Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 16
There is No Fast Track to Identify Fast Decliners in Alpha-1 Antitrypsin Deficiency by Spirometry: A Longitudinal Study of Repeated Measurements
Authors Stockley JA ![]() , Stockley RA
, Stockley RA ![]() , Sapey E
, Sapey E ![]()
Received 22 December 2020
Accepted for publication 14 March 2021
Published 29 March 2021 Volume 2021:16 Pages 835—840
DOI https://doi.org/10.2147/COPD.S298585
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Richard Russell
James A Stockley,1 Robert A Stockley,2 Elizabeth Sapey3
1Lung Function & Sleep Department, University Hospitals Birmingham NHS Foundation Trust, Birmingham, B15 2GW, UK; 2Respiratory Department, University Hospitals Birmingham NHS Foundation Trust, Birmingham, B15 2GW, UK; 3PIONEER Health Data Hub in Acute Care, Institute of Inflammation and Ageing, University of Birmingham, Birmingham, B15 2GW, UK
Correspondence: Elizabeth Sapey
PIONEER Health Data Hub in Acute Care, Institute of Inflammation and Ageing, University of Birmingham, Edgbaston, Birmingham, B15 2GW, UK
Email [email protected]
Background: It is known that lung function decline in Alpha-1 Antitrypsin Deficiency (AATD) varies. Those with a rapid decline are at highest risk of poorer outcomes but may benefit most from targeted treatments including augmentation therapy. Current evidence suggests rapid decliners can be identified after 3 years of serial follow-up. It would be advantageous to identify these patients over a shorter time period, especially in mild disease.
Methods: Post-bronchodilator spirometry was performed every 6 months for a total of 18 months (4 measurements) by PiZZ AATD patients (ex- or never-smokers) either without spirometric COPD or with mild COPD. Where possible, retrospective spirometry data were included. Decline was assessed using 2 (baseline and 6 month) or four measurements (including baseline, 6, 12 and 18 months) and compared to retrospective decline rates using annual measurements over 3 years.
Results: Seventy-two PiZZ AATD patients were included, with 27 having at least three years of retrospective, annual spirometry. 18-month progression obtained by linear regression showed variable degrees of change with 29 showing no decline, 8 showing slow decline and 35 showing rapid decline. Bland-Altman plots showed that there was no overall agreement between predicted rate of decline using data obtained over 6 months and that obtained over 18 months. Furthermore, there was no agreement between rate of decline from either 6 or 18 months’ data when compared to data collected over 3 years. The positive predictive value for rapid decline with 18 months of data compared to 3 years was only 50.0%.
Conclusion: This study suggests serial lung function over 18 months cannot identify AATD patients who have rapidly declining lung function. There is an urgent need for different biomarkers to help identify these patients at the earliest opportunity.
Keywords: lung function, decline, obstructive airways disease, alpha-1 antitrypsin deficiency
Plain Language Summary
Alpha-1 Anti-trypsin Deficiency is a highly variable lung disease. Some patients with Alpha-1 Antitrypsin Deficiency can experience a rapid decline in their lung health and some patients can remain stable and well. It is important to identify those who have rapidly deteriorating lung health, so treatments can be focused to prevent significant lung damage. Currently, this is achieved using breathing tests repeated annually each year for at least 3 years, meaning there is no quick means to identify those “high risk” rapid-declining patients.
This study sought to assess if performing breathing tests more frequently (every 6 months), over a shorter period of time (up to 18 months, providing 4 measurements) was as accurate as assessing the same patients over 3 years. Sadly, it was not. The measurements over 18 months could not identify rapid declining patients accurately. This study suggests we need new tools to identify these patients in a more timely manner.
