")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 16
Tildrakizumab: An Evidence-Based Review of Its Use in the Treatment of Moderate-to-Severe Chronic Plaque Psoriasis
Authors Näslund-Koch C , Zachariae C , Skov L
Received 8 July 2020
Accepted for publication 26 August 2020
Published 24 September 2020 Volume 2020:16 Pages 903—916
DOI https://doi.org/10.2147/TCRM.S227880
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Charlotte Näslund-Koch,1 Claus Zachariae,1,2 Lone Skov1,2
1Department of Dermatology and Allergy, Herlev and Gentofte Hospital, Copenhagen, Denmark; 2Faculty of Health and Medical Sciences, University of Copenhagen, Copenhagen, Denmark
Correspondence: Claus Zachariae Department of Dermatology and Allergy
University of Copenhagen, Herlev and Gentofte Hospital, Gentofte Hospitalsvej 15, Hellerup 2900, Denmark
Tel +4538673203
Fax +4538677615
Email [email protected]
Abstract: Psoriasis is a common immune-mediated chronic inflammatory disease, and observations have pointed toward the IL-23/Th17 cell axis as having a key role in the pathogenesis of psoriasis. This new immunological understanding of the pathogenesis has been translated into targeted and highly effective biologic therapies. Tildrakizumab is a humanized IgG1/k monoclonal antibody targeting the p19 unit of IL-23 and has been registered for the treatment of patients with moderate-to-severe chronic plaque psoriasis in adults since 2018. This review provides an overview of the efficacy and safety of tildrakizumab, focusing on the results from clinical trials. In both Phase II and III trials, tildrakizumab 100 and 200 mg was significantly more efficacious than both placebo and etanercept at week 12. The effect of tildrakizumab continued to increase until week 28. Long-term follow-up showed high levels of efficacy for up to 3 years. Despite no difference between 100 and 200 mg in Phase III studies, subgroup analyses showed better efficacy when treated with 200 mg in patients with bodyweight ≥ 90 kg. The overall drug safety was good, and besides discrete higher incidence of nasopharyngitis, the conducted clinical trials show that tildrakizumab was very well tolerated without any safety concerns. Compared to other IL-23p19 inhibitors, tildrakizumab seemed to have slightly lower efficacy. However, to determine its position in the treatment algorithm of psoriasis, head-to-head trials with other IL-17, IL-12/23, and IL-23 inhibitors and long-term real-world data are required.
Keywords: tildrakizumab, IL-23p19, biologics, psoriasis, safety, efficacy
Plain Language Summary (Optional)
Psoriasis is a common, inflammatory, chronic disease, that was considered to only affect the skin, but today is acknowledged as a systemic disease affecting both the skin and many other organs, especially the joints. Although the pathogenesis of psoriasis is not yet fully understood, we do know that the immune system plays a crucial role, triggering an immune response, which leads to increased proliferation of skin cells. The new immunological understanding of the disease has been translated into highly effective targeted therapies, called biologics. The first biological drug was approved for treatment of psoriasis in Europe in 2004, and since then, several drugs, each becoming more specific in the mechanism of action, have been registered. Tildrakizumab was approved in 2018 for the treatment of moderate-to-severe psoriasis in adults. Our review provides an overview of the efficacy and safety of tildrakizumab, focusing on the results from clinical trials. Tildrakizumab was significantly more efficacious than placebo after 12 weeks of treatment. The effect of tildrakizumab continued to increase until week 28, with long-term follow-up showing high levels of efficacy for up to 3 years. The overall drug safety was good, and besides the discrete higher risk of nasopharyngitis, the conducted clinical trials show no safety concerns. Compared to other biologics with the same mechanism of action, tildrakizumab seemed to have slightly lower efficacy. However, to determine its position in the treatment algorithm of psoriasis, further trials comparing the new biological treatments against one another are required.
Introduction
Psoriasis is an immune-mediated chronic inflammatory disease affecting 2–4% of the population in Europe and North America.1 Traditionally, psoriasis was thought to only involve the skin but today psoriasis is classified as a systemic inflammatory disease with extensive comorbidities.2 Psoriasis is associated with both physical and psychological burden and have severe effects on the quality of life, comparable to what is seen in cancer, heart disease, and depression.3
Several observations have pointed towards T helper (Th) type 17 cells, interleukin (IL)-17, and IL-23 as having key roles in the pathogenesis of psoriasis.4,5 This new immunological understanding of the pathogenesis of psoriasis has been translated into targeted and highly effective biologic therapies. Tildrakizumab is a specific IL-23 monoclonal antibody, registered for the treatment of moderate-to-severe chronic plaque psoriasis in adults. This review provides an overview of the efficacy and safety of tildrakizumab, focusing on the results from clinical trials and describes its use in the treatment of psoriasis.
Pathogenesis of Psoriasis
Psoriasis is a complex and multifactorial condition and the exact pathogenesis is not known. However, the involvement of the immune system primarily and subsequently, abnormal keratinocyte proliferation, is now widely accepted. It is a matter of dysregulated interactions between the innate and adaptive immune system that involves numerous proinflammatory cytokines.1 Dendritic cells play a major role in the initial stages of psoriasis.6 After the presentation of specific psoriasis autoantigens or certain environmental stimuli (triggers), activated dendritic cells produce, among other mediators, IL-12, IL-23, tumor necrosis factor-alpha (TNF-α), and IL-6 (Figure 1).1,4,6 TNF-α is produced by multiple cell types besides dendritic cells, and amplifies inflammation through several distinct pathways.1 Besides dendritic cells, several observations have pointed towards Th1 and Th17 cell activation and its associated interleukins as having key roles in the pathogenesis.4 The differentiation of Th cells into Th1 and Th17 cells is respectively mediated by IL-12 and IL-23 (Figure 1). Once activated, Th17 cells produce several mediators such as IL-17A-F, which induce keratinocyte proliferation.7
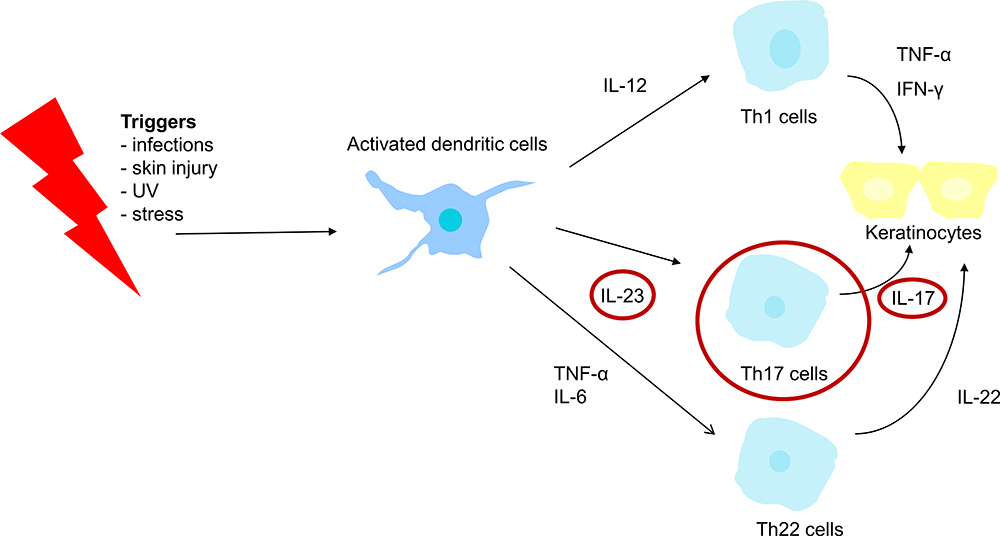 |
Figure 1 Simplified schematic figure illustrating the pathogenesis of psoriasis. |
Earlier, psoriasis was primarily characterized as a Th1-driven disease, but nowadays, the central importance of the IL-23/Th17 cell axis in the pathogenesis of psoriasis is growing.
