Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 12
Therapeutic targets of hypercholesterolemia: HMGCR and LDLR
Authors Ma S, Sun W, Gao L ![]() , Liu S
, Liu S ![]()
Received 11 June 2019
Accepted for publication 25 July 2019
Published 21 August 2019 Volume 2019:12 Pages 1543—1553
DOI https://doi.org/10.2147/DMSO.S219013
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ming-Hui Zou
Shizhan Ma,1,2 Wenxiu Sun,3 Ling Gao,1,4,5 Shudong Liu6
1Department of Endocrinology and Metabolism, Shandong Provincial Hospital Affiliated to Shandong First Medical University, Jinan 250021, People’s Republic of China; 2Department of Endocrinology and Metabolism, Shandong Provincial Hospital Affiliated to Shandong University, Jinan 250021, People’s Republic of China; 3Department of Pharmacy, Taishan Vocational College of Nursing, Taian 271000, People’s Republic of China; 4Scientific Center, Shandong Provincial Hospital Affiliated to Shandong First Medical University, Jinan 250021, People’s Republic of China; 5Scientific Center, Shandong Provincial Hospital Affiliated to Shandong University, Jinan 250021, People’s Republic of China; 6Department of Endocrinology, Shandong Rongjun General Hospital, Jinan 250013, People’s Republic of China
Correspondence: Ling Gao
Scientific Center, Shandong Provincial Hospital Affiliated to Shandong University, 324 Jing 5 Road, Jinan, Shandong Province 250021, People’s Republic of China
Tel +86 531 6877 6910
Email [email protected]
Shudong Liu
Department of Endocrinology, Shandong Rongjun General Hospital, 23 Jiefang Road, Jinan, Shandong Province 250013, People’s Republic of China
Tel +86 531 8238 2351
Email [email protected]
Abstract: Cholesterol homeostasis is critical and necessary for the body’s functions. Hypercholesterolemia can lead to significant clinical problems, such as cardiovascular disease (CVD). 3-Hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR) and low–density lipoprotein cholesterol receptor (LDLR) are major points of control in cholesterol homeostasis. We summarize the regulatory mechanisms of HMGCR and LDLR, which may provide insight for new drug design and development.
Keywords: cholesterol homeostasis, CVD, HMGCR, LDLR
Introduction
Cholesterol plays a key role in the regulation of the body’s essential functions. It is both one of the basal components of biological membranes and a precursor of a variety of physiologically active substances, such as bile acid, vitamin D, steroid hormones, ubiquinol and heme A, which are intermediate products of mevalonic cholesterol biosynthesis and exhibit pleiotropic effects on numerous diseases and energy metabolism.1–6 The main source of the body’s cholesterol is from mevalonic biosynthesis (de novo synthesis), particularly in the liver, where up to 1 g cholesterol can be synthesized, and in the extrahepatic tissues such as the small intestines and adrenal glands.7–11 3-Hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR) is the rate-limiting enzyme in the de novo synthesis of cholesterol and serves as a key regulatory enzyme controlling endogenous cholesterol synthesis.11,12
About 500 mg of total cholesterol is consumed each day in the bile acid synthesis pathway in the liver for lipid digestion and absorption, and 400–500 mg of dietary exogenous cholesterol is absorbed from the intestines each day.3,9,10 Additionally, hepatic cholesterol is transported in the blood by lipoproteins and utilized by peripheral tissues (Figure 1).13 The serum cholesterol is mainly metabolized through the low-density lipoprotein cholesterol (LDL-c) receptor (LDLR) pathway.14 In the peripheral tissues, LDLR on the cell surface membranes binds to plasma LDL-c particles transporting liver cholesterol, and LDL-c is then taken in by endocytosis and cleared.12,15
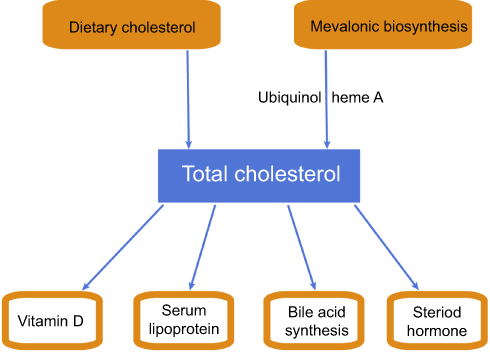 |
Figure 1 Schematic diagram of cholesterol metabolic pathway in the body. |
Hypercholesterolemia can lead to significant clinical problems such as cardiovascular disease (CVD), and hypocholesterolemic agents significantly reduce the risk of CVD events.16–22 Therefore, as key regulatory points in cholesterol metabolism, HMGCR and LDLR have been studied as targets for treating CVD and dyslipidemia in recent decades.23,24 In this review, we summarize the regulatory mechanisms of HMGCR and LDLR, which may provide insights for new drug design and development.
Regulatory mechanism of HMGCR
The human HMGCR gene (GeneID: 3156) is located on chromosome 5q12,25 and it encodes three isoforms (isoforms 1, 2 and 3) that are produced by alternative splicing and may lead to different responses to treatment with statin (an HMGCR inhibitor).26 HMGCR isoform 1 has been extensively investigated and is a membrane-bound glycoprotein comprising 888 amino acids that regulates mevalonate, which is an initial control point in the endogenous biosynthesis of cholesterol in the liver and small intestine.27 The activity and amount of HMGCR are regulated at multiple levels and through multiple mechanisms, such as negative feedback regulatory mechanisms mediated by sterols and nonsterol metabolites derived from mevalonate, posttranslational modification, degradation and hormone regulation.11,27–33 Different regulatory pathways interact to control cholesterol homeostasis (Figure 2).
