Back to Journals » Drug Design, Development and Therapy » Volume 12
Theaflavin alleviates inflammatory response and brain injury induced by cerebral hemorrhage via inhibiting the nuclear transcription factor kappa β-related pathway in rats
Authors Fu G, Wang H, Cai Y, Zhao H, Fu W ![]()
Received 1 February 2018
Accepted for publication 24 March 2018
Published 12 June 2018 Volume 2018:12 Pages 1609—1619
DOI https://doi.org/10.2147/DDDT.S164324
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Anastasios Lymperopoulos
This paper has been retracted.
Guanglei Fu,1 Hua Wang,2 Youli Cai,2 Hui Zhao,2 Wenjun Fu2
1Department of Neurology, The First Affiliated Hospital of Jinan University, Guangzhou, People’s Republic of China; 2School of Basic Medical Science, Guangzhou University of Chinese Medicine, Guangzhou, People’s Republic of China
Objective: Intracerebral hemorrhage (ICH) is one of the most common acute cerebrovascular diseases with high mortality. Numerous studies have shown that inflammatory response played an important role in ICH-induced brain injury. Theaflavin (TF) extracted from black tea has various biological functions including anti-inflammatory activity. In this study, we investigated whether TF could inhibit ICH-induced inflammatory response in rats and explored its mechanism.
Materials and methods: ICH rat models were induced with type VII collagenase and pretreated with TF by gavage in different doses (25 mg/kg–100 mg/kg). Twenty-four hours after ICH attack, we evaluated the rats’ behavioral performance, the blood–brain barrier (BBB) integrity, and the formation of cerebral edema. The levels of reactive oxygen species (ROS) and inflammatory cytokines were examined by 2',7'-dichlorofluorescin diacetate and enzyme-linked immunosorbent assay. Nissl staining and transferase dUTP nick end labeling (TUNEL) were aimed to detect the neuron loss and apoptosis, the mechanism of which was explored by Western blot.
Results: It was found that in the pretreated ICH rats TF significantly alleviated the behavioral defects, protected BBB integrity, and decreased the formation of cerebral edema and the levels of ROS as well as inflammatory cytokines (including interleukin-1 beta [IL-1β], IL-18, tumor nectosis factor-alpha, interferon-γ, transforming growth factor beta, and (C-X-C motif) ligand 1 [CXCL1]). Nissl staining and TUNEL displayed TF could protect against the neuron loss and apoptosis via inhibiting the activation of nuclear transcription factor kappa-β-p65 (NF-κβ-p65), caspase-1, and IL-1β. We also found that phorbol 12-myristate 13-acetate, a nonspecific activator of NF-κβ-p65, weakened the positive effect of TF on ICH-induced neural defects and neuron apoptosis by upregulating NF-κβ-related signaling pathway.
Conclusion: TF could alleviate ICH-induced inflammatory responses and brain injury in rats via inhibiting NF-κβ-related pathway, which may provide a new way for the therapy of ICH.
Keywords: cerebral hemorrhage, theaflavin, inflammatory response, NF-κβ-p65
Introduction
Intracerebral hemorrhage (ICH), caused by rupture of blood vessels in nontraumatic brain parenchyma, is one of the most common acute cerebrovascular diseases with high mortality.1 The extravasated blood accumulates and compresses the surrounding brain tissues forming hematoma, the components (especially the blood-derived leukocytes and neutrophils) of which infiltrate the brain parenchyma and break the blood–brain barrier (BBB) leading to inflammatory responses and cerebral edema as well as nerve damage. Numerous studies have shown that inflammatory response played an important role in ICH-induced brain injury.2–4 Inflammation is mediated by cellular components (such as leukocytes and microglia) and molecular components, including prostaglandins, chemokines, cytokines, extracellular proteases, and reactive oxygen species (ROS).5–7 The microglial cells, which can release pro-inflammatory cytokines and chemokines, were activated within minutes after ICH and recruited hematogenous inflammatory cells to the injury site triggering nuclear transcription factor kappa B (NF-κβ)-related inflammatory signaling pathway.8–10
NF-κβ, a key regulator of many pro-inflammatory cytokines such as tumor nectosis factor-alpha (TNF-α) and interleukin-1 beta (IL-1β), was without activity when combined with inhibitory κB proteins (IκBs) in no-stress conditions but activated as early as in 15 minutes and persisted for at least a week after ICH,10 and its activation was positively related to the progress of apoptotic in patients with ICH.11 ROS, also known as an important signaling molecular and released by neutrophils, vascular endothelium, and activated microglia/macrophages,7,12 could influence NF-κβ activation; meanwhile NF-κβ pathway could also influence the ROS level by increasing the expression of antioxidant proteins.13 Many researchers considered that ROS played an important role in ICH-induced brain injury,14,15 and excessive ROS was regarded as a hallmark of most brain damage.16 Maybe inhibiting the NF-κβ-related pathway and generation of excessive ROS could reduce the brain injury caused by ICH.
