Back to Journals » Journal of Blood Medicine » Volume 10
Thrombotic events with recombinant activated factor VII (rFVIIa) in approved indications are rare and associated with older age, cardiovascular disease, and concomitant use of activated prothrombin complex concentrates (aPCC)
Authors Rajpurkar M ![]() , Croteau SE, Boggio L, Cooper DL
, Croteau SE, Boggio L, Cooper DL ![]()
Received 15 June 2019
Accepted for publication 23 August 2019
Published 18 September 2019 Volume 2019:10 Pages 335—340
DOI https://doi.org/10.2147/JBM.S219573
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Martin Bluth
Madhvi Rajpurkar,1 Stacy E Croteau,2 Lisa Boggio,3 David L Cooper4
1Carman and Ann Adams Department of Pediatrics, Children’s Hospital of Michigan/Wayne State University, Detroit, MI, USA; 2Department of Pediatrics, Boston Children’s Hospital/Harvard Medical School, Boston, MA, USA; 3Hemophilia and Thrombophilia Center, Rush University Medical Center, Chicago, IL, USA; 4Clinical Development and Medical Affairs – Biopharm, Novo Nordisk Inc., Plainsboro, NJ, USA
Correspondence: Madhvi Rajpurkar
Carman and Ann Adams Department of Pediatrics, Children’s Hospital of Michigan, Wayne State University, 3901 Beaubien Blvd, Detroit, MI 48201, USA
Tel +1 313 745 5515
Fax +1 313 745 5237
Email [email protected]
Purpose: Recombinant activated factor VII (rFVIIa; NovoSeven® RT; Novo Nordisk A/S, Bagsvaerd, Denmark) is approved in the United States for the treatment of bleeding and perioperative management in congenital hemophilia with inhibitors (CHwI), acquired hemophilia (AH), congenital factor VII (FVII) deficiency, and Glanzmann’s thrombasthenia (GT) with refractoriness to platelets. The aim of the current analysis was to review clinical trials and registries pre- and post-licensure for each indication to establish the estimated rate of thrombosis and then to establish the association of all reported thrombotic events (TEs) with certain risk factors listed for many years in the prescribing information (PI).
Patients and methods: A retrospective safety assessment of both clinical trials and registries used to support licensure and postmarketing surveillance was performed. The rate of thrombosis was calculated in the 4 indicated disorders and an assessment of TE risk factors was conducted through a review of all narratives within those indications in the safety database.
Results: In clinical trials and registries used to support licensure and in postmarketing surveillance, the overall rate of thrombosis was 0.17% of 12,288 bleeding and surgical episodes. The specific risk by indication was 0.11% for CHwI, 0.82% for FVII deficiency, 0.19% for GT, and 1.77% for AH. The most common associated risk factor—“elderly” (29%), defined in the PI as age ≥65 years—was particularly prevalent in patients with AH. TE was also frequently reported with concomitant cardiac or vascular disease (18%) and use of activated prothrombin complex concentrates (18%).
Conclusion: Data show that the rate of TEs within the 4 licensed indications is low, as was originally described in the US PI from 1999 to 2009. It has remained stable over time during postapproval surveillance in multiple US and global registries with active surveillance for safety information across the 4 approved indications.
Keywords: postmarketing surveillance, acquired hemophilia, congenital hemophilia with inhibitors, congenital factor VII deficiency, Glanzmann’s thrombasthenia
Introduction
Recombinant activated factor VII (rFVIIa; NovoSeven® RT; Novo Nordisk A/S, Bagsvaerd, Denmark) is approved in the United States for the treatment of bleeding and perioperative management in congenital hemophilia with inhibitors (CHwI), acquired hemophilia (AH), congenital factor VII (FVII) deficiency, and Glanzmann’s thrombasthenia (GT) with refractoriness to platelets. The data supporting the development program for the current indications for rFVIIa since the first human dose in 1988 include an initial series of compassionate/emergency use studies, clinical trials (including pharmacokinetic, safety, and efficacy assessments), and national/international registries. Furthermore, safety data accumulated over the past 30 years encompass the literature (studies, case series, case reports) and spontaneous safety reporting.
