")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 18
Therapeutic Options for the Management of Pompe Disease: Current Challenges and Clinical Evidence in Therapeutics and Clinical Risk Management
Authors Bolano-Diaz C, Diaz-Manera J
Received 28 June 2022
Accepted for publication 21 November 2022
Published 13 December 2022 Volume 2022:18 Pages 1099—1115
DOI https://doi.org/10.2147/TCRM.S334232
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Garry Walsh
Carla Bolano-Diaz,1 Jordi Diaz-Manera1– 3
1The John Walton Muscular Dystrophy Research Center, Newcastle University Translational and Clinical Research Institute, Newcastle Upon Tyne, UK; 2Laboratori de Malalties Neuromusculars, Insitut de Recerca de l’Hospital de la Santa Creu i Sant Pau de Barcelona, Barcelona, Spain; 3Centro de Investigación Biomédica en Red en Enfermedades Raras (CIBERER), Barcelona, Spain
Correspondence: Jordi Diaz-Manera, The John Walton Muscular Dystrophy Research Center, Center for Life, Central Parkway, Newcastle Upon Tyne, NE13BZ, UK, Email [email protected]
Abstract: Pompe disease is a genetic disorder produced by mutations in the GAA gene leading to absence or reduced expression of acid alpha-glucosidase, an enzyme that metabolizes the breakdown of glycogen into glucose. There are two main phenotypes, the infantile consisting of early onset severe weakness and cardiomyopathy, and the adult which is characterized by slowly progressive skeletal and respiratory muscle weakness. Enzymatic replacement therapy (ERT) has been available for Pompe disease for more than 15 years. Although the treatment has improved many aspects of the disease, such as prolonged survival through improved cardiomyopathy and acquisition of motor milestones in infants and slower progression rate in adults, ERT is far from being a cure as both infantile and adult patients continue to progress. This fact has prompted the development of improved or new enzymes and other treatments such as gene therapy or substrate reduction strategies. Here, we review the data obtained from randomized clinical trials but also from open-label studies published so far that have assessed the advantages and limitations of this therapy. Moreover, we also review the new therapeutic strategies that are under development and provide our opinion on which are the unmet needs for patients with this disease.
Keywords: glycogen storage disease type II, Pompe disease, enzyme replacement therapy, recombinant human acid alpha-glucosidase, genetic therapy, clinical trials
Introduction to Pompe Disease
Definition and Clinical Phenotypes
Pompe disease is a genetic glycogen storage disorder with an autosomal recessive pattern of inheritance.1,2 The epidemiology of the disease is not clearly established although is suggested to be of almost 1:40,000 individuals.3 There is however demographic variability, from countries with just a few cases described to others with a higher prevalence.4–6 It is produced by mutations in the GAA gene localized to chromosome 17 which codifies for the enzyme acid alpha-glucosidase (AAG).7 AAG is responsible for the breakdown of glycogen into glucose inside the lysosomes of the cells. A deficient enzyme activity leads to the accumulation of glycogen within cells of many tissues. However, patients’ symptoms are mainly related to the dysfunction of skeletal, cardiac and smooth muscle and neurons, which are tissues with a high glucose demand.2
Two main clinical disease phenotypes have traditionally been described: the infantile (classic and non-classic) and the late onset Pompe phenotypes.2 Classic infantile onset Pompe disease (IOPD) typically presents early after birth with severe generalized muscle weakness, hypotonia, respiratory insufficiency and hypertrophic cardiomyopathy that, if left untreated, leads to death within the first year of life. IOPD patients can also show other associated symptoms such as hepatomegaly, feeding difficulties and macroglossia.2,8 These patients have zero or very reduced AAG protein expression, which can be detected using Western blot (WB) analysis, a technique commonly described as cross-reactive immunologic material (CRIM) in the scientific papers. Those patients who have some residual enzyme activity are labeled as CRIM positive while those with no residual activity are labeled CRIM negative.9 The term non-classic infantile onset Pompe disease is generally used to describe patients who have early onset of muscle weakness during the first two years of life with or without respiratory muscle dysfunction, but who do not develop cardiac involvement.2 These patients can present very early in life with severe muscle weakness, or a bit later with a delay in the acquisition of motor milestones. As these patients grow, they develop other complications such as scoliosis, dysphagia, hepatomegaly, irritable bowel disease and joint contractures.10
The term juvenile onset Pompe disease is used to describe patients who start presenting symptoms after 2 years of age, during childhood or adolescence.10 Nowadays, this group of patients is usually included within the most severe end of patients with late onset Pompe disease (LOPD) and the term juvenile onset is being progressively abandoned. The clinical presentation of LOPD is very heterogeneous as patients can present with isolated hyperCKemia, muscle weakness with a limb girdle pattern with or without respiratory involvement, predominant respiratory muscle weakness with no or minimal skeletal muscle weakness or predominant axial weakness.2 The heterogeneous phenotypes can be a challenge towards the diagnosis, especially in the first stages of the disease, but sooner or later most patients develop muscle weakness affecting the pelvic girdle, the axial and the respiratory muscles leading to a variable degree of disability ranging from patients requiring aids for walking to patients that are full-time wheelchair users. A large number of patients will need ventilatory support due to severe diaphragmatic weakness, this being the primary cause of death in this group. LOPD patients have a residual enzymatic activity lower than 30% but higher than 1%.2 There is not, however, a correlation between enzymatic activity and the age of onset or the severity of the disease in LOPD patients.
The Diagnostic Journey
The diagnostic process of Pompe disease has clearly improved in recent years mainly due to the growing use of the dried blood spot (DBS) test to measure AAG enzymatic activity and the access to next generation sequencing (NGS) technology in many hospitals, especially those within developed countries. Nowadays when facing a patient with a clinical suspicion of Pompe disease, enzymatic activity can be rapidly tested with DBS, and if low, later confirmed through genetic testing.11 Moreover, patients presenting with muscle weakness are more frequently being studied with gene panels that include the GAA gene, being diagnosed through this process without even requiring any enzymatic studies. Newborn screening, which is being implemented in many countries, is contributing to the identification of new cases, even in patients without any clinical symptoms.8 Other diagnostic tests can provide valuable information for the diagnosis of Pompe. Electromyography shows a myopathic pattern associated with myotonic discharges especially in the paraspinal muscles.11 Muscle MRI can identify fat replacement in many muscles with the tongue, the subscapularis, the axial, glutei and posterior thigh muscles being the more frequently affected.8 Muscle biopsy shows a variety of signs. In IOPD cases the common finding is massive accumulation of glycogen in the muscle fibers of patients which can be easily detected using Periodic Acid Schiff (PAS) staining, with or without autophagic vacuoles. In the case of LOPD patients, muscle biopsies can be normal or showing just mild myopathic but unspecific changes in many cases, especially if the muscle biopsied is not weak or affected on the MRI. In those biopsies showing changes the more frequent findings are muscle fibers with enlarged lysosomes containing glycogen, presence of glycogen free on the cytoplasm and autophagic vacuoles.12,13 The number of fibers showing these changes is low, ranging from 1 to 30% in most of the cases making the diagnosis difficult.14 Staining with acid phosphatase can reveal increased lysosomal activity in muscle fibers, which is useful for reaching the diagnosis. The definitive diagnosis is achieved when two pathogenic variants are found in the GAA gene. To date, more than 500 pathogenic variants have been reported that are collected and curated in the Pompe database.7 In some patients conventional DNA sequencing may only find one pathogenic variant or even none. These cases may be especially complex and require confirmation of low enzymatic activity in blood, skin fibroblasts or skeletal muscles and/or more sophisticated genetic studies sequencing RNA or assessing RNA copies in muscle samples. Moreover, some genetic variants lead to pseudodeficiency, which is characterized by patients having low enzymatic activity in blood but not producing any muscle damage.15
Therapeutic Options
The year 2006 was game changing for the Pompe community as enzyme replacement therapy (ERT) with alglucosidase alfa (Myozyme/Lumizyme®, Sanofi-Genzyme) was approved for marketing by the Food and Drug Administration (FDA) and the European Medical Agency (EMA).16 The first experiments with the aim to develop ERT for Pompe disease date as far back as 1963.17 In the 1990s, large-scale production of recombinant human alpha-glucosidase (rhGAA) was achieved. After this, in 2001, the first successful clinical trial on four IOPD patients with rhGAA from rabbit milk was published.18 This trial was followed by two trials performed in IOPD and LOPD published in 200719 and 2010,20 respectively. In the 16 years since the first approval of ERT, the field has evolved notably. A large number of open-label studies providing data on the progression of both IOPD and LOPD treated patients have been published, suggesting that although effective in improving some of the patients’ symptoms, a considerable number of patients still progress over time. These data have led to more research being done in the development of new therapeutic approaches, ranging from improved or new enzymatic formulations to gene therapy, drugs which are currently being tested in clinical trials.
In this paper, we review the evidence published on both IOPD (Table 1) and LOPD (Table 2) patients treated with ERT including all clinical trials published and observational studies providing new and relevant information (Figure 1). We report the main findings obtained and discuss the advantages and limitations of ERT. Finally, we provide an overview of the new therapies under development for Pompe disease.
