Back to Journals » Therapeutics and Clinical Risk Management » Volume 15
Therapeutic Challenges And Advances In The Management Of Systemic Sclerosis-Related Pulmonary Arterial Hypertension (SSc-PAH)
Authors Argula RG ![]() , Ward C
, Ward C ![]() , Feghali-Bostwick C
, Feghali-Bostwick C
Received 15 August 2019
Accepted for publication 6 October 2019
Published 13 December 2019 Volume 2019:15 Pages 1427—1442
DOI https://doi.org/10.2147/TCRM.S219024
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Rahul G Argula,1 Celine Ward,2 Carol Feghali-Bostwick2
1Department of Medicine, Division of Pulmonary and Critical Care Medicine, Medical University of South Carolina, Charleston, SC 29425, USA; 2Department of Medicine, Division of Rheumatology, Medical University of South Carolina, Charleston, SC 29425, USA
Correspondence: Rahul G Argula
Department of Medicine, Division of Pulmonary and Critical Care Medicine Medical University of South Carolina, 816 CSB, MSC 630, Jonathan Lucas Street, Charleston, SC 29425, USA
Tel +1 843 792 3167
Email [email protected]
Abstract: Systemic sclerosis (SSc) is a rare autoimmune disorder with multi-organ involvement. SSc-associated pulmonary arterial hypertension (SSc-PAH) is one of the leading causes of morbidity and mortality in the SSc population. With advances in our understanding of pulmonary arterial hypertension (PAH) diagnosis and treatment, outcomes for all PAH patients have significantly improved. While SSc-PAH patients have also benefited from these advances, significant challenges remain. Diagnosis of PAH is a challenging endeavor in SSc patients who often have many co-existing pulmonary and cardiac comorbidities. Given the significantly elevated prevalence and lifetime risk of PAH in the SSc population, screening for SSc-PAH is a critically useful strategy. Treatment with pulmonary arterial (PA) vasodilators has resulted in a dramatic improvement in the survival and quality of life of PAH patients. While therapy with PA vasodilators is beneficial in SSc-PAH patients, therapy effects appear to be attenuated when compared to responses in patients with idiopathic PAH (IPAH). This review attempts to chronicle and summarize the advances in our understanding of the optimal screening strategies to identify PAH in patients with SSc. The article also reviews the advances in the therapeutic and risk stratification strategies for SSc-PAH patients.
Keywords: systemic sclerosis, pulmonary arterial hypertension, screening, risk stratification, therapy, advances
Introduction
Systemic sclerosis (SSc) is a rare systemic disease characterized by chronic inflammation, autoimmune dysregulation, and microvascular endothelial dysfunction leading ultimately to fibrosis and excessive collagen deposition within the skin and various other organ systems.1 These pathologic changes lead to a characteristic skin thickening and significant dysfunction in the organs involved, resulting in the descriptive moniker for this disease. SSc affects around one in 10,000 people around the world.2 There are two major disease subtypes based on the extent of skin involvement: limited cutaneous SSc (lcSSc) and diffuse cutaneous SSc (dcSSc). Patients with lcSSc have skin involvement distal to the elbows and knees, with or without face and neck involvement. Patients with dcSSc have skin involvement extending to the proximal limbs and trunk.2 Currently, a diagnosis of SSc typically requires fulfilment of the 2013 American College of Rheumatology (ACR) and European League Against Rheumatism (EULAR) classification criteria.2 As with most connective tissue diseases, SSc disproportionately affects women, and lung involvement is a common cause of morbidity and mortality in SSc patients.3 While interstitial lung disease (ILD) is the commonest cause of death, pulmonary arterial hypertension (PAH) is the second most common cause of mortality in SSc patients.4
PAH is also a rare disease affecting the medium-to-small pulmonary arteries, resulting in a distinctive remodeling in the intimal, medial, and adventitial layers and causing significant narrowing of the pulmonary vascular lumen.5 These changes lead to a substantial elevation in the pulmonary vascular resistance (PVR) to blood flow.6 These progressive vessel changes also result in abnormal elevations in pulmonary vascular stiffness and reduced compliance.7 These abnormalities in vessel compliance in combination with a rising PVR lead to right ventricular (RV) hypertrophy, dysfunction, and failure, and ultimately result in death if untreated.8
The prevalence of PAH in the US is largely unknown. Using an insurance claims database, Kirson et al estimated the PAH prevalence at 109 (95% CI: 71–146) per million individuals (PMI) among the population under age 65 and 451 (95% CI: 384–519) PMI among the population aged 65 and over. Prior research has estimated PAH prevalence in Europe at ∼15–52 PMI.9 PAH can complicate several connective tissue diseases (CTDs) such as SSc, systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), and mixed CTD (MCTD) and is an important cause of morbidity and mortality in this group of patients.10 Among the CTDs, SSc has the highest known PAH prevalence (∼7–12%) and accounts for up to 60%–80% of all CTD-PAH in the US and Europe. PAH is one of the leading causes of death in SSc patients.11 The exact prevalence of PAH in the other CTDs is poorly described at present but thought to be no more than 1% in patients with SLE and between 20 and 50% in patients with MCTD.12,13
Survival in PAH patients has significantly improved over the past two decades owing to the advances in therapies, risk stratification, and our overall understanding of this debilitating disease. Unfortunately, this trend in improved survival has not been observed in all the sub-types of the WHO group I population. While idiopathic PAH (IPAH) patients have had significant improvements in their exercise capacity and quality of life due to the use of combination vasodilator therapies, SSc-PAH patients have not mirrored these trends.14,15
The most recent 1-, 3-, 5-, and 8-year survival estimates for SSc-PAH patients are at 95%, 75%, 63%, and 49%, respectively.16 While the short-term survival data in the modern era are comparable for IPAH and SSc-PAH patients, the long-term survival data put the SSc-PAH patients at a significant disadvantage.15 Data collated from clinical trials demonstrate clear differences between the SSc-PAH and IPAH groups even with regard to the response to PA vasodilator therapies, with the SSc-PAH patients experiencing a significantly lower improvement in 6MWD when compared to IPAH patients.17
The prevalence of PAH among SSc patients has been reported to be between 8 and 12% based on studies diagnosing PAH relying on right heart catheterization (RHC).18,19 The high prevalence and risk of development of PAH in the SSc population are the reasons why screening for PAH is recommended in this group. The differences in response to therapy between SSc-PAH and IPAH patients and the heterogeneity of treatment responses and the poor survival of the SSc-PAH population when compared to IPAH patients highlight the unique characteristics of the SSc-PAH phenotype. These clinico-epidemiological differences between SSc-PAH and IPAH lead us to the question: is SSc-PAH clinically distinct from IPAH and other forms of PAH? This review builds on the recent advances in the management of PAH in the SSc population, by focusing on two of the key aspects germane to the management of the SSc-PAH patient: screening and therapy. This article summarizes the advances in screening strategies for SSc-PAH and reviews the challenges and advances in the treatment of SSc-PAH.
Screening
While the gold standard modality for the diagnosis of PAH is right heart catheterization (RHC), 20 screening for PAH remains a difficult endeavor given the overall low prevalence in the general population. However, as highlighted in the introduction section of this review, the high prevalence of PAH in SSc patients makes screening for PAH a useful intervention in this population. Among the various manifestations of SSc, the limited SSc variant seems to have the strongest association with the development of PAH, although PAH has been known to occur with other SSc phenotypes as well.21 Hence, the principles of screening for PAH would be applicable to all patients with SSc.
The Role Of Screening For PAH In The SSc Population
Clinical features that suggest the presence of PAH include symptoms of dyspnea, inability to tolerate exercise, fatigue, lower extremity swelling, palpitations, among others.22,23 In the normal adult human being, there exists a significant redundancy with regard to the pulmonary vascular surface area, and this vascular redundancy provides for the ability to accommodate significantly increased pulmonary blood flows during exercise, of up to six times the cardiac output with no elevation in pulmonary vascular resistance (PVR). Elevation of PA pressures above 25mm Hg at rest is thought to result from the loss of up to 50% of the normal pulmonary vascular bed!24 This implies that by the time a patient is symptomatic, there has already been a significant loss of pulmonary vascular surface area. While in advanced cases of SSc-PAH, patients can present with severe dyspnea, hypoxemia, and pre-syncopal and sometimes syncopal episodes associated with exertion, up to 22% of SSc-PAH patients are asymptomatic at the time of their RHC-determined diagnosis of pulmonary hypertension.25
Since PAH has such a high prevalence in the SSc population and considering that SSc-PAH is a progressive disease and patients in the early stages of the disease are asymptomatic, a targeted screening strategy looking for PAH in the SSc population would be expected to have a significant clinical impact. The early identification of the PAH phenotype within the SSc population could prevent delays in therapeutic intervention with PA vasodilator therapies and prevent the development of RV dysfunction. Another potential advantage of such a screening strategy would be the identification of the SSc-PAH patient at an early stage of this “pulmonary vascular drop out”, setting the stage for an intervention with a novel, targeted therapy that could be used as an adjunct to PA vasodilators.
Clinical Features
Clinical features associated with the development of PAH in the SSc patients include older age, female sex, post-menopausal status, longer disease duration, having the limited cutaneous variant of SSc, number and size of cutaneous telangiectasias, and digital ulcers.26,27 While the presence of these features in an SSc patient should heighten the suspicion for underlying PAH, these clinical characteristics have had limited utility for the development of a quantitative screening tool for PAH in SSc. In fact, one study reported no difference with regard to the prevalence of dyspnea as a reported symptom between the SSc-PAH patients and SSc patients without PAH.28 Other practical challenges include the difficulty surrounding the ascertainment of some of these characteristics (e.g. time bias in determining the disease duration, quantifying the burden of telangiectasias, etc.) in a clinical setting. The lack of a consistent “dose–response” association between some of these clinical characteristics and the risk of development of PAH (e.g. age, digital ulcers, etc.) also adds to the uncertainty of employing a combination of these features as a practical screening tool for PAH in the SSc population.
