation studies. Therefore, intratracheal instillation studies might be useful for ranking the harmful effects of nanomaterials.34,37,38
In the present study, we observed Ti in the liver and kidney in the 0.2 g/kg and 1 g/kg groups after intratracheal injection of Nano-TiO2, in which the accumulated deposition in the lung was equal to about 26.74 days or 4.3 months exposure in human (8 hours per day, 0.3 mg/m3, limitation by NIOSH) for Nano-TiO2 calculated by the MPPD model.29,39
It was shown that ultrafine TiO2 particles could induce oxidative bronchial epithelial cell DNA damage.40 Besides, TiO2 could cause DNA double-strand breaks via the generation of a continued increased level of ROS.41 Proven pulmonary carcinogen could induce greater levels of ROS, leading to enhanced DNA damage and apoptosis. Amounts of evidence implicate ROS-induced DNA double-strand breaks as a signal, which activates transcription factors and leads to cell proliferation and carcinogenesis. The markers of oxidative stress in exhaled breath condensate samples were significantly higher in TiO2 production workers than in unexposed controls.42 Furthermore, several lipid oxidative markers (including malondialdehyde) were elevated in TiO2 production workers relative to the controls.1 Zhao et al43 found oxidative stress markers (SOD and MDA), and inflammation markers (IL-8, IL-6, IL-1β, TNF-α, and IL-10) were associated with occupational exposure to Nano-TiO2. The toxicity of TiO2 nanoparticles could mainly depend on the structural characteristics.44 Zhang et al45 detected the cytotoxicity of different-sized TiO2 nanoparticles. They found that the 25-nm anatase-phase TiO2 induced stronger cytotoxicity and oxidative stress than those of 5- and 100- nm anatase-phase TiO2. The 5- and 100-nm anatase-phase TiO2 had similar toxicity. Besides, anatase particles caused higher toxicity than rutile particles. Anatase-phase TiO2 induce observable DNA stress and membrane damage.46 Xiong et al47 found the correlation between cytotoxicity of TiO2 nanoparticles and particle size. This is because smaller particles have larger specific surface area, which could absorb more biomolecules in the environment. The biological effects might associate with particle size, dry particle size, and surface area.48
Nano-TiO2 usually enters the body through the skin, respiration, or digestion. TiO2 suspension (5 g/kg body weight) was given to mice by a syringe via gastrointestinal tract for 2 weeks, and TiO2 could accumulate in the liver, spleen, kidney, and lung tissues.49 Shinohara et al50 reported 94%, 2.0%, 0.17%, and 0.023% Ti were observed in the liver, spleen, lung, and kidney at 6 hours after intratracheal injection of Nano-TiO2, respectively. After 2, 10, and 50 mg/m3 Nano-TiO2 inhalation for 6 hours/day and continuing for 5 days, Nano-TiO2 burdens in the lung were 118.4, 544.9, and 1635 μg, respectively. After 16-day recovery, Nano-TiO2 burdens decreased to 25, 144.5, and 295 μg in the lung, respectively.51 Oberdörster et al52 reported that retention half times were 117 days for those exposed to fine particles (250 nm TiO2), and 541 days for those exposed to ultrafine particles (20 nm TiO2). In the 1 g/kg group of the present study, there was a significant increase in titanium content in the liver and kidney tissues with time, which might be related to the longer half-life period of TiO2. Our data suggested that slowly clearance of Ti and low particle translocation might partly explain that titanium was higher in lung tissue compared with liver and kidney. Accumulation of titanium in lung tissue caused continuous damage to the respiratory system. The titanium content in liver tissue increased, resulting in decreased liver detoxification ability. The accumulation of titanium in the kidney causes kidney damage and reduces excretion.