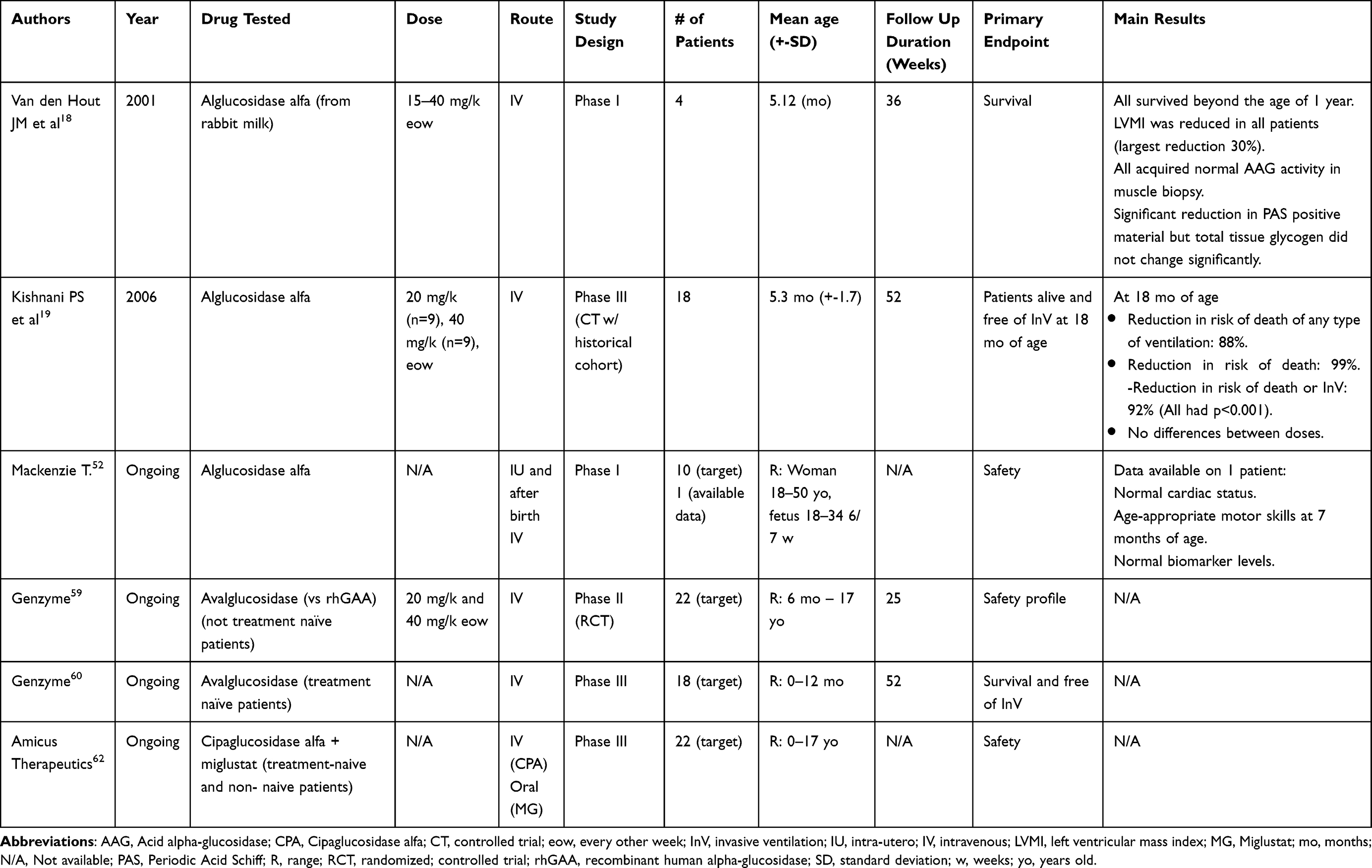 |
Table 1 Summary of Clinical Trials for Different Treatments for Infantile Onset Pompe Disease |
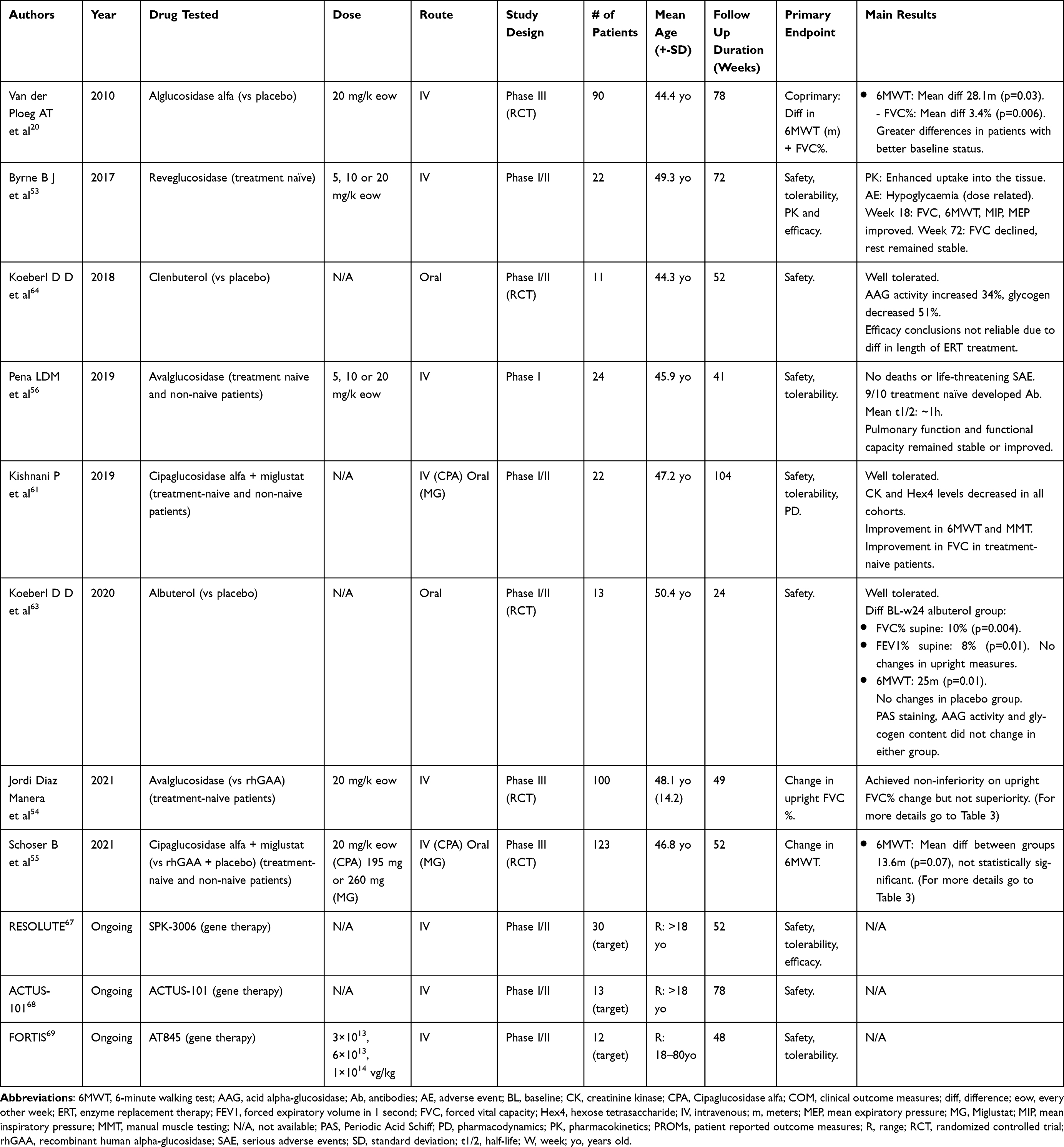 |
Table 2 Summary of Clinical Trials for Different Treatments for Late Onset Pompe Disease |
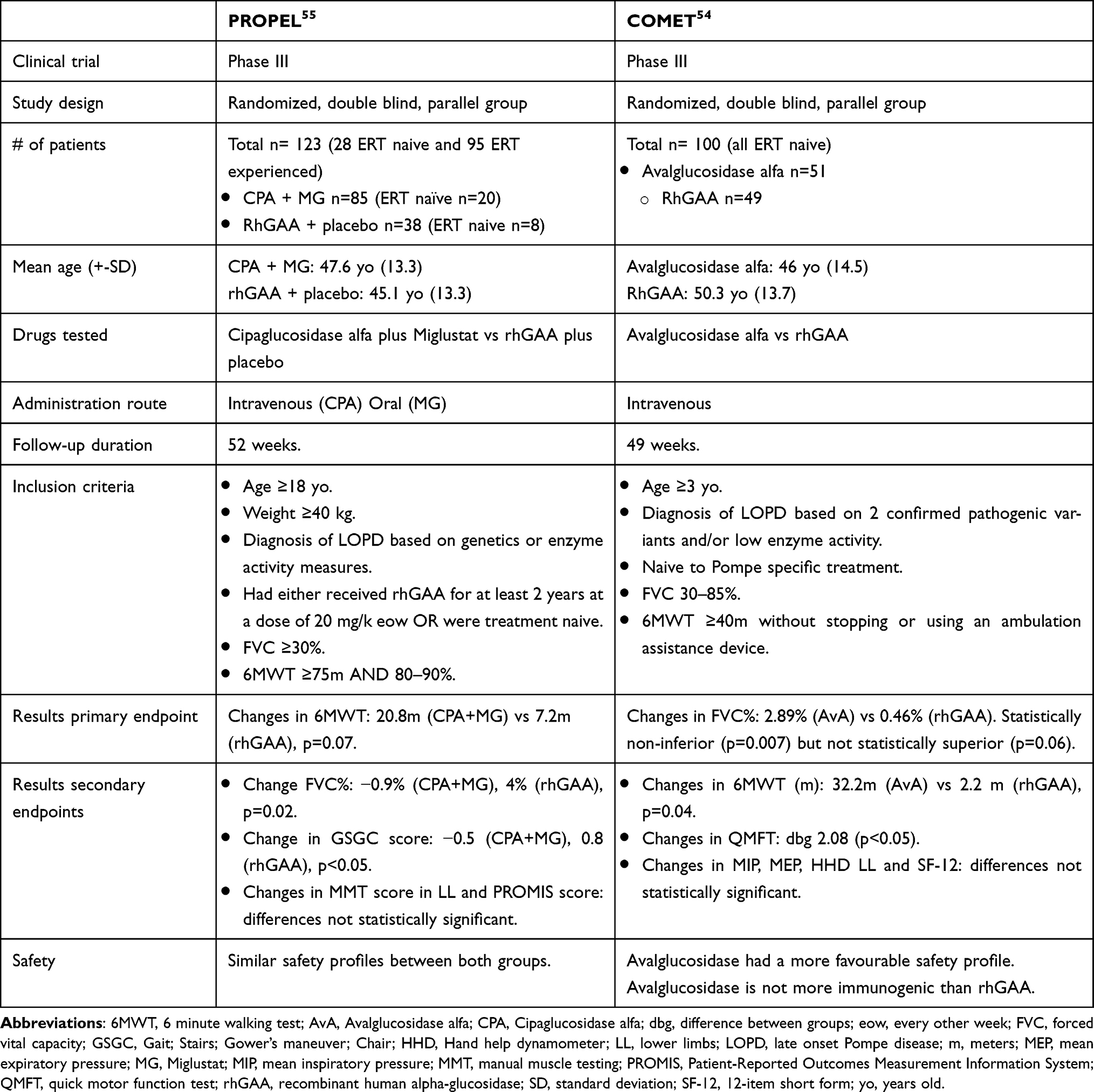 |
Table 3 PROPEL and COMET Clinical Trials |
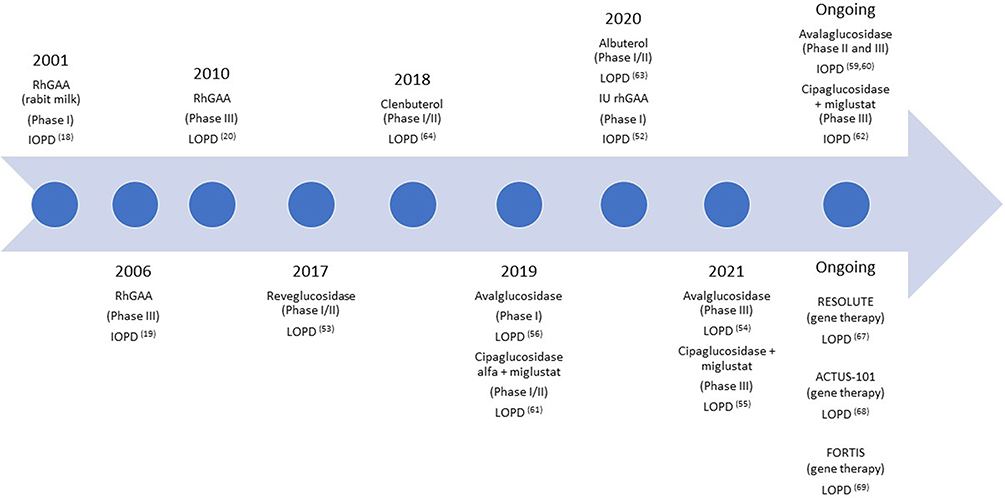 |
Figure 1 Timeline showing the different clinical trials published since 2001. Abbreviations: rhGAA, Recombinant human glucosidase alpha; IOPD, Infantile onset Pompe disease; LOPD, Late onset Pompe disease; IU, Intrauterine. |
Enzyme Replacement Therapy: Effects and Challenges
Infantile Onset Pompe Disease
Results of Clinical Trials in IOPD Patients
The first evidence on the safety and efficacy of rhGAA from rabbit milk in IOPD patients was published in 2001 when four classical IOPD patients were included in an open-label pilot study and received different doses of ERT ranging from 15–40 mg/kg every other week (eow).18 Authors reported an increase in AAG activity and a decrease in PAS positive material in muscle biopsies associated with significant changes in muscle strength, cardiac function and life expectancy. After this first trial, a new open-label study included 18 IOPD patients that were randomized 1:1 to receive either 20 or 40 mg/kg eow of rhGAA and their functional results were compared with an historical control group of 62 IOPD patients.19 At 18 months, none of the patients under ERT died compared with 61 out of 62 in the historical cohort, although eight patients required ventilatory support. A significant reduction in left ventricular mass (LVM) index was observed in all treated patients. Muscle function results were more heterogeneous: 13 patients obtained consistent motor and functional gains on the Alberta Infant Motor Scale (AIMS), seven patients being able to walk. AAG activity markedly increased in muscle biopsies and glycogen muscle content was stable or decreased compared with baseline in 15 out of 18 patients. Interestingly, none of the three patients whose glycogen levels increased from baseline showed any meaningful motor improvement. There were no differences in clinical responses when comparing the two doses tested. This cohort of 18 patients was followed-up for three years.21 In that period, five patients died and four became ventilator dependent. Seven patients were still able to walk, four were sitting independently and seven made only minimal motor gains at the time of their final AIMS assessment. Based on these results, ERT with Myozyme was approved in 2006. These first studies already showed that ERT has a positive impact on IOPD by prolonging life expectancy through improving cardiac hypertrophy. However, the effects on skeletal and respiratory muscle function were very variable, ranging from patients with a clear improvement to others with just a limited benefit19,21,22 and raising questions about the reasons for these different responses.
Reasons for the Heterogenic Response to the Treatment
The heterogenic response to ERT is still not completely understood and different factors are thought to contribute to it. Several open-label studies have assessed the long-term outcome of IOPD in the last 10 years confirming that patients do have a variable response to ERT, but also providing clues for some basic conclusions.