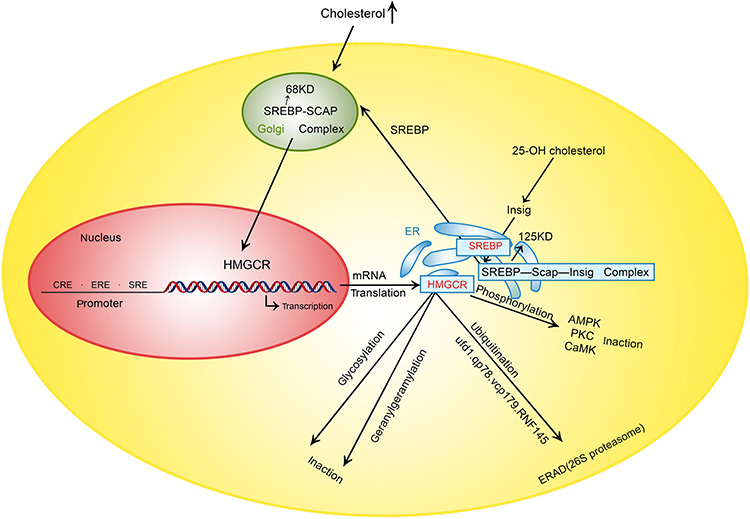 |
Figure 2 Control point of HMGCR. Abbreviations: HMGCR, 3-hydroxy-3-methylglutaryl coenzyme A reductase; SRE, Sterol regulatory element; ER, endoplasmic reticulum; SREBP, sterol regulatory element binding protein; SCAP, SREBP cleavage activating protein; AMPK, AMP-activated protein kinase; PKC, Protein kinase C; CaMK, Ca2+/calmodulin-dependent protein kinase; ERAD, ER-associated degradation; 25-OH cholesterol, 25-hydroxycholesterol; RNF145, ring finger protein 145. |
Negative feedback regulation
Negative feedback regulation is the most important way to control cholesterol synthesis. The cholesterol synthesis process is very complex, involving over 30 enzymatic reactions. HMGCR, the primary rate-limiting enzyme in the process, is a known target of feedback regulation whose concentration directly influences the amount of cholesterol synthesized.34 An increase in the concentration of cholesterol or 25-hydroxycholesterol (25-OH cholesterol) suppresses the synthesis of HMGCR and leads to a marked decrease in HMGCR; furthermore, cholesterol accelerates HMGCR degradation by facilitating HMGCR ubiquitination. These two mechanisms result in a synergistic effect, ultimately leading to a decrease in both the HMGCR concentration and cholesterol biosynthesis to decrease the concentration of cholesterol.28,30,31,35 The suppression of HMGCR synthesis is associated with a type of nuclear transcription factor termed sterol regulatory element-binding proteins (SREBPs).36
SREBPs, including SREBP-1a (GeneID: 6720) and SREBP-1c (GeneID: 6720), which are produced from the same gene by alternative splicing, and SREBP-2 (GeneID: 6721), are key lipogenic transcription regulators. SREBP-1a is a potent activator of all SREBP-responsive genes, and SREBP-1c activates genes involved in fatty acid and triglyceride synthesis. SREBP-2 preferentially regulates genes involved in the cholesterol synthesis pathway. SREBP-2 activates the transcription of genes by binding sterol regulatory element 1 (SRE-1; 5ʹ-ATCACCCCAC-3ʹ) in the LDLR and HMGCR promoters.36
SREBP-2 is initially formed as a 125 kDa precursor in the endoplasmic reticulum (ER), and its nuclear mature form (68 kDa) can enter the nucleus to activate the transcription of target genes only after proteolytic processing (which involves removal of its carboxyl terminus in the Golgi apparatus). In the Golgi apparatus, SREBP can interact with SREBP cleavage activating protein (SCAP). SCAP can combine with a pair of ER membrane protein insulin-induced genes (Insig1 and Insig2) to form the SREBP/SCAP/Insig complex. This complex becomes fixed on the ER after combining with Insig.36–38 SCAP (GeneID: 22937) contains 1279 amino acids and possesses a sterol-sensing domain (SSD);39 cholesterol can bind to the SSD of SCAP and thus change the conformation of SCAP. The SREBP/SCAP complex is retained in the ER when the sterol concentration is high, leading to the suppression of SREBP-mediated transcription and HMGCR product. When the cholesterol in cells is depleted, SCAP dissociates from the SREBP/SCAP/Insig complex and SREBP enters the Golgi complex to be processed, and the synthesis of cholesterol subsequently increases due to SREBP-mediated transcription.38,40–42 There is an obvious distinction between 25-OH cholesterol and cholesterol in the feedback regulation of cholesterol synthesis, and a recent study demonstrated that 25-OH cholesterol inhibits the synthesis of cholesterol by binding to Insig, not SCAP, although cholesterol is thought to bind to SCAP.38 Recent evidence demonstrated that sulfation of 25-OH cholesterol might inhibit lipid synthesis and inflammatory responses, and it may be a target for CVD prevention.43,44
Mevalonate-derived products participating in the feedback regulation of cholesterol synthesis are closely associated with the ER-associated degradation (ERAD) of HMGCR and can act synergistically with sterols to augment HMGCR degradation by facilitating both the ubiquitination of HMGCR and its dislocation out of the ER, which is involved in the geranylgeranylation of proteins (geranylgeranyl pyrophosphate (GGPP).45 The prevention of geranylgeranylation may be another mechanism by which statins lower cholesterol.46,47
Posttranslational modification of HMGCR
Importantly, the posttranslational modification of HMGCR influences its regulatory function, which depends on cellular energy conditions. Two forms of HMGCR exist: phosphorylated (inactive) and dephosphorylated (active) forms.33,48–50 The phosphorylation of human HMGCR at serine 872 by AMP-activated protein kinase (AMPK) reduces its enzymatic activity, and the dephosphorylation of HMGCR by protein phosphatase restores its enzymatic activity.48,51 Metformin, the most widely used hypoglycemic drug that acts by activating AMPK, can regulate lipid metabolism.52–54 Protein kinase C (PKC) and Ca2+/calmodulin-dependent protein kinase (CaMK) are also involved in the phosphorylation of HMGCR, but the specific phosphorylated residue in HMGCR has not been identified.55,56
The ubiquitination of HMGCR mainly influences its stability and facilitates its degradation to regulate cholesterol production. When cellular sterol accumulates, HMGCR binds to Insig1 and gp78, which is an E3 ubiquitin ligase, and interacts with ATPase valosin-containing protein (VCP/p97) in the ER to facilitate the ubiquitination (at lysine 248) and ERAD of HMGCR by the cytosolic 26S proteasome,31,57,58 while Ufd1, a gp78 cofactor, enhances and accelerates the ERAD of HMGCR.35 In liver-specific gp78 knockout mice, SREBP was decreased and this was accompanied by elevated levels of Insig1/2, leading to decreased cholesterol synthesis. However, the degradation of HMGCR was decreased, suggesting that gp78 may play an important role in the degradation of SREBP.59 Small-molecule compounds have shown a beneficial therapeutic effect in dyslipidemia by inhibiting the SREBP pathway, which suggests that SREBP may be a new drug target in the future.60,61 Ring finger protein 145 (RNF145) is a ubiquitin ligase involved in the degradation of HMGCR that was recently identified based on small-scale short hairpin RNA (shRNA) screening, and its cysteine 537 residue is critical for its function.62 Glycosylation is also involved in the negative regulation of HMGCR activity, which induces HMGCR localization to the ER.63 Therefore, another way to control cholesterol metabolism is by influencing the level of posttranslational modification of HMGCR.
Hormone regulation and genetic polymorphisms
The HMGCR promoter region contains a cyclic AMP response element (CRE) and an estrogen response element (ERE) in addition to a sterol regulatory element (SRE). These elements are involved in the regulation of HMGCR transcriptional activity.64,65 Cellular cholesterol levels are vital for the regulation of glial cell development and myelination by neuregulin, and the control of cholesterol synthesis by neuregulin was shown to be partly mediated by a CRE sequence in the HMGCR promoter.64 Estrogen transactivated HMGCR expression via binding to the ERE (AGTCCcatCGACC) in the HMGCR promoter, which induced an elevation in the total cholesterol and LDL-c levels in the newborns of pregnant women with high estradiol levels.65
Studies have shown that genetic polymorphisms of HMGCR are associated with its function and phenotype. Akadam-Teker et al have demonstrated that the total cholesterol and LDL-c levels are higher among male coronary heart disease patients aged <55 years carrying the HMGCR CC genotype (rs3761740) than those carrying CA + AA genotypes, which indicates an association between HMGCR polymorphisms and CVD.66 However, the A allele of the HMGCR genotype increased the risk of Alzheimer’s disease (AD) in an Italian study.67 Conversely, the rs3761740 variant of HMGCR was not associated with AD in a Swedish case-control study.68 Another HMGCR polymorphism (rs3846662) may increase the mRNA and protein levels of HMGCR by affecting alternative splicing, and thereby contribute to the onset and progression of AD.69–72 Additionally, gene polymorphisms affect the cholesterol-lowering response to statin treatment.73–75
Regulatory mechanism of LDLR
LDLR, which is the receptor for LDL-c, plays a critical role in cholesterol transport and clearance from the plasma to the cytoplasm, and its functional insufficiency can lead to familial hypercholesterolemia (FH) and increase the risk of CVD.76–80 LDLR functioning is regulated through various means, such as genetic variants, feedback regulation associated with gene transcription, posttranslational modifications and degradation (Figure 3). LDLR knockout mice are a well-established model of atherosclerosis that aid clinical research into the treatment of human atherosclerosis.53,81–84
 |
Figure 3 Control point of LDLR. Abbreviations: LDLR, low–density lipoprotein cholesterol receptor; SREBP, sterol regulatory element binding protein; IDOL, inducible degrader of the LDLR; PCSK9, proprotein convertase subtilisin/kexin type 9; LXR, Liver X receptors; R1, repeat 1 sequence; R2, repeat 2 sequence; R3, repeat 3 sequence; ERα, estrogen receptor α. |
Genetic variants
The LDLR gene (Gene ID: 3949), located on chromosome 19p13.2, encodes six isoforms produced by alternative splicing. The canonical sequence (isoform 1) contains 860 amino acids but undergoes glycosylation in the ER and is changed into a mature 839 amino acid form in the Golgi apparatus, which is then transported to the cell membrane.78,85,86 LDLR includes an extracellular domain (amino acids 22–788), a transmembrane domain (amino acids 789–810) and a cytoplasmic domain (amino acids 811–860). Natural variants and mutations of the LDLR gene have been reported to influence its function and were involved in FH, which was characterized by high plasma LDL-c levels, dyslipidemia and CVD.87–90 For example, a single-nucleotide variation (rs767618089) of the LDLR gene at position 300 in the protein sequence of the extracellular domain did not affect LDLR expression but resulted in a reduction in LDL-c binding activity and LDL-c uptake.91 A mutation at position 828 in the cytoplasmic domain did not affect binding activity but reduced LDLR internalization.92 To date, more than 2000 LDLR genetic variants have been described, and many have not been demonstrated to have pathological significance.87,93,94 Interestingly, there is a decreased risk of type 2 diabetes in patients with FH,95,96 but these findings need to be investigated for the further prevention of diabetes.