Pulmonary Function Testing (PFT)
Before the widespread availability of echocardiography, PFT was the only available means for evaluating the health of the pulmonary vascular bed. The diffusing capacity for carbon monoxide (DLCO) and the DLCO/VA (DLCO corrected for the alveolar volume) are good surrogates for pulmonary vascular function. Both DLCO and DLCO/VA are reduced in PAH patients. The reduction in these parameters is thought to be due to a) reduction in the pulmonary capillary blood volume in PAH patients and, more recently, b) a reduction in the diffusing capacity of the alveolar capillary membrane.29 While SSc patients with a low DLCO and DLCO/VA have a higher risk for the development of PAH, given the high prevalence of interstitial lung disease in the SSc population, they cannot in isolation be used to make the diagnosis of PAH in these patients. For instance, in the DETECT cohort, only 31% of SSc patients with DLCO less than 60% predicted had RHC-determined pulmonary hypertension.28 A disproportionate reduction in the DLCO for a given decline in the predicted FVC on the other hand is highly suggestive of the presence of pulmonary vascular disease in that patient. Indeed, the FVC (% predicted)/DLCO (% predicted) performs better as a screening tool for identifying the SSc-PAH patient.28
Trans-Thoracic Echocardiography (TTE)
Echocardiography is an important screening modality for SSc-PAH. Annual echocardiographic screening is recommended for SSc patients at high risk for developing PAH.30,31 The 2015 ESC/ERS guidelines make an important distinction between screening for SSc-PAH vs PAH in other CTDs. They recommend annual echocardiographic screening in asymptomatic SSc patients while they reserve echocardiograms for symptomatic CTD patients in whom PAH is suspected.30 While trans-thoracic echocardiography determined systolic pulmonary artery pressures (sPAP) are good estimates of the mean pulmonary artery pressures (mPAP), in the DETECT study, up to 37% and 18% of patients with normal sPAP (<35 mm Hg) were identified as having RHC-determined PH and PAH, respectively.28 The yearly rate of increase in the tricuspid regurgitant jet velocity (TRV) is another parameter that appears to identify SSc patients developing PAH.32
While the variability in assessment of the tricuspid regurgitant jet velocity (TRV) can adversely affect the predictive power of echocardiography as a screening tool for PAH in the SSc population, the ESC/ERS guidelines on the diagnosis and treatment of PH provide a practical framework for utilizing echocardiography in the diagnostic evaluation of a PH patient (Tables 1a and 1b). The absence or presence of an elevated TRV in combination with other echocardiographic features suggestive of PH place the patient into three degrees (low, intermediate, and high) of probabilities with regard to PH. In patients being evaluated for PAH, the guidelines recommend confirmatory testing via RHC in the high probability group and to consider RHC in the intermediate probability group.30
 |
Table 1a Echocardiographic Probability Of Pulmonary Hypertension (PH) In Symptomatic Patients With A Suspicion Of Pulmonary Hypertension. |
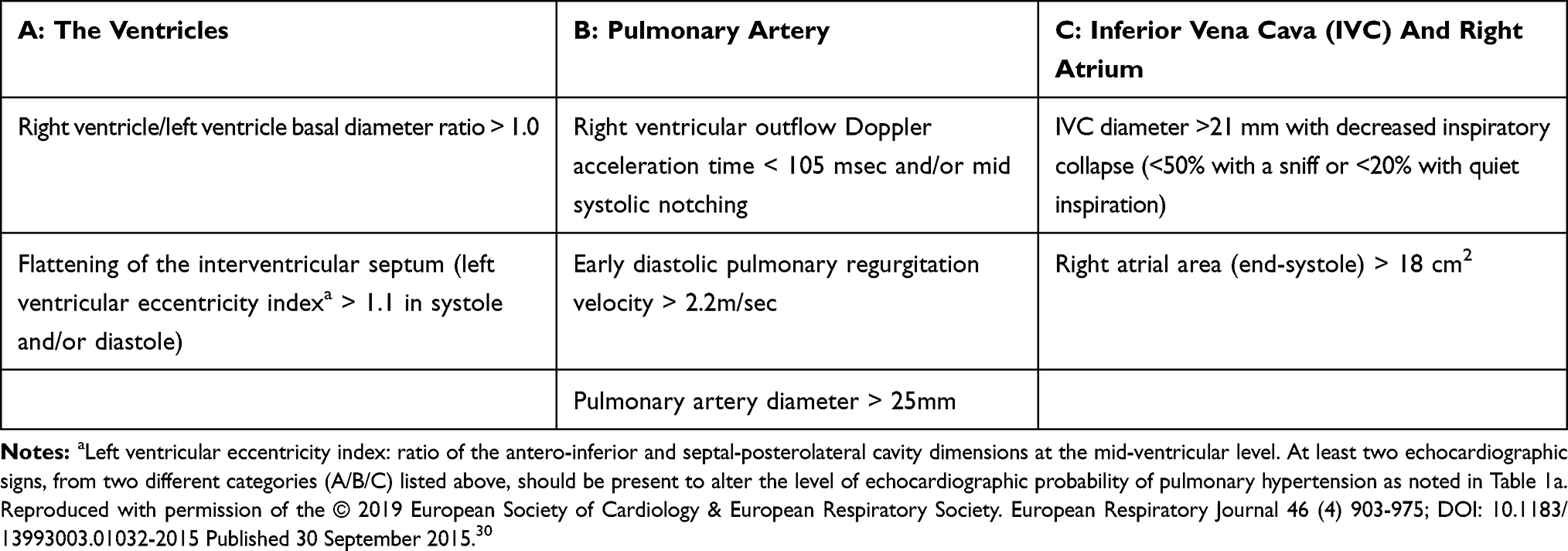 |
Table 1b Echocardiographic Signs Used To Assess The Probability Of Pulmonary Hypertension In Addition To The TR Velocity Measurement As Detailed In Table 1a. |
Serum Biomarkers
Autoantibodies
Various SSc-specific autoantibodies (ab) have been associated with SSc-PAH. High anti-centromere ab (ACA) titers and a nucleolar pattern of ANA were previously described in SSc-PAH patients.33 A South-Australian cohort reported the association of SSc-PAH with the presence of anti-U1-RNP and Th/To antibodies.34 However, in a study by Steen, none of the afore-mentioned antibodies could differentiate SSc-PAH and SSc without PAH patients. They did report that in a smaller subset, a significantly higher number of SSc-PAH patients tested positive for anti-U3-RNP when compared to the SSc without PAH patients.35 The largest North American, incident SSc-PAH (patients enrolled within 6 months of diagnosis of SSc-PAH) cohort analysis from the PHAROS (Pulmonary Hypertension Assessment and Recognition of Outcomes in Scleroderma) registry confirmed these associations and showed that the most common antibodies seen in SSc-PAH patients were ACA, anti-nucleolar abs, while the less common antibodies included anti-Scl-70, U1-RNP, and RNA polymerase antibodies.36 There have been reported associations between anticardiolipin antibodies (aCL), anti-beta-2 glycoprotein antibodies, and SSc-PAH in a French cohort.37 However, the pathophysiological significance of this finding is uncertain, and a lupus overlap syndrome in SSc patients could have also led to this finding. While AECAs (anti-endothelial cell antibodies) held promise as antibodies that were specific to SSc-mediated vascular pathobiology, there have not been any phenotype-specific associations within SSc.38,39 Despite the association of these antibodies with SSc-PAH, their utility with regard to screening has been limited. Researchers reported in a Greek SSc cohort that the presence of ACA abs was associated with a more rapid increase in the echocardiogram determined TRV over time.40 However, in the PHAROS cohort, none of the above-mentioned antibodies could predict the risk for the development of PH.41
Two intriguing antibodies with pulmonary vascular implications in the SSc population are the anti-endothelin receptor type A antibodies (ETAR) and anti-angiotensin receptor type I antibodies (AT1R). In one study, ETAR and AT1R levels were significantly higher in the SSc-PAH patients when compared to other WHO group I PH and CTEPH patients, suggesting that they might be useful in differentiating SSc-PAH from other forms of PAH. Another potential application might be with regard to screening for PAH, as the same group of researchers showed that SSc patients with a high AT1R and ETAR titer had a significantly higher risk of developing PAH when followed for a period of 6 years. In their cohort of SSc patients, high AT1R levels conferred a 4.3-fold higher risk (95% CI: 2.2–8.4) and high ETAR levels conferred a 3.5-fold higher risk (95% CI: 1.5–5.6) of developing PAH.42 The presence of these antibodies also seemed to predict death, further corroborating the mechanistic importance of these antibodies with regard to pulmonary vascular remodeling in the SSc-PAH patients.