Age at start of treatment seems to influence the treatment response considerably. In a retrospective study assessing response to treatment of IOPD patients seen in different countries, a group of Taiwanese CRIM-positive patients diagnosed through newborn screening and treated early after birth achieved the best functional results consisting of an increased probability to remain ventilator-free during early childhood and achieve independent walking at a near normal age.23 Based on these results, treatments are usually started as soon as possible in IODP patients with the aim to obtain a better and more sustained response over time.
Immune reaction to treatment is another determining factor influencing response to ERT.9 A retrospective study comparing the response to ERT between 21 CRIM positive and 11 CRIM negative IOPD patients9 showed after 27 months of treatment that all CRIM negative patients had either died (n=5) or were invasively ventilated (n=6), while in the CRIM positive cohort only one had died and three required invasive ventilation. Although LVM decreased considerably in both groups, the mass of the surviving CRIM negative patients was higher than the CRIM positive group. Gross motor development followed the same tendency favoring CRIM positive patients. All CRIM negative patients and 18 out of 20 CRIM positive patients developed IgG rhGAA antibodies, however antibody titers in CRIM negative patients were sustained at higher levels for a longer period than in CRIM positive patients. This first observation has been confirmed by several groups and this finding has led to the premise that CRIM status should be obtained as soon as possible after diagnosis as CRIM negative patients invariably develop a strong immunologic response, characterized by high antibody titers against ERT resulting in a less effective and sustained response to the treatment, while CRIM positive patients develop a milder immune reaction with a considerably better clinical response.9 In order to control this immune response, immunosuppression regimens have been successfully tested improving the outcome of CRIM negative patients.24 Different regimens have been reported, including a prophylactic tolerance induction using rituximab (RTX) and methotrexate (MTX) naïve to ERT and a therapeutic tolerance induction for patients with established antibodies using RTX, MTX and intravenous immunoglobulin (IVIG).25 This treatment can ameliorate the response of CRIM negative patients to ERT who may show improvement in the motor and respiratory function, ranging from being ventilator free and able to walk independently to still requiring ventilation and not being able to walk. Unfortunately, this immunosuppression regimen is not always able to induce an improvement, and some patients still response very poorly and die despite treatment. Moreover, little is known about the long-term prognosis of CRIM negative patients treated with these immunosuppression regimens. Another important point is that these regimens might not induce immune tolerance once immunosuppression is stopped.26 Antibody titers can increase coinciding with the recovery of B cell count, suggesting that immunosuppression therapy can induce temporary suppression of antibody formation but fails to induce immune tolerance. Moreover, CRIM status as a predictor of worse outcome might be more complex than we think, as even though CRIM negative patients have higher antibody titers and worse clinical outcome, there is a subgroup of CRIM positive patients who are at risk of developing high antibody titers as well.26 In recent years a group of investigators created a personalized immune tolerance-adjusted immunogenicity prediction tool for CRIM positive patients.27 Ultimately this algorithm would allow clinicians to calculate the patient’s relative risk of developing anti-drug antibodies through analysis of individual HLA-DR haplotype and GAA pathogenic variants, enhancing clinical decision-making prior to initiating treatment with ERT.