Feedback regulation and SREBP-2
LDLR expression is controlled by the negative feedback regulation of intracellular cholesterol via the SREBP-2 pathway due to an SRE-1 sequence in the LDLR promoter.36,97–101 When the cellular sterol level is low, LDLR expression is transcriptionally stimulated by nuclear SREBP and the specific transcription factor Sp1, which binds to the cis-acting element (repeat 3 sequence, R3) of the LDLR promoter with a synergistic effect.102,103 Sp1 phosphorylation at Thr453 and Thr739 is critical for Sp1 binding to the LDLR promoter to stimulate LDLR expression.103 Additionally, Sp1 is required for the increase of LDLR transcription by 17β-estradiol, in which 17β-estradiol binds to the estrogen receptor α (ERα)/Sp1 complex to trans-activate LDLR promoter activity in HepG2 cells.104,105 The heterogeneous nuclear ribonucleoprotein K (hnRNP K) protein, a heterogeneous nuclear ribonucleoprotein, controls LDLR transcriptional activity by specifically interacting with R3 in the LDLR promoter, which is also the binding site of Sp1, though the relationship between hnRNP K and Sp1 remains unknown.101
Posttranslational modification of LDLR
The regulation of LDLR also involves histone modifications and extensive posttranslational modifications that influence the stability or activity of nucleic acids and functional proteins. PKC induced the phosphorylation of histone H3 Ser10 at the LDLR promoter and stimulated the expression of LDLR.106 In addition, the level of histone acetylation at the LDLR promoter affected its transcriptional activity.101
N- and O-glycosylation play crucial roles in protein processing from the Golgi apparatus to the ER, which influences membrane protein folding and stability, cell signal transduction, ligand binding, immunological defense and organ development.107–109 The ligand-binding domain of LDLR is O-glycosylated, which increases its affinity for LDL-c by ∼5-fold.108,109
The ubiquitination of a lysine residue is related to the degradation of LDLR depending on inducible degrader of the LDLR (IDOL), which is an E3 ubiquitin ligase, in a liver X receptor (LXR)-induced regulatory manner. IDOL catalyzes the polylysine 63-linked ubiquitination of LDLR, thereby promoting LDLR lysosomal degradation and decreasing LDL-c clearance, and this regulatory process is not dependent on the SREBP-2 pathway.110–114 Studies of IDOL-knockout mice have reported an improvement in metabolic dysfunction, including a decrease in circulating cholesterol, triglyceride and glucose levels and hepatosteatosis and fat mass; these effects suggest that the inhibition of IDOL may be a future therapeutic strategy to combat dyslipidemia and/or CVD.115,116
Proprotein convertase subtilisin/kexin type 9 (PCSK9) regulatory pathway and LDLR degradation
PCSK9, a secretory serine protease, is involved in the degradation of LDLR.117–119 Ten phosphorylated serine residues have been identified on LDLR based on high-throughput screening using mass spectrometry, but it is not clear whether these phosphorylated serine residues are related to PCSK9.120,121 PCSK9 facilitated LDLR degradation in lysosomes by binding to ligand-binding repeats in the LDLR extracellular domain and led to a dramatic increase in plasma LDL-c, which was reversed by the loss of PCSK9 function and other anti-PCSK9 strategies.118,119 Alirocumab and evolocumab, novel PCSK9 inhibitors, exhibited a beneficial effect and improved dyslipidemia without an increase in diabetes risk in clinical studies.24,122–127 PCSK9 is also a downstream protein of SREBP-2 because of the SRE sequence in the PCSK9 promoter.128 A recent study showed that triciribine, a specific AKT inhibitor, inhibited PCSK9 expression by SREBP-2 transcriptional regulation accompanied by a decrease in HMGCR expression. In addition, triciribine induced LDLR mRNA stability and increased LDLR protein levels in a phosphorylated AKT-extracellular signal-regulated kinase (ERK)-dependent manner. Therefore, triciribine exerts beneficial and overlapping hypocholesterolemic effects, which make it an attractive potential drug target for hypercholesterolemia prevention.129
LDLR gene transcription was activated by the proinflammatory cytokine oncostatin M (OM), which induced a 3.8-fold maximal increase in LDLR mRNA levels in HepG2 cells, and this effect was mediated by the Janus kinase (JAK)-signal transducer and activator of transcription (STAT) and mitogen-activated protein kinase 1(MEK1)/ERK-PCSK9 pathways.130–132 OM downregulated PCSK9 transcription, which was accompanied by an elevation in LDLR and a significant decrease in plasma total cholesterol.130 The next works need to elucidate whether these findings have an important implication for future drug development. Studies demonstrated that an OM-induced complex involving CCAAT/enhancer binding protein (c/EBP), cAMP-responsive element binding protein (CBP) and early growth response gene 1 (Egr1) was required to bind to the sterol-independent regulatory element (SIRE) in the PCSK9 promoter and suppress PCSK9 expression.133–135 Different transcriptional factors were recruited and interacted as a complex to regulate downstream gene expression, and the complex also involved PKC or other regulators, demonstrating complex regulation.136
Perspectives
Intracellular cholesterol homeostasis depends on the balance between supply, including the intestinal intake of dietary cholesterol and its intracellular synthesis, and demand, including the bile acid pathway, the synthesis of steroid hormones and the clearance of cholesterol by the low-density lipoprotein (LDL)-LDL receptor (LDLR) pathway. Hypercholesterolemia, an imbalanced and pathologic state of cholesterol homeostasis, is a major risk factor for cardiovascular disease (CVD), which is the leading cause of mortality worldwide.14,137 Up to now, the ezetimibe and bile acid sequestrants as second-line cholesterol lowering agents were used to inhibit the cholesterol intake from the intestine. Up to now, the ezetimibe and bile acid sequestrants as second-line cholesterol lowering agents were used to inhibit the cholesterol intake from the intestine.138,139 The 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR) and proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors (statins, alirocumab and evolocumab) have been widely used to treat hypercholesterolemia in clinical settings. These four classes of cholesterol-lowering agents acts in different ways (Figure 4), but clinical studies demonstrated that these drugs need to be improved to achieve more effective outcomes.140,141 Therefore, for hypercholesterolemia and CVD prevention, further research is necessary to develop specific agents targeted at the HMGCR and LDLR regulatory pathway.