Studies have shown that Nano-TiO2 can cause significant inflammatory damage and pathological changes in the lungs and extra pulmonary organs. Nano-TiO2 inhalation resulted in alveolar macrophage and neutrophil infiltration in rats.51 On the 7th day after 5 nm TiO2 particles treatment via tracheal instillation, proliferation of macrophages was observed, as well as massive particulate deposition in gaps and the alveolar cavity of lung tissue in rats were found.53 Similarly, in the present study, we found inflammatory cells infiltrated in the lungs. Bronchus and the alveolar wall became thickened. Tracheal perfusion of Nano-TiO2 for 4 weeks showed pathological damage to lung tissue,54 and renal fibrosis.9 However, in our study, we did not find renal fibrosis, which might be due to the acute exposure by one-time intratracheal instillation. In addition, we found the disordered arrangement of liver cells and inflammatory cells in livers. Similarly, Nano-TiO2 caused serious damage to the liver, and the indicators of liver function, such as alkaline phosphatase, alanine aminotransferase, leucine acid peptide, pseudocholinesterase, total protein, and albumin level, were enhanced significantly.32,55
Our previous studies30 showed that Nano-TiO2 could induce liver and kidney function damage in mice, which is closely related to the increase of ROS and the decline of antioxidant capacity. The increase of ROS might lead to an imbalance in the oxidative metabolism of the cell and cause tissue damage.56,58 In the present study, we found that after treatment with Nano-TiO2, there was a dose-dependent ROS increase in the lung and liver. In kidney, only the 1 g/kg group was statistically significant compared with the control. This may be due to the fact that most of the ROS had been eliminated by the cellular antioxidant system,59 or low (0.2 g/kg) concentrations of Nano-TiO2 were not sufficient to cause mitochondrial damage, thereby failing to increase the ROS. After being ingested by cells, the nanoparticles enter the mitochondria,60 interfering with the antioxidant defense mechanism of the cells or increasing the content of reactive oxygen species inside the mitochondria, but low levels of oxidative stress induce protective responses. Studies have shown that the reduction of antioxidant enzymes (such as glutathione peroxidase, superoxide dismutase, catalase) is related to oxidative damage in the body.61
After Nano-TiO2 treatment by subcutaneous injection, the MDA content in the liver of mice increased, whereas SOD and GSH-Px activity decreased.62 The results of our study showed that the levels of MDA in liver, kidney, and lung tissues of rats were significantly increased, and the activities of SOD and GSH-Px were significantly decreased. On the 3th day and 7th day in the 0.2 g/kg and 1 g/kg groups, the activity of SOD, GSH-Px, and MDA in rats recovered partially, suggesting that the antioxidant system of the body played its own protective role. Thus, the observed Nano-TiO2-induced genotoxicity in the present study could be attributed to the accumulation of ROS generated by Nano-TiO2, as indicated by the significant elevations of MDA levels that depleted cellular GSH and resisted the defensive effects of cellular antioxidant enzymes including SOD and GPx in a dose-dependent manner leading to oxidative stress that causes lipid peroxidation and can damage DNA (Figure 7).
 |
Figure 7 A proposed action model for the PI3K/AKT/FoxO3a pathway via ROS regulating DNA damage in the lung, liver, and kidney induced by Nano-TiO2 intratracheal administration. |
It has been documented that Nano-TiO2 induced oxidative stress63,65 and DNA damage in mouse brain, liver, and bone marrow cells66 and human kidney embryo cells.67 The oxidative stress can induce oxidative DNA lesions such as DNA strand breaks.68,69 In this study, the contents of OTM and Tail DNA in the liver, kidney, and lung tissues of the rats in each dose group were significantly increased compared with the control group, indicating that Nano-TiO2 damaged the DNA, which was consistent with the above results. There was no difference of the OTM and Tail DNA between the 1st, 3rd, and 7th days, suggesting that the damage induced by Nano-TiO2 could be irreversible, which might have contributed to the dysfunction of repair ability.
When cells are under oxidative stress, the expression of FOXO3a was significantly increased, and further activated downstream signaling pathways for anti-oxidation and DNA repair functions.70 FOXO3a resist oxidative stress through phosphorylation at T32, S253, and S315, which is activated by the PI3K/AKT pathway.71 By catalyzing the phosphorylation of FOXO3a, AKT blocks FOXO3a-dependent transcription primarily by promoting the exclusion of FOXO3a from the nucleus, rendering the transcription factor unable to activate its target gene.71 Previous studies suggested that ROS could activate the PI3K/AKT pathway,20,72,73 leading to cell proliferation, translocation, and activation of serine/threonine kinases.74,75 Our results showthat the PI3K and p-AKT expression increased whereas the ratio of p-FOXO3a/FOXO3a decreased after Nano-TiO2 tracheal instillation in a dose-dependent manner. These data suggested that Nano-TiO2 could inhibit the activity of FOXO3a protein by activating the PI3K/AKT signaling pathway.