Serious arterial and venous thrombotic events (TEs) have been reported in clinical trials and postmarketing surveillance; however, the incidence of this risk (rate of thrombosis) is considered to be low when rFVIIa is used within labeled indications.1–4 TEs have been reported more frequently in AH than in other indications due to the older age of patients and the presence of comorbidities including cardiac and cardiovascular disease. The risks of TE associated with the use of rFVIIa in patients without bleeding disorders (outside of licensed indications) have been extensively studied.5–9
The aim of the current analysis was to review clinical trials and registries pre- and post-licensure for each of the 4 approved indications to establish the estimated rate of thrombosis and then to establish the association of reported TEs with certain risk factors listed for many years in the prescribing information (PI).
Materials and methods
A retrospective safety assessment of both clinical trials and registries used to support licensure and postmarketing surveillance was performed. The rate of thrombosis was calculated in the 4 indicated disorders: CHwI, AH, FVII deficiency, and GT.
Analysis considered all postmarketing TE case reports in the Novo Nordisk safety database through March 2017, including those from registries, spontaneous (unsolicited) reports, and the literature; isolated cases of catheter occlusion were not included. Event narratives were assessed to identify any of the risk factors listed in the PI and as a sensitivity analysis for additional risk factors associated with TE where there was a temporal relationship to rFVIIa use, which was defined as within 48 hrs, given the 2–3-hr half-life. Although manufacturer safety databases, including spontaneous reports, are not publicly available, TE data reported to the Federal Drug Administration (FDA) are available through the FDA’s Adverse Events Reporting System database.
Given the retrospective nature of these analyses, neither institutional review board nor ethics committee approval was required. All of the cited trials and registries were performed under the oversight of institutional ethics boards or national ethics committees.
Results