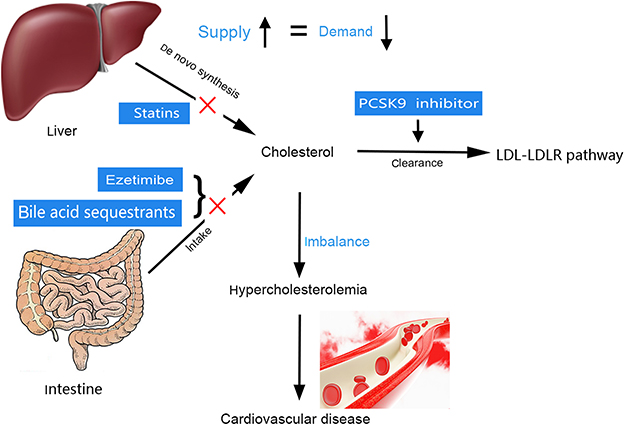 |
Figure 4 Schematic diagram of the hypercholesterolemia and current therapeutic drug. PCSK9 inhibitor: proprotein convertase subtilisin/kexin type 9 inhibitor. Abbreviations: LDL, low–density lipoprotein; LDLR, low–density lipoprotein cholesterol receptor. |
Acknowledgments
This work was funded from the project ZR2019BH023 supported by Shandong Provincial Natural Science Foundation and National Natural Science Foundation of China (grant number:81900716). The sponsors had no role in any of the stages from study design to submission of the manuscript for publication.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Mazein A, Watterson S, Gibbs HC, et al. Regulation and feedback of cholesterol metabolism. Nature Precedings. 2011;59(2):473–474.
2. Payne AH, Hales DB. Overview of steroidogenic enzymes in the pathway from cholesterol to active steroid hormones. Endocr Rev. 2004;25(6):947–970. doi:10.1210/er.2003-0030
3. Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem. 2003;72:137–174. doi:10.1146/annurev.biochem.72.121801.161712
4. Deichmann R, Lavie C, Andrews S. Coenzyme q10 and statin-induced mitochondrial dysfunction. Ochsner J. 2010;10(1):16–21.
5. Garrido-Maraver J, Cordero MD, Oropesa-Avila M, et al. Coenzyme q10 therapy. Mol Syndromol. 2014;5(3–4):187–197. doi:10.1159/000360101
6. Kaminska J, Grabinska K, Kwapisz M, et al. The isoprenoid biosynthetic pathway in Saccharomyces cerevisiae is affected in a maf1-1 mutant with altered tRNA synthesis. FEMS Yeast Res. 2002;2(1):31–37. doi:10.1111/j.1567-1364.2002.tb00066.x
7. Young NL, Lopez DR, McNamara DJ. Contributions of absorbed dietary cholesterol and cholesterol synthesized in small intestine to hypercholesterolemia in diabetic rats. Diabetes. 1988;37(8):1151–1156. doi:10.2337/diab.37.8.1151
8. Balasubramaniam S, Goldstein JL, Brown MS. Regulation of cholesterol synthesis in rat adrenal gland through coordinate control of 3-hydroxy-3-methylglutaryl coenzyme A synthase and reductase activities. Proc Natl Acad Sci U S A. 1977;74(4):1421–1425. doi:10.1073/pnas.74.4.1421
9. Hrydziuszko O, Wrona A, Balbus J, Kubica K. Mathematical two-compartment model of human cholesterol transport in application to high blood cholesterol diagnosis and treatment. Electron Notes Theor Comput Sci. 2014;306:19–30. doi:10.1016/j.entcs.2014.06.012
10. Grundy SM. Absorption and metabolism of dietary cholesterol. Annu Rev Nutr. 1983;3:71–96. doi:10.1146/annurev.nu.03.070183.000443
11. Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343(6257):425–430. doi:10.1038/343425a0
12. Goedeke L, Fernandez-Hernando C. Regulation of cholesterol homeostasis. Cell Mol Life Sci. 2012;69(6):915–930. doi:10.1007/s00018-011-0857-5
13. Wang HH, Garruti G, Liu M, Portincasa P, Wang DQ. Cholesterol and lipoprotein metabolism and atherosclerosis: recent advances in reverse cholesterol transport. Ann Hepatol. 2017;16 Suppl 1:S27–S42. doi:10.5604/01.3001.0010.5495
14. Masana L, Girona J, Ibarretxe D, et al. Clinical and pathophysiological evidence supporting the safety of extremely low LDL levels-The zero-LDL hypothesis. J Clin Lipidol. 2018;12(2):292–299 e293. doi:10.1016/j.jacl.2017.12.018
15. Zhang Y, Ma KL, Ruan XZ, Liu BC. Dysregulation of the low-density lipoprotein receptor pathway is involved in lipid disorder-mediated organ injury. Int J Biol Sci. 2016;12(5):569–579. doi:10.7150/ijbs.14027
16. Baigent C, Keech A, Kearney PM, et al.. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366(9493):1267–1278. doi:10.1016/S0140-6736(05)67394-1
17. Martin MJ, Hulley SB, Browner WS, Kuller LH, Wentworth D. Serum cholesterol, blood pressure, and mortality: implications from a cohort of 361,662 men. Lancet. 1986;2(8513):933–936. doi:10.1016/s0140-6736(86)90597-0
18. Shepherd J, Cobbe SM, Ford I, et al; West of Scotland Coronary Prevention Study Group. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. N Engl J Med. 1995;333(20):1301–1307. doi:10.1056/NEJM199511163332001
19. Expert Panel on Detection E, Treatment of High Blood Cholesterol in A. Executive summary of the third report of the National Cholesterol Education Program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III). JAMA. 2001;285(19):2486–2497. doi:10.1001/jama.285.19.2486
20. Steinberg D. Atherogenesis in perspective: hypercholesterolemia and inflammation as partners in crime. Nat Med. 2002;8(11):1211–1217. doi:10.1038/nm1102-1211
21. Maxfield FR, Tabas I. Role of cholesterol and lipid organization in disease. Nature. 2005;438(7068):612–621. doi:10.1038/nature04399
22. Grundy SM, Stone NJ, Bailey AL, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: executive summary: a report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines. J Am Coll Cardiol. 2019;73(24):3168–3209. doi: 10.1016/j.jacc.2018.11.002
23. Cholesterol Treatment Trialists C, Baigent C, Blackwell L, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376(9753):1670–1681. doi:10.1016/S0140-6736(10)61350-5
24. Sabatine MS, Leiter LA, Wiviott SD, et al. Cardiovascular safety and efficacy of the PCSK9 inhibitor evolocumab in patients with and without diabetes and the effect of evolocumab on glycaemia and risk of new-onset diabetes: a prespecified analysis of the FOURIER randomised controlled trial. Lancet Diabetes Endocrinol. 2017;5(12):941–950. doi:10.1016/S2213-8587(17)30313-3
25. Humphries SE, Tata F, Henry I, et al. The isolation, characterisation, and chromosomal assignment of the gene for human 3-hydroxy-3-methylglutaryl coenzyme A reductase, (HMG-CoA reductase). Hum Genet. 1985;71(3):254–258. doi:10.1007/bf00284585
26. Medina MW, Gao F, Ruan W, Rotter JI, Krauss RM. Alternative splicing of 3-hydroxy-3-methylglutaryl coenzyme A reductase is associated with plasma low-density lipoprotein cholesterol response to simvastatin. Circulation. 2008;118(4):355–362.