Cardiac Biomarkers
B-type natriuretic peptide (BNP) is a 32-amino acid peptide that is released by the cardiac atria and ventricles, predominantly in response to myocardial stretch. The precursor to BNP is a 134-amino acid pre-prohormone that undergoes a two-step cleavage process ultimately resulting in the active peptide BNP and an inactive 76-amino acid fragment: NT-Pro BNP (N terminal Pro B-type natriuretic peptide).43 Serum levels of BNP and NT-Pro BNP have good correlations with mean pulmonary artery pressure (mPAP), PVR, and RV strain in patients with PAH and chronic thromboembolic pulmonary hypertension (CTEPH).44,45 Because these biomarkers have a very high sensitivity and have a strong correlation with myocardial stretch, they have significant utility in screening for PAH in the SSc population. In a study by Allanore et al, elevated NT-Pro BNP levels could discriminate SSc patients with echo-determined PAH (RVSP >40mm Hg) from those without PAH. The negative predictive value for NT-Pro BNP as a screening tool for echo-determined SSc-PAH in their study was 96%.46 These associations were subsequently validated in RHC-confirmed SSc-PAH cohorts where an elevated NT-Pro BNP level (>395 pg/mL) had high specificity (95%) and positive predictive value in identifying SSc-PAH.47,48
Another biomarker that appears to have the potential to differentiate SSc-PAH from SSc without PAH patients is growth differentiation factor(GDF)-15. GDF-15 belongs to the transforming growth factor-β cytokine superfamily, is released from cardiac tissue under conditions of stress and in response to tissue damage, and appears to have a cardioprotective effect.49,50 Meadows et al, in a single-institutional, cross-sectional analysis, showed that SSc-PAH patients had significantly higher serum levels of GDF-15 when compared to SSc patients without PAH. In their study, GDF-15 levels had plausible correlations with parameters that correlate with the severity of pulmonary vascular disease such as DLCO, FVC/DLCO, and NT-Pro BNP and had a reasonably good discriminant power to differentiate SSc-PAH patients from those without PAH.51 We do not yet know if GDF-15 enhances the predictive power of BNP and NT-Pro BNP in diagnosing PAH, considering that both these biomarkers appear to have ontological similarities with regard to their production and that they are both present in the sera of patients with cardiopulmonary dysfunction.
A Practical Approach To Screening For PAH In The SSc Population
Considering the limitations of relying on one “tool” to identify PAH in the SSc population, a “multimodality approach” to screening that has a very high sensitivity is critical to minimize “missed diagnoses”. Two such strategies to screen for SSc-PAH are the DETECT and the ASIG (Australian Scleroderma Interest Group) algorithms. The DETECT algorithm was derived from a multicenter, international study evaluating SSc patients for PAH.28 The screening algorithm involves a two-step approach with the first step utilizing non-echocardiographic data which would be readily available to a rheumatology/primary care provider. If a patient’s cumulative score generated from this step is greater than 300 points, he/she would move to the second step which relies on echocardiographic data. Patients scoring below 300 points would not require further workup as the risk of PAH would be very low. In the second step, which is based on echocardiographic data, patients scoring greater than 35 points would be referred to RHC (Figure 1). The ASIG algorithm, on the other hand, uses just PFT data (DLCO <70% + FVC/DLCO >1.8 - Component A) and a serum NT-Pro BNP level > 210pg/mL (Component B) to determine eligibility for further workup of PAH. Patients with either a positive component A and/or B would require further workup with an echocardiogram, 6MWT, HRCT (high-resolution computed tomogram) of the chest and, ultimately, an RHC if suspicion for PAH remains high (Figure 2).52
 |
Figure 1 The DETECT algorithm to screen for pulmonary arterial hypertension in the systemic sclerosis patient.Note: Adapted by permission from BMJ Publishing Group Limited. [Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: the DETECT study. Coghlan JG, Denton CP, Grunig E, et al. Ann Rheum Dis. 73(7):1340–1349, copyright 2014].28 |
 |
Figure 2 The ASIG (Australian Scleroderma Interest Group) algorithm to screen for pulmonary arterial hypertension in a patient with systemic sclerosisNote: Data from Thakkar et al.52 |
While both these approaches have prospectively been shown to have a very high sensitivity and negative predictive value (NPV) – critical attributes required for a successful SSc-PAH screening approach – they have a poor positive predictive value (PPV), meaning that 40–45% of the patients (from the derivative cohorts used to develop these tools) ultimately referred to RHC did not have PAH.53 It is important to note that the PPV for these algorithms in the real world could be as low as 20% (8 out of 10 patients referred to RHC would not have PAH), as the prevalence for PAH in the SSc population is thought to be close to 10–15%, much lower than that in the derivative cohorts used to develop these algorithms.53 This is especially critical in resource-poor environments where such a screening strategy for SSc-PAH may not be economically feasible. Further research is needed to reduce this false-positive rate. It may be possible to improve the PPV and reduce the rate of unnecessary RHC referrals by utilizing the 2015 ESC/ERS echocardiographic criteria in combination with the ASIG algorithm for example and refer patients to RHC if they at least meet the intermediate probability criteria for PAH on echocardiography.30 Cardiac MRI which has a very high specificity and a high signal-to-noise ratio may help with improving this predictive accuracy.54
It is important to note that the screening strategies listed above have used the previous hemodynamic definitions for PAH (mPAP of ≥ 25 mm Hg, a PVR> 3 wood units, and normal left-sided filling pressures). The newer hemodynamic definitions for PAH proposed during the sixth world symposium on PH use an mPAP ≥ 20mm Hg, a PVR>3 wood units, and normal left-sided filling pressures to diagnose PAH.55 Currently, there are limited data with regard to the impact of vasodilator therapies on patients classified as having PAH using the new definitions. However, when these guidelines are widely implemented, it will be important to prospectively develop screening tools designed to identify PAH using the new definitions.
Therapy And Prognosis
Therapy
Pulmonary Arterial Vasodilators
The advent of pulmonary arterial vasodilator therapies has dramatically reduced the morbidity and mortality among patients with PAH. While PA vasodilator therapies have been validated for use in all WHO group I PAH patients, there is not a treatment strategy outlined specifically for SSc-PAH patients. The level of evidence and the strength/class of recommendations for the use of individual drugs in the PAH patient have been very well summarized in the 2015 ESC/ERS guidelines, and we would refer the interested reader to this publication.30 The various PA vasodilator therapies approved for use in PAH patients are listed in Table 2. The three important vasoactive biologic pathways that can be manipulated to achieve pulmonary arterial vasodilation and offload the RV are a) the nitric oxide pathway, b) endothelin pathway, and c) prostacyclin pathway.56 Achieving maximal pulmonary arterial vasodilation synergistically using the various vasoactive pathways is the cornerstone of the therapeutic strategy for managing a PAH patient.
 |
Table 2 FDA-Approved Therapies For Pulmonary Arterial Hypertension |
Calcium-Channel Blockers
Based on the preliminary studies by Rich and Brundage, high-dose calcium-channel blocker therapy seemed to benefit a subset of patients with primary pulmonary hypertension (as IPAH was known back then).57 While only a small number of patients with IPAH seemed to respond to calcium-channel blockers, these “responders” had a substantially better 5-year survival in comparison to the “non-responders” as noted in a prospective study.58 In the present day, vasoreactivity to inhaled nitric oxide or other vasodilators determined acutely during right heart catheterization is used as a surrogate marker to identify the “responder” group. Vasoreactivity testing is routinely performed during RHC as part of the diagnostic evaluation of a PAH patient. Although there were reports of acute and short-term improvement in hemodynamics with exposure to oral nifedipine in SSc-PAH patients, 59,60 no long-term calcium-channel blocker trials were undertaken. Given that the majority of SSc-PAH patients, not unlike the IPAH patients, are calcium-channel blocker non-responders, 61 and with the availability of more potent vasodilator therapies, and more importantly, considering the potential risks of the negative inotropic effects of high-dose calcium-channel blocker therapy, these drugs are no longer recommended for treatment of SSc-PAH. Considering the lack of therapeutic benefit associated with calcium-channel blocker use in SSc-PAH patients, vasoreactivity testing during RHC is not required in a patient with previously established SSc. However, it is important to note that calcium-channel blockers are still used for the treatment of Raynaud phenomenon associated with SSc.
Prostacyclin Analogues
After calcium-channel blockers, the next therapy that emerged for PAH patients was the use of prostacyclin analogues or prostanoids. The two common prostanoids delivered via a continuous infusion pump for the treatment of PAH in the US are epoprostenol and treprostinil. The first successful demonstration of the long-term use of continuous infusion IV epoprostenol was in primary pulmonary hypertension patients.62 Shortly after that, one of the earliest demonstrations of the utility of long-term infusions of IV prostacyclins in SSc-PAH patients produced mixed results. De La Mata and colleagues used continuously infused IV iloprost to treat three SSc-PAH patients in a prospective open-labeled fashion.63 Although they demonstrated an acute reduction in PA pressures and PVR, the 32-week hemodynamics worsened again. Unfortunately, the mean dose used in these patients was 2.3ng/kg/min – which is the starting dose for modern-day prostacyclin infusion therapy. It was not until a few years later that the first-ever, randomized, placebo-controlled end-point-driven study evaluating epoprostenol in PAH patients was completed. In this study, patients randomized to the epoprostenol arm demonstrated a significant reduction in the PVR and an improvement in the 6-minute walk distance (6MWD) at 12 weeks when compared to the placebo group. All fatalities that occurred in this study were in patients assigned to the placebo arm.64 Shortly after this landmark study, reports of improvements in hemodynamics with IV prostanoids in SSc-PAH patients were published.65 Four years later, the first, randomized, open-label controlled trial with IV epoprostenol in SSc-PAH patients was completed. SSc-PAH patients receiving epoprostenol had a significant improvement in exercise capacity and cardiopulmonary hemodynamics at 12 weeks.66 To this day, IV prostanoids remain the first choice for SSc-PAH patients presenting with advanced disease or WHO/NYHA class IV symptoms and the last resort when SSc-PAH patients receiving other therapies experience disease progression.