Rate of thrombosis
In clinical trials and registries used to support licensure and in postmarketing surveillance, the overall rate of thrombosis was 0.17% of 12,288 bleeding and surgical episodes (Table 1). Twenty-one TEs were identified (12 CHwI, 3 FVII deficiency, 1 GT, and 5 AH). The specific risk by indication was 0.11% for CHwI (11,121 episodes), 0.82% for FVII deficiency (367 episodes), 0.19% for GT (518 episodes), and 1.77% for AH (282 episodes). An additional Japanese postmarketing study in 132 patients with AH with 371 bleeding episodes reported 3 TEs and a thrombosis rate of 0.8%. This additional analysis reduced the calculated TE rate in AH from 1.77% (Table 1) to 1.2%.
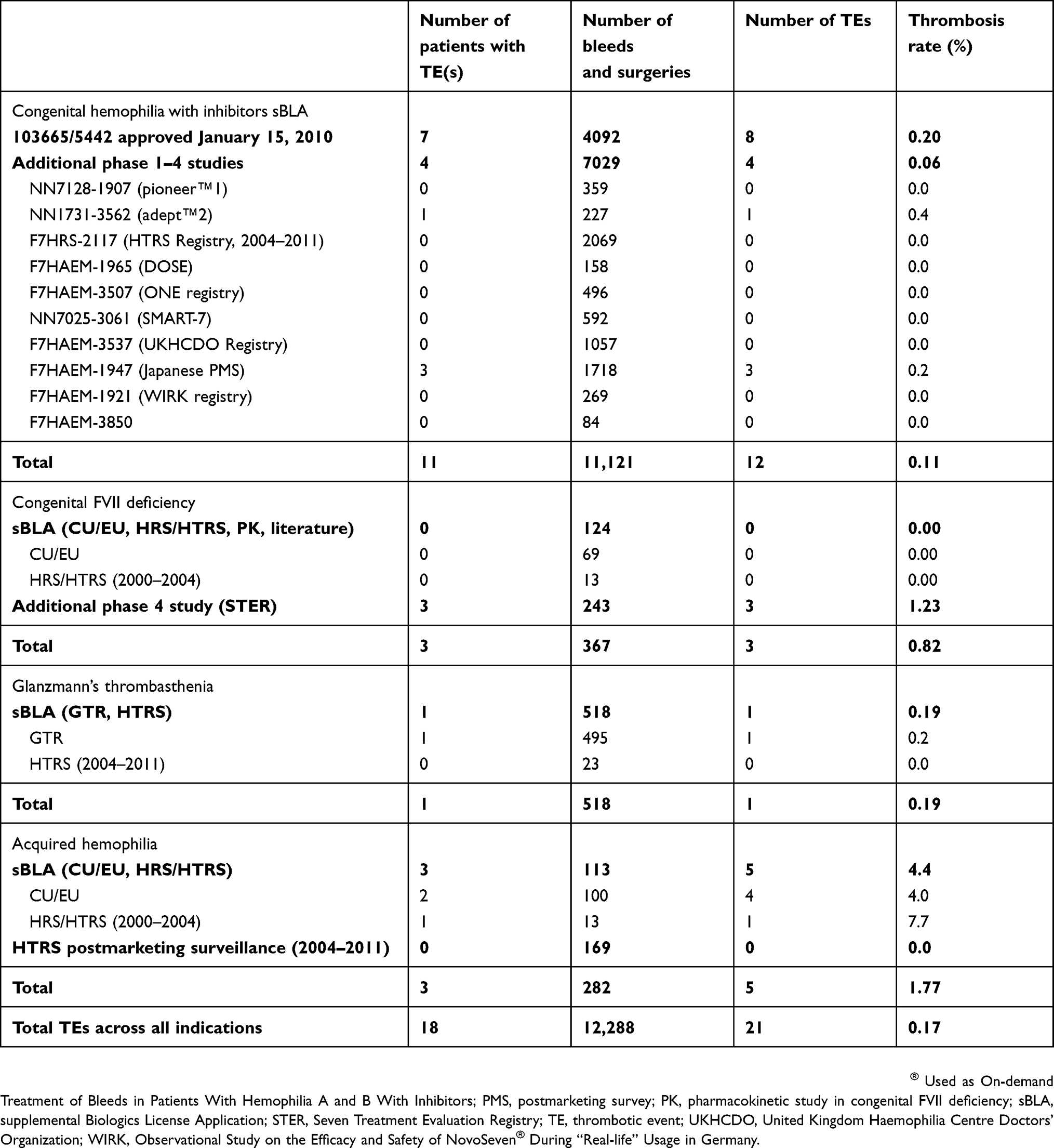 |
Table 1 Thrombotic events in clinical studies |
Risk factors for thrombosis
Analysis of all postmarketing databases revealed 213 TEs (88 CHwI, 61 AH, 53 FVII deficiency, 11 GT; Table 2). The majority were reported spontaneously (unsolicited) to the manufacturer (141), with fewer reported in the literature (47) or solicited through safety reporting in registries or investigator-sponsored studies (25) (Table 2). Of 213 TE cases, temporal relationship was assessed as plausible in 188 (88.3%) cases, in which rFVIIa use was reported within 48 hrs prior to TE or the timing was unknown; timing was confirmed to be more than 48 hrs in 25 (11.7%) cases. Overall, the most common associated risk factor—“elderly,” defined in the PI as age ≥65 years (28.6%)—was particularly prevalent in patients with AH and TE (67.2%; Table 3). Another common factor was concomitant use of activated prothrombin complex concentrates (aPCC), identified in 17.8% of all TEs and 34.1% of TEs in CHwI. Broadly defined cardiovascular disease (including cardiac arrhythmias) was noted in 17.8% of all TEs (36.1% AH, 15.1% FVII deficiency, 9.1% CHwI) and was much more common than specific note of atherosclerotic disease (1.9%). Other risk factors listed in the US PI (through October 2017) were uncommon, and in some cases, never identified in TE reports (eg, postoperative immobilization, sepsis, liver disease, disseminated intravascular coagulation, neonates, pregnancy, postpartum hemorrhage, crush injury).