27. Brown MS, Goldstein JL. Multivalent feedback regulation of HMG CoA reductase, a control mechanism coordinating isoprenoid synthesis and cell growth. J Lipid Res. 1980;21(5):505–517.
28. Faust JR, Luskey KL, Chin DJ, Goldstein JL, Brown MS. Regulation of synthesis and degradation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase by low density lipoprotein and 25-hydroxycholesterol in UT-1 cells. Proc Natl Acad Sci U S A. 1982;79(17):5205–5209. doi:10.1073/pnas.79.17.5205
29. Beg ZH, Stonik JA, Brewer HB
30. Hwang S, Hartman IZ, Calhoun LN, et al. Contribution of accelerated degradation to feedback regulation of 3-Hydroxy-3-methylglutaryl coenzyme A reductase and cholesterol metabolism in the liver. J Biol Chem. 2016;291(26):13479–13494. doi:10.1074/jbc.M116.728469
31. Song BL, Sever N, DeBose-Boyd RA. Gp78, a membrane-anchored ubiquitin ligase, associates with Insig-1 and couples sterol-regulated ubiquitination to degradation of HMG CoA reductase. Mol Cell. 2005;19(6):829–840. doi:10.1016/j.molcel.2005.08.009
32. Istvan ES, Palnitkar M, Buchanan SK, Deisenhofer J. Crystal structure of the catalytic portion of human HMG-CoA reductase: insights into regulation of activity and catalysis. Embo J. 2000;19(5):819–830. doi:10.1093/emboj/19.5.819
33. Omkumar RV, Darnay BG, Rodwell VW. Modulation of Syrian hamster 3-hydroxy-3-methylglutaryl-CoA reductase activity by phosphorylation. Role of serine 871. J Biol Chem. 1994;269(9):6810–6814.
34. Ness GC, Holland RC, Lopez D. Selective compensatory induction of hepatic HMG-CoA reductase in response to inhibition of cholesterol absorption. Exp Biol Med (Maywood). 2006;231(5):559–565. doi:10.1177/153537020623100510
35. Cao J, Wang J, Qi W, et al. Ufd1 is a cofactor of gp78 and plays a key role in cholesterol metabolism by regulating the stability of HMG-CoA reductase. Cell Metab. 2007;6(2):115–128. doi:10.1016/j.cmet.2007.07.002
36. Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89(3):331–340. doi:10.1016/s0092-8674(00)80213-5
37. Nohturfft A, Yabe D, Goldstein JL, Brown MS, Espenshade PJ. Regulated step in cholesterol feedback localized to budding of SCAP from ER membranes. Cell. 2000;102(3):315–323. doi:10.1016/s0092-8674(00)00037-4
38. Gao Y, Zhou Y, Goldstein JL, Brown MS, Radhakrishnan A. Cholesterol-induced conformational changes in the sterol-sensing domain of the Scap protein suggest feedback mechanism to control cholesterol synthesis. J Biol Chem. 2017;292(21):8729–8737. doi:10.1074/jbc.M117.783894
39. Hua X, Nohturfft A, Goldstein JL, Brown MS. Sterol resistance in CHO cells traced to point mutation in SREBP cleavage-activating protein. Cell. 1996;87(3):415–426. doi:10.1016/s0092-8674(00)81362-8
40. Yang T, Goldstein JL, Brown MS. Overexpression of membrane domain of SCAP prevents sterols from inhibiting SCAP.SREBP exit from endoplasmic reticulum. J Biol Chem. 2000;275(38):29881–29886. doi:10.1074/jbc.M005439200
41. Yang T, Espenshade PJ, Wright ME, et al. Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell. 2002;110(4):489–500. doi:10.1016/s0092-8674(02)00872-3
42. Theesfeld CL, Pourmand D, Davis T, Garza RM, Hampton RY. The sterol-sensing domain (SSD) directly mediates signal-regulated endoplasmic reticulum-associated degradation (ERAD) of 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase isozyme Hmg2. J Biol Chem. 2011;286(30):26298–26307. doi:10.1074/jbc.M111.244798
43. Xu L, Bai Q, Rodriguez-Agudo D, et al. Regulation of hepatocyte lipid metabolism and inflammatory response by 25-hydroxycholesterol and 25-hydroxycholesterol-3-sulfate. Lipids. 2010;45(9):821–832. doi:10.1007/s11745-010-3451-y
44. Bai Q, Xu L, Kakiyama G, et al. Sulfation of 25-hydroxycholesterol by SULT2B1b decreases cellular lipids via the LXR/SREBP-1c signaling pathway in human aortic endothelial cells. Atherosclerosis. 2011;214(2):350–356. doi:10.1016/j.atherosclerosis.2010.11.021
45. Leichner GS, Avner R, Harats D, Roitelman J. Metabolically regulated endoplasmic reticulum-associated degradation of 3-hydroxy-3-methylglutaryl-CoA reductase: evidence for requirement of a geranylgeranylated protein. J Biol Chem. 2011;286(37):32150–32161. doi:10.1074/jbc.M111.278036
46. Alarcon VB, Marikawa Y. Statins inhibit blastocyst formation by preventing geranylgeranylation. Mol Hum Reprod. 2016;22(5):350–363. doi:10.1093/molehr/gaw011
47. Zhong WB, Wang CY, Chang TC, Lee WS. Lovastatin induces apoptosis of anaplastic thyroid cancer cells via inhibition of protein geranylgeranylation and de novo protein synthesis. Endocrinology. 2003;144(9):3852–3859. doi:10.1210/en.2003-0098
48. Zhang X, Song Y, Feng M, et al. Thyroid-stimulating hormone decreases HMG-CoA reductase phosphorylation via AMP-activated protein kinase in the liver. J Lipid Res. 2015;56(5):963–971. doi:10.1194/jlr.M047654
49. Beg ZH, Brewer HB
50. Beg ZH, Stonik JA, Brewer HB
51. Clarke PR, Hardie DG. Regulation of HMG-CoA reductase: identification of the site phosphorylated by the AMP-activated protein kinase in vitro and in intact rat liver. Embo J. 1990;9(8):2439–2446.
52. Zang M, Zuccollo A, Hou X, et al. AMP-activated protein kinase is required for the lipid-lowering effect of metformin in insulin-resistant human HepG2 cells. J Biol Chem. 2004;279(46):47898–47905. doi:10.1074/jbc.M408149200