While initial attempts at using oral prostacyclin analogs to treat Raynaud symptoms in SSc patients were unsuccessful, 67 their use in the treatment of SSc-PAH met with more success. Oral treprostinil and the novel IP receptor analog selexipag both have applications in the treatment of PAH in SSc patients.68,69 A recent sub-group analysis of the GRIPHON study showed beneficial effects of selexipag in patients with SSc-PAH, with patients taking selexipag less likely to experience disease progression when compared to those receiving placebo.70 Despite the advances in the development of oral prostacyclin analogs, these drugs can be difficult to tolerate, with significant gastrointestinal side effects including nausea, vomiting, and diarrhea.68,70 A significant number of patients are unable to tolerate doses high enough to reach serum levels that are equivalent to parenterally administered prostanoids. Over the long term, a significant number of patients initiated on oral prostacyclins eventually end up transitioning to IV prostacyclins due to disease progression.71 With improvements in oral and inhaled drug formulations, we might be able to achieve a higher dose delivery through these routes in the future, but until then, parenterally delivered prostacyclins will remain one of the most potent vasodilator options for SSc-PAH patients.
Endothelin Receptor Antagonists
Endothelins (ET-1, ET-2, ET-3) are vasoactive proteins that regulate vascular tone. ET-1 is produced in the endothelial cells and has an affinity for ET-A and ET-B, both G-protein-coupled receptors. The ET-A receptor is predominantly expressed on vascular smooth muscle cells and mediates vasoconstriction.72 The ET-B receptor is expressed on endothelial cells and on vascular smooth muscle and is responsible for clearing circulating ET-1 levels but can contribute to vasoconstrictive effects in pathologic conditions such as pulmonary hypertension.73 Drugs selectively blocking the ET-A receptor (ambrisentan) and both the ET-A and ET-B receptors (bosentan and macitentan) are important therapeutic options for management of the PAH patient.
SSc is characterized by microvascular destruction, and the initiating event is thought to be endothelial cell injury.74 Endothelial injury can result in the release of ET-1, and ET-1-induced vasoconstriction coupled with the loss of vasodilatory checks and balances could be an important contributor to the vasculopathy that characterizes SSc.75 Indeed, elevated levels of circulating ET-1 levels have been noted in SSc patients with Raynaud phenomenon.76 Use of dual (ET-A + ET-B) and single (ET-A) endothelin receptor antagonists has been associated with a reduction in the rate of new digital ulcer formation77,78 and Raynaud phenomenon in SSc patients without PAH.79
Endothelin receptor blockade has been one of the therapeutic mainstays for SSc-PAH. Bosentan, ambrisentan, and more recently macitentan have all been associated with improvements in 6MWD, functional class, patient-reported quality of life and have been shown to reduce morbidity in various clinical trials for PAH patients.80–82 Although these drugs benefit SSc-PAH patients, sub-group analyses seem to demonstrate a reduced efficacy of endothelin receptor antagonists in patients with connective tissue disease-associated PAH (CTD PAH) (a significant number of patients had SSc-PAH in these studies), and the reasons for this differential treatment response are unclear.17,83
Phosphodiesterase Inhibitors
Sildenafil and tadalafil are phosphodiesterase type V inhibitors (PDE5i) that are approved for use in patients with PAH. PDE5 inhibition prevents degradation of cyclic GMP (cGMP), which relaxes vascular smooth muscle. The pivotal clinical trials that led to sildenafil – SUPER84 – and tadalafil – PHIRST85 – being approved for therapy both had a significant number of SSc-PAH patients. While no sub-group analyses focused on the SSc-PAH patients from the PHIRST study population are available, in a post-hoc analysis of the SUPER study, the 84 CTD PAH patients (43% SSc-PAH) in the sildenafil arm were noted to have experienced an improvement in exercise capacity, functional class, and hemodynamic measures.86 Sildenafil and tadalafil are widely used as first-line agents for PAH patients due to their good tolerability profile when compared to the other PA vasodilators.
Soluble Guanylate Cyclase (sGC) Stimulator
The oral sGC stimulator riociguat is a very potent alternative to PDE5is for the treatment of PAH. In the PATENT-1 study, PAH patients receiving riociguat had a significant improvement in exercise capacity, NT-Pro BNP levels, WHO functional class, and quality of life when compared to patients receiving placebo.87 Subsequently, a post-hoc subset analysis of the PATENT-1 and PATENT-2 (long-term extension study) data looking specifically at CTD PAH patients showed that CTD PAH patients receiving riociguat experienced an improvement in exercise capacity and WHO functional class and also improved hemodynamic parameters. However, the SSc-PAH population in this subset had an attenuated improvement in exercise capacity when compared to the overall CTD PAH group.88
A significant number of PAH patients might have reduced cGMP levels, and in these patients, PDE5 inhibition may not be an efficacious means of pulmonary vasodilation.89 Further, there appears to be a reduced expression of endothelial nitric oxide (NO) synthase in some PAH patients.90 Riociguat could address the deficient NO and cGMP signaling in these PAH patients by synergistically increasing the sensitivity of sGC to NO and also by the direct stimulation of sGC, thereby increasing cGMP levels.91 The RESPITE study, a multi-center, open-label uncontrolled study, evaluated switching PAH patients in WHO functional class III, 6MWD less than 440 m, a cardiac index <3L/min/m2, and PVR > 400 dynes/sec/cm5 PDE5is to riociguat.92 In this study, 54% of patients who were switched from PDE5is to riociguat had a significant improvement in 6MWD and WHO functional class and a reduction in NT-Pro BNP. This again seems to suggest that there is a subset of PAH patients that benefits from direct sGC stimulation as compared to PDE5 inhibition. It should be noted that none of the patients in this study had SSc-PAH.
Combination Therapy
With the availability of many PA vasodilator agents, combination therapy exploits the synergy of the different vasodilatory pathways to achieve optimal “RV offloading” in the SSc-PAH patient. The proceedings from the sixth world symposium on pulmonary hypertension from Nice, France, summarize the current strategy for managing patients with PAH while progressively escalating the intensity of therapy using different combinations of PA vasodilators.93
Until the earlier part of this decade, there was very little evidence to suggest that starting multiple agents “upfront” was superior to monotherapy or a sequential addition of therapies for the management of PAH. With the AMBITION study, an upfront combination therapy using tadalafil and ambrisentan proved to be superior to monotherapy with either of these agents in preventing disease progression, improving 6MWD, and reducing serum NT-Pro BNP levels.94 A post-hoc subgroup analysis of the primary analysis set of the AMBITION study population showed that CTD PAH (n=187, 63% SSc-PAH) patients who were randomized to the “upfront combination therapy” arm were significantly less likely to experience disease progression when compared to the pooled monotherapy arm. Importantly, the incidence of adverse effects was no different between the combination therapy and the pooled monotherapy arms.95 In an open-label, prospective study in incident (diagnosed within the previous 6 months) SSc-PAH patients by Hassoun et al, an upfront combination therapy with ambrisentan and tadalafil resulted in a significant improvement in RV function and WHO functional class when followed over a 36-week period.96
In building upon the principle of exploiting this “multi-pathway synergy” in the naive PAH patient, we currently do not have evidence to support a triple-upfront combination therapy using all three pathways. The TRITON study evaluating the use of initial triple oral (tadalafil, macitentan, selexipag) vs dual oral (tadalafil, macitentan) combination therapy in the treatment of newly diagnosed patients with PAH is currently underway (NCT02558231) and results should become available in the year 2020. In a subgroup analysis of the GRIPHON study that included both prevalent and incident PAH patients, Coghlan et al demonstrated that prevalent PAH patients on dual background therapy benefited from the addition of selexipag. The patients randomized to selexipag had a lower risk for meeting the composite primary endpoint of morbidity or mortality when compared to the placebo group.97 Although this is a post-hoc analysis of data from a clinical trial, results from such analyses corroborate the validity of the “progressive escalation approach” with regard to the addition of PA vasodilators in the PAH patient.
Currently, it is our practice to treat newly diagnosed, therapy-naive SSc-PAH patients with a combination of two oral therapies, usually a PDE5i/sGC stimulator in combination with an ERA (ambrisentan/macitentan). Our practice is to follow a “close sequential” up-titration of therapies given the adverse effect profile of these medications. We start with one drug and add a second agent in a week or two with up-titration of each agent in a sequential fashion in the following weeks. This approach has been helpful in associating a particular adverse effect with a drug and might potentially help improve adherence to therapy. Change in therapy or addition of another PA vasodilator is usually decided after assessing the improvement (or lack thereof) in clinical symptoms and exercise tolerance at the follow-up visit.
Immunosuppressive And Anti-Inflammatory Agents
In the past two decades, there has been an increasing amount of evidence implicating the role of immunity and inflammatory pathways in the genesis and progression of PAH.98 Understanding the role of inflammation is especially germane in SSc-PAH. Many different antibodies directed against antigens in the endothelium and fibroblasts have been detected in both IPAH and SSc-PAH patients. Immune responses directed against the endothelium could promote injury and apoptotic cell death and result in the varied clinical manifestations in SSc including PAH.98 While anti-inflammatory therapy seems to ameliorate PAH in other forms of CTD PAH, it does not appear to improve disease symptoms or hemodynamics in patients with SSc-PAH.99 The multi-center randomized placebo-controlled trial evaluating the use of rituximab in SSc-PAH patients (NCT01086540) has finished enrollment and is expected to be completed at the end of 2019. Bardoxolone methyl, an Nrf2-activator, showed promising results in a Phase II study – LARIAT (NCT02036970), and patients with IPAH and CTD PAH treated with the drug showed an improvement in 6MWD (results presented in abstract form). The multicenter, placebo-controlled Phase III study evaluating bardoxolone in CTD PAH patients – CATALYST (NCT02657356) – is nearing completion, and results from this study should be able to inform us about the role of adjunctive therapies in CTD PAH who are already being treated with PA vasodilators.