FOXO can regulate DNA repair by activating downstream GADD45α and forms the FOXO3a/GADD45α complex, which further initiates the process of downstream DNA damage repair to protect damaged cells.18 GADD45α is a member of the GADD45 gene family and can be rapidly induced by a variety of injury factors, playing an important role in the cellular response to stress at the G2-M checkpoint and maintaining genomic stability. Low doses of Nano-TiO2 up-regulated the expression of GADD45α in HepG2 cells.76 GADD45α was increased in mice after oxidative stress,77,78 and DNA damage repair was abnormal in GADD45α knockout mice.79 Our results indicated that the expression of GADD45α increased after Nano-TiO2 exposure, suggesting that Nano-TiO2 induced the expression of GADD45α in the lung, liver, and kidney, thereby promoting the DNA damage repair function. The formation of DNA strand breaks in turn activates a number of downstream targets, including histone H2AX, as well as the cellular check-point kinase Chk2.80,81 DNA damage consequently activated the ATM–H2AX/Chk2–p53 pathway.82 Nano-TiO2 can activate the human fibroblast DNA damage response to the ATM-ChK2 pathway, thereby promoting ChK2 expression.83 The cell cycle monitoring point regulatory gene ChK2 participated in the repair of DNA damage through the regulation of XRCC1.84 XRCC1 is a key gene in the base excision repair (BER) pathway and is highly reactive to a variety of DNA damaging agents, including oxidants, alkylating agents, radiation, etc. Reduced XRCC1 function or XRCC1 deficiency might exacerbate genomic instability and increased the risk of asbestos-related malignancy.85 Our previous study found that XRCC1 deficiency sensitizes HepG2 cells to cisplatin, leading to cell cycle disorder and decreased DNA repair capacity.86 In the present study, the expression of ChK2 and XRCC1 in the lung, liver, and kidney increased in dose-dependent manners after intratracheal instillation of Nano-TiO2, suggesting that Nano-TiO2 could induce DNA damage by ChK2 related cell-cycle arrest.
Conclusion
Ti could distribute in main tissues of rats after intratracheal instillation of Nano-TiO2. Nano-TiO2 could produce oxidative stress in the lung, liver, and kidney, and then caused DNA damage, which could not recover within 7 days. Nano-TiO2 activated the PI3K/AKT/FOXO3a pathway and increased the expression of GADD45α, ChK2, and XRCC1 in the lung, liver, and kidney of rats responding to DNA damage. This study might help to deepen the understanding of the regulation mechanism of DNA damage caused by Nano-TiO2.
Abbreviations
ROS, reactive oxygen species; FOXO, Fork head O; PI3K, phosphatidylinositol 3-kinase; AKT, protein kinase B; GADD45α, growth arrest and DNA damage α; ChK2, checkpoint kinase; ATM, ataxia telangiectasia mutation; XRCC1, x-ray repair cross complementing gene 1; TEM, transmission electron microscopy; SEM, scanning electron microscopy; HE, hematoxylin and eosin; GSH-Px, glutathione peroxidase; SOD, superoxide dismutase; MDA, malonaldehyde; DCFH-DA, 2ʹ, 7ʹ-dichlorofluorescein diacetate; OTM, olive tail moment; PMSF, phenylmethylsulfonyl fluoride; PVDF, polyvinylidene fluoride; ANOVA, one-way analysis of variance; BER, base excision repair.
Ethics Approval
The animal use protocol has been reviewed and approved by the Laboratory Animal Ethical and Welfare Committee of Hebei Medical University, Shijiazhuang, China. Approval No. is IACUC-Hebmu-20,160,048.
Acknowledgments
Skilled technical assistance from Tianxu Liu and Chunfang Zhao is greatly appreciated.
Author Contributions
All authors contributed to data analysis, drafting, or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
We declare that we do not have any commercial or associative interest that represents a conflict of interest in connection with the work submitted. The authors report no conflicts of interest for this work.