Theaflavin (TF), first found by Roberts et al17 from black tea, has been demonstrated as having various biological functions, such as radical scavenging, antimutagenicity, hypolipidemic, anticancers, antiviral, as well as anti-inflammatory activity.18 Kim et al19 demonstrated that TFs significantly reduced the mRNA level of the lipopolysaccharide (LPS)-induced IL-6, monocyte chemoattractant protein-1, and intercellular adhesion molecule-1 in bone marrow-derived macrophages, inhibited the cytosolic IκBα protein degradation, and blocked the nuclear translocation of RelA (NF-κβ-p65), suggesting that inhibition of pro-inflammatory mediator production by TFs occurred via blocking of NF-κβ signaling pathway. Ukil et al20 also found that pretreatment with TF-3,3′-digallate markedly inhibited trinitrobenzene sulfonic acid (TNBS)-induced increases in nuclear localization of NF-κβ, cytosolic IkappaB kinase activity, and preserved IκBα in colon tissue. Another study showed that TF administration ameliorated the brain’s infarct as well as edema volume in rat model of middle cerebral artery occlusion and protected neurons from cerebral ischemia–reperfusion injury through its anti-inflammatory effect.21 In this study, we investigated whether TF could inhibit the inflammatory response caused by ICH in rats and explored its mechanism.
Materials and methods
Animals administration and ICH induction
One hundred and sixty-five adult Sprague–Dawley rats (240 g–260 g), obtained from Charles River Laboratories (Wilmington, MA, USA) were accommodated in a temperature-controlled reverse light cycle environment (23°C ± 2°C, 12-hour light/dark cycle) without limited access of food and water. All experiment procedures complied with the National Institute Rules for the Care and Use of Laboratory Animals and were approved by the Guangzhou University of Chinese Medicine Animal Committee. As previously described,22,23 ICH was induced as follows: the rats were anesthetized and fixed in a stereotactic frame in a prone position, then a 1 mm burr hole was made and a 26-gauge needle was inserted into the right basal ganglia. The stereotaxic coordinates were 0.2 mm anterior, 3 mm right lateral to the bregma, and 6 mm ventral to the skull. The ICH model was constructed by type VII collagenase (0.5 U in 2 μL saline per rat, C0773, Sigma-Aldrich Co., St Louis, MO, USA) at a flow rate of 0.4 μL/min. The sham group rats were perforated at the same position and infused in an equal volume of saline into the brain.
Drug administration
For TF (Sigma-Aldrich Co., purity >80%) administration, the rats were treated with TF by gavage at the given dose (25 mg/kg–100 mg/kg) 1 day before ICH. For the sham group and ICH group, the animals were given the same volume of saline. NF-κβ could be activated by a wide variety of inflammatory stimuli, including TNF, IL-1, phorbol 12-myristate 13-acetate (PMA), H2O2, ceramide, LPS, and ceramide.24–26 Here, we used PMA (ab120297, Abcam, Cambridge, UK) as a NF-κβ activator and dissolved it in dimethyl sulfoxide (D8418, Sigma-Aldrich Co.), which was intracerebroventricularly injected (100 μg/kg) just before ICH attach by making another burr hole as previously described.27–31
Behavioral testing
Behavioral tests including corner test and paw placement test were performed 24 hours after ICH induction. The scores were recorded by an experienced researcher who was unacquainted with the experiment conditions.