Anti-Proliferative Agents
PAH is a disease characterized by an obliterative vasculopathy resulting from unchecked endothelial and pulmonary vascular smooth muscle proliferation.100 Since this is a progressive disease not unlike a neoplastic process, there has been an interest in the role of anti-proliferative agents in preventing the progression of the vasculopathy underlying PAH. Platelet-derived growth factor (PDGF) and c-KIT signaling appear to play an important role in vascular smooth muscle proliferation and have been identified as potential therapeutic targets in PAH.101,102 The tyrosine kinase inhibitor (TKI) – imatinib mesylate, which in addition to being an inhibitor of the BCR-ABL tyrosine kinase in CML (chronic myelogenous leukemia) patients, was noted to have inhibitory effects on the PDGF receptors (PDGFR) and C-Kit (a receptor tyrosine kinase). Given its record of safety in the treatment of CML patients, it seemed reasonable to repurpose it for PAH treatment. Therapeutic interest in imatinib mesylate increased after anecdotal reports showed some promise in the management of patients with advanced and often, end-stage PAH, not responding to maximal therapy with PA vasodilators.103 The IMPRES trial was a multicenter Phase III study that evaluated the use of imatinib for patients with IPAH and CTD PAH with 6MWD as the primary outcome measure. In this study, patients randomized to imatinib therapy had a significantly greater improvement in 6MWD when compared to the placebo arm. There was some hemodynamic improvement in the imatinib arm, but serious adverse events including sub-arachnoid hemorrhages were noted in the patients treated with imatinib.104 The risk of intracranial hemorrhages was again noted in a long-term observational study that included patients with both IPAH (n=13) and CTD PAH (n=2).105 Thus, despite some clinical and hemodynamic improvements noted with imatinib use, the risk of serious adverse events such as intra-cranial hemorrhages has cast questions on its viability as a safe adjunctive therapy in PAH patients.
Another anti-proliferative agent with therapeutic potential in the PAH arena appears to be tacrolimus (FK-506). FK-506 is thought to exert its anti-proliferative effects on the endothelial cells by the activation of BMPR2 (bone morphogenic protein receptor 2). BMPR2 signaling, which is important for endothelial cell regulation, is dysfunctional and/or downregulated in PAH patients and activating BMPR2 could ameliorate endothelial proliferation.106 Some positive clinical experiences with tacrolimus in end-stage PAH patients107 paved the way for a Phase IIa clinical trial. Low-dose tacrolimus (2–5ng/mL) was found to be well tolerated in PAH patients (including CTD PAH patients) without major adverse reactions for a period of 16 weeks in the Phase IIa study. A subset of patients did demonstrate an increase in BMPR2 levels.108 Future, large, event-driven, outcomes-based studies in the future will determine the utility of tacrolimus as an adjunctive therapy in patients with PAH.
While intravenous prostacyclin analogs remain the earliest developed and the most potent therapies for the management of PAH, we now have an armamentarium of PA vasodilator therapies spanning all the different routes of administration (Table 2). There is now considerable clinical evidence to direct early and upfront combination of vasodilator therapy for the management of all PAH patients, including SSc-PAH patients. The most recent guidelines from the sixth world symposium on pulmonary hypertension underscore the need for aggressive therapy with vasodilators for WHO group I PH patients and SSc-PAH patients are no exception.93 Results from ongoing human studies using anti-proliferative, immunosuppressive, and novel anti-inflammatory agents will determine their utility as adjunctive therapies for PAH patients.
Prognosis And Risk Stratification
In the last decade, there have been significant advances with regard to the risk stratification of PAH patients who are already on therapy. The two well-validated risk stratification algorithms that serve as useful guides for providers caring for PAH patients are the REVEAL 2.0 risk calculator109 and the 2015 ESC/ERS risk stratification guidelines.30 Broadly speaking, PAH risk stratification tools delineate the mortality risk in a PAH patient based on clinical parameters such as exercise capacity, RV function, serum cardiac biomarkers, and hemodynamic indicators from RHC.93 The REVEAL 2.0 risk calculator was derived from the REVEAL (Registry to Evaluate Early and Long-Term PAH Disease Management) registry which was an industry-sponsored observational study that enrolled patients with WHO group I PAH from different regions within the US. This registry was established in 2006 and completed enrollment in 2012 and was originally designed to provide longitudinal follow-up data for a period of 5 years.110 The calculator was derived from a Bayesian analysis of prospectively collected mortality data over a period of 1 year for all the patients in the registry. The resultant risk score assigns a low (<5%), intermediate (5–10%), and high (>10%) 1-year mortality risk.109 The ESC/ERS risk stratification tool, on the other hand, utilizes a collection of clinical parameters and threshold values for those parameters, most of which are based upon expert opinion, to stratify patients into low (<5%), intermediate (5–10%), and high (>10%) 1-year mortality risk categories.30
While both the ESC/ERS risk stratification tool and the REVEAL 2.0 calculator classify patients into low-, intermediate-, and high-risk categories, one advantage of the REVEAL calculator over other risk stratification tools, especially with regard to SSc, could be that the REVEAL registry contained a significantly larger number of SSc-PAH patients when compared to other registries from around the world. In fact, over 25% of patients in this registry had connective tissue disease-associated PAH, with a significant number having SSc-PAH.109 The REVEAL calculator appears to have reasonably good validity for the risk stratification of incident SSc-PAH patients, as demonstrated by the study by Mullin and colleagues in the PHAROS population.111 Risk stratification tools help the clinical provider add or titrate vasodilator therapies with a goal to prospectively achieve the lowest possible risk category. Risk stratification also helps identify patients unresponsive to therapies and consider lung transplantation in a timely fashion.
Historical cohort analyses comparing the survival trends between IPAH and SSc-PAH patients have ascribed a survival disadvantage to the SSc-PAH population.14 With advances in risk stratification, improvements in the awareness and the diagnostic process, and more available combination therapies for the treatment of PAH, there have been some improvements in the short-term survival in the incident SSc-PAH patients.16,112 While the 1- and 3-year survival among incident SSc-PAH patients appears comparable to IPAH patients in the modern era, the overall median survival of incident SSc-PAH patients is still significantly worse when compared to IPAH patients.15,112,113 The reasons for this are incompletely understood at this time. Poor RV adaptation to pulmonary vascular load in the SSc-PAH patients vs the IPAH patients is thought to be one reason.114–116 Another driver of this difference could be that SSc-PAH appears to be a distinct pulmonary vasculopathy when compared to IPAH. Histopathological analyses of explanted lungs from SSc-PAH and IPAH patients seem to corroborate some of these findings.117 In a pulmonary vascular histo-morphometric analysis comparing IPAH with SSc-PAH lung specimens, we identified differences between the two diseases. In our sample, we did not note any plexiform lesions in the SSc-PAH lungs, while 33% of IPAH lungs had plexiform lesions. There was a significantly greater amount of interstitial cellularity and fibrosis noted in the SSc-PAH lung specimens when compared to the IPAH lungs (unpublished data – manuscript in preparation).
Another important difference between SSc-PAH patients and IPAH patients is with regard to their clinical response to PA vasodilator therapies. While patients with IPAH have had a significant improvement in morbidity with use of single and combination PA vasodilator therapies, these responses have not uniformly translated into all the WHO group I phenotypes and especially to the SSc-PAH population. Rhee and colleagues analyzed data from various therapeutic clinical trials over the last decade and compared treatment responses between the CTD PAH patients (56% had SSc-PAH) and IPAH patients.17 Their retrospective multi-cohort analysis showed that PA vasodilator therapies were less efficacious in CTD PAH patients with regard to improvement in 6MWD and in preventing disease progression when compared to the IPAH patients. The treatment heterogeneity between IPAH and SSc-PAH again brings into the spotlight the inherent differences between these two disease entities. Across various cohort analyses (for both incident and prevalent PAH patients) comparing the incident SSc-PAH with IPAH patients, the SSc-PAH patients, despite having lower mean PA pressures and PVR values when compared to the IPAH patients, have significantly lower DLCO values when compared to their IPAH counterparts. DLCO depends on both pulmonary capillary blood volume and membrane conductance – both of which are likely affected in SSc-PAH patients (possibly from subclinical interstitial fibrosis as noted in our histo-morphometric analyses). Perhaps, SSc-PAH patients have a severely reduced pulmonary capillary surface area (aggressive vasculopathy with additional capillary loss from interstitial fibrosis) when compared to IPAH patients, and so these patients have a very low “recruitable” capillary population, and this could probably explain the attenuated treatment responses. In our clinical practice, we often see patients with significant restriction noted on lung function testing with minimal to no ILD noted on high-resolution computed tomogram of the chest evaluation. Histo-morphometric analyses from our group, as previously noted, seem to demonstrate a significantly higher burden of interstitial cellularity and fibrosis in end-stage/advanced SSc-PAH patients when compared to IPAH patients with advanced disease (unpublished data – manuscript in preparation).
It is important to understand these differences to be able to develop therapies tailored to SSc-PAH patients. The role of antifibrotic therapies and immunomodulator therapies as adjuncts to PA vasodilator-based treatment in the SSc-PAH population is still unclear. Results from some of the ongoing clinical trials looking at immunosuppression and novel inflammatory modulation in the CTD PAH population will further inform our strategies to manage these challenging patients better. In Summary, SSc-PAH is a challenging disease that is a leading cause of mortality in patients with SSc. With advances in screening strategies, greater availability of PA vasodilator combinations, and improvements in risk stratification tools, there have been improvements in short-term survival for incident SSc-PAH patients. However, overall survival still remains worse in SSc-PAH when compared to IPAH patients, and importantly, SSc-PAH patients appear to be at a disadvantage when compared to IPAH patients with regard to treatment responses to PA vasodilators. With advances in our understanding of the pulmonary vasculopathy and RV adaptation in SSc-PAH, availability of novel immunomodulatory and anti-inflammatory therapies, we hope to be able to develop a therapeutic strategy “personalized” for SSc-PAH patients in the near future.