Severity of Psoriasis
The severity of the disease is generally determined on the bases of some objective and subjective measures. As for objective measures, Body Surface Area (BSA), Psoriasis Area and Severity Index (PASI) and Physician´s Global Assessment (PGA) have been used as the gold standard. Dermatology Life Quality Index (DLQI) is a subjective tool, assessing the impact of skin disease on the quality of life of the affected person. The tool is often used for psoriasis despite not being specifically designed for psoriasis. Moderate-to-severe psoriasis is often defined as BSA involvement >10% or PASI >10, or DLQI >10.8 This has also been known as “the rule of ten”. In clinical trials, PASI and PGA are the most frequently reported psoriasis efficacy measures; it can either be the number of patients obtaining a 50%, a 75%, a 90%, or a 100% reduction in PASI from baseline, also called PASI50, PASI75, PASI90, and PASI100. Regarding PGA, it can either be the number of patients obtaining at least a reduction of 2 or the proportion of patients reaching a score of 0 or 1, at a given time point, normally 12 or 16 weeks after treatment initiation.9 PASI75 used to be accepted as the primary outcome, but after the introduction of more effective treatments, PASI90 is the new gold standard. As many patients switch directly from one treatment to another, new treatment guidelines increasingly focus on residual disease and actual PASI instead of relative improvement. An absolute PASI of less than 2 or 3 is now been considered as desirable.10
Current Treatments of Psoriasis
Systemic treatment for psoriasis consists of conventional systemic agents (eg acitretin, ciclosporin, and methotrexate), small molecules (eg apremilast and dimethyl fumarate), and biological treatments. Biologics are treatments that use substances made from living organisms or synthetic versions, to target the immune system and block the action of the proinflammatory cytokines described above.11 The development of biological therapies has revolutionized the management of psoriasis. Over the last two decades, numerous biologics have been approved for the treatment of psoriasis.12 They are divided according to the mechanism of action in anti-TNF, anti-IL-12/23, anti-IL-17, and anti-IL-23 (Figure 2).11 Infliximab, etanercept, adalimumab, and certolizumab pegol are all TNF inhibitors and are considered first-generation biologics, all approved for psoriasis and psoriatic arthritis. They have a relatively nonspecific mode of action as they act several places in the inflammation cascade. The search for more effective treatments for psoriasis with fewer side effects through an even more targeted immunological treatment led to the development of second-generation biologics, such as monoclonal antibodies targeting the IL-23 and the IL-17 pathways. IL-23 is composed of two subunits, p19 and p40. The p40 unit is shared with IL-12. Ustekinumab is a human monoclonal antibody that binds to the p40 subunit of both IL-12 and IL-23. However, IL-23 signaling seems to be more important than IL-12, and this understanding has led to the development of the IL-23p19 inhibitors guselkumab, tildrakizumab, and most recently risankizumab. Secukinumab, a human IL-17A antibody, was approved as the first biological agent blocking IL-171 and now ixekizumab, a humanized IL-17A antibody, and brodalumab, a human IL-17 receptor antagonist (IL-17RA), are also approved for treatment of psoriasis (Figure 2).12 Secukinumab and ixekizumab have also been approved for the treatment of psoriatic arthritis.
 |
Figure 2 Biologics on the market. |
The above-mentioned biologics are different with regards to efficacy, safety, and side effects.12 It is crucial for dermatologists and other physicians using these biologics to understand the major benefits and limitations of each of these biological therapies to be able to choose the best treatment for the individual patient. Tildrakizumab, a monoclonal antibody targeting IL-23p19, was approved in 2018 by both the United States Food and Drug Administration (US FDA)13 and the European Medicines Agency (EMA)14 for the treatment of patients with moderate-to-severe chronic plaque psoriasis. US FDA and EMA currently approve the following dosing regimen for tildrakizumab; 100 mg injection subcutaneously (SC) at week 0 and 4, followed by 100 mg injection every 12 weeks thereafter.13,14 The EU label also accepts dosage with 200 mg in certain conditions, such as high disease burden and bodyweight ≥90 kg.14 Furthermore, the EU label acknowledges that patients with a partial response may subsequently improve with continued treatment past 28 weeks.
Methods
A literature review was undertaken by the authors, searching the PubMed database for clinical trials published in English until 1 June, 2020, using the following combinations of MeSH search terms: ”psoriasis” and ”tildrakizumab”. Subgroup analysis was subsequently searched for using the MeSH terms “psoriasis”, “tildrakizumab”, and “subgroup”. Further relevant literature was searched for manually in the reference lists of the included papers.
Pharmacokinetics of Tildrakizumab
In 2008 to 2010, Khalilieh et al performed two randomized, placebo-controlled Phase I trials to assess the pharmacokinetics, bioavailability, and safety of single ascending doses tildrakizumab after intravenous (IV) and SC dosing in healthy subjects.15 After both IV and SC dosing, tildrakizumab exhibited slow systemic clearance, limited volume of distribution and long half-life (t1/2), ranging from 26.8 to 32.4 days. Furthermore, the maximum concentration (Cmax) and the area under the curve (AUC) increased proportionally with dose over a range from 0.1 to 10 mg/kg (IV) and 50–200 mg (SC), respectively. Subcutaneously administered 50 mg tildrakizumab, had a bioavailability of 80% (similar to 0.5 mg/kg IV) and 200 mg of 73% (similar to 3 mg/kg IV).
In 2008 to 2011, Kopp et al performed a three-part, randomized, placebo-controlled, rising multiple-dose phase I trial in patients with moderate-to-severe psoriasis to evaluate the clinical activity, safety, and pharmacokinetics of tildrakizumab.16 Tildrakizumab demonstrated similar results in this phase I trial, as the study by Khalilieh et al showed slow systemic clearance and a long t1/2, with mean clearance ranging from 1.57 to 2.50 mL/day/kg and mean t1/2 ranging from 20.2 to 26.9 days. No dose trends were observed for these pharmacokinetic parameters. In part 2 of the trial the AUC and Cmax were dose proportional for tildrakizumab 3 and 10 mg/kg.
Another phase I trial, also conducted by Khalilieh et al in 2015 to 2016,17 evaluated whether tildrakizumab could alter the Cytochrome P450 (CYP) metabolism in subjects with moderate-to-severe psoriasis. Treatment with two doses of tildrakizumab 200 mg did not have any clinically meaningful effect on CYP metabolism in subjects with moderate-to-severe psoriasis. The potential for clinically meaningful drug–drug interactions in patients with psoriasis treated with tildrakizumab therefore seems low.
Furthermore, the pharmacokinetics of tildrakizumab seems to be unaffected by ethnic variability, with comparable Cmax, and t1/2 in Japanese, Chinese, and Caucasian healthy subjects.18
New population–pharmacokinetic analysis across Phase I, II, and III of tildrakizumab showed that subjects with bodyweight >90 kg had a lower area under the plasma concentration–time curve at steady state vs those with bodyweight ≤90 kg.19
Conclusively, in both healthy subjects and patients with moderate-to-severe psoriasis, tildrakizumab demonstrated a t1/2 of approximately 3 weeks, the AUC and Cmax were dose-proportional and unaffected by ethnic variability.