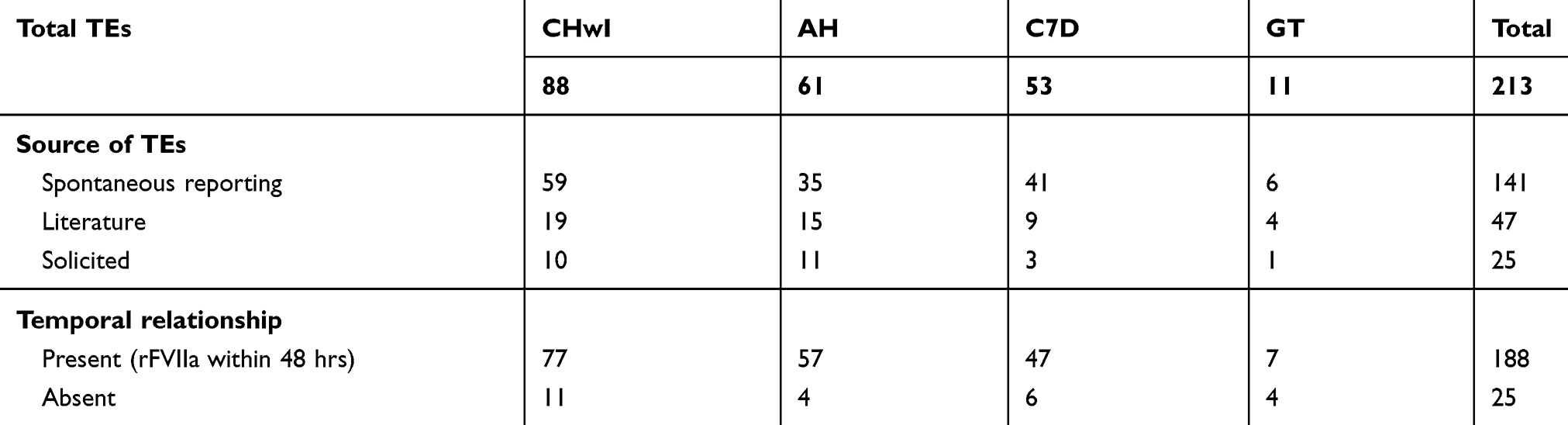 |
Table 2 Thrombotic events in safety reports |
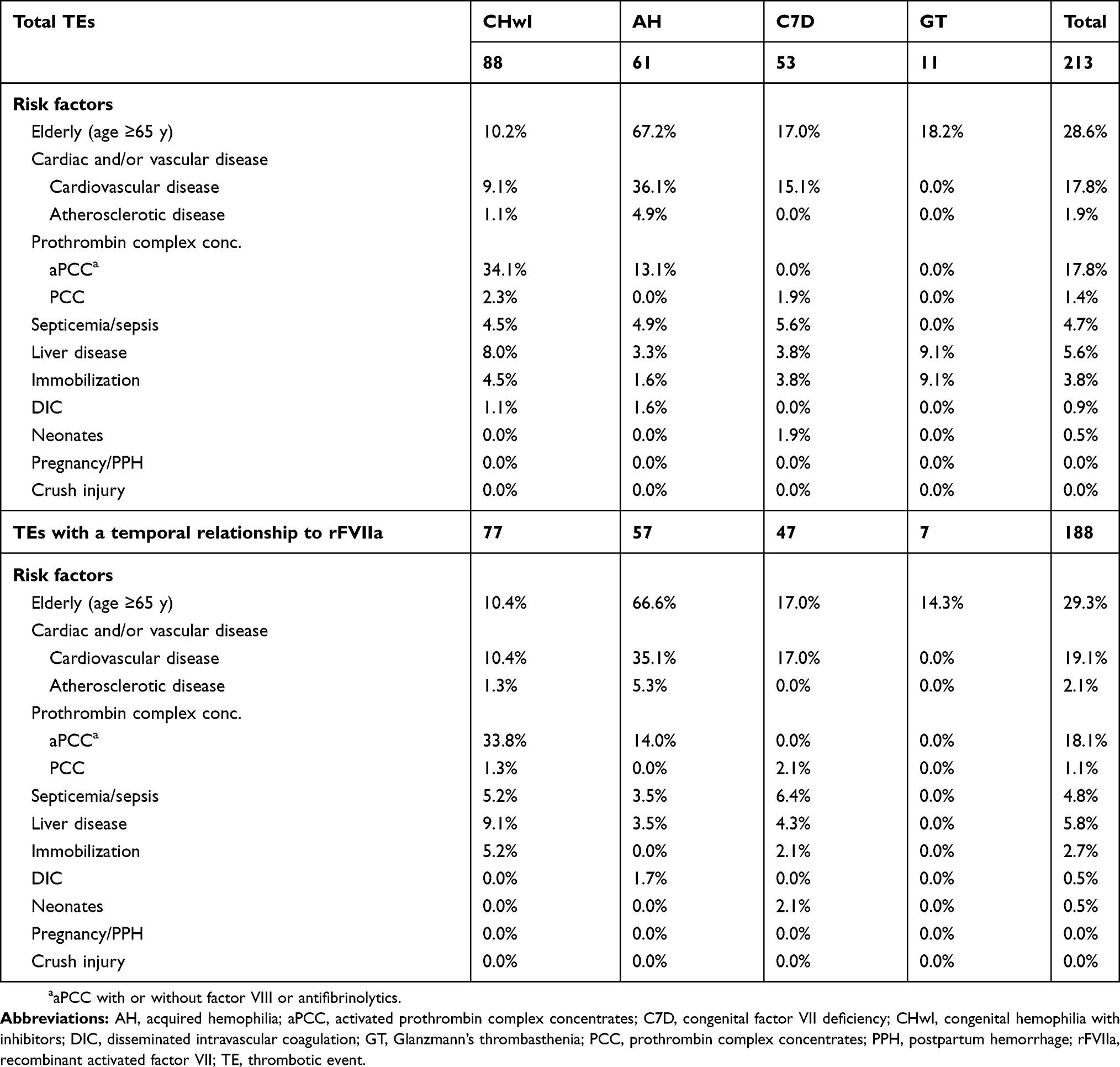 |
Table 3 Risk factors for thrombotic events |
Based on the 2–3-hr half-life of rFVIIa, the event was considered to have a temporal relationship to the treatment if the time between the last dose of rFVIIa and the event onset was within 48 hrs or if the timing was unknown. Sensitivity analysis of TE cases with a possible temporal relationship (event onset within 48 hrs of last administration or unknown) demonstrated similar results (Table 3).
Discussion
Safety data collected over the past 30 years have demonstrated a good safety profile for rFVIIa within the 4 indicated disorders. In the current retrospective safety analysis of both extensive clinical trials and registries used to support licensure and postmarketing surveillance, 21 TEs were identified in 18 patients treated for 12,288 bleeding and surgical episodes, generating a TE rate of 0.17%. Previous retrospective studies of rFVIIa postmarketing surveillance identified varying rates of TEs due to differences in the sources of data (eg, registries, clinical trials, literature reviews, spontaneous [unsolicited] reports) and time frames of each analysis, but overall, the incidence of TEs with rFVIIa use within the 4 licensed indications was very low.