Disclosure
Dr Rahul G Argula serves in the Scientific Advisory Board at Bayer Pharmaceuticals. He also serves in the Physician Advisory Board at United Therapeutics, Gilead Pharmaceuticals, and Actelion Pharmaceuticals. He received educational grant from REATA pharmaceuticals and owns stocks from Gilead pharmaceuticals, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Chaisson NF, Hassoun PM. Systemic sclerosis-associated pulmonary arterial hypertension. Chest. 2013;144(4):1346–1356. doi:10.1378/chest.12-2396
2. Denton CP, Khanna D. Systemic sclerosis. Lancet. 2017;390(10103):1685–1699. doi:10.1016/S0140-6736(17)30933-9
3. Steen VD, Medsger TA. Changes in causes of death in systemic sclerosis, 1972-2002. Ann Rheum Dis. 2007;66(7):940–944. doi:10.1136/ard.2006.066068
4. Schoenfeld SR, Castelino FV. Evaluation and management approaches for scleroderma lung disease. Ther Adv Respir Dis. 2017;11(8):327–340. doi:10.1177/1753465817713680
5. Tuder RM, Stacher E, Robinson J, Kumar R, Graham BB. Pathology of pulmonary hypertension. Clin Chest Med. 2013;34(4):639–650. doi:10.1016/j.ccm.2013.08.009
6. Stacher E, Graham BB, Hunt JM, et al. Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;186(3):261–272. doi:10.1164/rccm.201201-0164OC
7. Sanz J, Kariisa M, Dellegrottaglie S, et al. Evaluation of pulmonary artery stiffness in pulmonary hypertension with cardiac magnetic resonance. JACC Cardiovasc Imaging. 2009;2(3):286–295. doi:10.1016/j.jcmg.2008.08.007
8. D’Alonzo GE, Barst RJ, Ayres SM, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med. 1991;115(5):343–349. doi:10.7326/0003-4819-115-5-343
9. Kirson NY, Birnbaum HG, Ivanova JI, Waldman T, Joish V, Williamson T. Prevalence of pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension in the United States. Curr Med Res Opin. 2011;27(9):1763–1768. doi:10.1185/03007995.2011.604310
10. Hassoun PM. Pulmonary arterial hypertension complicating connective tissue diseases. Semin Respir Crit Care Med. 2009;30(4):429–439. doi:10.1055/s-0029-1233312
11. Fischer A, Bull TM, Steen VD. Practical approach to screening for scleroderma-associated pulmonary arterial hypertension. Arthritis Care Res (Hoboken). 2012;64(3):303–310. doi:10.1002/acr.20693
12. Schreiber BE, Connolly MJ, Coghlan JG. Pulmonary hypertension in systemic lupus erythematosus. Best Pract Res Clin Rheumatol. 2013;27(3):425–434. doi:10.1016/j.berh.2013.07.011
13. Shahane A. Pulmonary hypertension in rheumatic diseases: epidemiology and pathogenesis. Rheumatol Int. 2013;33(7):1655–1667. doi:10.1007/s00296-012-2659-y
14. Rubenfire M, Huffman MD, Krishnan S, Seibold JR, Schiopu E, McLaughlin VV. Survival in systemic sclerosis with pulmonary arterial hypertension has not improved in the modern era. Chest. 2013;144(4):1282–1290. doi:10.1378/chest.12-0653
15. Ramjug S, Hussain N, Hurdman J, et al. Idiopathic and systemic sclerosis-associated pulmonary arterial hypertension: a comparison of demographic, hemodynamic, and mri characteristics and outcomes. Chest. 2017;152(1):92–102. doi:10.1016/j.chest.2017.02.010
16. Kolstad KD, Li S, Steen V, Chung L, PHAROS Investigators. Long-term outcomes in systemic sclerosis-associated pulmonary arterial hypertension from the Pulmonary Hypertension Assessment and Recognition of Outcomes in Scleroderma Registry (PHAROS). Chest. 2018;154(4):862–871. doi:10.1016/j.chest.2018.05.002
17. Rhee RL, Gabler NB, Sangani S, Praestgaard A, Merkel PA, Kawut SM. Comparison of treatment response in idiopathic and connective tissue disease-associated pulmonary arterial hypertension. Am J Respir Crit Care Med. 2015. doi:10.1164/rccm.201507-1456OC
18. Hachulla E, Gressin V, Guillevin L, et al. Early detection of pulmonary arterial hypertension in systemic sclerosis: a French nationwide prospective multicenter study. Arthritis Rheum. 2005;52(12):3792–3800. doi:10.1002/art.21433
19. Avouac J, Airo P, Meune C, et al. Prevalence of pulmonary hypertension in systemic sclerosis in European Caucasians and metaanalysis of 5 studies. J Rheumatol. 2010;37(11):2290–2298. doi:10.3899/jrheum.100245
20. Frost A, Badesch D, Gibbs JSR, et al. Diagnosis of pulmonary hypertension. Eur Respir J. 2019;53(1). doi:10.1183/13993003.01904,2018 Print 2019 Jan.
21. Hachulla E, de Groote P, Gressin V, et al. The three-year incidence of pulmonary arterial hypertension associated with systemic sclerosis in a multicenter nationwide longitudinal study in France. Arthritis Rheum. 2009;60(6):1831–1839. doi:10.1002/art.24525
22. Rich S, Dantzker DR, Ayres SM, et al. Primary pulmonary hypertension. A national prospective study. Ann Intern Med. 1987;107(2):216–223. doi:10.7326/0003-4819-107-2-216
23. Badesch DB, Raskob GE, Elliott CG, et al. Pulmonary arterial hypertension: baseline characteristics from the REVEAL Registry. Chest. 2010;137(2):376–387. doi:10.1378/chest.09-1140
24. Naeije R. Pulmonary vascular function. In: Peacock AJ, Naeije R, Rubin LJ, editors. Pulmonary Circulation: Disease and Their Treatment.
25. Chung L, Domsic RT, Lingala B, et al. Survival and predictors of mortality in systemic sclerosis-associated pulmonary arterial hypertension: outcomes from the pulmonary hypertension assessment and recognition of outcomes in scleroderma registry. Arthritis Care Res (Hoboken). 2014;66(3):489–495. doi:10.1002/acr.22121
26. Hinchcliff M, Fischer A, Schiopu E, Steen VD, PHAROS Investigators. Pulmonary Hypertension Assessment and Recognition of Outcomes in Scleroderma (PHAROS): baseline characteristics and description of study population. J Rheumatol. 2011;38(10):2172–2179. doi:10.3899/jrheum.101243
27. Shah AA, Wigley FM, Hummers LK. Telangiectases in scleroderma: a potential clinical marker of pulmonary arterial hypertension. J Rheumatol. 2010;37(1):98–104. doi:10.3899/jrheum.090697
28. Coghlan JG, Denton CP, Grunig E, et al. Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: the DETECT study. Ann Rheum Dis. 2014;73(7):1340–1349. doi:10.1136/annrheumdis-2013-203301
29. Farha S, Laskowski D, George D, et al. Loss of alveolar membrane diffusing capacity and pulmonary capillary blood volume in pulmonary arterial hypertension. Respir Res. 2013;14(1):6. doi:10.1186/1465-9921-14-19
30. Galie N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: the joint task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J. 2016;37(1):67–119. doi:10.1093/eurheartj/ehv317
31. Khanna D, Gladue H, Channick R, et al. Recommendations for screening and detection of connective tissue disease-associated pulmonary arterial hypertension. Arthritis Rheum. 2013;65(12):3194–3201. doi:10.1002/art.38172
32. Shah AA, Chung S, Wigley FM, Wise RA, Hummers LK. Changes in estimated right ventricular systolic pressure predict mortality and pulmonary hypertension in a cohort of scleroderma patients. Ann Rheum Dis. 2013;72(7):1136–1140. BMJ Publishing Group Ltd and European League Against Rheumatism. doi:10.1136/annrheumdis-2012-201861
33. Steen VD. Autoantibodies in systemic sclerosis. Semin Arthritis Rheum. 2005;35(1):35–42. doi:10.1016/j.semarthrit.2005.03.005
34. Graf SW, Hakendorf P, Lester S, et al. South Australian Scleroderma Register: autoantibodies as predictive biomarkers of phenotype and outcome. Int J Rheum Dis. 2012;15(1):102–109. doi:10.1111/j.1756-185X.2011.01688.x
35. Steen V, Medsger TA
36. Hinchcliff M, Khanna S, Hsu VM, et al. Survival in systemic sclerosis–pulmonary arterial hypertension by serum autoantibody status in the Pulmonary Hypertension Assessment and Recognition of Outcomes in Scleroderma (PHAROS) registry. Semin Arthritis Rheum. 2015;45(3):309–314. doi:10.1016/j.semarthrit.2015.06.011
37. Assous N, Allanore Y, Batteux F, et al. Prevalence of antiphospholipid antibodies in systemic sclerosis and association with primitive pulmonary arterial hypertension and endothelial injury. Clin Exp Rheumatol. 2005;23(2):199–204.
38. Negi VS, Tripathy NK, Misra R, Nityanand S. Antiendothelial cell antibodies in scleroderma correlate with severe digital ischemia and pulmonary arterial hypertension. J Rheumatol. 1998;25(3):462–466.