53. Li Y, Xu S, Mihaylova MM, et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011;13(4):376–388.
54. Salpeter SR, Buckley NS, Kahn JA, Salpeter EE. Meta-analysis: metformin treatment in persons at risk for diabetes mellitus. Am J Med. 2008;121(2):149–157 e142. doi:10.1016/j.amjmed.2007.09.016
55. Beg ZH, Stonik JA, Brewer HB
56. Beg ZH, Stonik JA, Brewer HB
57. Sever N, Song BL, Yabe D, Goldstein JL, Brown MS, DeBose-Boyd RA. Insig-dependent ubiquitination and degradation of mammalian 3-hydroxy-3-methylglutaryl-CoA reductase stimulated by sterols and geranylgeraniol. J Biol Chem. 2003;278(52):52479–52490. doi:10.1074/jbc.M310053200
58. Morris LL, Hartman IZ, Jun DJ, Seemann J, DeBose-Boyd RA. Sequential actions of the AAA-ATPase valosin-containing protein (VCP)/p97 and the proteasome 19 S regulatory particle in sterol-accelerated, endoplasmic reticulum (ER)-associated degradation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase. J Biol Chem. 2014;289(27):19053–19066. doi:10.1074/jbc.M114.576652
59. Liu TF, Tang JJ, Li PS, et al. Ablation of gp78 in liver improves hyperlipidemia and insulin resistance by inhibiting SREBP to decrease lipid biosynthesis. Cell Metab. 2012;16(2):213–225. doi:10.1016/j.cmet.2012.06.014
60. Milne JC, Lambert PD, Schenk S, et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007;450(7170):712–716. doi:10.1038/nature06261
61. Walker AK, Yang F, Jiang K, et al. Conserved role of SIRT1 orthologs in fasting-dependent inhibition of the lipid/cholesterol regulator SREBP. Genes Dev. 2010;24(13):1403–1417. doi:10.1101/gad.1901210
62. Jiang LY, Jiang W, Tian N, et al. Ring finger protein 145 (RNF145) is a ubiquitin ligase for sterol-induced degradation of HMG-CoA reductase. J Biol Chem. 2018;293(11):4047–4055. doi:10.1074/jbc.RA117.001260
63. Hartman IZ, Liu P, Zehmer JK, et al. Sterol-induced dislocation of 3-hydroxy-3-methylglutaryl coenzyme A reductase from endoplasmic reticulum membranes into the cytosol through a subcellular compartment resembling lipid droplets. J Biol Chem. 2010;285(25):19288–19298. doi:10.1074/jbc.M110.134213
64. Pertusa M, Morenilla-Palao C, Carteron C, Viana F, Cabedo H. Transcriptional control of cholesterol biosynthesis in Schwann cells by axonal neuregulin 1. J Biol Chem. 2007;282(39):28768–28778. doi:10.1074/jbc.M701878200
65. Meng Y, Lv PP, Ding GL, et al. High maternal serum estradiol levels induce dyslipidemia in human newborns via a hepatic HMGCR estrogen response element. Sci Rep. 2015;5:10086. doi:10.1038/srep10086
66. Akadam-Teker B, Kurnaz O, Coskunpinar E, et al. The effects of age and gender on the relationship between HMGCR promoter-911 SNP (rs33761740) and serum lipids in patients with coronary heart disease. Gene. 2013;528(2):93–98. doi:10.1016/j.gene.2013.07.056
67. Porcellini E, Calabrese E, Guerini F, et al. The hydroxy-methyl-glutaryl CoA reductase promoter polymorphism is associated with Alzheimer’s risk and cognitive deterioration. Neurosci Lett. 2007;416(1):66–70. doi:10.1016/j.neulet.2007.01.046
68. Keller L, Murphy C, Wang HX, et al. A functional polymorphism in the HMGCR promoter affects transcriptional activity but not the risk for Alzheimer disease in Swedish populations. Brain Res. 2010;1344:185–191. doi:10.1016/j.brainres.2010.04.073
69. Chang XL, Tan L, Tan MS, et al. Association of HMGCR polymorphism with late-onset Alzheimer’s disease in Han Chinese. Oncotarget. 2016;7(16):22746–22751. doi:10.18632/oncotarget.8176
70. Leduc V, Theroux L, Dea D, Dufour R, Poirier J. Effects of rs3846662 variants on HMGCR mRNA and protein levels and on markers of Alzheimer’s disease pathology. J Mol Neurosci. 2016;58(1):109–119. doi:10.1007/s12031-015-0666-7
71. Burkhardt R, Kenny EE, Lowe JK, et al. Common SNPs in HMGCR in micronesians and whites associated with LDL-cholesterol levels affect alternative splicing of exon13. Arterioscler Thromb Vasc Biol. 2008;28(11):2078–2084. doi:10.1161/ATVBAHA.108.172288
72. Cao L, Wang HF, Tan L, et al. Alzheimer’s Disease Neuroimaging I. Effect of HMGCR genetic variation on neuroimaging biomarkers in healthy, mild cognitive impairment and Alzheimer’s disease cohorts. Oncotarget. 2016;7(12):13319–13327. doi:10.18632/oncotarget.7797
73. Ying S, Sun YM, Liu XM, An CY, Gao YY. Effect of ScrF I polymorphism in the 2nd intron of the HMGCR gene on lipid-lowering response to simvastatin in Chinese diabetic patients. Biochem Biophys Res Commun. 2007;363(2):395–398. doi:10.1016/j.bbrc.2007.08.182
74. Leduc V, Bourque L, Poirier J, Dufour R. Role of rs3846662 and HMGCR alternative splicing in statin efficacy and baseline lipid levels in familial hypercholesterolemia. Pharmacogenet Genomics. 2016;26(1):1–11. doi:10.1097/FPC.0000000000000178
75. Cuevas A, Fernandez C, Ferrada L, et al. HMGCR rs17671591 SNP determines lower plasma LDL-C after atorvastatin therapy in Chilean individuals. Basic Clin Pharmacol Toxicol. 2016;118(4):292–297. doi:10.1111/bcpt.12493
76. Abifadel M, Rabes JP, Jambart S, et al. The molecular basis of familial hypercholesterolemia in Lebanon: spectrum of LDLR mutations and role of PCSK9 as a modifier gene. Hum Mutat. 2009;30(7):E682–E691. doi:10.1002/humu.21002
77. Tichy L, Freiberger T, Zapletalova P, Soska V, Ravcukova B, Fajkusova L. The molecular basis of familial hypercholesterolemia in the Czech Republic: spectrum of LDLR mutations and genotype-phenotype correlations. Atherosclerosis. 2012;223(2):401–408. doi:10.1016/j.atherosclerosis.2012.05.014
78. Benito-Vicente A, Uribe KB, Jebari S, Galicia-Garcia U, Ostolaza H, Martin C. Validation of LDLr activity as a tool to improve genetic diagnosis of familial hypercholesterolemia: a retrospective on functional characterization of LDLr variants. Int J Mol Sci. 2018;19(6):1676. doi:10.3390/ijms19061676
79. Benito-Vicente A, Alves AC, Etxebarria A, Medeiros AM, Martin C, Bourbon M. The importance of an integrated analysis of clinical, molecular, and functional data for the genetic diagnosis of familial hypercholesterolemia. Genet Med. 2015;17(12):980–988. doi:10.1038/gim.2015.14
80. Beheshti S, Madsen CM, Varbo A, Benn M, Nordestgaard BG. Relationship of familial hypercholesterolemia and high low-density lipoprotein cholesterol to ischemic stroke. Circulation. 2018;138(6):578–589. doi:10.1161/CIRCULATIONAHA.118.033470
81. Nakaya H, Summers BD, Nicholson AC, Gotto AM
82. Ishibashi S, Brown MS, Goldstein JL, Gerard RD, Hammer RE, Herz J. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J Clin Invest. 1993;92(2):883–893. doi:10.1172/JCI116663
83. Krishna SM, Moxon JV, Jose RJ, et al. Anionic nanoliposomes reduced atherosclerosis progression in Low Density Lipoprotein Receptor (LDLR) deficient mice fed a high fat diet. J Cell Physiol. 2018;233(10):6951–6964. doi:10.1002/jcp.26610
84. Mehta JL, Sanada N, Hu CP, et al. Deletion of LOX-1 reduces atherogenesis in LDLR knockout mice fed high cholesterol diet. Circ Res. 2007;100(11):1634–1642. doi:10.1161/CIRCRESAHA.107.149724
85. Brown MS, Herz J, Goldstein JL. LDL-receptor structure. Calcium cages acid baths and recycling receptors. Nature. 1997;388(6643):629–630.