At the moment, there are three IL-23p19 inhibitors registered for the treatment of moderate-to-severe plaque psoriasis in adults (Table 1); tildrakizumab (a humanized IgG1/k monoclonal antibody),13 guselkumab (a human IgG1λ monoclonal antibody),20 and risankizumab (a humanized IgG1 monoclonal antibody).21 They are all high-affinity antibodies with an equilibrium dissociation constant (Kd) in the picomolar range (pM),22–24 even though tildrakizumab shows the highest KD corresponding to the lowest affinity of the three antibodies (Table 1). Similar to tildrakizumab, guselkumab and risankizumab also exhibit dose-proportional plasma concentrations. Guselkumab has the shortest t1/2, ranging from 15 to 18 days,20 while risankizumab demonstrates the longest with t1/2 = 28 days.21 The absolute bioavailability of risankizumab is similar to tildrakizumab, while for guselkumab it is estimated to be approximately 49% in healthy subjects. The peak concentration of guselkumab is similar to tildrakizumab, though demonstrated in healthy subjects, and for risankizumab the peak concentrations is reached from 3 to 14 days (Table 1).13,20,21
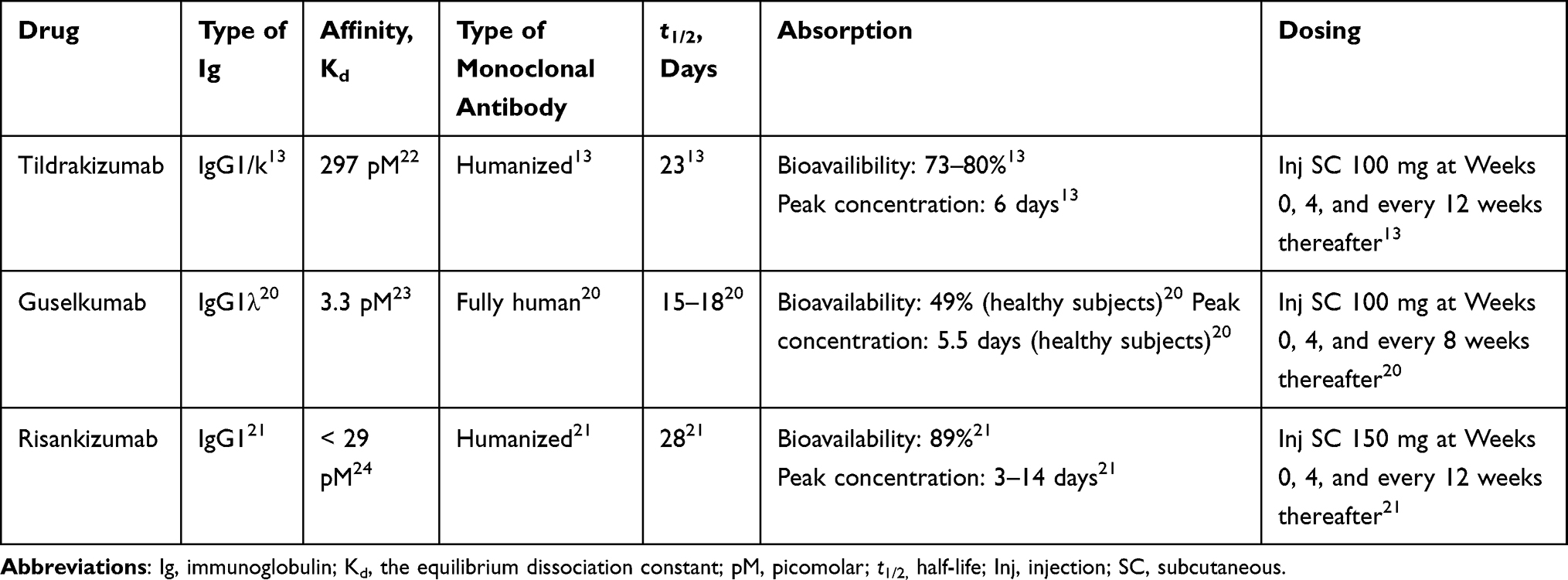 |
Table 1 Overview of the IL-23p19 Inhibitors |
Therapeutic Efficacy of Tildrakizumab
The efficacy of SC tildrakizumab for the treatment of moderate-to-severe chronic plaque psoriasis in adults has been evaluated in one phase IIb trial25 and two phase III trials (Table 2).26
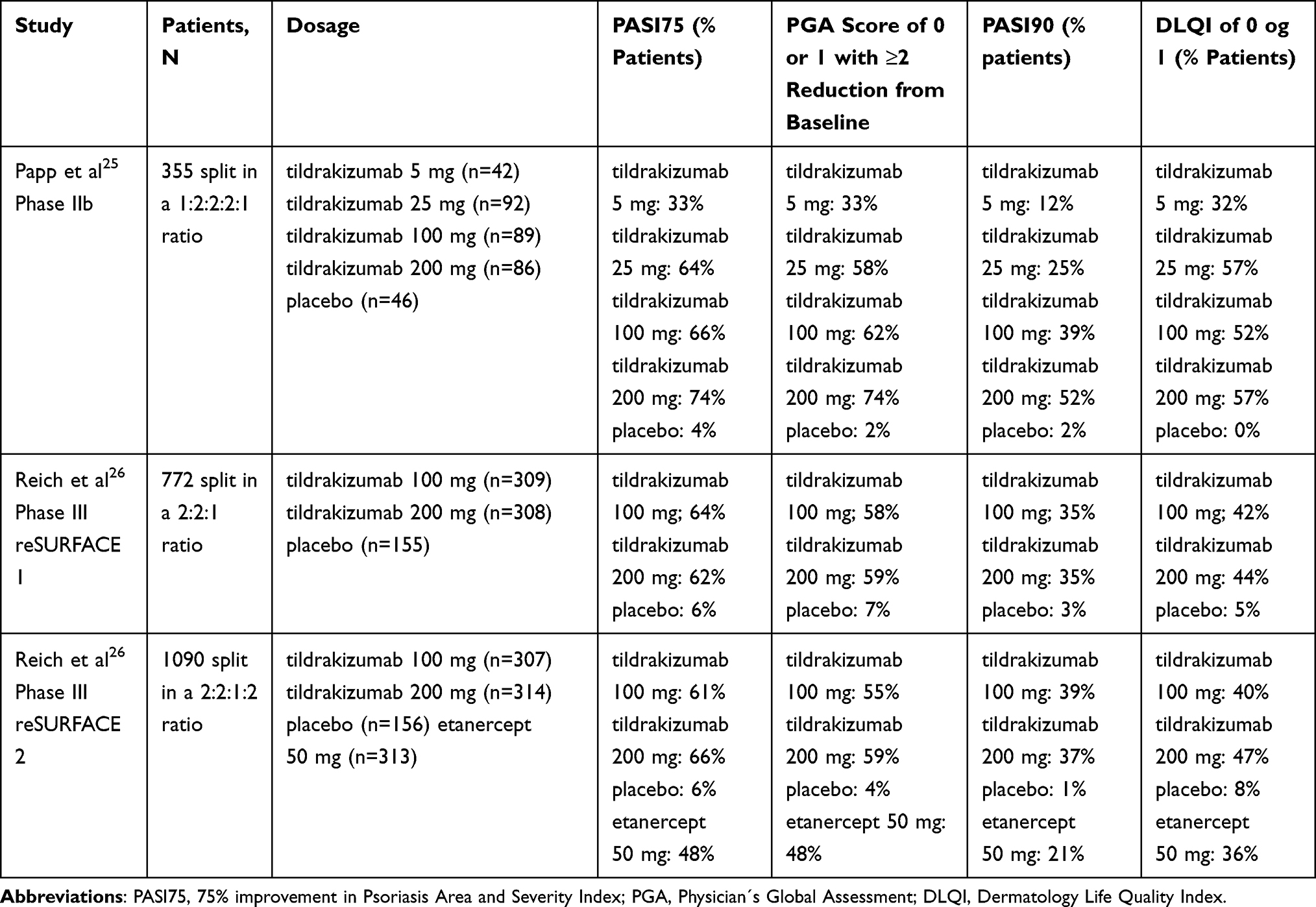 |
Table 2 Summary of Phase II and III Trials with Main Results from Initiation of Treatment to Week 12 or 16 |
Phase II Trials
Papp et al reported data from a randomized, double-blind, placebo-controlled, multinational, dose-finding phase IIb trial.25 Eligible patients (n = 355) were adults (age 18 or older), with moderate-to-severe chronic plaque psoriasis, who were candidates for phototherapy or systemic therapy. Participants were randomized to five treatment arms (SC tildrakizumab; 5, 25, 100, or 200 mg), or placebo (Table 2). Tildrakizumab was administered every 4 weeks in part 1 (week 0–16), and thereafter every 12 weeks in part 2 (week 16–52). Re-randomization occurred at week 16, based on responder status. PASI75 responders who had received tildrakizumab 5 or 25 mg continued the respective doses. Responders who had received tildrakizumab 100 or 200 mg were re-randomized to continue the same dose or on a reduced dose (100 to 25 mg and 200 to 100 mg). Nonresponders at week 16 were re-randomized to an increased dose; participants from the placebo, 5 and 25 mg groups to receive 100 mg, and participants from the 100 mg group to receive 200 mg. Nonresponders in the 200 mg tildrakizumab group remained on this dose throughout part 2. Study participants discontinued treatment at week 52, and entered a 20-week follow-up period, namely part 3 (week 52–72).
The primary endpoint was the proportion of participants achieving PASI75 at week 16. Secondary endpoints included the proportion of participants with a PGA score of 0 and 1 at week 16, PASI90 at week 16, and the mean change from baseline in DLQI at week 16.