39. Lewandowska K, Ciurzynski M, Gorska E, et al. Antiendothelial cells antibodies in patients with systemic sclerosis in relation to pulmonary hypertension and lung fibrosis. Adv Exp Med Biol. 2013;756:147–153. doi:10.1007/978-94-007-4549-0_19
40. Kampolis C, Plastiras SC, Vlachoyiannopoulos PG, Moyssakis I, Tzelepis GE. The presence of anti‐centromere antibodies may predict progression of estimated pulmonary arterial systolic pressure in systemic sclerosis. Scand J Rheumatol. 2008;37(4):278–283. doi:10.1080/03009740801978871
41. Hsu VM, Chung L, Hummers LK, et al. Risk factors for mortality and cardiopulmonary hospitalization in systemic sclerosis patients at risk for pulmonary hypertension, in the PHAROS registry. J Rheumatol. 2019;46(2):176–183. doi:10.3899/jrheum.180018
42. Becker MO, Kill A, Kutsche M, et al. Vascular receptor autoantibodies in pulmonary arterial hypertension associated with systemic sclerosis. Am J Respir Crit Care Med. 2014;190(7):808–817. doi:10.1164/rccm.201403-0442OC
43. Casserly B, Klinger JR. Brain natriuretic peptide in pulmonary arterial hypertension: biomarker and potential therapeutic agent. Drug Des Devel Ther. 2009;29(3):269–287.
44. Nagaya N, Nishikimi T, Okano Y, et al. Plasma brain natriuretic peptide levels increase in proportion to the extent of right ventricular dysfunction in pulmonary hypertension. J Am Coll Cardiol. 1998;31(1):202–208. doi:10.1016/s0735-1097(97)00452-x
45. Souza R, Jardim C, Julio Cesar Fernandes C, Silveira LM, Rabelo R, Humbert M. NT-proBNP as a tool to stratify disease severity in pulmonary arterial hypertension. Respir Med. 2007;101(1):69–75. doi:10.1016/j.rmed.2006.04.014
46. Allanore Y, Borderie D, Meune C, et al. N-terminal pro-brain natriuretic peptide as a diagnostic marker of early pulmonary artery hypertension in patients with systemic sclerosis and effects of calcium-channel blockers. Arthritis Rheum. 2003;48(12):3503–3508. doi:10.1002/art.11345
47. Mukerjee D, St George D, Knight C, et al. Echocardiography and pulmonary function as screening tests for pulmonary arterial hypertension in systemic sclerosis. Rheumatology (Oxford). 2004;43(4):461–466. doi:10.1093/rheumatology/keh067
48. Williams MH, Handler CE, Akram R, et al. Role of N-terminal brain natriuretic peptide (N-TproBNP) in scleroderma-associated pulmonary arterial hypertension. Eur Heart J. 2006;27(12):1485–1494. doi:10.1093/eurheartj/ehi891
49. Xu J, Kimball TR, Lorenz JN, et al. GDF15/MIC-1 functions as a protective and antihypertrophic factor released from the myocardium in association with SMAD protein activation. Circ Res. 2006;98(3):342–350. doi:10.1161/01.RES.0000202804.84885.d0
50. Kempf T, Eden M, Strelau J, et al. The transforming growth factor-beta superfamily member growth-differentiation factor-15 protects the heart from ischemia/reperfusion injury. Circ Res. 2006;98(3):351–360. doi:10.1161/01.RES.0000202805.73038.48
51. Meadows CA, Risbano MG, Zhang L, et al. Increased expression of growth differentiation factor-15 in systemic sclerosis-associated pulmonary arterial hypertension. Chest. 2011;139(5):994–1002. doi:10.1378/chest.10-0302
52. Thakkar V, Stevens WM, Prior D, et al. N-terminal pro-brain natriuretic peptide in a novel screening algorithm for pulmonary arterial hypertension in systemic sclerosis: a case-control study. Arthritis Res Ther. 2012;14(3):R143. doi:10.1186/ar3876
53. Hao Y, Thakkar V, Stevens W, et al. A comparison of the predictive accuracy of three screening models for pulmonary arterial hypertension in systemic sclerosis. Arthritis Res Ther. 2015;17:
54. Addetia K, Bhave NM, Tabit CE, et al. Sample size and cost analysis for pulmonary arterial hypertension drug trials using various imaging modalities to assess right ventricular size and function end points. Circ Cardiovasc Imaging. 2014;7(1):115–124. doi:10.1161/CIRCIMAGING.113.000932
55. Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019;53(1). doi:10.1183/13993003.01913,2018 Print 2019 Jan.
56. Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med. 2004;351(14):1425–1436. doi:10.1056/NEJMra040291
57. Rich S, Brundage BH. High-dose calcium channel-blocking therapy for primary pulmonary hypertension: evidence for long-term reduction in pulmonary arterial pressure and regression of right ventricular hypertrophy. Circulation. 1987;76(1):135–141. doi:10.1161/01.cir.76.1.135
58. Rich S, Kaufmann E, Levy PS. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N Engl J Med. 1992;327(2):76–81. doi:10.1056/NEJM199207093270203
59. Sfikakis PP, Kyriakidis MK, Vergos CG, et al. Cardiopulmonary hemodynamics in systemic sclerosis and response to nifedipine and captopril. Am J Med. 1991;90(5):541–546.
60. Alpert MA, Pressly TA, Mukerji V, et al. Acute and long-term effects of nifedipine on pulmonary and systemic hemodynamics in patients with pulmonary hypertension associated with diffuse systemic sclerosis, the CREST syndrome and mixed connective tissue disease. Am J Cardiol. 1991;68(17):1687–1691. doi:10.1016/0002-9149(91)90330-n
61. Humbert M, Sitbon O, Chaouat A, et al. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med. 2006;173(9):1023–1030. doi:10.1164/rccm.200510-1668OC
62. Rubin LJ, Mendoza J, Hood M, et al. Treatment of primary pulmonary hypertension with continuous intravenous prostacyclin (epoprostenol). Results of a randomized trial. Ann Intern Med. 1990;112(7):485–491. doi:10.7326/0003-4819-112-7-485
63. de la Mata J, Gomez-Sanchez MA, Aranzana M, Gomez-Reino JJ. Long-term iloprost infusion therapy for severe pulmonary hypertension in patients with connective tissue diseases. Arthritis Rheum. 1994;37(10):1528–1533. doi:10.1002/art.1780371018
64. Barst RJ, Rubin LJ, Long WA, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med. 1996;334(5):296–301. doi:10.1056/NEJM199602013340504
65. Menon N, McAlpine L, Peacock AJ, Madhok R. The acute effects of prostacyclin on pulmonary hemodynamics in patients with pulmonary hypertension secondary to systemic sclerosis. Arthritis Rheum. 1998;41(3):466–469. doi:10.1002/1529-0131(199803)41:3<466::AID-ART13>3.0.CO;2-O
66. Badesch DB, Tapson VF, McGoon MD, et al. Continuous intravenous epoprostenol for pulmonary hypertension due to the scleroderma spectrum of disease. A randomized, controlled trial. Ann Intern Med. 2000;132(6):425–434. doi:10.7326/0003-4819-132-6-200003210-00002
67. Wigley FM, Korn JH, Csuka ME, et al. Oral iloprost treatment in patients with Raynaud’s phenomenon secondary to systemic sclerosis: a multicenter, placebo-controlled, double-blind study. Arthritis Rheum. 1998;41(4):670–677. doi:10.1002/1529-0131(199804)41:4<670::AID-ART14>3.0.CO;2-I
68. Jing ZC, Parikh K, Pulido T, et al. Efficacy and safety of oral treprostinil monotherapy for the treatment of pulmonary arterial hypertension: a randomized, controlled trial. Circulation. 2013;127(5):624–633. doi:10.1161/CIRCULATIONAHA.112.124388
69. Sitbon O, Channick R, Chin KM, et al. Selexipag for the treatment of pulmonary arterial hypertension. N Engl J Med. 2015;373(26):2522–2533. doi:10.1056/NEJMoa1503184
70. Gaine S, Chin K, Coghlan G, et al. Selexipag for the treatment of connective tissue disease-associated pulmonary arterial hypertension. Eur Respir J. 2017;50(2). doi:10.1183/13993003.02493,2016 Print 2017 Aug.
71. Chin KM, Ruggiero R, Bartolome S, et al. Long-term therapy with oral treprostinil in pulmonary arterial hypertension failed to lead to improvement in important physiologic measures: results from a single center. Pulm Circ. 2015;5(3):513–520. doi:10.1086/682224
72. Rubin LJ. Endothelin receptor antagonists for the treatment of pulmonary artery hypertension. Life Sci. 2012;91(13–14):517–521. doi:10.1016/j.lfs.2012.07.033
73. Dupuis J, Jasmin JF, Prie S, Cernacek P. Importance of local production of endothelin-1 and of the ET(B)Receptor in the regulation of pulmonary vascular tone. Pulm Pharmacol Ther. 2000;13(3):135–140. doi:10.1006/pupt.2000.0242
74. Campbell PM, LeRoy EC. Pathogenesis of systemic sclerosis: a vascular hypothesis. Semin Arthritis Rheum. 1975;4(4):351–368.