Another important factor that could influence the response to ERT is pretreatment muscle pathology, including the presence of extra lysosomal glycogen, altered autophagy and distribution of type I and II fibers. As previously mentioned, the most prominent features of IOPD muscle biopsies are massive glycogen accumulation and autophagic vacuoles.12,13 Incremental enlargement of glycogen-filled lysosomes results in lysosomal rupture and release of glycogen and toxic substances into the sarcoplasm. This, added to glycogen accumulation in the sarcoplasm, leads to the destruction of the contractile elements and muscle fiber death. ERT has shown, in some patients, to reduce glycogen content in repeated muscle biopsies, although progressive autophagic buildup has been observed even in those patients with good response to ERT.12,13 Neither extra lysosomal glycogen accumulation nor the autophagic vacuoles are targeted by ERT, and therefore the existence of these two pathologic features at baseline biopsy may hamper the response to treatment.28,29 Autophagic vacuoles are mainly seen in the glycolytic type II muscle fibers, while they are rarely seen in the oxidative type I fibers which could potentially respond better to treatment.30 A high proportion of type I fibers in the muscle biopsy has been proposed as a good prognostic factor to treatment response.12
Even though the pivotal clinical trials which led to the approval of rhGAA19,20 were not able to find any significant difference in treatment response when analyzing different doses, with the widespread use of ERT more evidence arose in favor that a higher dose would be more effective than the standard dose stated in the guidelines (20 mg/kg eow). Nevertheless, it was not until 2021 when an observational multicenter cohort study assessing the effect of different ERT regimens in IOPD patients was published that this difference in efficacy between different dosing regimens was shown.31 The analysis of survival rate and motor milestones of 116 IOPD patients treated with ERT under 39 different regimens led authors to conclude that patients under continuous treatment with high dosing (40 mg/kg per week) had a better survival rate at 5 years than patients on regular dosing (20 mg/kg eow) which was not influenced by CRIM status. However, no statistically significant differences were found when analyzing walking ability, even though 53% of patients treated with a standard dose learned to walk compared with 93% of patients on a high dose regimen. In our opinion, although there are some data supporting a weekly treatment at higher doses, this question remains open and should be clarified in a controlled randomized clinical trial.
Infantile Onset Pompe Disease: A New Reality with a Distinct Phenotype
New standards of care for IOPD patients including early treatment with rhGAA, immunosuppression and improved respiratory and nutritional support are changing the natural history of IOPD mainly through prolonging life expectancy. However, this is leading to a new phenotype characterized by multisystemic involvement that is focusing the interest of researchers working in this field. Central nervous system (CNS) involvement is a rising concern within the scientific community, as ERT is unable to cross the blood–brain barrier.32 In 2018 a cohort of 11 IOPD patients aged between 4 and 17 years who had started ERT treatment between 1999 and 2009 was analyzed using brain MRIs and intelligence tests.32 Progressive white matter abnormalities affecting first the centrum semiovale, then the subcortical areas and finally the infratentorial white matter were associated to stabilization or decline in neuropsychological functions over time. In these patients, brain involvement seemed to be independent of motor functioning. Another follow-up study done in IOPD patients treated with ERT33 showed that cognitive function improved only between baseline and 12 weeks of treatment, whereas motor function improved throughout the whole study compared with baseline. Sensorineural hearing loss, which can be found in a considerable number of patients, could also increase the complexity of CNS involvement, as glycogen has been shown to accumulate in the cochlea of knock-out Pompe mice models.34,35
Additionally, patients initially responding to ERT can develop new weakness during follow up, affecting not only the limb girdle but also distal muscles in hands and feet. The severe muscle involvement can lead to scoliosis, bilateral ptosis, severe facial weakness and dysphagia and dysarthria produced by tongue weakness, velopharyngeal incompetence and dysfunction of the esophageal sphincter.36–39
In summary, although there is enough evidence supporting a positive effect of ERT on patients with IOPD including prolonged survival, improved cardiac structure, improvement of muscle weakness leading to the achievement of motor milestones and reduced need of ventilatory support, the truth is that the response is very heterogeneous among patients and influenced by several factors, such as age when treatment was started, CRIM status and clinical and pathologic features. Long-term response to the drug seems to be very variable as well and patients may progress, developing further muscular and extramuscular complications. There is a huge need for either improved or new medications for more effective long-term symptom control and to reduce the burden produced by CNS involvement.
Late Onset Pompe Disease
Results of Clinical Trials and Open-Label Studies in LOPD Patients
ERT was first tested in LOPD patients through an international multicenter clinical trial that aimed at evaluating the safety and efficacy of alglucosidase alfa compared with placebo.20 This study included 90 patients who were assigned on a 2:1 ratio to 20 mg/kg of ERT eow or placebo for 78 weeks. The study showed an increase of 25 meters in the distance covered in the six minutes walking test (6MWT) that was statistically higher than the three-meter increase observed in the placebo group. Patients receiving alfa-glucosidase increased by 1.2% their forced vital capacity (FVC) while patients in the placebo group decreased FVC by 2.2%, this difference being statistically significant. For both outcome measures, there was an initial improvement during the first weeks of treatment that reached a plateau around week 26 and was maintained throughout the study. Other outcome measures, such as muscle strength measured using handheld dynamometry (HHD), maximum inspiratory pressure (MIP) and maximum expiratory pressure (MEP) showed a similar pattern of response, but in those cases the differences between both groups did not reach statistical significance. These patients were followed in an open-label study until week 104 showing stabilization of both the motor and respiratory functions.40 However, patients who previously were in the placebo group and started receiving rhGAA did not show any improvement or decline during this extended follow-up.40
Alphaglucosidase did not induce any safety issues and produced a modest improvement in motor and respiratory function, that is why it was approved. Since LOPD is a disorder characterized by slow but invariably progressive weakness, a modest improvement or even a stabilization in muscle function should be interpreted as a positive outcome, especially if maintained over time. Several open-label studies have tried to assess the effects of alpha-glucosidase on LOPD in real world settings. These studies differ in their methodology, most of them being retrospective and not controlled. A detailed analysis of all these studies is out of the scope of this paper. A meta-analysis reviewed 19 of these studies, including a total of 438 patients followed on average for 45.7 months.16 The authors reported a mean improvement of 43 meters on the 6MWT after 12 months of treatment compared with untreated patients. Moreover, treated patients rapidly improved FVC during the first 2 months of treatment by 1.4% and gradually returned to baseline by year 3, followed by a slight decline. These results altogether impacted the mortality rate, that was 5-fold lower than in untreated patients. This meta-analysis already showed that even though during the first months of treatment there is an improvement in clinical outcome measures, that improvement eventually turns into a plateau and can be followed by a progressive decline in the long term.
Long-Term Response of LOPD Patients
Now that several LOPD patients have been treated for longer, open-label studies collecting data of patients treated for 5 or even 10 years have been published.7,41–43 These studies confirm the first observation that the initial improvement in the 6MWT and FVC observed during the first months of treatment is followed by a plateau that can last for some years before patients start declining. A recent study analyzing disease progression for 10 years41 revealed that although 93% of patients had at least a positive effect on the 6MWT and FVC after starting ERT, 76% showed a secondary decline. However, at their last measurement, 52% of patients had the same or better scores in 6MWT and/or FVC compared with baseline while in 48% of the cases these outcome measures showed worse results than at baseline. The question about what could have been the progression of these patients if untreated remains open. This study tried to answer this question by predicting what disease progression could have been, as some of the patients were already followed up longitudinally in their clinics before ERT was started. The authors observed that even if muscle function worsened, most of the patients were still better than expected according to Pompe’s natural disease progression. Unfortunately, the decline in muscle function that patients suffer has a negative impact on their quality of life and the number of patients being completely or partially wheelchair dependent or needing non-invasive ventilation (NIV) increases with time. However, the data published so far show that there is a huge variability in how the disease progresses from one patient to the other, ranging from patients who start declining very soon after starting ERT to others who remain stable for many years, and therefore long-term trajectories can be different between these cases.