Corner test
As Xi et al described,22 the rats were placed at a 30° corner and the number of right turns were recorded. Each rat was allowed 10 attempts (n =5 rats/group). The score of corner test was calculated as number of right turns ÷ total turns × 100 and expressed as percentage.
Paw placement test
Also as Xi et al described,22 the rats’ bodies were held lightly paralleled up to a table board, then moved slowly and vertically until the vibrissae on one side touched the table surface. The time of the ipsilateral paw’s forward movements was also recorded: 0 point represented no placing of the paw, 1 point indicated delayed placing (>2 s), and 2 points meant immediate placing. The test was repeated 10 times (n =5 rats/group).
Evans blue extravasation assay
Evans blue was used to assess the BBB permeability 24 hours after ICH.22 The method was as follows: 2% Evans blue dye (2 mL/kg, E104208) injected into the tail vein and maintained for 2 hours; the brains from the euthanized rats were taken out for weighing, then treated with formamide for 24 hours at 37°C, and the samples were centrifuged at 2,000 ×g for 10 minutes to collect the supernatant. The 632 nm wave length was used to measure the samples’ optical density.
Brain water content measurement
Brain water content measurement32 was used to evaluate the formation of brain edema. Each brain was immediately weighed as the wet weight after being divided into two halves (the ICH half and the uninjured half), then was dried at 100°C for 24 hours and weighed again as the dry weight. The brain water content was calculated as (wet weight – dry weight) ÷ wet weight × 100 and expressed as percentage (n =5 rats/group).
Measurement of ROS production
As Yuan previously described,16 the ROS levels in the brain were measured by 2′,7′-dichlorofluorescin diacetate (DCFH-DA, Beyotime Institute of Biotechnology, Haimen, People’s Republic of China). The tissues were homogenized and treated with 5 mM DCFH-DA in phosphate buffered saline for 30 minutes at room temperature. For the detection of the fluorescence, the excitation splitter and the emission splitter were 484 nm and 530 nm, respectively. Set the sham group to 100% to calculate the ROS content (n =5 rats/group).
Nissl staining
Procedures for Nissl staining33 were as follows. Briefly, after dehydration in 30% sugar solution, the tissues were sliced with a freezing microtome (Leica Microsystems, Wetzlar, Germany). The sections were stained with toluidine blue (89640, Sigma-Aldrich Co.) and covered with 50% glycerin. Images were taken with a light microscope (Olympus Corporation, Tokyo, Japan) and a digital camera (Olympus Corporation) (n =3 rats/group).
Apoptosis assay
Cellular apoptosis in the frozen sections were assessed by in situ cell death detection kit (based on terminal-deoxynucleoitidyl transferase mediated nick end labeling) (TUNEL, Hoffman-La Roche Ltd., Basel, Switzerland) according to the manufacturer’s protocol. The nuclei were stained with 4′,6-diamidino-2-phenylindole and the images were taken using a fluorescent microscope (n =3 rats/group).
Quantitative reverse transcription-polymerase chain reaction (qRT-PCR)
As the manufacturer described, total RNA was extracted from frozen brain with RNAiso Plus (9108, Takara, Otsu, Japan) and first-strand cDNA was synthesized from 1 μL total RNA using PrimeScript™ RT reagent kit with gDNA Eraser (RR047A, Takara). qRT-PCR was performed according to the procedures of SYBR® Premix Ex Taq™ II (Tli RNaseH Plus) kit (RR820L, Takara). Target gene expression was normalized by the endogenous control, β-actin. The primers were as follows: β-actin sense 5′-CGTAAAGACCTCTATGCCAA-3′, β-actin antisense 5′-AGCCATGCCAAATGTGTCAT-3′; NF-κβ-p65 sense 5′-ACGATCTGTTTCCCCTCATCT-3′, NF-κβ-p65 antisense 5′-TGCTTCTCTCCCCAGGAATA-3′; IL-1β sense 5′-CACCTCTCAAGCAGAGCACAG-3′, IL-1β antisense 5′-GGGTTCCATGGTGAAGTCAAC-3′; Caspase-1 sense 5′-CCAGAGCACAAGACTTCTGAC-3′, caspase-1 antisense 5′-TGGTGTTGAAGAGCAGAAAGC-3′ (n =3 rats/group).