86. Ota T, Suzuki Y, Nishikawa T, et al. Complete sequencing and characterization of 21,243 full-length human cDNAs. Nat Genet. 2004;36(1):40–45. doi:10.1038/ng1285
87. Usifo E, Leigh SE, Whittall RA, et al. Low-density lipoprotein receptor gene familial hypercholesterolemia variant database: update and pathological assessment. Ann Hum Genet. 2012;76(5):387–401. doi:10.1111/j.1469-1809.2012.00724.x
88. ArulJothi KN, Suruthi Abirami B, Devi A. Genetic spectrum of low density lipoprotein receptor gene variations in South Indian population. Clin Chim Acta. 2018;478:28–36. doi:10.1016/j.cca.2017.12.024
89. Paththinige CS, Rajapakse J, Constantine GR, et al. Spectrum of low-density lipoprotein receptor (LDLR) mutations in a cohort of Sri Lankan patients with familial hypercholesterolemia - a preliminary report. Lipids Health Dis. 2018;17(1):100. doi:10.1186/s12944-018-0763-z
90. Hobbs HH, Brown MS, Goldstein JL. Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum Mutat. 1992;1(6):445–466. doi:10.1002/humu.1380010602
91. Etxebarria A, Benito-Vicente A, Stef M, Ostolaza H, Palacios L, Martin C. Activity-associated effect of LDL receptor missense variants located in the cysteine-rich repeats. Atherosclerosis. 2015;238(2):304–312. doi:10.1016/j.atherosclerosis.2014.12.026
92. Davis CG, Lehrman MA, Russell DW, et al. mutation in familial hypercholesterolemia: amino acid substitution in cytoplasmic domain impedes internalization of LDL receptors. Cell. 1986;45(1):15–24. doi:10.1016/0092-8674(86)90533-7
93. Benito-Vicente A, Siddiqi H, Uribe KB, et al. p. (Asp47Asn) and p. (Thr62Met): non deleterious LDL receptor missense variants functionally characterized in vitro. Sci Rep. 2018;8(1):16614. doi:10.1038/s41598-018-34715-x
94. Chora JR, Medeiros AM, Alves AC, Bourbon M. Analysis of publicly available LDLR, APOB, and PCSK9 variants associated with familial hypercholesterolemia: application of ACMG guidelines and implications for familial hypercholesterolemia diagnosis. Genet Med. 2018;20(6):591–598. doi:10.1038/gim.2017.151
95. Besseling J, Kastelein JJ, Defesche JC, Hutten BA, Hovingh GK. Association between familial hypercholesterolemia and prevalence of type 2 diabetes mellitus. JAMA. 2015;313(10):1029–1036. doi:10.1001/jama.2015.1206
96. Besseling J, Defesche J, Kastelein JJ, Hutten BA, Hovingh GK. Abstract 13703: patients with familial hypercholesterolemia are protected against type II diabetes - a cross-sectional study in 63,000 individuals tested for the presence of LDL receptor mutations. Circulation. 2014;130(suppl_2):A13703–A13703.
97. Smith JR, Osborne TF, Goldstein JL, Brown MS. Identification of nucleotides responsible for enhancer activity of sterol regulatory element in low density lipoprotein receptor gene. J Biol Chem. 1990;265(4):2306–2310.
98. Horton JD, Shimomura I, Brown MS, Hammer RE, Goldstein JL, Shimano H. Activation of cholesterol synthesis in preference to fatty acid synthesis in liver and adipose tissue of transgenic mice overproducing sterol regulatory element-binding protein-2. J Clin Invest. 1998;101(11):2331–2339. doi:10.1172/JCI2961
99. Hua X, Yokoyama C, Wu J, et al. SREBP-2, a second basic-helix-loop-helix-leucine zipper protein that stimulates transcription by binding to a sterol regulatory element. Proc Natl Acad Sci U S A. 1993;90(24):11603–11607. doi:10.1073/pnas.90.24.11603
100. Van Rooyen DM, Farrell GC. SREBP-2: a link between insulin resistance, hepatic cholesterol, and inflammation in NASH. J Gastroenterol Hepatol. 2011;26(5):789–792. doi:10.1111/j.1440-1746.2011.06704.x
101. Li H, Liu J. Identification of heterogeneous nuclear ribonucleoprotein K as a transactivator for human low density lipoprotein receptor gene transcription. J Biol Chem. 2010;285(23):17789–17797. doi:10.1074/jbc.M109.082057
102. Sanchez HB, Yieh L, Osborne TF. Cooperation by sterol regulatory element-binding protein and Sp1 in sterol regulation of low density lipoprotein receptor gene. J Biol Chem. 1995;270(3):1161–1169. doi:10.1074/jbc.270.3.1161
103. Ochiai A, Miyata S, Iwase M, Shimizu M, Inoue J, Sato R. Kaempferol stimulates gene expression of low-density lipoprotein receptor through activation of Sp1 in cultured hepatocytes. Sci Rep. 2016;6:24940. doi:10.1038/srep24940
104. Li C, Briggs MR, Ahlborn TE, Kraemer FB, Liu J. Requirement of Sp1 and estrogen receptor alpha interaction in 17beta-estradiol-mediated transcriptional activation of the low density lipoprotein receptor gene expression. Endocrinology. 2001;142(4):1546–1553. doi:10.1210/endo.142.4.8096
105. Bruning JC, Lingohr P, Gillette J, et al. Estrogen receptor-alpha and Sp1 interact in the induction of the low density lipoprotein-receptor. J Steroid Biochem Mol Biol. 2003;86(2):113–121.