Factors Influencing Long-Term Response of LOPD Patients
It is not yet fully understood why the response to ERT in LOPD patients worsens through time and why this response is so heterogeneous. Even though immune response and CRIM status play a key role in IOPD patients, there is no evidence suggesting that it has any influence on LOPD patients.20,44,45 The more affected fibers, characterized by accumulation of glycogen free in the sarcoplasm and autophagic vacuoles, seem to have a reduced response to ERT, as suggested by a study which analyzed 18 patients’ muscle biopsies before and after starting treatment.46 This study showed that ERT had an overall positive effect on skeletal muscle pathology, reducing the accumulation of glycogen in muscle. However, fibers accumulating autophagosomes did not respond to treatment as well as the rest, as autophagosomes were still present in the post-treatment biopsy. Interestingly, some of the patients who had increased autophagy process were the ones that also showed the highest levels of enzymatic activity, suggesting that presence of autophagic vacuoles in the sarcoplasm is associated with impaired endocytic transport from muscle membrane to the lysosomes resulting in a lack of effect of the enzyme as it does not or only partially reaches its target organelle. RhGAA is introduced into the lysosomes via receptor-mediated uptake from the cell surface and acts more efficiently under the acidic pH in these structures, and therefore rhGAA is probably only able to clear the glycogen that is accumulated inside the lysosomes.12
Another factor influencing response to treatment seems to be baseline clinical status. Those who start treatment when they are less affected should expect better long-term outcomes related to respiratory function7,47 and to overall survival rate,43 probably because the primary cause of death of these patients is respiratory insufficiency. However, no clear evidence has been observed between baseline status and motor response to treatment, even though it has been suggested that male gender and having a 6MWT results higher than 50% of predicted by age are factors associated with better response to ERT.41 This correlation between baseline clinical status and disease progression was also found in MRI studies following patients longitudinally.48 It has been shown that an increase in fat fraction over a short period of time, an indirect measure of disease activity and loss of muscle tissue, is higher in patients with worse muscle function and higher fat fraction (FF) at baseline MRI. Indeed, this study also demonstrated that patients treated for longer periods with ERT were the ones who progressed the most, confirming a kind of loss of effect over time of the treatment as has been previously discussed.48,49 Age of patients at baseline and their capacity to regenerate muscle fibers once the treatment has been started could also influence disease progression and response to the drug, although this has never been demonstrated in any of the studies performed.
When to Start ERT in LOPD Patients?
Nowadays, LOPD patients are sometimes diagnosed before being symptomatic because of family history, laboratory findings such as hyperCKemia and elevated liver enzymes or non-specific symptoms such as pain and cramps. Moreover, newborn screening is identifying many babies with LOPD who are asymptomatic but could develop symptoms later in life and will need to be followed up in clinics. This brings about another controversial aspect of ERT treatment which is when it should be started. According to the European consensus for starting and stopping enzyme replacement therapy in adult patients with Pompe disease published in 201750 treatment should be started in all symptomatic LOPD patients with a confirmed diagnosis who have residual skeletal and respiratory muscle function. In these guidelines, being symptomatic was defined as presence of skeletal or respiratory muscle weakness identified in the clinical examination and/or spirometry. Unfortunately, many patients are not aware of any clinical symptoms until muscle pathology is already diffuse and they have a large amount of their muscles replaced by fat,49 which has been associated with worse treatment response and disease progression as previously discussed. Previous studies have shown that fat replacement can be identified in asymptomatic LOPD patients before they notice any muscle weakness using MRI,49 especially in the paraspinal and adductor magnus muscles.48 Therefore, MRI could be useful to detect patients that have already started to lose muscle mass but remain asymptomatic in order to start treatment earlier.8 Unfortunately, there are no available data showing if early treatment with ERT in these asymptomatic LOPD patients could be associated with a delay in symptom onset or to a milder disease progression. Such a study, although relevant for the LOPD population, is difficult to plan because for asymptomatic patients many years might need to go by before muscle loss affects their activities of daily living.
New Therapies
Even though the approval of ERT for the treatment of Pompe disease has been a turning event in the development of an effective treatment against this disease, there are still many aspects to it that are far from perfect and need improvement, as has been discussed previously. Therefore, new therapies are continuously being investigated and developed, some following the same principles as ERT and others having a completely different approach (Figure 2). These new treatments should be able to tackle some of the challenges of the disease including an increase in the distribution of the drug associated with better uptake, reaching a larger number of fibers in a more consistent way and earlier access to treatment to prevent glycogen accumulation in lysosomes, which is responsible for the autophagic build-up and the presence of glycogen in the sarcoplasm. New drugs should also be able to cross the blood–brain barrier to reduce the neuronal dysfunction observed in IOPD surviving patients.
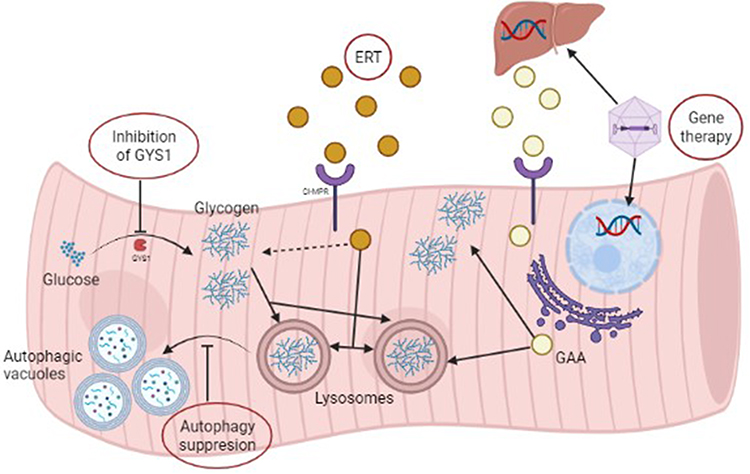 |
Figure 2 Treatment strategies for Pompe disease. This figure shows the different available/under research treatment strategies for Pompe disease. RhGAA’s uptake is mediated by CI-MPR and, once inside the muscle fiber, is delivered into the cytoplasm and lysosomes where it breaks down glycogen. There are two different gene therapy strategies: one directed towards the liver and one towards the muscle fiber. In the first one GAA is synthesized by the liver, released into the bloodstream and later delivered to the muscle fiber. In the second one GAA is synthesized by the muscle fibre and acts locally. Autophagy suppression would inhibit the formation of autophagic vacuoles and consequently cell death. Inhibition of GYS1 would be a substrate reduction strategy. Created with BioRender.com. Abbreviations: CI-MPR, cation-independent mannose-6-phosphate receptor; ERT, Enzyme replacement therapy; GAA, alglucosidase alfa; GYS1, glycogen synthase 1; rhGAA, Recombinant human alglucosidase alfa. |
Intra-utero ERT Treatment
As a general rule, the less symptomatic IOPD patients are at the beginning of ERT, the better the long-term response.19,21,22,51 Therefore, a phase I clinical trial is being carried out from April 2021 at Duke University aiming to determine the maternal and fetal safety and feasibility of in utero fetal enzyme replacement therapy in fetuses with Pompe disease.52 At the WORLD Symposium 2022 this research group shared the results of the first patient treated with in utero ERT in the fetal period. The patient was treated with intra-uterine injections starting at week 24 of gestation, receiving one infusion eow, until she was born at 37 weeks with no complications. Postnatally she continued to receive biweekly infusions and received immune tolerance induction (ITI) at birth. The patient treated intrauterine achieved a normal cardiac status (never showed any signs of cardiomyopathy before or after birth), age-appropriate motor skills and assessments, normal CK values and responded better to ITI treatment as antibody titers descended faster than in the historic control patients. Moreover, no adverse safety events were reported.