Western blotting analysis
The whole cell and cytoplasmic-nuclear proteins were extracted according to the manufacturer’s instruction (Beyotime Institute of Biotechnology). For Western blotting analysis, 40 μg/lane proteins were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA, USA). The blots were probed with primary antibodies as described below, followed by incubation with horseradish peroxidase-conjugated goat anti-rabbit IgG as the secondary antibody (ab6721) (1:3,000 dilution). The rabbit anti-NF-κβ-p65 (ab16502) (1:200 dilution) and anti-caspase-1 antibody (ab108362) (1:1,000 dilution) were from Abcam. The rabbit anti-IL-1β antibody (sc-7884) (1:1,000 dilution) and β-actin (sc-47778) (1:1,000 dilution) were from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). The proteins were visualized using enhanced chemiluminescence reagents (P0018, Beyotime Institute of Biotechnology), the signals of which were collected by the motored molecular imaging system (Tanon-5500, Tanon Science & Technology Co. Ltd., Shanghai, People’s Republic of China). Densitometric analysis was performed with the ImageJ software (n =3 rats/group).
Enzyme-linked immunosorbent assay (ELISA)
According to the manufacturer’s protocols, the post-ICH brain tissue homogenate was analyzed for inflammatory cytokines including IL-1β (EK0393), IL-18 (EK0592), TNF-α (EK0526), interferon gamma (INF-γ) (EK0374), transforming growth factor beta (TGF-β) (EK0514), as well as the chemokine CXCL1 (EK0724) using ELISA kits (Boster Biological Technology, Pleasanton, CA, USA). The cytokine concentration was normalized by the total protein concentration, which was tested by a BCA protein assay kit (PC0020, Solarbio Life Sciences, Beijing, People’s Republic of China) (n =5 rats/group).
Statistical analysis
Statistical analysis was carried out with analysis of variance followed by Bonferron’s multiple t-tests. A p-value <0.05 was considered to be statistically significant.
Results
TF treatment protected the rats from ICH-induced brain injury
There was no dead rat in the sham group and in the TF group. However, three rats died after ICH attack and another three were supplied. The effect of TF on the rats’ behavioral performance was investigated by corner test and paw placement test 24 hours after ICH. As shown as Figure 1A and B, the ICH group animals showed a higher frequency of right turns and a lower paw placement score than the sham group ones while administration of TF greatly reduced the frequency of right turns and improved the paw placement score, which proved that TF could significantly improve the behavioral performance after ICH.