Introduction
Alpha-1 antitrypsin deficiency (AATD) is a widely recognised genetic defect that predisposes to serine proteinase-induced lung disease, classically associated with basal panacinar emphysema. The clinical features develop at an earlier age than usual (non-AATD) emphysema and are accelerated by smoking.1 Because the deficiency explains the pathophysiology of the disease,2 augmentation of the plasma (and hence lung) AAT level with purified protein is offered in some but not all countries.3
Augmentation therapy generally involves weekly infusions of 60mg/kg (or its equivalent regimen). This is both expensive4 and inconvenient to the patient, although evidence suggests an impact on disease progression. For instance, observational studies show a reduced decline in lung function as measured by the forced expired volume in 1 second (FEV1)5,6 and lung densitometry indicated a general slowing of emphysema progression in placebo-controlled trials.7–9
However, lung disease and its progression is highly variable in AATD with many patients showing relative disease stability (even with severely deficient plasma AAT levels) and only a proportion of never-smokers demonstrate physiological progression that exceeds the normal age-related decline.10 Furthermore, even smokers with clearly established airflow obstruction of varying severity can stabilise once they stop smoking.10 This is an important observation since augmentation therapy is contraindicated in current smokers and may be inappropriate or ineffective for those with stable lung disease.
In view of the healthcare implications and costs, it is logical to consider stratifying augmentation therapy to those with evidence of continued decline. At present, augmentation therapy is mainly restricted to those with established chronic obstructive pulmonary disease (COPD) in the FEV1 range ≥30% and ≤60% predicted with evidence of emphysema. However, there is interest in considering augmentation therapy in those with milder disease who are rapidly declining, in order to slow progression. Similarly, novel treatments may have the greatest effect in rapid decliners, including mild disease.
There is interest in identifying AATD patients with a rapid decline in lung function at the earliest possible juncture, to ensure smoking cessation and consider augmentation therapy. Also, by identifying the individual rate of decline, it may be possible to serially assess if this has slowed with appropriate interventions.
Previous studies from our group indicated that 4 annual FEV1 measurements over at least 3 years would be necessary to identify patients who are stable or rapidly declining, with a degree of confidence.10 There is significant interest in assessing if serial lung function, over shorter time periods can identify rapid decliners with similar accuracy.
The purpose of the present study was to determine if lung function decline can be accurately identified over a shorter time span in non-smoking AATD patients by monitoring spirometry (including the Forced Expiratory Volume in 1 Second (FEV1) and Forced Vital Capacity (FVC)) every 6 months (for a total of 18 months). The results were compared to longer follow-up (≥3 years) prior to the 18-month trial period.
Materials and Methods
Study Subjects
The current study was a single-centre prospective study, approved by the South Birmingham Research and Ethics Committee (reference 14/WM/1243) and was conducted in accordance with the Declaration of Helsinki. Participants were all ex- or never-smokers with the severe PiZZ AATD genotype, either without spirometric COPD (FEV1/FVC ≥ 70%) or with mild COPD (FEV1/FVC < 70%, FEV1 ≥ 80% predicted).
The spirometry data prior to the current study was performed under the Antitrypsin Deficiency Assessment and Programme for Treatment (ADAPT), which is a Birmingham-based registry for UK individuals with AATD. The ADAPT programme was approved by the South Birmingham Research and Ethics Committee (Ref number; 3359a). Participants for the current study were originally identified from the ADAPT register and all provided written, informed consent for the additional study.
Methods
Patients participating in the study performed post-bronchodilator (5.0 mg salbutamol sulphate plus 0.5 mg ipratropium bromide by nebulisation) spirometry every 6 months for a total of 18 months (4 measurements). Spirometry was performed on Jaeger Masterscreen Pro lung function system (Jaeger Ltd, Hochberg, Germany) according to the Association for Respiratory Technology and Physiology (ARTP)/British Thoracic Society (BTS) guidelines for quality control.11 Data was collected from 2015 to 2019.
Spirometry data included FEV1, FVC, and FEV1/FVC. FEV1 and FVC were expressed as % predicted, to standardise for height, age, sex and race and allow for the differentiation of decline due to natural ageing (where % predicted remains stable) and decline due to disease progression (where % predicted decreases). The FEV1/FVC ratio was expressed in the conventional manner as a percentage.
Retrospective spirometry data for the 3 years comparison were collected under the ADAPT programme, and at the start of this data collection, predicted values were calculated from the European Community for Steel and Coal (ECSC) reference equations.12 Therefore, predicted values for the current study were also calculated from the ECSC reference equations for consistency, despite more up-to-date reference equations being available.13
Analysis
The main outcome measure was the FEV1 decline expressed as % predicted. For parametric data, t-tests were used to compare categories and Pearson’s correlations were used to determine the strength of any relationship. For non-parametric data, Mann–Whitney U-tests were used to compare categories and Spearman’s Rank correlations were used to determine the strength of any relationship. Chi-Squared tests were used to compare categorical data between groups. A p value of <0.05 was taken to be statistically significant.