PASI75 response rates at week 16 was 33%, 64%, 66%, and 74% for the participants in the tildrakizumab 5, 25, 100, and 200 mg groups, respectively, compared to 4% for placebo (Table 2). The PGA response of 0 or 1 at week 16 were 33%, 58%, 62%, and 74% for the participants in the tildrakizumab 5, 25, 100, and 200 mg groups, respectively, compared to 2% for placebo (Table 2). PASI90 response rates at week 16 were 12%, 25%, 39%, and 52% for the participants on the tildrakizumab 5, 25, 100, and 200 mg groups, respectively, compared to 2% for placebo (Table 2). Improvements in DLQI were observed in all groups receiving tildrakizumab compared to placebo (Table 2).
PASI75 responders who continued to receive doses of 100 or 200 mg tildrakizumab after week 16, still experienced efficacy at week 52. More than 90% maintained PASI75 at week 52, compared to only 70% of the PASI75 responders who received a reduction in dose at week 16 from 100 to 25 mg. For PASI75 nonresponders, who received an increased dose at week 16, PASI75 tended to increase with time. For nonresponders, who remained on 200 mg tildrakizumab at week 16, no increase in PASI75 was seen in part 2.
Data from part 3, shows that 96% and 93% of the participants receiving doses of 100 and 200 mg tildrakizumab, respectively, maintained PASI75 at week 72, despite drug discontinuation at week 52. This indicates a long-lasting effect after the treatment has been stopped.
Conclusively, this phase IIb trial demonstrated significant dose-related efficacy evaluated by PASI75, PASI90, and PGA response compared to placebo. Doses of 100 and 200 mg showed the most optimal outcomes, with a discrete higher efficacy of 200 mg, and this especially in attaining PASI90. PASI75 response was generally maintained through 72 weeks, and reduction of dose from 100 to 25 mg led to significant loss of efficacy.
Phase III Trials
The phase III trials were two large, randomized, double-blind, placebo-controlled, multinational, parallel-group studies; reSURFACE 1 and reSURFACE 2.26 The doses evaluated here were based on results from the previous phase IIb study.25 Eligible patients were adults (age 18 or older), with moderate-to-severe chronic plaque psoriasis, who were candidates for phototherapy or systemic therapy.
reSURFACE 1 was conducted from December 2012 to October 2015. In part 1 of the trial (week 0–12), 772 participants split in a 2:2:1 ratio were randomized to tildrakizumab 200 mg, tildrakizumab 100 mg, or placebo. In part 2 (week 12–28), those in the placebo group were re-randomized to either tildrakizumab 200 or 100 mg. The rest of the participants remained in the original groups. In part 3 (week 28–64), responders (PASI ≥75) were re-randomized to continue on the same dose tildrakizumab or to receive a placebo. Partial responders (PASI ≥50 and PASI ≤75) to tildrakizumab 200 mg continued on the same dose, while partial responders to 100 mg were re-randomized to 200 or 100 mg tildrakizumab.27
Tildrakizumab was administered SC at weeks 0 and 4 during part 1 and then every 12 weeks subsequently. Re-randomized placebo patients received tildrakizumab at weeks 12 and 16.
reSURFACE 2 was conducted from February 2013 to September 2015. In this trial, an active comparator arm (etanercept) was incorporated. In part 1 (week 0–12), 1090 participants split in a 2:2:1:2 ratio were randomized to tildrakizumab 200 mg, tildrakizumab 100 mg, placebo, or etanercept 50 mg. In reSURFACE 2 tildrakizumab was administered in the same way as in reSURFACE 1, and etanercept was given twice weekly in part 1 and once weekly in part 2. In part 2 (week 12–28), participants from the placebo group was re-randomized to either tildrakizumab 200 or 100 mg. The rest of the participants remained in the original groups. In part 3 (week 28–52), responders (PASI ≥75) to tildrakizumab 200 mg were re-randomized to tildrakizumab 200 or 100 mg, while responders to tildrakizumab 100 mg maintained on the same dose. Partial responders to tildrakizumab 200 mg continued on the same dose, while partial responders to 100 mg were re-randomized to 200 or 100 mg tildrakizumab. Partial and nonresponders of etanercept were crossed over to tildrakizumab 200 mg after a 4-week washout period.
The primary endpoint for both studies was the proportion of patients achieving at least PASI75 and PGA score of 0 or 1 with ≥2-grade score reduction from baseline at week 12. Secondary endpoints were PASI90, PASI100 at week 12, and proportion of participants with a DLQI score of 0 or 1 at week 12 and 28.
At week 12 in reSURFACE 1, 64% of the patients treated with tildrakizumab 100 mg and 62% of the patients treated with tildrakizumab 200 mg attained PASI75, compared to 6% in the placebo group (Table 2). PGA response of 0/1 was achieved by 58% of the patients treated with tildrakizumab 100 mg and 59% of the patients treated with tildrakizumab 200 mg, compared to 7% in the placebo group (Table 2). PASI90 was achieved among 35% of the patients receiving 100 mg tildrakizumab and 35% of the patients receiving 200 mg tildrakizumab, compared to 3% in the placebo group (Table 2). The proportion of participants with a DLQI of 0 or 1 at week 12 was 42%, 44%, and 5% in the group of tildrakizumab 100 mg, tildrakizumab 200 mg, and placebo, respectively (Table 2).
At week 12 in reSURFACE 2, 61% of the patients treated with tildrakizumab 100 mg and 66% of the patients treated with tildrakizumab 200 mg attained PASI75, compared to 48% of the patients treated with etanercept and 6% in the placebo group (Table 2). PGA response of 0/1 was achieved by 55% of the patients treated with tildrakizumab 100 mg and 59% of the patients treated with tildrakizumab 200 mg, compared to 48% of the patients treated with etanercept and 4% in the placebo group (Table 2). PASI90 was achieved by 39% of the patients receiving 100 mg tildrakizumab and 37% of the patients receiving 200 mg tildrakizumab, compared to 21% of the patients treated with etanercept and 1% in the placebo group. The proportion of participants with a DLQI of 0 or 1 was 40%, 47%, 36%, and 8% in the group of tildrakizumab 100 mg, tildrakizumab 200 mg, etanercept 50 mg, and placebo, respectively.
At week 28 in reSURFACE 1, 77% of the patients treated with 100 mg tildrakizumab, and 79% of the patients treated with 200 mg tildrakizumab achieved PASI75 (nonresponder imputation, NRI). Similar results were seen in the re-randomized placebo groups that had received tildrakizumab from week 12 until week 28. For PASI90, the proportions were 49% and 57% for tildrakizumab 100 mg and 200 mg, respectively (NRI). Similar results were seen in the re-randomized placebo groups that had received tildrakizumab from week 12 until week 28. The proportion of patients achieving DLQI 0 or 1 at week 28 was 57% among tildrakizumab 200 mg and 52% among tildrakizumab 100 mg (observed data, OD). Similar results were seen in the re-randomized placebo group that had received tildrakizumab from week 12 until week 28.
At week 28 in reSURFACE 2, 73% of the patients treated with both 100 and 200 mg tildrakizumab achieved PASI75 (NRI). Among participants treated with etanercept, 54% achieved PASI75 (NRI). In the re-randomized placebo groups who had received tildrakizumab 100 or 200 mg from week 12 until week 28, 54% and 55% achieved PASI75, respectively. For PASI90, the proportion of patients was 55%, 57%, and 29%, for tildrakizumab 100 mg, tildrakizumab 200 mg, and etanercept, respectively (NRI). In the re-randomized placebo group, that had received tildrakizumab 100 mg the proportion was 38%, and in the group who received tildrakizumab 200 mg, 46%. The proportion of patients achieving DLQI 0 or 1 at week 28 was 65% for tildrakizumab 200 mg and 54% for tildrakizumab 100 mg (OD). Among patients that were re-randomized from placebo to tildrakizumab 200 mg, the proportion was 57%, and from placebo to tildrakizumab 100 mg, 38%. In comparison, 39% of the patients treated with etanercept from week 0 to 28 attained a DLQI of 0 or 1.
Conclusively, in both phase III trials investigating tildrakizumab 100 and 200 mg for moderate-to-severe psoriasis in adult patients, tildrakizumab was significantly more efficacious than both placebo and etanercept at week 12. The effect of tildrakizumab continued to increase until week 28, and in phase III studies, there was no difference between tildrakizumab 100 and 200 mg.