References
1. Pelclova D, Zdimal V, Kacer P, et al. Markers of lipid oxidative damage in the exhaled breath condensate of nano TiO2 production workers. Nanotoxicology. 2017;11(1):52–63. doi:10.1080/17435390.2016.1262921
2. Bhattacharya K, Davoren M, Boertz J, Schins RP, Hoffmann E, Dopp E. Titanium dioxide nanoparticles induce oxidative stress and DNA-adduct formation but not DNA-breakage in human lung cells. Part Fibre Toxicol. 2009;6:17. doi:10.1186/1743-8977-6-17
3. Brezová V, Gabčová S, Dvoranová D, Staško A. Reactive oxygen species produced upon photoexcitation of sunscreens containing titanium dioxide (an EPR study). J Photochem Photobiol B. 2005;79(2):121–134. doi:10.1016/j.jphotobiol.2004.12.006
4. Dunford R, Cai LSN, Horikoshi S, Hidaka HKJ, Salinaro A. Chemical oxidation and DNA damage catalysed by inorganic sunscreen ingredients. FEBS Lett. 1997;418(2):87–90. doi:10.1016/S0014-5793(97)01356-2
5. Yu KN, Sung JH, Lee S, et al. Inhalation of titanium dioxide induces endoplasmic reticulum stress-mediated autophagy and inflammation in mice. Food Chem Toxicol. 2015;85:106–113. doi:10.1016/j.fct.2015.08.001
6. Stapleton PA, Hathaway QA, Nichols CE, et al. Maternal engineered nanomaterial inhalation during gestation alters the fetal transcriptome. Part Fibre Toxicol. 2018;15(1):3. doi:10.1186/s12989-017-0239-8
7. Shinohara N, Oshima Y, Kobayashi T, et al. Dose-dependent clearance kinetics of intratracheally administered titanium dioxide nanoparticles in rat lung. Toxicology. 2014;325(1):1–11. doi:10.1016/j.tox.2014.08.003
8. Husain M, Wu D, Saber AT, et al. Intratracheally instilled titanium dioxide nanoparticles translocate to heart and liver and activate complement cascade in the heart of C57BL/6 mice. Nanotoxicology. 2015;9(8):1013–1022. doi:10.3109/17435390.2014.996192
9. Huang KT, Wu CT, Huang KH, et al. Titanium nanoparticle inhalation induces renal fibrosis in mice via an oxidative stress upregulated transforming growth factor-beta pathway. Chem Res Toxicol. 2015;28(3):354–364. doi:10.1021/tx500287f
10. Ze Y, Zheng L, Zhao X, et al. Molecular mechanism of titanium dioxide nanoparticles-induced oxidative injury in the brain of mice. Chemosphere. 2013;92(9):1183–1189. doi:10.1016/j.chemosphere.2013.01.094
11. Berquist BR, Iii DMW. Pathways for repairing and tolerating the spectrum of oxidative DNA lesions. Cancer Lett. 2012;327(1–2):61–72. doi:10.1016/j.canlet.2012.02.001
12. Shukla RK, Vyom S, Pandey AK, Shashi S, Sarwat S, Alok D. ROS-mediated genotoxicity induced by titanium dioxide nanoparticles in human epidermal cells. Toxicol in Vitro. 2011;25(1):231–241. doi:10.1016/j.tiv.2010.11.008
13. Kops GJPL, Dansen TB, Polderman PE, et al. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419(6904):316–321. doi:10.1038/nature01036
14. Nemoto S, Finkel T. Redox regulation of forkhead proteins through a p66shc-dependent signaling pathway. Science. 2002;295(5564):2450–2452. doi:10.1126/science.1069004
15. Naoya M, Hiroto I, Rie M, et al. Nipradilol and timolol induce Foxo3a and peroxiredoxin 2 expression and protect trabecular meshwork cells from oxidative stress. Invest Ophthalmol Vis Sci. 2009;50(6):2777. doi:10.1167/iovs.08-3061
16. Wu D, Liang M, Dang H, Fang F, Xu F, Liu C. Hydrogen protects against hyperoxia-induced apoptosis in type II alveolar epithelial cells via activation of PI3K/Akt/Foxo3a signaling pathway. Biochem Biophys Res Commun. 2018;495(2):1620–1627. doi:10.1016/j.bbrc.2017.11.193
17. Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene. 2005;24(50):7410–7425. doi:10.1038/sj.onc.1209086
18. Tran H, Brunet A, Grenier JM, et al. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science. 2002;296(5567):530–534. doi:10.1126/science.1068712