The change in FEV1% predicted was determined by linear regression and its agreement over different time scales (6 months, 18 months and 3+ years) was assessed using Bland-Altman plots. In addition, test characteristics (including positive and negative predictive values) were analysed for 6-month and 18-month data with 3+ years as the reference standard.
Role of the Funding Source
This study was funded by the Alpha-1 Foundation.
Results
A total of 72 ex- or never-smoking PiZZ AATD patients completed the spirometry measurements over 18 months. Of these, 23 patients had mild spirometric COPD (FEV1 > 80% predicted and FEV1/FVC < 70%), with the other 49 having an FEV1/FVC > 70%. Of the 27 patients who had annual historical spirometry data obtained previously for 3 years or more, 10 were classified as “rapid decliners” with a loss of FEV1 ≥ 1% predicted per year.12 Baseline demographic data and smoking history were no different between rapid and non-rapid decliners. There was no difference in baseline FEV1% predicted or FEV1/FVC, although FVC % predicted was significantly lower in the non-rapid decline group (p = 0.027) and, by definition, FEV1 decline was significantly greater in the rapid decline group (p < 0.0001). The demographic and lung function data are shown in Table 1.
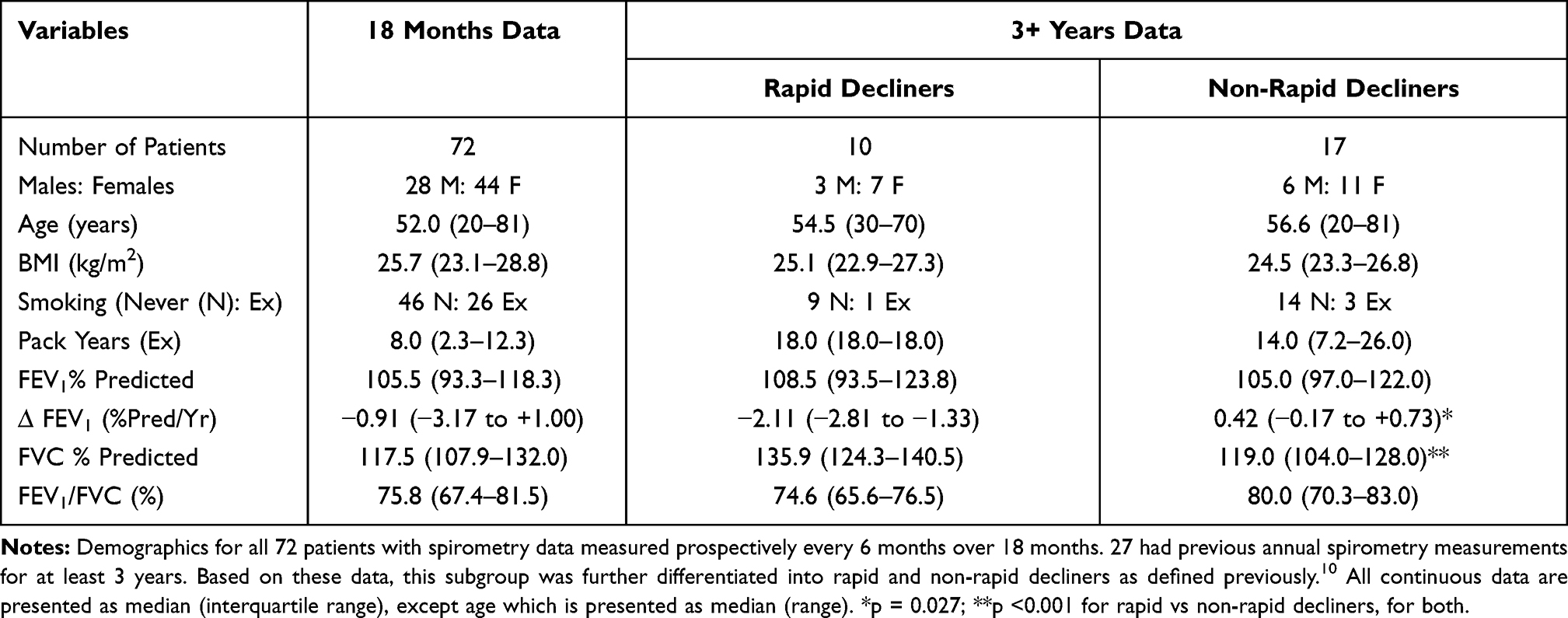 |
Table 1 Baseline Patient Demographics |
Individual patient data for the 18-month progression obtained by linear regression showed variable degrees of change in FEV1 over time, with 29 patients showing no decline, 8 showing slow decline and 35 showing rapid decline, with the latter ranging from −1.1 to −7.9% predicted/year and the individual r2 values ranged from 0.12 to 0.98.