Improved or New Enzymatic Replacement Therapies
Another of the challenges ERT faces is that its delivery into the muscle fibers is low because rhGAA uptake is mediated by the cation-independent mannose-6-phosphate receptor (CI-MPR) which is not expressed in large amounts on the myofibers’ membrane. In order to overcome this problem new rhGAA molecules have been developed with a better intracellular delivery rate, such as reveglucosidase,53 avalglucosidase54 and cipaglucosidase.55 Reveglucosidase is an AAG analog tagged to a novel insulin-like growth factor 2 with theoretical improved lysosomal uptake, avalglucosidase is an improved version of the old alpha-glucosidase molecule that has more mannose-6-phosphate residues in the surface and therefore is theoretically able to enter in the myofiber with higher efficiency and cipaglucosidase is a novel rhGAA administered with a chaperone (miglustat) that extends its half-life in the serum, allowing myofibers to be exposed to the drug for longer before it is eliminated from the bloodstream.53–55 Reveglucosidase, which was only administered to ERT-naïve patients, showed an improvement in respiratory function with a limited effect on walking tests but associated with a high risk of hypoglycemia which led to a discontinuation of its clinical development.53 Avalglucosidase is being tested in clinical trials in LOPD and IOPD patients. Safety, pharmacokinetics and short- and long-term efficacy of avalglucosidase have been studied in LOPD in phase I/II (NEO and NEO-ext)56,57 and phase III (COMET)54 (Table 3) clinical trials. Overall, the data collected by these studies suggest that avalglucosidase is not inferior to alpha-glucosidase in the short term (49 weeks), improving muscle and respiratory function of patients treated slightly more than alpha-glucosidase.54,58 This improvement is maintained over time, as shown in the open-label extension of COMET where patients have been followed by 155 weeks54 and by the NEO-ext study, that has shown stabilization of the disease during a period of 4.5 years of follow up.58 However, data coming from the NEO-ext study needs to be interpreted with care as it was obtained from only 17 patients. Interestingly, patients who switched from alpha-glucosidase to avalglucosidase in the COMET study did not show any additional benefit,54 which suggests that glycogen clearance already happens early after starting treatment, regardless of what enzyme is prescribed, and this clearance has a limited impact in muscle function in LOPD, which could be more dependent on the remaining muscle that is not lost and replaced by fat. However, an effective and sustained clearance of glycogen could also be reflected as a slowing or stabilization of disease progression over time. In the case of IOPD, avalglucosidase is also being tested in a phase II59 and a phase III60 clinical trial, although so far to our knowledge data on efficacy in these cohorts has not been published. Cipaglucosidase has been tested in one phase I/II61 and one phase III (PROPEL)55 (Table 3) clinical trial in LOPD patients. This drug has shown to improve muscle function safely in treated patients. The results of the phase III study were, in our opinion, complex to analyze, as they were obtained from a cohort of patients which included treatment-naive patients but also patients who were already treated with alpha-glucosidase for a variable period of time, from two to eight years. The main result of PROPEL is that cipaglucosidase is not superior to alpha-glucosidase when administered to naive LOPD patients but seems to induce a higher improvement in clinical outcome measures in the short term when administered to LOPD patients who were previously under treatment with alpha-glucosidase.55 To our knowledge, there is not a large amount of data available in the long-term results for cipaglucosidase to achieve any conclusion yet. Cipaglucosidase is also being tested in a phase III clinical trial in IOPD patients, although data from this study have not been released.62
Combined Therapies
Other trials have administered alpha-glucosidase in association with molecules that enhance the expression of CI-MPR on the muscle cell’s membrane, such as the beta-agonists albuterol63 and clenbuterol.64 With both drugs there was a statistically significant improvement in some of the assessed parameters (for albuterol FVC%, forced expiratory volume in 1 second (FEV1) when lying down and 6MWT and for clenbuterol the 6MWT and mean inspiratory pressure (MIP)), however there is no data on long-term benefit, as albuterol’s effect was measured at 6 months and clenbuterol’s was measured at one year’s time.63,64
Gene Therapy
Being a monogenic disease, the premise behind gene therapy is to deliver a healthy copy of the GAA gene inside the cell’s nuclei through an adeno-associated virus (AAV) vector and for this delivered gene to be translated into AAG using the cell’s intrinsic machinery.65 In 2017, a first in human trial with an intramuscular injection in the diaphragm of nine IOPD patients was performed using AAV1.66 The procedure-related adverse events (pneumothorax, pericardial and pleural effusion, lung contusion) reflected the need to change the delivery of the drug from local to systemic. To this day, there are three ongoing gene therapy clinical trials. The molecules SPK-3006 (RESOLUTE trial, NCT04093349, Spark Therapeutics)67 and ACTUS-101 (NCT03533673, Asklepios Biopharmaceutical, Inc.)68 are directed towards the liver, their main aim being to induce AAG production within the liver and for that enzyme to reach the skeletal muscle through the bloodstream. The other molecule, AT845 (FORTIS trial, NCT04174105, Astellas Gene Therapies),69 has the skeletal muscle as its main target, directly inducing AAG synthesis within the muscle. These are phase I/II studies currently recruiting and only limited data have been communicated in congresses related to their early results. If successful, gene therapy has an intrinsic advantage over ERT, in the fact that once delivered a continuous expression of the enzyme is achieved.70 However, there are still many questions regarding the use of gene therapy to treat muscle diseases that need to be answered, also involving Pompe disease. One major limitation is satisfactory targeting, as skeletal muscle is the most abundant tissue in the body and is formed by muscle fibers, which are a syncytium of cells containing thousands of nuclei. AAV should be able to reach as many nuclei as possible and deliver one healthy copy of the gene to effectively change the concentration of AAG in the muscles. Immune response is another limiting factor, as patients already having antibodies anti-AAV are not treatable with this strategy, limiting the number of patients that could benefit from this approach. As AAV does not integrate the gene transported into the chromosomes, this genetic material is lost when the cell divides. In the case of muscle, there is a physiologic renewal of muscle fibers which is increased in the case of a disease because of continuous muscle damage, resulting in a reduction of the number of nuclei expressing the gene. The need for reinfusion has been extensively discussed, but it will require either a change in the type of virus used or immunosuppression to limit the immune response. Finally, there is growing concern about the adverse effects produced by this type of treatment that has led to stopping some of the ongoing trials and investigate the causes. Among the factors related to them, the new protein expressed and the capsid of the virus can become an antigen for the patient’s immune response.70 Another strategy is testing ex-vivo hematopoietic stem cell gene therapy in preclinical animal models and this has shown an increased expression of AAG in several tissues including skeletal muscle. A clinical trial with this strategy is under development.71
Substrate Reduction Strategies
Another newer therapeutic strategy under development is the reduction of the substrate through glycogen synthase 1 (GYS1) inhibitors. Glycogen synthase 1 “is the rate limiting enzyme in muscle glycogen biosynthesis”72 and its genetic ablation in Pompe mouse models lowered the amount of glycogen accumulation and muscle pathology.72 A group of scientists from Maze Therapeutics in the USA developed an orally bioavailable small molecule (MZE001) which inhibits this enzyme and has already been proven to be a potent and selective inhibitor of GYS1 in biochemical, cell and tissue-based data.72 A phase I randomized, double-blind, placebo-controlled trial to assess the safety, tolerability, pharmacokinetics and pharmacodynamics of this molecule in healthy volunteers was started in February 2022 (NCT05249621, Maze Therapeutics).73
Autophagy Inhibition
Inhibition of autophagy has already been demonstrated to be useful to slow down disease progression in preclinical animal models several years ago. The group of Dr. Nina Raben has worked extensively in this topic. They developed double knock-out mice for GAA and for AGT7, a protein involved in the autophagic build-up, that showed a better response to ERT than the single GAA knock-out, with a reduced content of autophagic vacuoles.74 Recently, they were able to reduce autophagy by enhancing mTOR activity using different approaches.75 In both cases, the combination of ERT with inhibited autophagy led to better results than ERT alone. These are encouraging results suggesting that combined therapy in patients could lead to better results.
Conclusions
Even though Pompe disease is a monogenic pathology, the clinical spectrum is far from homogeneous, ranging from patients who develop muscle weakness early after birth to patients who present clinical manifestations late into adulthood, including patients who only present an increase in CK levels with minimal muscle weakness. This heterogeneity makes generalizing treatment guidelines a complex task. ERT has impacted disease progression considerably, especially in the case of IOPD. Despite all its limitations, many infants benefit from this therapy, even though they can develop complications involving many different tissues which is leading to a new phenotype with new unmet needs. The progressive accumulation of autophagic vacuoles in the muscles causes new and sustained muscle weakness and the lack of expression of AAG in the brain, a tissue not targeted by ERT yet, is associated with CNS complications. It is clear that new therapies are needed, but we are hesitant as to whether an increase in the dose of Myozyme or the improved ERT under development will be able to provide a final solution for these two issues. In the case of LOPD patients the response to treatment, although variable, is not complete, and is associated with a continued progression of muscle degeneration leading to an increase in the muscle weakness. The increase in the concentration of AAG in muscle fibers, especially in early stages of the disease, could lead to a delaying of muscle degeneration, but if the treatment is administered later on the disease, when many muscle fibers already have large autophagic build-up, new ERT drugs will probably result in the same pattern of progression observed with alpha-glucosidase. In this sense, gene therapy, if effective and safe, could lead to a change in progression of the disease but, by concept, it will need to be indicated early in the disease progression, to avoid the new generated enzyme to be trapped in the autophagic vacuoles. Considering all these findings, the indication about when to start the treatment needs to be reconsidered and new outcome measures to identify early changes in muscle function or structure such as digital technologies or muscle MRI should be included in the equation. We are convinced that the combination of a more effective therapy, which could be gene therapy, with earlier access to treatment, could provide a real change in the natural progression of the disease.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Hicks J, Wartchow E, Mierau G. Glycogen storage diseases: a brief review and update on clinical features, genetic abnormalities, pathologic features, and treatment. Ultrastruct Pathol. 2011;35(5):183–196. doi:10.3109/01913123.2011.601404
2. Lim JA, Li L, Raben N. Pompe disease: from pathophysiology to therapy and back again. Front Aging Neurosci. 2014;6:177. doi:10.3389/fnagi.2014.00177
3. Ausems MG, Verbiest J, Hermans MP, et al. Frequency of glycogen storage disease type II in The Netherlands: implications for diagnosis and genetic counselling. Eur J Hum Genet. 1999;7(6):713–716. doi:10.1038/sj.ejhg.5200367
4. Gutiérrez-Rivas E, Bautista J, Vílchez JJ, et al. Dried blood spot for screening for late-onset pompe disease: a Spanish cohort. J Neuromuscul Dis. 2015;2(s1):S42. doi:10.3233/JND-159038
5. Alonso-Pérez J, Segovia S, Domínguez-González C, et al. Spanish Pompe registry: baseline characteristics of first 49 patients with adult onset of Pompe disease. Med Clin. 2020;154(3):80–85. doi:10.1016/j.medcli.2019.03.036
6. Lukacs Z, Cobos PN, Wenninger S, et al. Prevalence of Pompe disease in 3076 patients with hyperCKemia and limb-girdle muscular weakness. Neurology. 2016;87(3):295–298. doi:10.1212/WNL.0000000000002758
7. Gutschmidt K, Musumeci O, Díaz-Manera J, et al. STIG study: real-world data of long-term outcomes of adults with Pompe disease under enzyme replacement therapy with alglucosidase alfa. J Neurol. 2021;268(7):2482–2492. doi:10.1007/s00415-021-10409-9
8. Díaz-Manera J, Walter G, Straub V. Skeletal muscle magnetic resonance imaging in Pompe disease. Muscle Nerve. 2021;63(5):640–650. doi:10.1002/mus.27099
9. Kishnani PS, Goldenberg PC, DeArmey SL, et al. Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants. Mol Genet Metab. 2010;99(1):26–33. doi:10.1016/j.ymgme.2009.08.003
10. Zapata-Aldana E, McMillan HJ, Rupar T, et al. Muscle problems in juvenile-onset acid maltase deficiency (Pompe disease). Paediatr Child Health. 2019;24(4):270–271. doi:10.1093/pch/pxy153
11. Taverna S, Cammarata G, Colomba P, et al. Pompe disease: pathogenesis, molecular genetics and diagnosis. Aging. 2020;12(15):15856–15874. doi:10.18632/aging.103794
12. Thurberg BL, Lynch Maloney C, Vaccaro C, et al. Characterization of pre- and post-treatment pathology after enzyme replacement therapy for Pompe disease. Lab Invest. 2006;86(12):1208–1220. doi:10.1038/labinvest.3700484
13. Schänzer A, Kaiser AK, Mühlfeld C, et al. Quantification of muscle pathology in infantile Pompe disease. Neuromuscul Disord. 2017;27(2):141–152. doi:10.1016/j.nmd.2016.10.010
14. Carrasco-Rozas A, Fernández-Simón E, Suárez-Calvet X, et al. BNIP3 is involved in muscle fiber atrophy in late-onset pompe disease patients. Am J Pathol. 2022;192(8):1151–1166. doi:10.1016/j.ajpath.2022.05.003
15. Thuriot F, Gravel E, Hodson K, et al. Molecular diagnosis of pompe disease in the genomic era: correlation with acid alpha-glucosidase activity in dried blood spots. J Clin Med. 2021;10:3868. doi:10.3390/jcm10173868
16. Schoser B, Stewart A, Kanters S, et al. Survival and long-term outcomes in late-onset Pompe disease following alglucosidase alfa treatment: a systematic review and meta-analysis. J Neurol. 2017;264(4):621–630. doi:10.1007/s00415-016-8219-8