75. Katsumoto TR, Whitfield ML, Connolly MK. The pathogenesis of systemic sclerosis. Annu Rev Pathol. 2011;6:509–537. doi:10.1146/annurev-pathol-011110-130312
76. Zamora MR, O’Brien RF, Rutherford RB, Weil JV. Serum endothelin-1 concentrations and cold provocation in primary Raynaud’s phenomenon. Lancet. 1990;336(8724):1144–1147. doi:10.1016/0140-6736(90)92766-b
77. Korn JH, Mayes M, Matucci Cerinic M, et al. Digital ulcers in systemic sclerosis: prevention by treatment with bosentan, an oral endothelin receptor antagonist. Arthritis Rheum. 2004;50(12):3985–3993. doi:10.1002/art.20676
78. Matucci-Cerinic M, Denton CP, Furst DE, et al. Bosentan treatment of digital ulcers related to systemic sclerosis: results from the RAPIDS-2 randomised, double-blind, placebo-controlled trial. Ann Rheum Dis. 2011;70(1):32–38. doi:10.1136/ard.2010.130658
79. Parisi S, Peroni CL, Lagana A, et al. Efficacy of ambrisentan in the treatment of digital ulcers in patients with systemic sclerosis: a preliminary study. Rheumatology (Oxford). 2013;52(6):1142–1144. doi:10.1093/rheumatology/ket019
80. Rubin LJ, Badesch DB, Barst RJ, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med. 2002;346(12):896–903. doi:10.1056/NEJMoa012212
81. Galie N, Olschewski H, Oudiz RJ, et al. Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation. 2008;117(23):3010–3019. doi:10.1161/CIRCULATIONAHA.107.742510
82. Pulido T, Adzerikho I, Channick RN, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369(9):809–818. doi:10.1056/NEJMoa1213917
83. Girgis RE, Mathai SC, Krishnan JA, Wigley FM, Hassoun PM. Long-term outcome of bosentan treatment in idiopathic pulmonary arterial hypertension and pulmonary arterial hypertension associated with the scleroderma spectrum of diseases. J Heart Lung Transplant. 2005;24(10):1626–1631. doi:10.1016/j.healun.2004.12.113
84. Galie N, Ghofrani HA, Torbicki A, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005;353(20):2148–2157. doi:10.1056/NEJMoa050010
85. Galie N, Brundage BH, Ghofrani HA, et al. Tadalafil therapy for pulmonary arterial hypertension. Circulation. 2009;119(22):2894–2903. doi:10.1161/CIRCULATIONAHA.108.839274
86. Badesch DB, Hill NS, Burgess G, et al. Sildenafil for pulmonary arterial hypertension associated with connective tissue disease. J Rheumatol. 2007;34(12):2417–2422.
87. Ghofrani HA, Galie N, Grimminger F, et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med. 2013;369(4):330–340. doi:10.1056/NEJMoa1209655
88. Humbert M, Coghlan JG, Ghofrani H-A, et al. Riociguat for the treatment of pulmonary arterial hypertension associated with connective tissue disease: results from PATENT-1 and PATENT-2. Ann Rheum Dis. 2017;76(2):422–426. doi:10.1136/annrheumdis-2015-209087
89. Chockalingam A, Gnanavelu G, Venkatesan S, et al. Efficacy and optimal dose of sildenafil in primary pulmonary hypertension. Int J Cardiol. 2005;99(1):91–95. doi:10.1016/j.ijcard.2003.12.023
90. Giaid A, Saleh D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med. 1995;333(4):214–221. doi:10.1056/NEJM199507273330403
91. Stasch JP, Pacher P, Evgenov OV. Soluble guanylate cyclase as an emerging therapeutic target in cardiopulmonary disease. Circulation. 2011;123(20):2263–2273. doi:10.1161/CIRCULATIONAHA.110.981738
92. Hoeper MM, Simonneau G, Corris PA, et al. RESPITE: switching to riociguat in pulmonary arterial hypertension patients with inadequate response to phosphodiesterase-5 inhibitors. Eur Respir J. 2017;50(3). doi:10.1183/13993003.02425,2016 Print 2017 Sep.
93. Galie N, Channick RN, Frantz RP, et al. Risk stratification and medical therapy of pulmonary arterial hypertension. Eur Respir J. 2019;53(1). doi:10.1183/13993003.01889,2018 Print 2019 Jan.
94. Galie N, Barbera JA, Frost AE, et al. Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. N Engl J Med. 2015;373(9):834–844. doi:10.1056/NEJMoa1413687
95. Coghlan JG, Galie N, Barbera JA, et al. Initial combination therapy with ambrisentan and tadalafil in connective tissue disease-associated pulmonary arterial hypertension (CTD-PAH): subgroup analysis from the AMBITION trial. Ann Rheum Dis. 2017;76(7):1219–1227. doi:10.1136/annrheumdis-2016-210236
96. Hassoun PM, Zamanian RT, Damico R, et al. Ambrisentan and tadalafil up-front combination therapy in scleroderma-associated pulmonary arterial hypertension. Am J Respir Crit Care Med. 2015;192(9):1102–1110. doi:10.1164/rccm.201507-1398OC
97. Coghlan JG, Channick R, Chin K, et al. Targeting the prostacyclin pathway with selexipag in patients with pulmonary arterial hypertension receiving double combination therapy: insights from the randomized controlled GRIPHON study. Am J Cardiovasc Drugs. 2018;18(1):37–47. doi:10.1007/s40256-017-0262-z
98. Rabinovitch M, Guignabert C, Humbert M, Nicolls MR. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ Res. 2014;115(1):165–175. doi:10.1161/CIRCRESAHA.113.301141
99. Sanchez O, Sitbon O, Jais X, Simonneau G, Humbert M. Immunosuppressive therapy in connective tissue diseases-associated pulmonary arterial hypertension. Chest. 2006;130(1):182–189. doi:10.1378/chest.130.1.182
100. Morrell NW, Adnot S, Archer SL, et al. Cellular and molecular basis of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54(1 Suppl):S20–31. doi:10.1016/j.jacc.2009.04.018
101. Montani D, Perros F, Gambaryan N, et al. C-kit-positive cells accumulate in remodeled vessels of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2011;184(1):116–123. doi:10.1164/rccm.201006-0905OC
102. Perros F, Montani D, Dorfmuller P, et al. Platelet-derived growth factor expression and function in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2008;178(1):81–88. doi:10.1164/rccm.200707-1037OC
103. Ghofrani HA, Seeger W, Grimminger F. Imatinib for the treatment of pulmonary arterial hypertension. N Engl J Med. 2005;353(13):1412–1413. doi:10.1056/NEJMc051946
104. Hoeper MM, Barst RJ, Bourge RC, et al. Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: results of the randomized IMPRES study. Circulation. 2013;127(10):1128–1138. doi:10.1161/CIRCULATIONAHA.112.000765
105. Speich R, Ulrich S, Domenighetti G, et al. Efficacy and safety of long-term imatinib therapy for pulmonary arterial hypertension. Respiration. 2015;89(6):515–524. doi:10.1159/000381923
106. Spiekerkoetter E, Tian X, Cai J, et al. FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J Clin Invest. 2013;123(8):3600–3613. doi:10.1172/JCI65592
107. Spiekerkoetter E, Sung YK, Sudheendra D, et al. Low-dose FK506 (tacrolimus) in end-stage pulmonary arterial hypertension. Am J Respir Crit Care Med. 2015;192(2):254–257. doi:10.1164/rccm.201411-2061LE
108. Spiekerkoetter E, Sung YK, Sudheendra D, et al. Randomised placebo-controlled safety and tolerability trial of FK506 (tacrolimus) for pulmonary arterial hypertension. Eur Respir J. 2017;50(3). doi:10.1183/13993003.02449,2016 Print 2017 Sep.
109. Benza RL, Gomberg-Maitland M, Elliott CG, et al. Predicting survival in patients with pulmonary arterial hypertension: the REVEAL risk score calculator 2.0 and comparison with ESC/ERS-based risk assessment strategies. Chest. 2019. doi:10.1016/j.chest.2019.02.004
110. McGoon MD, Krichman A, Farber HW, et al. Design of the REVEAL registry for US patients with pulmonary arterial hypertension. Mayo Clin Proc. 2008;83(8):923–931. doi:10.4065/83.8.923
111. Mullin CJ, Khair RM, Damico RL, et al. Validation of the REVEAL prognostic equation and risk score calculator in incident systemic sclerosis-associated pulmonary arterial hypertension. Arthritis Rheumatol. 2019. doi:10.1002/art.40918
112. Humbert M, Sitbon O, Chaouat A, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation. 2010;122(2):156–163. doi:10.1161/CIRCULATIONAHA.109.911818
113. Morrisroe K, Stevens W, Huq M, et al. Survival and quality of life in incident systemic sclerosis-related pulmonary arterial hypertension. Arthritis Res Ther. 2017;19(1):
114. Tedford RJ, Mudd JO, Girgis RE, et al. Right ventricular dysfunction in systemic sclerosis-associated pulmonary arterial hypertension. Circ Heart Fail. 2013;6(5):953–963. doi:10.1161/CIRCHEARTFAILURE.112.000008
115. Hsu S, Kokkonen-Simon KM, Kirk JA, et al. Right ventricular myofilament functional differences in humans with systemic sclerosis-associated versus idiopathic pulmonary arterial hypertension. Circulation. 2018;137(22):2360–2370. doi:10.1161/CIRCULATIONAHA.117.033147
116. Argula RG, Karwa A, Lauer A, et al. Differences in right ventricular functional changes during treatment between systemic sclerosis-associated pulmonary arterial hypertension and idiopathic pulmonary arterial hypertension. Ann Am Thorac Soc. 2017;14(5):682–689. doi:10.1513/AnnalsATS.201608-655OC
117. Overbeek MJ, Vonk MC, Boonstra A, et al. Pulmonary arterial hypertension in limited cutaneous systemic sclerosis: a distinctive vasculopathy. Eur Respir J. 2009;34(2):371–379. doi:10.1183/09031936.00106008
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.