Rates of TEs are easily calculated from registries in which the number of TEs and the total number of bleeding episodes are collected, and in which safety data are required to be reported (eg, mandatory field affirming presence/absence of adverse reactions). For example, an analysis of Hemostasis and Thrombosis Research Society registry data from 2004 to 2008 found 0 TEs in 2041 rFVIIa-treated bleeding events, yielding a rate of 0.00%.10
Other postmarketing surveillance analyses that included spontaneous and clinical trial reports and registry data found 25 TEs between 1996 and 20031 and 30 TEs between 2003 and 2006.7 The approaches used in these analyses allow for the determination of the number of TEs. Using an estimation of the number of standard (90 mcg/kg) rFVIIa doses given during that time frame, an approximation of the rate of TEs can be calculated. An additional review of safety data from 1996 to 2013 found 195 TEs among the estimated 4 million rFVIIa doses.3 The most recent cumulative review identified 217 TEs between 1996 and 2016, a period covered by an estimated 5.4 million standard doses.4
Spontaneous reports to manufacturers can be subject to poor-quality data lacking detail, limiting their usefulness in safety analyses. In addition, as these reports are unsolicited, it is possible that the number of TEs relayed through spontaneous reports is underestimated; however, this underestimation is likely balanced by an under-reporting of rFVIIa usage. Clinical trials are a more reliable source of data because they have more stringent safety reporting (eg, mandated affirmations of presence/absence of adverse events for each episode) and follow-up compared with spontaneous reports. However, there are limitations to using only clinical trials for safety analyses. For example, many trials have exclusion criteria that limit the population of eligible patients and therefore do not reflect the real-world population; compassionate/emergency use studies and registries are more inclusive of real-world populations and constitute the majority of rFVIIa clinical data. Therefore, the 21 TEs within the 12,288 bleeds and surgeries accumulated in compassionate/emergency use studies, clinical trials, and real-world registries performed over 30 years throughout the world are a better reflection of the true incidence of adverse events in a controlled setting in terms of mandatory safety reporting. In the current analysis, the addition of spontaneous reports of TEs with a varied patient population is best used to help identify safety signals such as the risk factors for TEs.
This study highlights the importance of understanding the risk factors for thrombosis in patients with hemophilia. For more than a decade, risk factors typically associated with thrombosis in patients without an underlying bleeding disorder were included in the PI as warnings, perhaps due in part to previous clinical trials with rFVIIa outside the 4 currently indicated disorders. However, the present analysis suggests that older age, cardiac and cardiovascular disease (beyond atherosclerosis only), and concomitant use of aPCC are the most common associated risk factors for thrombosis among patients with hemophilia. The validity of the current analytical approach to risk factor identification is supported by the inclusion of this new information in the 2018 PI update to the warnings around TE risk.
As new generations of treatments, such as non-factor products and gene therapy, become available and are combined with factor products and bypassing agents for breakthrough bleeds and surgeries, the risk of TE must be considered. Prescribers will require detailed information about factors that may predict risk. It is essential to balance the degree of hemostasis with the desire to avoid any bleeding and possible product/molecule-specific interactions with acute hemostatic treatment. Importantly, thus far, no safety concerns with concomitant use of rFVIIa and emicizumab have been identified.4,11,12
Conclusion
These data demonstrate that the rate of TEs within the 4 licensed indications is low (0.17%), as was originally described in the US PI from 1999 to 2009. It has remained stable over time during postapproval surveillance in multiple US and global registries with active surveillance for safety information across the 4 approved indications. Based on an assessment of postmarketing safety reports, patients with CHwI receiving concomitant treatment with aPCC, older patients, particularly those with AH who are receiving other hemostatic agents, and patients with a history of cardiac and cardiovascular disease may have an increased risk of developing TEs.
Abbreviations
AH, acquired hemophilia; aPCC, activated prothrombin complex concentrates; CHwI, congenital hemophilia with inhibitors; FVII, factor VII; GT, Glanzmann’s thrombasthenia; PI, prescribing information; rFVIIa, recombinant activated factor VII; TEs, thrombotic events.
Acknowledgments
This retrospective analysis was funded by Novo Nordisk, Inc., Plainsboro, New Jersey. Writing assistance was provided by Amy Ross, PhD, of ETHOS Health Communications in Yardley, Pennsylvania, and was supported financially by Novo Nordisk Inc., in compliance with international Good Publication Practice guidelines. The abstract and select data from this paper were presented at the 2018 American Society of Hematology Conference as a poster presentation. The poster’s abstract was published in “Abstracts and Meeting Program” in Blood 2018 132:1203: http://www.bloodjournal.org/content/132/Suppl_1/1203.