19. Arden KC. Multiple roles of FOXO transcription factors in mammalian cells point to multiple roles in cancer. Exp Gerontol. 2006;41(8):709–717. doi:10.1016/j.exger.2006.05.015
20. Papaiahgari S, Zhang QS, Cho HY, Reddy SP. Hyperoxia stimulates an Nrf2-ARE transcriptional response via ROS-EGFR-PI3K-Akt/ERK MAP kinase signaling in pulmonary epithelial cells. Antioxid Redox Signal. 2006;8(2):43–52. doi:10.1089/ars.2006.8.43
21. Sundaresan NR, Madhu G, Rajamohan SB, Ayman I, Gupta MP. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J Clin Invest. 2009;119(9):2758–2771. doi:10.1172/JCI39162
22. Siafakas AR, Richardson DR. Growth arrest and DNA damage-45 alpha (GADD45α). Int J Biochem Cell Biol. 2009;41(5):986–989. doi:10.1016/j.biocel.2008.06.018
23. Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432(7015):316–323. doi:10.1038/nature03097
24. Oner D, Ghosh M, Bove H, et al. Differences in MWCNT- and SWCNT-induced DNA methylation alterations in association with the nuclear deposition. Part Fibre Toxicol. 2018;15(1):11. doi:10.1186/s12989-018-0244-6
25. Wang S, Gong Z, Chen R, et al. JWA regulates XRCC1 and functions as a novel base excision repair protein in oxidative-stress-induced DNA single-strand breaks. Nucleic Acids Res. 2009;37(6):1936–1950. doi:10.1093/nar/gkp054
26. Avanti K, Mcneill DR, Marc G, Mattson MP, Wilson DM. XRCC1 protects against the lethality of induced oxidative DNA damage in nondividing neural cells. Nucleic Acids Res. 2008;36(15):5111–5121. doi:10.1093/nar/gkn480
27. Niu Y, Zhang X, Zheng Y, Zhang R. XRCC1 deficiency increased the DNA damage induced by γ-ray in HepG2 cell: involvement of DSB repair and cell cycle arrest. Environ Toxicol Pharmacol. 2013;36(2):311–319. doi:10.1016/j.etap.2013.04.009
28. King J, Deboisblanc BP, Mason CM, et al. Effect of granulocyte colony-stimulating factor on acute lung injury in the rat. Am J Respir Crit Care Med. 1995;151(2 Pt 1):302–309. doi:10.1164/ajrccm.151.2.7531097
29. Asgharian B, Anjilvel S. A multiple-path model of fiber deposition in the rat lung. Toxicol Sci. 1998;44(1):80–86. doi:10.1093/toxsci/44.1.80
30. Zhang R, Niu Y, Li Y, et al. Acute toxicity study of the interaction between titanium dioxide nanoparticles and lead acetate in mice. Environ Toxicol Pharmacol. 2010;30(1):0–60. doi:10.1016/j.etap.2010.03.015
31. Wang H, Joseph JA. Quantifying cellular oxidative stress by dichlorofluorescein assay using microplate reader 1. Free Radic Biol Med. 1999;27(5–6):612–616. doi:10.1016/S0891-5849(99)00107-0
32. Shi H, Magaye R, Castranova V, Zhao J. Titanium dioxide nanoparticles: a review of current toxicological data. Part Fibre Toxicol. 2013;10:15. doi:10.1186/1743-8977-10-15
33. Kobayashi T, Oshima Y, Tsubokura Y, et al. rats. Regul Toxicol Pharmacol. 2016;81:233–241. doi:10.1016/j.yrtph.2016.08.018
34. Baisch BL, Corson NM, Wade-Mercer P, et al. Equivalent titanium dioxide nanoparticle deposition by intratracheal instillation and whole body inhalation: the effect of dose rate on acute respiratory tract inflammation. Part Fibre Toxicol. 2014;11:5. doi:10.1186/1743-8977-11-5
35. Prodan AM, Ciobanu CS, Popa CL, Iconaru SL, Predoi D. rats. Biomed Res Int. 2014;2014:134260. doi:10.1155/2014/134260
36. Yoshiura Y, Izumi H, Oyabu T, et al. Pulmonary toxicity of well-dispersed titanium dioxide nanoparticles following intratracheal instillation. J Nanopart Res. 2015;17(6):241. doi:10.1007/s11051-015-3054-x
37. Morimoto Y, Izumi H, Yoshiura Y, Fujishima K, Yatera K, Yamamoto K. Usefulness of intratracheal instillation studies for estimating nanoparticle-induced pulmonary toxicity. Int J Mol Sci. 2016;17(2).