Subsequently, pooled subgroup analysis of the above phase IIb and phase III trials showed discrete higher proportions of PASI75 and PASI90 at week 12 among participants <65 years of age, bodyweight ≤90 kg, and without psoriatic arthritis.28 Similarly, further subgroup analyses showed higher PASI and PGA responses in the group receiving tildrakizumab 200 mg compared to 100 mg in patients with a higher bodyweight.29 Pooled exposure-response analysis confirmed this tendency, and demonstrated that high bodyweight (>90 kg) was correlated with decreased tildrakizumab exposure (measured as average concentrations of tildrakizumab) and decreased PASI response.30 PASI90, PASI100, and PGA responses were generally higher in patients without prior biologics exposure.28,29 However, efficacy did not seem to differ with respect to sex, race, or number of prior failed conventional systemic treatments.28
Subgroup analysis of reSURFACE 1 and 2 also showed that participants with < PASI50 at week 28 could be identified at week 8, suggesting that patients that are unlikely to respond to treatment can be identified as early as treatment week 8.31 The same pooled analysis demonstrated that week 28 PASI improvement level correlated with improvement in DLQI. Furthermore, greater improvement in PASI was associated with a greater likelihood of achieving DLQI 0 or 1.31
In real-life settings, treatment interruption and dose adjustments are common. Newly published subgroup analyses from reSURFACE 1 and 232 covering this matter are therefore interesting. In part 3 in reSURFACE 1, a subgroup of the responders and partial responders from tildrakizumab 200 and 100 mg were at week 28 re-randomized to placebo. The median time to relapse (defined as a 50% reduction in maximum PASI response) was 24 weeks in both groups. Approximately 85% of the patients that reinitiated tildrakizumab for at least 12 weeks had achieved PASI75 by week 64, indicating that the efficacy of tildrakizumab can be regained after treatment interruption and reinitiation. Dose adjustment analysis revealed that patients who were responders to tildrakizumab 200 mg at week 28, and were re-randomized to tildrakizumab 100 mg, experienced little impact of efficacy demonstrated by the proportion of PASI75 responders that remained consistent throughout week 52. Partial responders to tildrakizumab 100 mg that were re-randomized to 200 mg at week 28, continued to improve but without meaningful difference compared to those who continued the dosage of 100 mg.32
Several new systematic reviews and meta-analyses comparing the efficacy of biologics, conventional systemic therapies, and placebo, showing that biologics outperform placebo33,34 and conventional systemic therapies,11,35 have been published in recent years. New generation biologics (anti-IL-17 and anti-IL-23 therapies) have shown better efficacy than the first biologics registered for treatment for psoriasis. Recently, all the IL-17 inhibitors and the IL-23 inhibitors guselkumab, and risankizumab were found to be more efficacious than tildrakizumab, ustekinumab, and the TNF inhibitors adalimumab, certolizumab pegol, and etanercept measured with all levels of PASI response.34
A network meta-analysis from Cochrane, comparing systemic pharmacological treatments for chronic plaque psoriasis, concluded that in terms of achieving PASI90, all of the anti-IL-17 drugs (ixekizumab, secukinumab, bimekizumab, and brodalumab) and the anti-IL-23 drugs (risankizumab and guselkumab, but not tildrakizumab) were significantly more effective than ustekinumab, adalimumab, certolizumab pegol, and etanercept.11 Some of the evaluated drugs have only been investigated in a few trials, and results should be interpreted with caution. Similar results were seen in another recent and comprehensive meta-analyses and systematic review from Armstrong et al, comparing treatments for moderate-to-severe plaque psoriasis.35 Brodalumab, guselkumab, ixekizumab, and risankizumab were associated with the highest rate of PASI90, both in the short and long term. A single systematic review and meta-analysis from Xu et al claimed that guselkumab was the most efficient treatment of the three IL-23p19 inhibitors.36
To directly compare and discriminate between the different biologics, there is a need for head-to-head randomized clinical trials and real-life data from patients with psoriasis on biologics.
Maintenance of Disease Control
Psoriasis requires an effective long-term treatment considering the chronic nature of the disease. Therefore, data on long-term maintenance of disease control is crucial. Newly published results from a 3-year follow-up of reSURFACE 1 and 2 showed high levels of efficacy after treatment with tildrakizumab for up to 3 years.27 For responders (≥ PASI75 at week 28) who remained on tildrakizumab 100 mg, approximately 73% attained PASI75 and 54% PASI90 at the end of the follow-up at week 148, and for responders who remained on 200 mg approximately 80% attained PASI75 and 60% PASI90.
The efficacy of tildrakizumab in long-term studies were slightly lower than data published on other IL-23p19 inhibitors, even though follow-up period was shorter for risankizumab. For guselkumab, a three-year follow-up of the phase III trial VOYAGE 1 and VOYAGE 2 showed maintenance of clinical response through 3 years.37 Approximately 72% (NRI) of the patients on continuous treatment with guselkumab attained PASI90 at week 156. Risankizumab, the most recent approved IL-23p19 inhibitor, showed slightly higher PASI90 responses but at week 52 in the phase III trials UltIMMa-1, UltIMMa-2, and IMMhance, with approximately 80–85% of the patients attaining PASI90.38 Studies on long-term efficacy of risankizumab are ongoing.39 Real-life data on anti-IL-23 are still lacking.
Safety and Adverse Events (AEs)
AEs in Early Clinical Trials
Phase I trials did not show any concerning AEs, with a similar incidence of adverse effects across treatment groups.15,16 In the phase IIb trial, hypertension was reported more often in treatment groups (9 of 308) compared to placebo (0 of 45).25 Most of these patients had hypertension or borderline hypertension at baseline, and none of the participants discontinued the treatment because of hypertension. Few serious AEs (SAEs) were identified.15–17,25 From Kopp et al,16 only one of the SAEs were considered possibly related to the treatment. Seventeen days after dosing with tildrakizumab 10 mg/kg, one elderly man experienced convulsion. Several confounding factors could possibly explain the convulsion; sleep deprivation, alcohol consumption, and use of benzodiazepine. From Khalilieh et al,17 one participant discontinued treatment due to an infected dermal cyst considered to be related to the treatment. From the phase IIb trial,25 six SAEs were assessed as possibly related to tildrakizumab. SAEs reported in part 1 of the trial were bacterial arthritis and lymphoedema, and in part 2 melanoma, stroke, epiglottitis, and knee infection. One patient died, during part 1, but not related to the study drug. Regarding prespecified AEs of clinical interest (serious infections, major cardiovascular events, malignancies, and drug-related hypersensitivity reactions), there were no significant differences observed between treatment groups and placebo (Table 3).25