106. Huang W, Mishra V, Batra S, Dillon I, Mehta KD. Phorbol ester promotes histone H3-Ser10 phosphorylation at the LDL receptor promoter in a protein kinase C-dependent manner. J Lipid Res. 2004;45(8):1519–1527. doi:10.1194/jlr.M400088-JLR200
107. Hassinen A, Pujol FM, Kokkonen N, et al. Functional organization of Golgi N- and O-glycosylation pathways involves pH-dependent complex formation that is impaired in cancer cells. J Biol Chem. 2011;286(44):38329–38340. doi:10.1074/jbc.M111.277681
108. Wang S, Mao Y, Narimatsu Y, et al. Site-specific O-glycosylation of members of the low-density lipoprotein receptor superfamily enhances ligand interactions. J Biol Chem. 2018;293(19):7408–7422. doi:10.1074/jbc.M117.817981
109. Pedersen NB, Wang S, Narimatsu Y, et al. Low density lipoprotein receptor class A repeats are O-glycosylated in linker regions. J Biol Chem. 2014;289(25):17312–17324. doi:10.1074/jbc.M113.545053
110. Zelcer N, Hong C, Boyadjian R, Tontonoz P. LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science. 2009;325(5936):100–104. doi:10.1126/science.1168974
111. Sorrentino V, Zelcer N. Post-transcriptional regulation of lipoprotein receptors by the E3-ubiquitin ligase inducible degrader of the low-density lipoprotein receptor. Curr Opin Lipidol. 2012;23(3):213–219. doi:10.1097/MOL.0b013e3283532947
112. Scotti E, Hong C, Yoshinaga Y, et al. Targeted disruption of the idol gene alters cellular regulation of the low-density lipoprotein receptor by sterols and liver x receptor agonists. Mol Cell Biol. 2011;31(9):1885–1893. doi:10.1128/MCB.01469-10
113. Sorrentino V, Scheer L, Santos A, Reits E, Bleijlevens B, Zelcer N. Distinct functional domains contribute to degradation of the low density lipoprotein receptor (LDLR) by the E3 ubiquitin ligase inducible Degrader of the LDLR (IDOL). J Biol Chem. 2011;286(34):30190–30199. doi:10.1074/jbc.M111.249557
114. Sorrentino V, Nelson JK, Maspero E, et al. The LXR-IDOL axis defines a clathrin-, caveolae-, and dynamin-independent endocytic route for LDLR internalization and lysosomal degradation. J Lipid Res. 2013;54(8):2174–2184. doi:10.1194/jlr.M037713
115. van Loon NM, Ottenhoff R, Kooijman S, et al. Inactivation of the E3 ubiquitin ligase IDOL attenuates diet-induced obesity and metabolic dysfunction in mice. Arterioscler Thromb Vasc Biol. 2018;38(8):1785–1795. doi:10.1161/ATVBAHA.118.311168
116. Sorrentino V, Fouchier SW, Motazacker MM, et al. Identification of a loss-of-function inducible degrader of the low-density lipoprotein receptor variant in individuals with low circulating low-density lipoprotein. Eur Heart J. 2013;34(17):1292–1297. doi:10.1093/eurheartj/ehs472
117. Steinberg D, Witztum JL. Inhibition of PCSK9: a powerful weapon for achieving ideal LDL cholesterol levels. Proc Natl Acad Sci U S A. 2009;106(24):9546–9547. doi:10.1073/pnas.0904560106
118. Della Badia LA, Elshourbagy NA, Mousa SA. Targeting PCSK9 as a promising new mechanism for lowering low-density lipoprotein cholesterol. Pharmacol Ther. 2016;164:183–194. doi:10.1016/j.pharmthera.2016.04.011
119. Deng SJ, Alabi A, Gu HM, Adijiang A, Qin S, Zhang DW. Identification of amino acid residues in the ligand binding repeats of LDL receptor important for PCSK9 binding. J Lipid Res. 2019;60(3):516–527. doi:10.1194/jlr.M089193
120. Sharma K, D’Souza RC, Tyanova S, et al. Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep. 2014;8(5):1583–1594. doi:10.1016/j.celrep.2014.07.036
121. Palacios-Moreno J, Foltz L, Guo A, et al. Neuroblastoma tyrosine kinase signaling networks involve FYN and LYN in endosomes and lipid rafts. PLoS Comput Biol. 2015;11(4):e1004130. doi:10.1371/journal.pcbi.1004130
122. Sattar N, Preiss D, Robinson JG, et al. Lipid-lowering efficacy of the PCSK9 inhibitor evolocumab (AMG 145) in patients with type 2 diabetes: a meta-analysis of individual patient data. Lancet Diabetes Endocrinol. 2016;4(5):403–410. doi:10.1016/S2213-8587(16)00003-6
123. Reyes-Soffer G, Pavlyha M, Ngai C, et al. Effects of PCSK9 inhibition with alirocumab on lipoprotein metabolism in healthy humans. Circulation. 2017;135(4):352–362. doi:10.1161/CIRCULATIONAHA.116.025253
124. Farnier M, Jones P, Severance R, et al. Efficacy and safety of adding alirocumab to rosuvastatin versus adding ezetimibe or doubling the rosuvastatin dose in high cardiovascular-risk patients: the ODYSSEY OPTIONS II randomized trial. Atherosclerosis. 2016;244:138–146. doi:10.1016/j.atherosclerosis.2015.11.010
125. Colhoun HM, Ginsberg HN, Robinson JG, et al. No effect of PCSK9 inhibitor alirocumab on the incidence of diabetes in a pooled analysis from 10 ODYSSEY Phase 3 studies. Eur Heart J. 2016;37(39):2981–2989. doi:10.1093/eurheartj/ehw292
126. Tavori H, Giunzioni I, Fazio S. PCSK9 inhibition to reduce cardiovascular disease risk: recent findings from the biology of PCSK9. Curr Opin Endocrinol Diabetes Obes. 2015;22(2):126–132. doi:10.1097/MED.0000000000000137
127. Fitzgerald G, Kiernan T. PCSK9 inhibitors and LDL reduction: pharmacology, clinical implications, and future perspectives. Expert Rev Cardiovasc Ther. 2018;16(8):567–578. doi:10.1080/14779072.2018.1497975
128. Jeong HJ, Lee HS, Kim KS, Kim YK, Yoon D, Park SW. Sterol-dependent regulation of proprotein convertase subtilisin/kexin type 9 expression by sterol-regulatory element binding protein-2. J Lipid Res. 2008;49(2):399–409. doi:10.1194/jlr.M700443-JLR200
129. Bjune K, Wierod L, Naderi S. Triciribine increases LDLR expression and LDL uptake through stabilization of LDLR mRNA. Sci Rep. 2018;8(1):16174. doi:10.1038/s41598-018-34237-6
130. Cao A, Wu M, Li H, Liu J. Janus kinase activation by cytokine oncostatin M decreases PCSK9 expression in liver cells. J Lipid Res. 2011;52(3):518–530. doi:10.1194/jlr.M010603
131. Li C, Kraemer FB, Ahlborn TE, Liu J. Induction of low density lipoprotein receptor (LDLR) transcription by oncostatin M is mediated by the extracellular signal-regulated kinase signaling pathway and the repeat 3 element of the LDLR promoter. J Biol Chem. 1999;274(10):6747–6753. doi:10.1074/jbc.274.10.6747
132. Liu J, Streiff R, Zhang YL, Vestal RE, Spence MJ, Briggs MR. Novel mechanism of transcriptional activation of hepatic LDL receptor by oncostatin M. J Lipid Res. 1997;38(10):2035–2048.
133. Zhang F, Ahlborn TE, Li C, Kraemer FB, Liu J. Identification of Egr1 as the oncostatin M-induced transcription activator that binds to sterol-independent regulatory element of human LDL receptor promoter. J Lipid Res. 2002;43(9):1477–1485. doi:10.1194/jlr.m200126-jlr200
134. Heinrich PC, Behrmann I, Muller-Newen G, Schaper F, Graeve L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J. 1998;334(Pt 2):297–314. doi:10.1042/bj3340297
135. Zhang F, Lin M, Abidi P, Thiel G, Liu J. Specific interaction of Egr1 and c/EBPbeta leads to the transcriptional activation of the human low density lipoprotein receptor gene. J Biol Chem. 2003;278(45):44246–44254. doi:10.1074/jbc.M305564200
136. Kapoor GS, Golden C, Atkins B, Mehta KD. pp90RSK- and protein kinase C-dependent pathway regulates p42/44MAPK-induced LDL receptor transcription in HepG2 cells. J Lipid Res. 2003;44(3):584–593. doi:10.1194/jlr.M200302-JLR200
137. Expert Panel on Integrated Guidelines for Cardiovascular H, Risk Reduction in C, Adolescents, National Heart L, Blood I. Expert panel on integrated guidelines for cardiovascular health and risk reduction in children and adolescents: summary report. Pediatrics. 2011;128 Suppl 5:S213–S256. doi:10.1542/peds.2009-2107C
138. Cannon CP, Blazing MA, Giugliano RP, et al; Investigators I-I. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med. 2015;372(25):2387–2397. doi:10.1056/NEJMoa1410489
139. Insull W
140. Gu HM, Zhang DW. Hypercholesterolemia, low density lipoprotein receptor and proprotein convertase subtilisin/kexin-type 9. J Biomed Res. 2015;29(5):356–361. doi:10.7555/JBR.29.20150067
141. Bellosta S, Paoletti R, Corsini A. Safety of statins: focus on clinical pharmacokinetics and drug interactions. Circulation. 2004;109(23 Suppl 1):III50–III57. doi:10.1161/01.CIR.0000131519.15067.1f
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.