38. Morimoto Y, Izumi H, Yoshiura Y, Fujisawa Y, Fujita K. Significance of intratracheal instillation tests for the screening of pulmonary toxicity of nanomaterials. J UOEH. 2017;39(2):123–132. doi:10.7888/juoeh.39.123
39. Gangwal S, Brown JS, Wang A, et al. Informing selection of nanomaterial concentrations for ToxCast in vitro testing based on occupational exposure potential. Environ Health Perspect. 2011;119(11):1539–1546. doi:10.1289/ehp.1103750
40. Hart GA, Hesterberg TW. In vitro toxicity of respirable-size particles of diatomaceous earth and crystalline silica compared with asbestos and titanium dioxide. J Occup Environ Med. 1998;40(1):29–42. doi:10.1097/00043764-199801000-00008
41. Msiska Z, Pacurari M, Mishra A, Leonard SS, Castranova V, Vallyathan V. DNA double-strand breaks by asbestos, silica, and titanium dioxide: possible biomarker of carcinogenic potential? Am J Respir Cell Mol Biol. 2010;43(2):210–219. doi:10.1165/rcmb.2009-0062OC
42. Pelclova D, Zdimal V, Fenclova Z, et al. Markers of oxidative damage of nucleic acids and proteins among workers exposed to TiO2 (nano) particles. Occup Environ Med. 2016;73(2):110–118. doi:10.1136/oemed-2015-103161
43. Zhao L, Zhu Y, Chen Z, et al. Cardiopulmonary effects induced by occupational exposure to titanium dioxide nanoparticles. Nanotoxicology. 2018;12(2):169–184. doi:10.1080/17435390.2018.1425502
44. Zhang X, Li W, Yang Z. Toxicology of nanosized titanium dioxide: an update. Arch Toxicol. 2015;89(12):2207–2217. doi:10.1007/s00204-015-1594-6
45. Zhang J, Song W, Guo J, et al. Cytotoxicity of different sized TiO2 nanoparticles in mouse macrophages. Toxicol Ind Health. 2013;29(6):523–533. doi:10.1177/0748233712442708
46. Gou N, Gu AZ. A new transcriptional effect level index (TELI) for toxicogenomics-based toxicity assessment. Environ Sci Technol. 2011;45(12):5410–5417. doi:10.1021/es200455p
47. Xiong S, George S, Yu H, et al. Size influences the cytotoxicity of poly (lactic-co-glycolic acid) (PLGA) and titanium dioxide (TiO(2)) nanoparticles. Arch Toxicol. 2013;87(6):1075–1086. doi:10.1007/s00204-012-0938-8
48. Thai SF, Wallace KA, Jones CP, et al. Differential genomic effects of six different TiO2 nanomaterials on human liver HepG2 cells. J Biochem Mol Toxicol. 2016;30(7):331–341. doi:10.1002/jbt.21798
49. Jiangxue W, Guoqiang Z, Chunying C, et al. Acute toxicity and biodistribution of different sized titanium dioxide particles in mice after oral administration. Toxicol Lett. 2007;168(2):176–185. doi:10.1016/j.toxlet.2006.12.001
50. Shinohara N, Danno N, Ichinose T, et al. Tissue distribution and clearance of intravenously administered titanium dioxide (TiO2) nanoparticles. Nanotoxicology. 2014;8(2):132–141. doi:10.3109/17435390.2012.763001