Bland-Altman plots showed that there was no overall agreement between decline data obtained over 6 months and that obtained over 18 months or in those who had previously determined data over 3 or more years (Figure 1). Indeed, comparison between data obtained over 6 months versus 18 months and 6 months versus 3+ years showed proportional bias. Moreover, there was no correlation between the 18 months and 3+ years’ data (Figure 2).
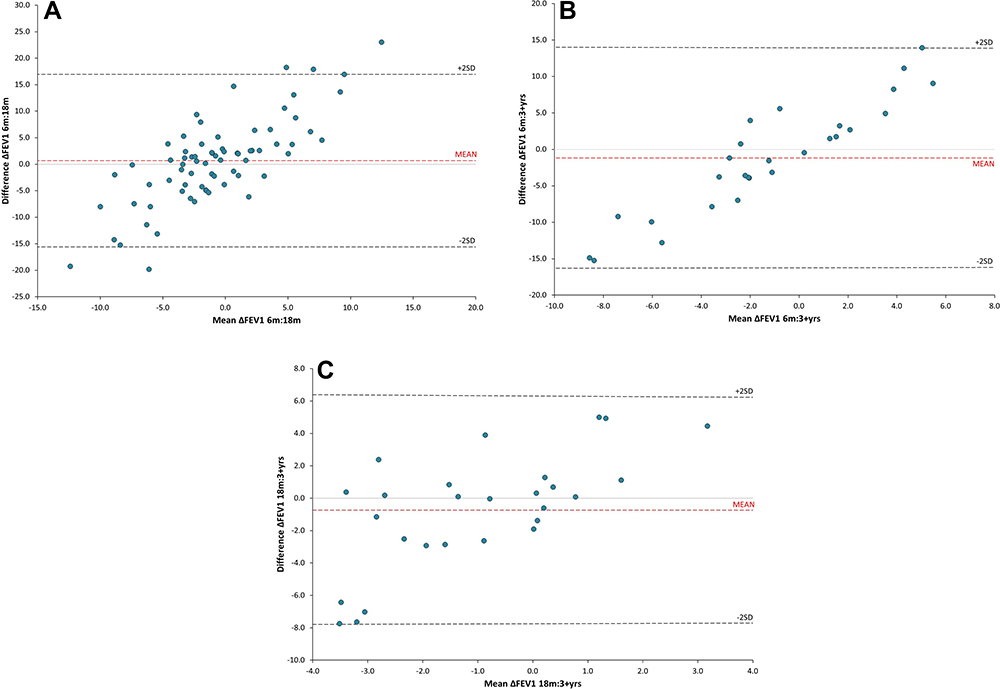 |
Figure 1 Bland-Altman plots comparing the annualised change in FEV1% predicted (∆FEV1%predicted/year) calculated by linear regression between data collected over (A) 6 months versus 18 months (n = 72), (B) 6 months versus 3+ years (n = 27), and (C) 18 months versus 3+ years (n = 27). Data collected over 3+ years was used as the reference standard. All comparisons show that there is no agreement between any timeframe, with comparisons of 6 months versus 18 months and 6 months versus 3+ years showing proportional bias. |
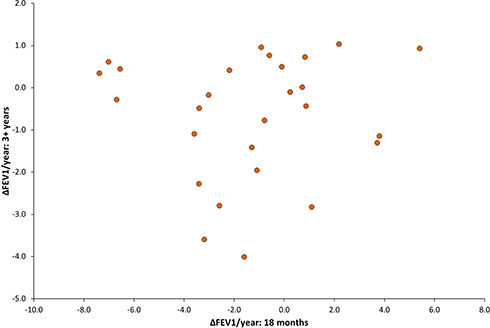 |
Figure 2 A scatter plot comparing the change (∆) in FEV1% predicted (calculated by linear regression) between data collected over 18 months versus data collected over 3+ years (n=27). There is no correlation between the predicted rate of FEV1 change over the two timeframes. |
The positive predictive value for rapid decline with 18 months’ data compared to 3 or more years was only 50.0% but, for those who did not show rapid decline over 18 months, the positive predictive value for similar lung function stability over 3 or more years was 76.9%.