Disclosure
MR has served as an advisory board member for Novo Nordisk, HEMA Biologics, Spark Therapeutics and Takeda. MR reports personal fees from Novo Nordisk and Spark Therapeutics, outside the submitted work. SEC has consulted for Pfizer, Shire, Bayer, CSL-Behring, Genentech, Novo Nordisk, and Octapharma. She has received grant and honorarium from Pfizer and Octopharma, outside the submitted work. LB has served as an advisory board member for Novo Nordisk, Baxalta, CSL, and Octapharma, outside the submitted work. DLC is an employee of Novo Nordisk Inc. The authors report no other conflicts of interest in this work.
References
1. Abshire T, Kenet G. Recombinant factor VIIa: review of efficacy, dosing regimens and safety in patients with congenital and acquired factor VIII or IX inhibitors. J Thromb Haemost. 2004;2(6):899–909. doi:10.1111/j.1538-7836.2004.00759.x
2. Abshire T, Kenet G. Safety update on the use of recombinant factor VIIa and the treatment of congenital and acquired deficiency of factor VIII or IX with inhibitors. Haemophilia. 2008;14(5):898–902. doi:10.1111/j.1365-2516.2008.01829.x
3. Neufeld EJ, Negrier C, Arkhammar P, et al. Safety update on the use of recombinant activated factor VII in approved indications. Blood Rev. 2015;29 Suppl 1:S34–S41. doi:10.1016/S0268-960X(15)30006-0
4. Neufeld EJ, Negrier C, Benchikh El Fegoun S, Cooper DL, Rojas-Rios A, Seremetis S. Recombinant activated factor VII in approved indications: update on safety. Haemophilia. 2018;24(4):e275–e277. doi:10.1111/hae.13547
5. Levi M, Levy JH, Andersen HF, Truloff D. Safety of recombinant activated factor VII in randomized clinical trials. N Engl J Med. 2010;363(19):1791–1800. doi:10.1056/NEJMoa1006221
6. Hardy JF, Belisle S, Van der Linden P. Efficacy and safety of recombinant activated factor VII to control bleeding in nonhemophiliac patients: a review of 17 randomized controlled trials. Ann Thorac Surg. 2008;86:1038–1048. doi:10.1016/j.athoracsur.2008.05.013
7. Hsia CC, Zurawska JH, Tong MZY, Eckert K, McAlister VC, Chin-Yee IH. Recombinant activated factor VII in the treatment of non-haemophilia patients: physician under-reporting of thromboembolic adverse events. Transfus Med. 2009;19:43–49. doi:10.1111/j.1365-3148.2009.00913.x
8. O’Connell KA, Wood JJ, Wise JJ, Lozier JN, Braun MM. Thromboembolic adverse events after use of recombinant human coagulation factor VIIa. JAMA. 2006;295:293–298. doi:10.1001/jama.295.3.293
9. Lin Y, Stanworth S, Birchall J, Doree C, Hyde C. Recombinant factor VIIa for the prevention and treatment of bleeding in patients without haemophilia. Cochrane Database Syst Rev. 2007; 2: Art. No.: CD005011.
10. Neufeld EJ, Saxena K, Kessler CM, Cooper DL, HTRS Investigators. Dosing, efficacy, and safety of recombinant factor VIIa (rFVIIa) in pediatric versus adult patients: the experience of the Hemostasis and Thrombosis Research Society (HTRS) Registry (2004-2008). Pediatr Blood Cancer. 2013;60(7):1178–1183. doi:10.1002/pbc.24472
11. Oldenburg J, Mahlangu JN, Kim B, et al. Emicizumab prophylaxis in hemophilia A with inhibitors. N Engl J Med. 2017;377(9):809–818. doi:10.1056/NEJMoa1703068
12. Levy GG, Asikanius E, Kuebler P, Benchikh El Fegoun S, Esbjerg S, Seremetis S. Safety analysis of rFVIIa with emicizumab dosing in congenital hemophilia A with inhibitors: experience from the HAVEN clinical program. J Thromb Haemost. 2019. Epub ahead of print. doi:10.1111/jth.14491
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.