51. Ma-Hock L, Burkhardt S, Strauss V, et al. Development of a short-term inhalation test in the rat using nano-titanium dioxide as a model substance. Inhal Toxicol. 2009;21(2):102–118. doi:10.1080/08958370802361057
52. Oberdorster G, Ferin J, Lehnert BE. Correlation between particle size, in vivo particle persistence, and lung injury. Environ Health Perspect. 1994;102(Suppl 5):173–179. doi:10.1289/ehp.102-1567252
53. Tang M, Zhang T, Xue Y, et al. Metabonomic studies of biochemical changes in the serum of rats by intratracheally instilled TiO2 nanoparticles. J Nanosci Nanotechnol. 2011;11(4):3065–3074. doi:10.1166/jnn.2011.3604
54. Tang M, Zhang T, Xue Y, et al. Dose dependent in vivo metabolic characteristics of titanium dioxide nanoparticles. J Nanosci Nanotechnol. 2010;10(12):8575–8583. doi:10.1166/jnn.2010.2482
55. Liu H, Ma L, Zhao J, et al. Biochemical toxicity of nano-anatase TiO2 particles in mice. Biol Trace Elem Res. 2009;129(1–3):170–180. doi:10.1007/s12011-008-8285-6
56. Maziere C, Floret S, Santus R, Morliere P, Marcheux V, Maziere JC. Impairment of the EGF signaling pathway by the oxidative stress generated with UVA. Free Radic Biol Med. 2003;34(6):629–636. doi:10.1016/S0891-5849(02)01329-1
57. Kawanishi S, Oikawa S, Inoue S, Nishino K. Distinct mechanisms of oxidative DNA damage induced by carcinogenic nickel subsulfide and nickel oxides. Environ Health Perspect. 2002;110(Suppl 5):789–791. doi:10.1289/ehp.02110s5789
58. Gottschling BC, Maronpot RR, Hailey JR, et al. rats. Toxicol Sci. 2001;64(1):28–40. doi:10.1093/toxsci/64.1.28
59. AshaRani PV, Low Kah Mun G, Hande MP, Valiyaveettil S. Cytotoxicity and genotoxicity of silver nanoparticles in human cells. ACS Nano. 2009;3(2):279–290. doi:10.1021/nn800596w
60. Li N, Sioutas C, Cho A, et al. Ultrafine particulate pollutants induce oxidative stress and mitochondrial damage. Environ Health Perspect. 2003;111(4):455–460. doi:10.1289/ehp.6000
61. Michiels C, Raes M, Toussaint O, Remacle J. Importance of Se-glutathione peroxidase, catalase, and Cu/Zn-SOD for cell survival against oxidative stress. Free Radic Biol Med. 1994;17(3):235–248. doi:10.1016/0891-5849(94)90079-5
62. Shakeel M, Jabeen F, Qureshi NA, Fakhr EAM. Toxic effects of titanium dioxide nanoparticles and titanium dioxide bulk salt in the liver and blood of male sprague-dawley rats assessed by different assays. Biol Trace Elem Res. 2016;173(2):405–426. doi:10.1007/s12011-016-0677-4
63. Aitken RJ, Buckingham D, West K, Wu FC, Zikopoulos K, Richardson DW. Differential contribution of leucocytes and spermatozoa to the generation of reactive oxygen species in the ejaculates of oligozoospermic patients and fertile donors. J Reprod Fertil. 1992;94(2):451. doi:10.1530/jrf.0.0940451
64. Tamburrino L, Marchiani S, Montoya M, et al. Mechanisms and clinical correlates of sperm DNA damage. Asian J Androl. 2012;14(1):24–31. doi:10.1038/aja.2011.59