Discussion
The decision to instigate augmentation therapy with AAT depends on the confidence that the patient will continue to decline rapidly without intervention. The sooner decline can be determined, the better for patient management but our previous study suggested this should be over at least 3 years.10
This study was conducted to determine if a shorter period of more intense data collection (6-monthly for 18 months, with 4 measurements) could identify rapid decliners with sufficient accuracy. The results demonstrate that 18 months of follow-up is not sufficient to achieve this goal. Furthermore, in this group of patients, there were no distinctive clinical characteristics that are captured routinely (such as exacerbation frequency, change in smoking status or change in therapies) which might identify those who were declining quickly from those who were not (although symptom scores were not captured in the current study and were found to be different in fast decliners previously14).
The data shows that post bronchodilator spirometry over 18 months shows variable change both for the cohort and in the r2 values for the slope for individuals. Two data points over 6 months do not relate to the 18-month estimate. Furthermore, the 18-month data relates poorly to data collected over 3 or more years from a subgroup where this data was previously available.10 In that previous study, the positive predictive value of 3 or more years of data was good to moderate for predicting long-term (11 years) decline, especially in those whose physiology remained stable.10
Here, the best prediction of long-term decline was for those whose physiology remained stable over 18 months, although with only a positive predictive value of 76.9%. Identification of rapid decliners using this timeframe was only 50.0%, which is less than previously reported using 3 years’ data.10
The current study has limitations. These include the limited patient recruitment, especially for comparison with longer term follow-up. This limitation largely reflects the rarity of AATD and the relative infrequency of identifying AATD patients with mild disease. Also, the AATD registry cohort potentially adds selection bias as this is not formed through full population screening (which is not available for AATD in the UK). Nevertheless, it represents a more natural history of the condition as none received augmentation therapy, both because of their good lung function and the lack of availability of this treatment in the UK. Clearly, longer term follow-up would be advisable to confirm these data with collaboration between specialised centres working to the same standard operating procedures.
The decline in FEV1% predicted is adjusted for sex, age and height, but was not adjusted for smoking history. The reasons for this included that there was no difference in pack years between rapid and non-rapid decliners, other studies have not modelled decline adjusting for smoking behaviour and this would not be included when designing a clinical study. However, larger datasets, including patients with a broader range of smoking behaviours and lung function measurements could include this modelling to see its effect, although this would be complex in a rare disease.
The patients included had no or mild COPD but all subjects had an FEV1 in the normal range and the patients included have relatively limited smoking histories. It is uncertain if the results could be extrapolated to those with a high pack-year history or with more significant lung disease. However, studies suggest that rapid lung function decline is not restricted to heavier smokers, and that a higher proportion of “rapid decliners” were seen in milder disease.10
The reason for the presence of proportional bias shown in some Bland Altman plots is unclear. Further, the finding that 6 months, 18 months and 3 years measurements of lung function decline are non-equivalent is not easily explained. This may represent the inherent variable in the FEV1 measurement or that lung disease may progress in a series of “sprints” depending on inflammatory burden, but this remain speculation. It is unclear is this finding can be extrapolated to non-AATD COPD, but there are on-going studies assessing early COPD with six-monthly lung function, which might provide more information on publication.
The implication of the current data is that even frequent measurements over 18 months are insufficient to accurately determine longer term spirometric outcome in AATD for an individual or a patient cohort. At least 3 years are required to have more confidence in identifying rapid decliners. This has successive implications for the design of studies that wish to focus on rapid decliners and clinical practice if one wishes to stratify augmentation therapy to rapid decliners only. Alternative approaches to identifying this group of patients with rapidly deteriorating lung function, through new biomarkers or other assessment of pathology, are urgently required.