17. Hers HG. a-glucosidase deficiency in generalized glycogen storage disease (Pompe’s disease). Biochem. 1963;86:11–16.
18. Van den Hout JM, Reuser AJ, de Klerk JB, Arts WF, Smeitink JA. Van der Ploeg AT. Enzyme therapy for pompe disease with recombinant human alpha-glucosidase from rabbit milk. J Inherit Metab Dis. 2001;24(2):266–274. doi:10.1023/A:1010383421286
19. Kishnani PS, Corzo D, Nicolino M, et al. Recombinant human acid [alpha]-glucosidase: major clinical benefits in infantile-onset Pompe disease. Neurology. 2007;68(2):99–109. doi:10.1212/01.wnl.0000251268.41188.04
20. van der Ploeg AT, Clemens PR, Corzo D, et al. A randomized study of alglucosidase alfa in late-onset Pompe’s disease. N Engl J Med. 2010;362(15):1396–1406. doi:10.1056/NEJMoa0909859
21. Kishnani PS, Corzo D, Leslie ND, et al. Early treatment with alglucosidase alpha prolongs long-term survival of infants with Pompe disease. Pediatr Res. 2009;66(3):329–335. doi:10.1203/PDR.0b013e3181b24e94
22. Nicolino M, Byrne B, Wraith JE, et al. Clinical outcomes after long-term treatment with alglucosidase alfa in infants and children with advanced Pompe disease. Genet Med. 2009;11(3):210–219. doi:10.1097/GIM.0b013e31819d0996
23. Chien YH, Lee NC, Chen CA, et al. Long-term prognosis of patients with infantile-onset Pompe disease diagnosed by newborn screening and treated since birth. J Pediatr. 2015;166(4):985–991. doi:10.1016/j.jpeds.2014.10.068
24. Desai AK, Li C, Rosenberg AS, et al. Immunological challenges and approaches to immunomodulation in Pompe disease: a literature review. Ann Transl Med. 2019;7(13):285. doi:10.21037/atm.2019.05.27
25. Messinger YH, Mendelsohn NJ, Rhead W, et al. Successful immune tolerance induction to enzyme replacement therapy in CRIM-negative infantile Pompe disease. Genet Med. 2012;14(1):135–142. doi:10.1038/gim.2011.4
26. Poelman E, Hoogeveen-Westerveld M, Kroos-de Haan MA, et al. High sustained antibody titers in patients with classic infantile pompe disease following immunomodulation at start of enzyme replacement therapy. J Pediatr. 2018;195:236–243.e3. doi:10.1016/j.jpeds.2017.11.046
27. De Groot AS, Desai AK, Lelias S, et al. Immune tolerance-adjusted personalized immunogenicity prediction for pompe disease. Front Immunol. 2021;12:636731. doi:10.3389/fimmu.2021.636731
28. Raben N, Ralston E, Chien YH, et al. Differences in the predominance of lysosomal and autophagic pathologies between infants and adults with Pompe disease: implications for therapy. Mol Genet Metab. 2010;101(4):324–331. doi:10.1016/j.ymgme.2010.08.001
29. Kishnani PS, Beckemeyer AA, Mendelsohn NJ. The new era of Pompe disease: advances in the detection, understanding of the phenotypic spectrum, pathophysiology, and management. Am J Med Genet C Semin Med Genet. 2012;160C(1):1–7. doi:10.1002/ajmg.c.31324
30. Takikita S, Schreiner C, Baum R, et al. Fiber type conversion by PGC-1α activates lysosomal and autophagosomal biogenesis in both unaffected and Pompe skeletal muscle. PLoS One. 2010;5(12):e15239. doi:10.1371/journal.pone.0015239
31. Ditters IAM, Huidekoper HH, Kruijshaar ME, et al. European Pompe Consortium project group on classic infantile Pompe disease. Effect of alglucosidase alfa dosage on survival and walking ability in patients with classic infantile Pompe disease: a multicentre observational cohort study from the European Pompe Consortium. Lancet Child Adolesc Health. 2022;6(1):28–37. doi:10.1016/S2352-4642(21)00308-4
32. Ebbink BJ, Poelman E, Aarsen FK, et al. Classic infantile Pompe patients approaching adulthood: a cohort study on consequences for the brain. Dev Med Child Neurol. 2018;60(6):579–586. doi:10.1111/dmcn.13740
33. Spiridigliozzi GA, Heller JH, Case LE, Jones HN, Kishnani PS. Early cognitive development in children with infantile Pompe disease. Mol Genet Metab. 2012;105(3):428–432. doi:10.1016/j.ymgme.2011.10.012
34. Kamphoven JH, de Ruiter MM, Winkel LP, et al. Hearing loss in infantile Pompe’s disease and determination of underlying pathology in the knockout mouse. Neurobiol Dis. 2004;16(1):14–20. doi:10.1016/j.nbd.2003.12.018
35. van Capelle CI, Goedegebure A, Homans NC, et al. Hearing loss in Pompe disease revisited: results from a study of 24 children. J Inherit Metab Dis. 2010;33(5):597–602. doi:10.1007/s10545-010-9144-0
36. van Gelder CM, van Capelle CI, Ebbink BJ, et al. Facial-muscle weakness, speech disorders and dysphagia are common in patients with classic infantile Pompe disease treated with enzyme therapy. J Inherit Metab Dis. 2012;35(3):505–511. doi:10.1007/s10545-011-9404-7
37. Swift G, Cleary M, Grunewald S, et al. Swallow prognosis and follow-up protocol in infantile onset pompe disease. JIMD Rep. 2017;33:11–17.