65. Sharma R, Masaki J, Agarwal A Sperm DNA Fragmentation Analysis Using the TUNEL Assay. 2013.
66. Noshy AEG, Galal A, Mohamed HR. Normalization of nano-sized TiO2-induced clastogenicity, genotoxicity and mutagenicity by chlorophyllin administration in mice brain, liver, and bone marrow cells. Toxicol Sci. 2014;142(1):21. doi:10.1093/toxsci/kfu157
67. Meena R, Rani M, Pal R, Rajamani P. Nano-TiO 2 -induced apoptosis by oxidative stress-mediated DNA damage and activation of p53 in human embryonic kidney cells. Appl Biochem Biotechnol. 2012;167(4):791–808. doi:10.1007/s12010-012-9699-3
68. Bonetta S, Gianotti V, Bonetta S, et al. DNA damage in A549 cells exposed to different extracts of PM(2.5) from industrial, urban and highway sites. Chemosphere. 2009;77(7):1030–1034. doi:10.1016/j.chemosphere.2009.07.076
69. Danielsen PH, Loft S, Kocbach A, Schwarze PE, Møller P. Oxidative damage to DNA and repair induced by Norwegian wood smoke particles in human A549 and THP-1 cell lines. Mutat Res Genet Toxicol Environ Mutagen. 2009;674(1):116–122. doi:10.1016/j.mrgentox.2008.10.014
70. Zuzana T, Ramya K, Huntly BJ, et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128(2):325–339. doi:10.1016/j.cell.2007.01.003
71. Brunet A, Bonni A, Zigmond MJ, et al. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell. 1999;96(6):857–868. doi:10.1016/S0092-8674(00)80595-4
72. Kwon J, Lee SR, Yang KS, et al. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc Natl Acad Sci U S A. 2004;101(47):16419–16424. doi:10.1073/pnas.0407396101
73. Leslie NR. The redox regulation of PI 3-kinase-dependent signaling. Antioxid Redox Signal. 2006;8(9–10):1765–1774. doi:10.1089/ars.2006.8.1765
74. Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408(6809):239–247. doi:10.1038/35041687
75. Kamata H, Hirata H. Redox regulation of cellular signalling. Cell Signal. 1999;11(1):1–14. doi:10.1016/S0898-6568(98)00037-0
76. Jana P, Bojana Z, Magdalena S, et al. DNA damage and alterations in expression of DNA damage responsive genes induced by TiO2 nanoparticles in human hepatoma HepG2 cells. Nanotoxicology. 2011;5(3):341–353. doi:10.3109/17435390.2010.507316
77. Hye Joung C, Ki Sung K, Masayuki F, Ting ZB. Critical role of the JNK-p53-GADD45α apoptotic cascade in mediating oxidative cytotoxicity in hippocampal neurons. Br J Pharmacol. 2015;162(1):175–192.
78. Srivastava AK, Srivastava PK, Al-Khedhairy AA, Musarrat J, Shukla Y. Allethrin-induced genotoxicity and oxidative stress in Swiss albino mice. Mutat Res. 2012;747(1):22–28. doi:10.1016/j.mrgentox.2012.03.003
79. Guillermo B, Andrea SF, Joachim M, et al. Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation. Nature. 2007;445(7128):671–675. doi:10.1038/nature05515
80. Tanaka T, Huang X, Halicka H, et al. Cytometry of ATM activation and histone H2AX phosphorylation to estimate extent of DNA damage induced by exogenous agents. Cytometry A. 2010;71A(9):648–661. doi:10.1002/cyto.a.20426
81. Yosef S. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer. 2003;3(3):155–168. doi:10.1038/nrc1011
82. Bartkova J, Horejsi Z, Koed K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434(7035):864–870. doi:10.1038/nature03482
83. Prasad RY, Chastain PD, Nana NF, Lisa S, Kaufmann WK, Fry RC. Titanium dioxide nanoparticles activate the ATM-Chk2 DNA damage response in human dermal fibroblasts. Nanotoxicology. 2013;7(6):1111–1119. doi:10.3109/17435390.2012.710659
84. Wen-Cheng C, Hui-Chun W, Fen-Hwa W, et al. Chk2-dependent phosphorylation of XRCC1 in the DNA damage response promotes base excision repair. EMBO J. 2014;27(23):3140–3150.
85. Pietruska JR, Tatiana J, Anatoly Z, Kane AB. XRCC1 deficiency sensitizes human lung epithelial cells to genotoxicity by crocidolite asbestos and Libby amphibole. Environ Health Perspect. 2010;118(12):1707–1713. doi:10.1289/ehp.1002312
86. Rong Z, Yujie N, Yikai Z. Increase the cisplatin cytotoxicity and cisplatin-induced DNA damage in HepG2 cells by XRCC1 abrogation related mechanisms. Toxicol Lett. 2010;192(2):108–114. doi:10.1016/j.toxlet.2009.10.012
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.