Acknowledgments
This work was funded by the Alpha-1 Foundation. Health data curation was supported by PIONEER, the Health data hub in acute care, which is funded by Health Data Research UK. We thank all the patients with Alpha-1 Antitrypsin Deficiency for contributing to this study.
Disclosure
Professor Robert A Stockley is steering committee for Vertex, advisory board for Z factor, and chair of Data Safety Monitoring Board for Kamada; he also reports grants and/or personal fees from CSL Behring, Mereo biopharma, took part in the laboratory analyses for Takeda, during the conduct of the study. Professor Elizabeth Sapey reports grants from Alpha-1 Foundation, during the conduct of the study; grants from HDR-UK, The Wellcome Trust, MRC, British Lung Foundation, NIHR, and EPSRC, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Evald T, Dirksen A, Keittelmann S, Viskum K, Kok-Jensen A. Decline in pulmonary function in patients with α1-antitrypsin deficiency. Lung. 1990;168(S1):579–585. doi:10.1007/BF02718180
2. Gadek JE, Klein HG, Holland PV, Crystal RG. Replacement therapy of alpha 1-antitrypsin deficiency. Reversal of protease-antiprotease imbalance within the alveolar structures of PiZ subjects. J Clin Invest. 1981;68(5):1158–1165. doi:10.1172/JCI110360
3. Wewers MD, Casolaro MA, Sellers SE, et al. Replacement therapy for Alpha 1-antitrypsin deficiency associated with emphysema. N Engl J Med. 1987;316(17):1055–1062. doi:10.1056/NEJM198704233161704
4. Stoller JK, Aboussouan LS. Alpha 1-antitrypsin deficiency 5: intravenous augmentation therapy: current understanding. Thorax. 2004;59:708. doi:10.1136/thx.2003.006544
5. Chapman KR, Stockley RA, Dawkins C, Wilkes MM, Navickis RJ. Augmentation therapy for α 1 antitrypsin deficiency: a meta-analysis. COPD. 2009;6(3):177–184. doi:10.1080/15412550902905961
6. Vreim CE, Wu M, Crystal RGet al. Survival and FEV1 decline in individuals with severe deficiency of α1-antitrypsin. Am J Respir Crit Care Med. 1998;158:49–59. doi:10.1164/ajrccm.158.1.9712017
7. Dirksen A, Dijkman JH, Madsen F, et al. A randomized clinical trial of α1-antitrypsin augmentation therapy. Am J Respir Crit Care Med. 1999;160:1468–1472. doi:10.1164/ajrccm.160.5.9901055
8. Dirksen A, Piitulainen E, Parr DG, et al. Exploring the role of CT densitometry: a randomised study of augmentation therapy in alpha 1-antitrypsin deficiency. Eur Respir J. 2009;33:1345. doi:10.1183/09031936.00159408
9. Chapman KR, Burdon JGW, Piitulainen E, et al. Intravenous augmentation treatment and lung density in severe alpha 1 antitrypsin deficiency (RAPID): a randomised, double-blind, placebo-controlled trial. Lancet. 2015;386(9991):360–368. doi:10.1016/S0140-6736(15)60860-1
10. Stockley RA, Edgar RG, Pillai A, Turner AM. Individualized lung function trends in alpha-1-antitrypsin deficiency: a need for patience in order to provide patient centered management? Int J Chron Obstruct Pulmon Dis. 2016;11:1745–1756. doi:10.2147/COPD.S111508
11. Sylvester KP, Clayton N, Cliff I, et al. ARTP statement on pulmonary function testing 2020. BMJ Open Respir Res. 2020;7(1):e000575. doi:10.1136/bmjresp-2020-000575
12. No authors listed. Standardized lung function testing. Report working party. Bull Eur Physiopathol Respir. 1983;19 Suppl 5:1–95.
13. Quanjer PH, Stanojevic S, Cole TJ, et al., Initiative ERSGLF. Multi-ethnic reference values for spirometry for the 3-95-yr age range: the global lung function 2012 equations. Eur Respir J. 2012;40(6):1324–1343. doi:10.1183/09031936.00080312
14. Stockley JA, Ismail AM, Hughes SM, Edgar R, Stockley Robert A, Sapey E. Maximal mid-expiratory flow detects early lung disease in α1-antitrypsin deficiency. Eur Respir J. 2017;49:1602055. doi:10.1183/13993003.02055-2016
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.