38. Hahn A, Schänzer A. Long-term outcome and unmet needs in infantile-onset Pompe disease. Ann Transl Med. 2019;7(13):283. doi:10.21037/atm.2019.04.70
39. van den Dorpel JJA, Poelman E, Harlaar L, et al. Distal muscle weakness is a common and early feature in long-term enzyme-treated classic infantile Pompe patients. Orphanet J Rare Dis. 2020;15(1):247. doi:10.1186/s13023-020-01482-w
40. van der Ploeg AT, Barohn R, Carlson L, et al. Open-label extension study following the Late-Onset Treatment Study (LOTS) of alglucosidase alfa. Mol Genet Metab. 2012;107(3):456–461. doi:10.1016/j.ymgme.2012.09.015
41. Harlaar L, Hogrel JY, Perniconi B, et al. Large variation in effects during 10 years of enzyme therapy in adults with Pompe disease. Neurology. 2019;93(19):e1756–e1767. doi:10.1212/WNL.0000000000008441
42. Kuperus E, Kruijshaar ME, Wens SCA, et al. Long-term benefit of enzyme replacement therapy in Pompe disease: a 5-year prospective study. Neurology. 2017;89(23):2365–2373. doi:10.1212/WNL.0000000000004711
43. Güngör D, Kruijshaar ME, Plug I, et al. Impact of enzyme replacement therapy on survival in adults with Pompe disease: results from a prospective international observational study. Orphanet J Rare Dis. 2013;8:49. doi:10.1186/1750-1172-8-49
44. Park JS, Kim HG, Shin JH, Choi YC, Kim DS. Effect of enzyme replacement therapy in late onset Pompe disease: open pilot study of 48 weeks follow-up. Neurol Sci. 2015;36(4):599–605. doi:10.1007/s10072-014-2000-5
45. Filosto M, Cotti Piccinelli S, Ravaglia S, et al. Assessing the role of Anti rh-GAA in modulating response to ERT in a late-onset pompe disease cohort from the Italian GSDII Study Group. Adv Ther. 2019;36(5):1177–1189. doi:10.1007/s12325-019-00926-5
46. Ripolone M, Violano R, Ronchi D, et al. Effects of short-to-long term enzyme replacement therapy (ERT) on skeletal muscle tissue in late onset Pompe disease (LOPD). Neuropathol Appl Neurobiol. 2018;44(5):449–462. doi:10.1111/nan.12414
47. Stockton DW, Kishnani P, van der Ploeg A, et al. Respiratory function during enzyme replacement therapy in late-onset Pompe disease: longitudinal course, prognostic factors, and the impact of time from diagnosis to treatment start. J Neurol. 2020;267(10):3038–3053. doi:10.1007/s00415-020-09936-8
48. Nuñez-Peralta C, Alonso-Pérez J, Llauger J, et al. Follow-up of late-onset Pompe disease patients with muscle magnetic resonance imaging reveals increase in fat replacement in skeletal muscles. J Cachexia Sarcopenia Muscle. 2020;11(4):1032–1046. doi:10.1002/jcsm.12555
49. Figueroa-Bonaparte S, Segovia S, Llauger J, et al. Muscle MRI findings in childhood/adult onset pompe disease correlate with muscle function. PLoS One. 2016;11(10):e0163493. doi:10.1371/journal.pone.0163493
50. van der Ploeg AT, Kruijshaar ME, Toscano A, et al. European Pompe Consortium. European consensus for starting and stopping enzyme replacement therapy in adult patients with Pompe disease: a 10-year experience. Eur J Neurol. 2017;24(6):768–e31. doi:10.1111/ene.13285
51. Li C, Desai AK, Gupta P, et al. Transforming the clinical outcome in CRIM-negative infantile Pompe disease identified via newborn screening: the benefits of early treatment with enzyme replacement therapy and immune tolerance induction. Genet Med. 2021;23(5):845–855. doi:10.1038/s41436-020-01080-y
52. ClinicalTrials.gov. In utero enzyme replacement therapy for lysosomal storage diseases - full text view. Clinicaltrials.gov; 2022. Available from: https://clinicaltrials.gov/ct2/show/NCT04532047.
53. Byrne BJ, Geberhiwot T, Barshop BA, et al. A study on the safety and efficacy of reveglucosidase alfa in patients with late-onset Pompe disease. Orphanet J Rare Dis. 2017;12(1):144. doi:10.1186/s13023-017-0693-2
54. Diaz-Manera J, Kishnani PS, Kushlaf H, et al. Safety and efficacy of avalglucosidase alfa versus alglucosidase alfa in patients with late-onset Pompe disease (COMET): a Phase 3, randomised, multicentre trial. Lancet Neurol. 2021;20(12):1012–1026. doi:10.1016/S1474-4422(21)00241-6
55. Schoser B, Roberts M, Byrne BJ, et al. Safety and efficacy of cipaglucosidase alfa plus miglustat versus alglucosidase alfa plus placebo in late-onset Pompe disease (PROPEL): an international, randomised, double-blind, parallel-group, phase 3 trial. Lancet Neurol. 2021;20(12):1027–1037. doi:10.1016/S1474-4422(21)00331-8
56. Pena LDM, Barohn RJ, Byrne BJ, et al. Safety, tolerability, pharmacokinetics, pharmacodynamics, and exploratory efficacy of the novel enzyme replacement therapy avalglucosidase alfa (neoGAA) in treatment-naïve and alglucosidase alfa-treated patients with late-onset Pompe disease: a phase 1, open-label, multicenter, multinational, ascending dose study. Neuromuscul Disord. 2019;29(3):167–186. doi:10.1016/j.nmd.2018.12.004
57. ClinicalTrials.gov. Avalglucosidase alfa extension study - full text view. Available from: https://clinicaltrials.gov/ct2/show/NCT02032524.
58. Dimachkie M, Barohn R, Byrne B, et al. Long-term safety and efficacy of avalglucosidase alfa in patients with late-onset pompe disease. Neurology. 2022;99(5):e536–e548. doi:10.1212/WNL.0000000000200746
59. ClinicalTrials.gov. A study to assess safety and efficacy of avalglucosidase alfa administered every other week in pediatric patients with infantile-onset pompe disease previously treated with alglucosidase alfa - full text view. Available from: https://clinicaltrials.gov/ct2/show/NCT03019406?cond=avalglucosidase&draw=2&rank=4.
60. ClinicalTrials.gov. Clinical study for treatment-naïve IOPD babies to evaluate efficacy and safety of ERT with avalglucosidase alfa - full text view. Available from: https://clinicaltrials.gov/ct2/show/NCT04910776?cond=avalglucosidase&draw=2&rank=3.
61. Kishnani P, Schoser B, Bratkovic D, et al. First-in-human study of advanced and targeted acid α-glucosidase (AT-GAA) (ATB200/AT2221) in patients with Pompe disease: preliminary functional assessment results from the ATB200-02 trial. Mol Genet Metab. 2019;126(2):S86. doi:10.1016/j.ymgme.2018.12.212
62. ClinicalTrials.gov. Rossella: a Study to evaluate the safety, PK, efficacy, PD and immunogenicity of cipaglucosidase alfa/miglustat in IOPD subjects aged 0 to <18 - Full text view. Available from: https://clinicaltrials.gov/ct2/show/NCT04808505?cond=cipaglucosidase&draw=2&rank=1.
63. Koeberl DD, Case LE, Desai A, et al. Improved muscle function in a phase I/II clinical trial of albuterol in Pompe disease. Mol Genet Metab. 2020;129(2):67–72. doi:10.1016/j.ymgme.2019.12.008
64. Koeberl DD, Case LE, Smith EC, et al. Correction of biochemical abnormalities and improved muscle function in a phase I/II clinical trial of clenbuterol in pompe disease. Mol Ther. 2018;26(9):2304–2314. doi:10.1016/j.ymthe.2018.06.023
65. Ronzitti G, Collaud F, Laforet P, Mingozzi F. Progress and challenges of gene therapy for Pompe disease. Ann Transl Med. 2019;7(13):287. doi:10.21037/atm.2019.04.67
66. Corti M, Liberati C, Smith BK, et al. Safety of intradiaphragmatic delivery of adeno-associated virus-mediated alpha-glucosidase (rAAV1-CMV-hGAA) gene therapy in children affected by pompe disease. Hum Gene Ther Clin Dev. 2017;28(4):208–218. doi:10.1089/humc.2017.146
67. ClinicalTrials.gov. A gene transfer study for late-onset pompe disease (RESOLUTE) - full text view. Clinicaltrials.gov; 2022. Available from: https://clinicaltrials.gov/ct2/show/NCT04093349.
68. ClinicalTrials.gov. AAV2/8-LSPhGAA (ACTUS-101) in late-onset pompe disease - full text view. Clinicaltrials.gov; 2022. Available from: https://clinicaltrials.gov/ct2/show/NCT03533673.
69. Clinicaltrials.gov. Gene transfer study in patients with late onset pompe disease - full text view. ClinicalTrials.gov; 2022. Available from: https://clinicaltrials.gov/ct2/show/NCT04174105.
70. Weber T. Anti-AAV antibodies in AAV gene therapy: current challenges and possible solutions. Front Immunol. 2021;12:658399. doi:10.3389/fimmu.2021.658399
71. Stok M, de Boer H, Huston MW, et al. Lentiviral hematopoietic stem cell gene therapy corrects murine pompe disease. Mol Ther Methods Clin Dev. 2020;4(17):1014–1025. doi:10.1016/j.omtm.2020.04.023
72. Choy R, Reiton D, Merritt H, et al. In-vitro characterization of MZE001, an orally active GYS1 inhibitor to treat Pompe disease. Mol Genet Metab. 2022;135(2):S32. doi:10.1016/j.ymgme.2021.11.067
73. ClinicalTrials.gov. A randomized phase 1 single and multiple ascending dose study of MZE001 - full text view. Clinicaltrials.gov; 2022. Available from: https://clinicaltrials.gov/ct2/show/NCT05249621.
74. Raben N, Schreiner C, Baum R, et al. Suppression of autophagy permits successful enzyme replacement therapy in a lysosomal storage disorder-murine Pompe disease. Autophagy. 2010;6(8):1078–1089. doi:10.4161/auto.6.8.13378
75. Lim JA, Li L, Shirihai OS, et al. Modulation of mTOR signaling as a strategy for the treatment of Pompe disease. EMBO Mol Med. 2017;9(3):353–370. doi:10.15252/emmm.201606547
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.