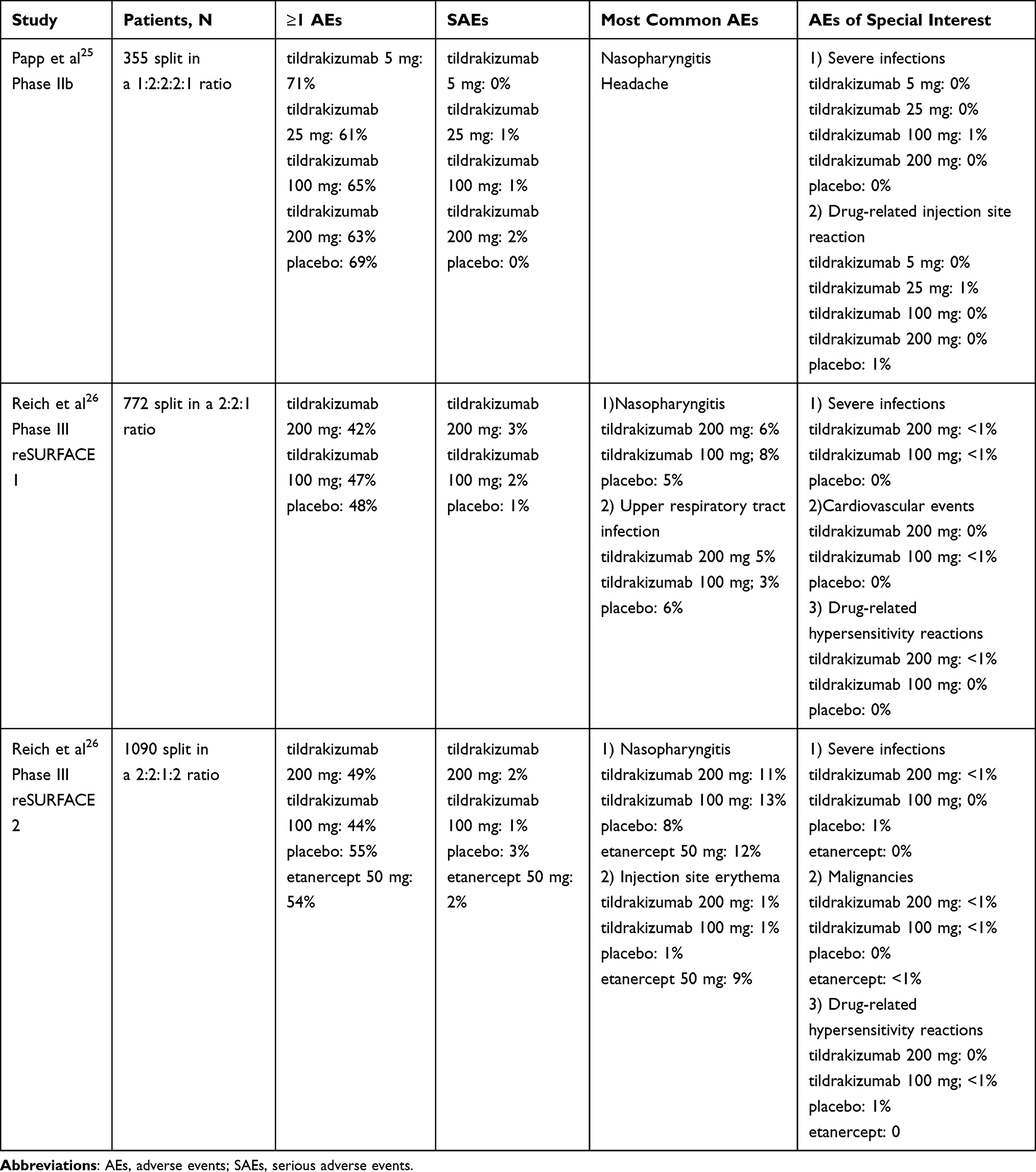 |
Table 3 Summary of Adverse Events in Phase II and III Trials During Week 0–12/16 |
AEs in Phase III Trials
The most common AEs in both reSURFACE 1 and reSURFACE 2 was nasopharyngitis, with a discrete higher incidence in the groups receiving active treatment compared to placebo (Table 3).26 In reSURFACE 2, injection-site erythema was also among the most common adverse events, however, most of these events were recorded in the group of patients treated with etanercept (Table 3). Discontinuations because of AEs were infrequent. From reSURFACE 1 in part 1 (week 0–12), five participants from tildrakizumab 200 mg discontinued the treatment because of AEs, and one participant from the placebo group. From reSURFACE 2 in part 1 (week 0–12), three participants from tildrakizumab 200 mg and 3 participants from tildrakizumab 100 mg discontinued the treatment because of AEs, compared to 6 participants from etanercept and 1 participant from the placebo group.
Major adverse cardiovascular events have been suggested to be increased when treated with briakinumab,40 a human monoclonal antibody that, like ustekinumab, directly targets the p40 subunit, shared by IL-12 and IL-23. In 2011, and the development of the drug was stopped before approval. With this background, the safety profile regarding major cardiovascular events are of special interest when investigating new biologic treatments, especially IL-12/IL-23 related. Data from a 3-year follow-up during and after treatment with tildrakizumab showed no increased risk of major cardiovascular events.27 Other AEs of special interest were AEs associated with other types of biologics (eg serious infections, tuberculosis, malignancies, candida infection, worsening of pre-existing inflammatory bowel disease, suicidal ideation and behavior),12,41,42 and no increased rates of these AEs were seen during treatment with tildrakizumab.
Data from the long-term follow-up after 148 weeks has recently been published.27 Safety was assessed in a pooled analysis from reSURFACE 1 and reSURFACE 2, and the most common treatment-emergent AE in all treatment groups was still nasopharyngitis. Other frequent AEs and AEs of special interest were all low and comparable across treatment groups. Nine deaths occurred in total, six during the base study, and three during the extension period. None of these deaths were considered related to study medication.
Safety results of tildrakizumab are similar to what has been reported from analyses of phase III trials regarding other IL-23p19 inhibitors.43 Rates of AEs and SAEs are also mostly comparable across groups in phase III studies of guselkumab and risankizumab.43 However, one systemic review and meta-analysis on IL-23p19 blockers indicated an increased incidence rate of infections when treating with guselkumab and risankizumab.44 The reported infections did not evolve to serious infections or other serious events. The same meta-analyses showed best overall tolerance for risankizumab, compared to tildrakizumab and guselkumab.44
Conclusively, besides a discrete higher incidence of nasopharyngitis, all clinical trials do not show any safety concerns regarding treatment with tildrakizumab for moderate-to-severe chronic plaque psoriasis in adults. However, real-life data from routine practice settings are not yet available and are needed to fully establish the safety profile.
Immunogenicity
Some patients experienced a lack and/or loss of efficacy during treatment with biologics. The development of antidrug antibodies (ADAs) may contribute to this phenomenon and can occur with all types of therapeutic proteins.45 Neutralizing ADAs negate the clinical benefit of the drug, and the non-neutralizing ADAs can also reduce efficacy, eg by affecting clearance, pharmacodynamics, and pharmacokinetics.46
Immunogenicity developing during treatment with tildrakizumab was not described in the original reports from reSURFACE 1 and reSURFACE 2.26 However, recently, Kimball et al evaluated the immunogenicity in both the phase IIb and III trials.47 At 12–16 weeks after initiation of treatment, 4% of the 1400 evaluable patients had positive treatment-emergent ADAs. At week 52–64, 7% of the 780 evaluable patients with continuous use of tildrakizumab had positive treatment-emergent ADAs. However, only 3% of all participants had neutralizing ADAs at week 52–64, and this subgroup had lower serum concentrations of tildrakizumab and showed reduced efficacy. No association between ADAs and AEs or discontinuation were observed.
The reported results of the development of ADAs during treatment with tildrakizumab are in the range observed for other IL-23p19 inhibitors. Among patients treated with guselkumab, approximately 6% developed ADAs up to week 52, and 7% of them had antibodies that were classified as neutralizing. ADAs to guselkumab were generally not associated with changes in clinical response.20 The number of patients developing ADAs on treatment with risankizumab was slightly higher, and by week 52, approximately 24% had developed ADAs, of which 57% were classified as neutralizing. Only 1% of the patients with ADAs had lower risankizumab concentrations and reduced clinical response.21
This wide range of development of ADAs was also observed among the monoclonal antibodies targeting IL-17 and IL-12/23 pathways.48–51 It is crucial to mention that the detection of ADAs depends on several factors, including assay methodology, sample handling, the timing of sample collection, concomitant medications, and underlying disease.47 Results from different trials can, therefore, be difficult to compare.
Discussion
The IL-23p19 inhibitor tildrakizumab was in clinical trials significantly more efficacious than both placebo and etanercept at week 12, and the efficacy continued to increase until week 28. Long term follow-up showed high levels of efficacy for up to 3 years. Furthermore, most patients discontinuing treatment with tildrakizumab later regained effect after reinitiating treatment. Importantly, efficacy measured by skin clearance (improvements in PASI and PGA) correlated with the impact of the quality of life. Besides discrete higher incidence of nasopharyngitis, the conducted clinical trials show no safety concerns and tildrakizumab seems to be very well tolerated. Furthermore, tildrakizumab are only administered every 3 months in the maintenance period, which is an advantage when treating a chronic disease.
In recent years, PASI90 has become the new gold standard of primary endpoints in clinical trials investigating biologics in the treatment of psoriasis. Therefore, it is remarkable that PASI75 is chosen as the primary endpoint in all the clinical trials investigating the efficacy of tildrakizumab. Furthermore, etanercept is used as the active comparator arm in the phase III trial, however, etanercept is not as effective as the other anti-TNF or newer biologics for psoriasis and seldom used as an active comparator in trials with psoriasis. Second-generation biologics often show higher efficacy compared to first-generation biologics11 and should therefore be compared with one another in head-to-head trials.
In previous meta-analyses, tildrakizumab seems to be the least efficacious IL-23p19 inhibitors.11,34,35 Even though all of the three IL-23p19 inhibitors are high-affinity monoclonal antibodies, tildrakizumab demonstrated the weakest affinity, which may be the reason for the lower efficacy (Table 1). One would therefore expect a higher effect of 200 mg than of 100 mg, and even though the phase IIb trial and the pooled pharmacokinetic analysis supports this theory, a dose-dependent efficacy could not be seen in the two phase III trials. Tildrakizumab are registered for SC injection with 100 mg, however, according to the EU label, dosing with 200 mg is acceptable in patients with high body weight.29
However, the suggestion that tildrakizumab may be less efficient than the other IL-23p19 inhibitors is based on an indirect comparison of PASI and should be interpreted with caution.
Selectively targeting the IL23p19 appears to avoid many of the adverse events associated with other biologics from the anti-TNF and anti-IL17 groups,43 perhaps explained by the more specific mechanism of action of the IL23p19 inhibitors. Tildrakizumab seems to have a very favorable safety profile, as also seen with guselkumab and risankizumab, only nasopharyngitis being more frequent among patients treated with tildrakizumab compared to placebo.
Certain biologics can be more suitable for a subset of patients with psoriasis. Patients that suffer from both psoriasis and psoriatic arthritis benefit the most from treatment with TNF blockers and IL-17 blockers.12 Overweight is another important issue, as 25–30% of the psoriasis patients in clinical trials have a bodyweight greater than 100 kg. These patients have shown to often have reduced clinical response to biologics that are administered in a fixed-dose regimen.52 As earlier described, tildrakizumab can be administered in a double dosage for patients with high bodyweight. Patients with a history of previous failure on biologic treatment will be an important issue in the future. For tildrakizumab, efficacy was generally higher in patients without prior biologics. Data of biologic switch, especially between second-generation biologics, is limited. Extension studies of reSURFACE 1 and 2, demonstrated that partial responders or nonresponders to etanercept, switching to tildrakizumab 200 mg at week 28, resulted in PASI75 responses in 8 out of every 10 patients after only 2 doses tildrakizumab.27 However, there is a need for data on biologic switch between the IL-23p19 antibodies and time will show where in the treatment algorithm tildrakizumab will be placed. Furthermore, investigations of the utility of tildrakizumab in certain subgroups of patients with psoriasis will also elucidate this.
In conclusion, the monoclonal IL-23p19 antibody tildrakizumab is an effective and very well-tolerated treatment for patients with moderate-to-severe psoriasis compared to placebo and etanercept. The efficacy and safety are maintained for up to 3 years, and under various changing treatment scenarios. The efficacy and safety compared to IL-17 and IL-12/23 inhibitors, and other selective IL-23 inhibitors, needs to be assessed in direct head-to-head randomized clinical trials and real-life data from clinical practice.
Disclosure
Charlotte Näslund-Koch reports no conflicts of interest in this work. Claus Zachariae has been a paid speaker for Eli Lilly, Novartis, and LEO Pharma, and has been a consultant or has served on Advisory Boards with AbbVie, Janssen Cilag, Novartis, Eli Lilly, LEO Pharma, Almirall, and CSL. He has served as an investigator for AbbVie, Sanofi, Janssen Cilag, Boehringer Ingelheim, AstraZenica, Eli Lilly, Novartis, Pfizer, Regeneron, and LEO Pharma. Lone Skov has been a paid speaker for AbbVie, Eli Lilly, Novartis, Sanofi, and LEO Pharma, and has been a consultant or has served on Advisory Boards with AbbVie, Janssen Cilag, Novartis, Eli Lilly, LEO Pharma, UCB, Almirall, and Sanofi. She has served as an investigator for AbbVie, Sanofi, Janssen Cilag, Boehringer Ingelheim, AstraZenica, Eli Lilly, Novartis, Pfizer, Regeneron, and LEO Pharma, and has received research and educational grants from Novartis, Sanofi, Janssen Cilag, and LEO Pharma. The authors report no other conflicts of interest in this work.
References
1. Boehncke WH, Schön MP. Psoriasis. Lancet. 2015;386(9997):983–994. doi:10.1016/S0140-6736(14)61909-7
2. WHO. World psoriasis day-document EB133.R2, agenda item 6.2. 2013. https://apps.who.int/gb/ebwha/pdf_files/EB133/B133_R2-en.pdf.
3. Rapp SR, Feldman SR, Exum ML, Fleischer AB, Reboussin DM. Psoriasis causes as much disability as other major medical diseases. J Am Acad Dermatol. 1999;41(3):401–407. doi:10.1016/S0190-9622(99)70112-X
4. Nestle FO, Kaplan DH, Barker J. Mechanisms of disease: psoriasis. N Engl J Med. 2009;361(5):5. doi:10.1056/NEJMra0804595
5. Boehncke WH. Etiology and Pathogenesis of Psoriasis. Rheum Dis Clin North Am. 2015;41(4):665–675. doi:10.1016/j.rdc.2015.07.013
6. Rendon A, Schäkel K. Psoriasis Pathogenesis and Treatment. Int J Mol Sci. 2019;20(6):6. doi:10.3390/ijms20061475
7. Gunter NV, Yap BJM, Chua CLL, Yap WH. Combining Understanding of Immunological Mechanisms and Genetic Variants Toward Development of Personalized Medicine for Psoriasis Patients. Front Genet. 2019;10:
8. Finlay AY. Current severe psoriasis and the Rule of Tens. Br J Dermatol. 2005;152(5):861–867. doi:10.1111/j.1365-2133.2005.06502.x
9. Gordon KB, Reich K, Crowley JJ, et al. Disease activity and treatment efficacy using patient-level Psoriasis Area and Severity Index scores from tildrakizumab Phase 3 clinical trials. J Dermatolog Treat. 2020:1–10. doi:10.1080/09546634.2020.1747590
10. Puig L, Dossenbach M, Berggren L, Ljungberg A, Zachariae C. Absolute and relative psoriasis area and severity indices (PASI) for comparison of the efficacy of ixekizumab to etanercept and placebo in patients with moderate-to-severe plaque psoriasis: an integrated analysis of UNCOVER-2 and UNCOVER-3 outcomes. Acta Derm Venereol. 2019;99(11):971–977. doi:10.2340/00015555-3245
11. Sbidian E, Chaimani A, Afach S, et al. Systemic pharmacological treatments for chronic plaque psoriasis: a network meta-analysis. Cochrane Database Syst Rev. 2020;1:CD011535. doi:10.1002/14651858.CD011535.pub3
12. Kim HJ, Lebwohl MG. Biologics and Psoriasis: the Beat Goes On. Dermatol Clin. 2019;37(1):29–36. doi:10.1016/j.det.2018.07.004
13. US Food and Drug Administration. ILUMYA (tildrakizumab-asmn).2018. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761067s000lbl.pdf.
14. Ilumetri | European Medicines Agency. https://www.ema.europa.eu/en/medicines/human/EPAR/ilumetri.
15. Khalilieh S, Hodsman P, Xu C, Tzontcheva A, Glasgow S, Montgomery D. Pharmacokinetics of Tildrakizumab (MK-3222), an Anti-IL-23 Monoclonal Antibody, After Intravenous or Subcutaneous Administration in Healthy Subjects. Basic Clin Pharmacol Toxicol. 2018;123(3):294–300. doi:10.1111/bcpt.13001
16. Kopp T, Riedl E, Bangert C, et al. Clinical improvement in psoriasis with specific targeting of interleukin-23. Nature. 2015;521(7551):222–226. doi:10.1038/nature14175
17. Khalilieh S, Hussain A, Montgomery D, et al. Effect of tildrakizumab (MK-3222), a high affinity, selective anti-IL23p19 monoclonal antibody, on cytochrome P450 metabolism in subjects with moderate to severe psoriasis. Br J Clin Pharmacol. 2018;84(10):2292–2302. doi:10.1111/bcp.13670
18. Zandvliet A, Glasgow S, Horowitz A, et al. Tildrakizumab, a novel anti-IL-23 monoclonal antibody, is unaffected by ethnic variability in Caucasian, Chinese, and Japanese subjects. Int J Clin Pharmacol Ther. 2015;53(2):139–146. doi:10.5414/CP202176
19. Jauslin P, Kulkarni P, Li H, et al. Population-Pharmacokinetic Modeling of Tildrakizumab (MK-3222), an Anti-Interleukin-23-p19 Monoclonal Antibody, in Healthy Volunteers and Subjects with Psoriasis. Clin Pharmacokinet. 2019;58(8):1059–1068. doi:10.1007/s40262-019-00743-7
20. US Food and Drug Administration. TREMFYA ® (guselkumab). https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761061s000lbl.pdf.
21. US Food and Drug Administration. SKYRIZI ® (risankizumab-rzaa). https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761105s000lbl.pdf.
22. US Food and Drug Administration. CENTER FOR DRUG EVALUATION AND RESEARCH. ILUMYA (Tildrakizumab). https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/761067Orig1s000MultdisciplineR.pdf.
23. US Food and Drug Administration. CENTER FOR DRUG EVALUATION AND RESEARCH. TREMFYA (Guselkumab). https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/761061Orig1s000MultidisciplineR.pdf.
24. Therapeutic Goods Administration. Australian Public Assessment Report for Risankizumab.; 2020.
25. Papp K, Thaçi D, Reich K, et al. Tildrakizumab (MK-3222), an anti-interleukin-23p19 monoclonal antibody, improves psoriasis in a phase IIb randomized placebo-controlled trial. Br J Dermatol. 2015;173(4):930–939. doi:10.1111/bjd.13932
26. Reich K, Papp KA, Blauvelt A, et al. Tildrakizumab versus placebo or etanercept for chronic plaque psoriasis (reSURFACE 1 and reSURFACE 2): results from two randomised controlled, phase 3 trials. Lancet. 2017;390(10091):276–288. doi:10.1016/S0140-6736(17)31279-5
27. Reich K, Warren RB, Iversen L, et al. Long-term efficacy and safety of tildrakizumab for moderate-to-severe psoriasis: pooled analyses of two randomized phase III clinical trials (reSURFACE 1 and reSURFACE 2) through 148 weeks. Br J Dermatol. 2020;182(3):605–617. doi:10.1111/bjd.18232
28. Poulin Y, Ramon M, Rosoph L, et al. Efficacy of tildrakizumab by patient demographic and disease characteristics across a phase 2b and 2 phase 3 trials in patients with moderate‐to‐severe chronic plaque psoriasis. J Eur Acad Dermatology Venereol. 2020:
29. Papp KA, Reich K, Blauvelt A, et al. Efficacy of tildrakizumab for moderate-to-severe plaque psoriasis: pooled analysis of three randomized controlled trials at weeks 12 and 28. J Eur Acad Dermatology Venereol. 2019;33(6):1098–1106. doi:10.1111/jdv.15400
30. Kerbusch T, Li H, Wada R, Jauslin PM, Wenning L. Exposure–response characterisation of tildrakizumab in chronic plaque psoriasis: pooled analysis of 3 randomised controlled trials. Br J Clin Pharmacol. 2020;86(9):1795–1806. doi:10.1111/bcp.14280
31. Blauvelt A, Sofen H, Papp K, et al. Tildrakizumab efficacy and impact on quality of life up to 52 weeks in patients with moderate‐to‐severe psoriasis: a pooled analysis of two randomized controlled trials. J Eur Acad Dermatology Venereol. 2019;33(12):2305–2312. doi:10.1111/jdv.15862
32. Kimball AB, Papp KA, Reich K, et al. Efficacy and safety of tildrakizumab for plaque psoriasis with continuous dosing, treatment interruption, dose adjustments and switching from etanercept: results from phase III studies. Br J Dermatol. 2019;182(4):6. doi:10.1111/bjd.18484
33. Bilal J, Berlinberg A, Bhattacharjee S, Trost J, Bin RI, Kurtzman DJB. A systematic review and meta-analysis of the efficacy and safety of the interleukin (IL)-12/23 and IL-17 inhibitors ustekinumab, secukinumab, ixekizumab, brodalumab, guselkumab and tildrakizumab for the treatment of moderate to severe plaque psoriasis. J Dermatolog Treat. 2018;29(6):569–578. doi:10.1080/09546634.2017.1422591
34. Sawyer LM, Malottki K, Sabry-Grant C, et al. Assessing the relative efficacy of interleukin-17 and interleukin-23 targeted treatments for moderate-to-severe plaque psoriasis: A systematic review and network meta-analysis of PASI response. PLoS One. 2019;14(8):8. doi:10.1371/journal.pone.0220868
35. Armstrong AW, Puig L, Joshi A, et al. Comparison of Biologics and Oral Treatments for Plaque Psoriasis: A Meta-analysis. JAMA Dermatology. 2020;156(3):258–269. doi:10.1001/jamadermatol.2019.4029
36. Xu S, Zhang X, Pan M, Shuai Z, Xu S, Pan F. Treatment of plaque psoriasis with IL-23p19 blockers: A systematic review and meta-analysis. Int Immunopharmacol. 2019;75. doi:10.1016/j.intimp.2019.105841
37. Reich K, Griffiths CEM, Gordon KB, et al. Maintenance of clinical response and consistent safety profile with up to 3 years of continuous treatment with guselkumab: results from the VOYAGE 1 and VOYAGE 2 trials. J Am Acad Dermatol. 2020;82(4):936–945. doi:10.1016/j.jaad.2019.11.040
38. Reddy V, Yang EJ, Myers B, Liao W. Clinical evaluation of risankizumab-rzaa in the treatment of plaque psoriasis. J Inflamm Res. 2020;13:53–60. doi:10.2147/JIR.S215196
39. AbbVie. A Study to Assess the Safety and Efficacy of Risankizumab for Maintenance in Moderate to Severe Plaque Type Psoriasis (LIMMITLESS). https://clinicaltrials.gov/ct2/show/NCT03047395.
40. Langley RG, Papp K, Gottlieb AB, et al. Safety results from a pooled analysis of randomized, controlled phase II and III clinical trials and interim data from an open-label extension trial of the interleukin-12/23 monoclonal antibody, briakinumab, in moderate to severe psoriasis. J Eur Acad Dermatology Venereol. 2013;27(10):1252–1261. doi:10.1111/j.1468-3083.2012.04705.x
41. Saunte DM, Mrowietz U, Puig L, Zachariae C. Candida infections in patients with psoriasis and psoriatic arthritis treated with interleukin-17 inhibitors and their practical management. Br J Dermatol. 2017;177(1):47–62. doi:10.1111/bjd.15015
42. Lebwohl MG, Papp KA, Marangell LB, et al. Psychiatric adverse events during treatment with brodalumab: analysis of psoriasis clinical trials. J Am Acad Dermatol. 2018;78(1):81–89.e5. doi:10.1016/j.jaad.2017.08.024
43. Crowley JJ, Warren RB, Cather JC. Safety of selective IL-23p19 inhibitors for the treatment of psoriasis. J Eur Acad Dermatology Venereol. 2019;33(9):1676–1684. doi:10.1111/jdv.15653
44. Xu S, Zhang X, Pan M, Shuai Z, Xu S, Pan F. Treatment of plaque psoriasis with IL-23p19 blockers: A systematic review and meta-analysis. Int Immunopharmacol. 2019;75:105841. doi:10.1016/j.intimp.2019.105841
45. Jullien D, Prinz JC, Nestle FO. Immunogenicity of biotherapy used in psoriasis: the science behind the scenes. J Invest Dermatol. 2015;135(1):31–38. doi:10.1038/jid.2014.295
46. Pratt K. Anti-Drug Antibodies: emerging Approaches to Predict, Reduce or Reverse Biotherapeutic Immunogenicity. Antibodies. 2018;7(2):19. doi:10.3390/antib7020019
47. Kimball AB, Kerbusch T, van Aarle F, et al. Assessment of the effects of immunogenicity on the pharmacokinetics, efficacy and safety of tildrakizumab. Br J Dermatol. 2020;182(1):180–189. doi:10.1111/bjd.17918
48. US Food and Drug Administration. COSENTYX ® (secukinumab). https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125504s013lbl.pdf.
49. US Food and Drug Administration. TALTZ (ixekizumab). https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/125521s004lbl.pdf.
50. US Food and Drug Administration. SILIQ (brodalumab). https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761032lbl.pdf.
51. US Food and Drug Administration. STELARA ® (ustekinumab). https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125261s147lbl.pdf.
52. Puig L. Obesity and psoriasis: body weight and body mass index influence the response to biological treatment. J Eur Acad Dermatology Venereol. 2011;25(9):1007–1011. doi:10.1111/j.1468-3083.2011.04065.x
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.