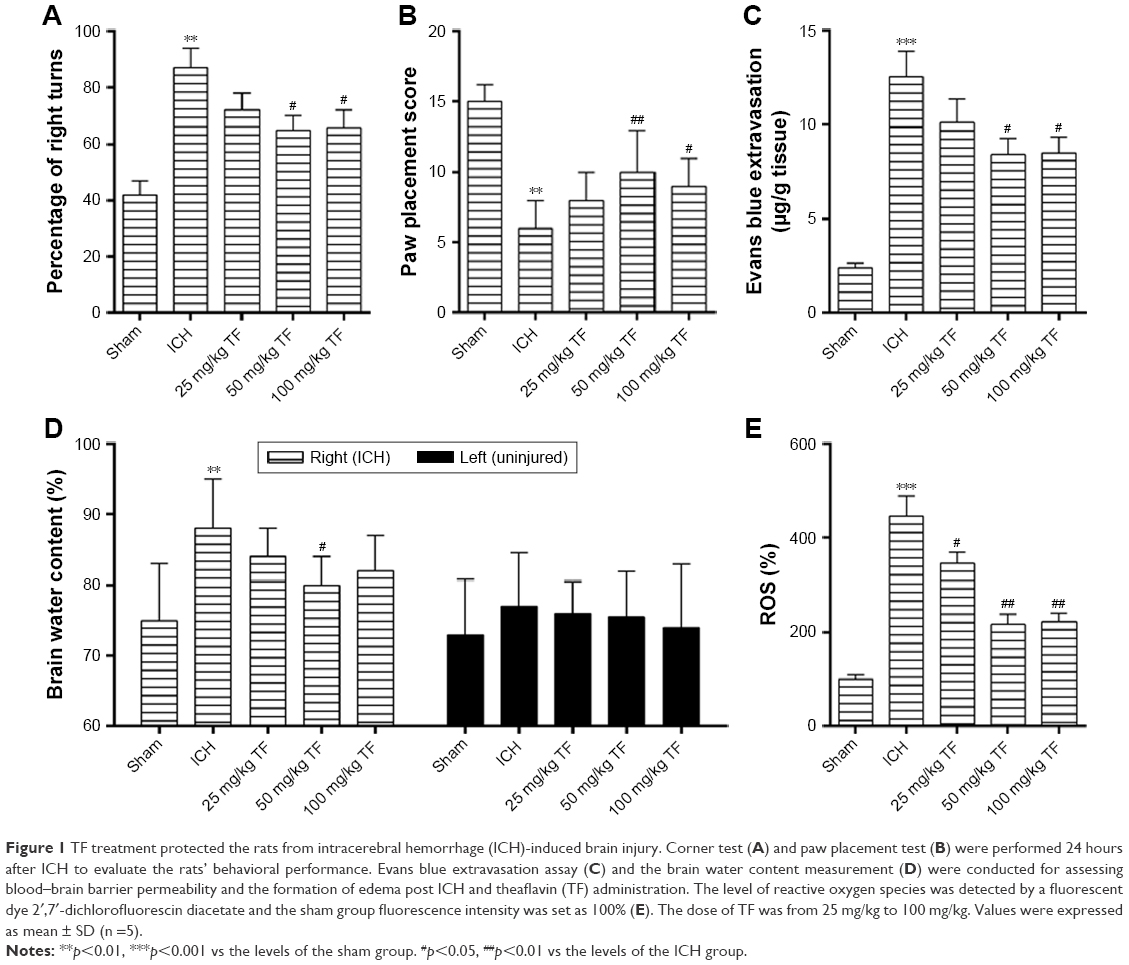 | Figure 1 TF treatment protected the rats from intracerebral hemorrhage (ICH)-induced brain injury. Corner test (A) and paw placement test (B) were performed 24 hours after ICH to evaluate the rats’ behavioral performance. Evans blue extravasation assay (C) and the brain water content measurement (D) were conducted for assessing blood–brain barrier permeability and the formation of edema post ICH and theaflavin (TF) administration. The level of reactive oxygen species was detected by a fluorescent dye 2′,7′-dichlorofluorescin diacetate and the sham group fluorescence intensity was set as 100% (E). The dose of TF was from 25 mg/kg to 100 mg/kg. Values were expressed as mean ± SD (n =5). |
Next, we examined other injury indicators induced by ICH. Compared to the sham group, Evans blue extravasation (Figure 1C), the brain water content of the right hemisphere (the ICH part, Figure 1D) and the ROS level (Figure 1E) were significantly high in the ICH group. However, pretreatment of the ICH rats with different doses of TF reduced all these three injury indicators to a large degree. But for the brain water content assay, we found that there were no significant differences among the groups in the left hemisphere (the uninjured part, Figure 1D). These results suggested that TF could decrease BBB permeability, inhibit the formation of edema, and scavenge the excessive ROS induced by ICH.
Taken together, TF could alleviate ICH injury and improve the neurological functions via maintaining the BBB integrity and preventing the formation of brain edema as well as scavenging the over-produced ROS. All the experiments showed that 50 mg/kg TF had the best effect.
TF treatment suppressed the inflammatory response induced by ICH
Increasing evidences suggest that inflammatory response plays a key role in the ICH-induced brain injury. Here, we tested whether TF could resist the release of inflammatory cytokines with ELISA 24 hours post ICH. As shown as Figure 2, the levels of IL-1β (Figure 2A), IL-18 (Figure 2B), TNF-α (Figure 2C), interferon-γ (IFN-γ) (Figure 2D), TGF-β (Figure 2E), CXCL1 (Figure 2F) in ICH brain homogenates were obviously increased compared to the ones in the sham group. Nevertheless, taking TF decreased the expressions of these factors, indicating that TF could suppress the ICH-induced inflammatory response. The data also showed that the dose of 50 mg/kg TF was the best choice.
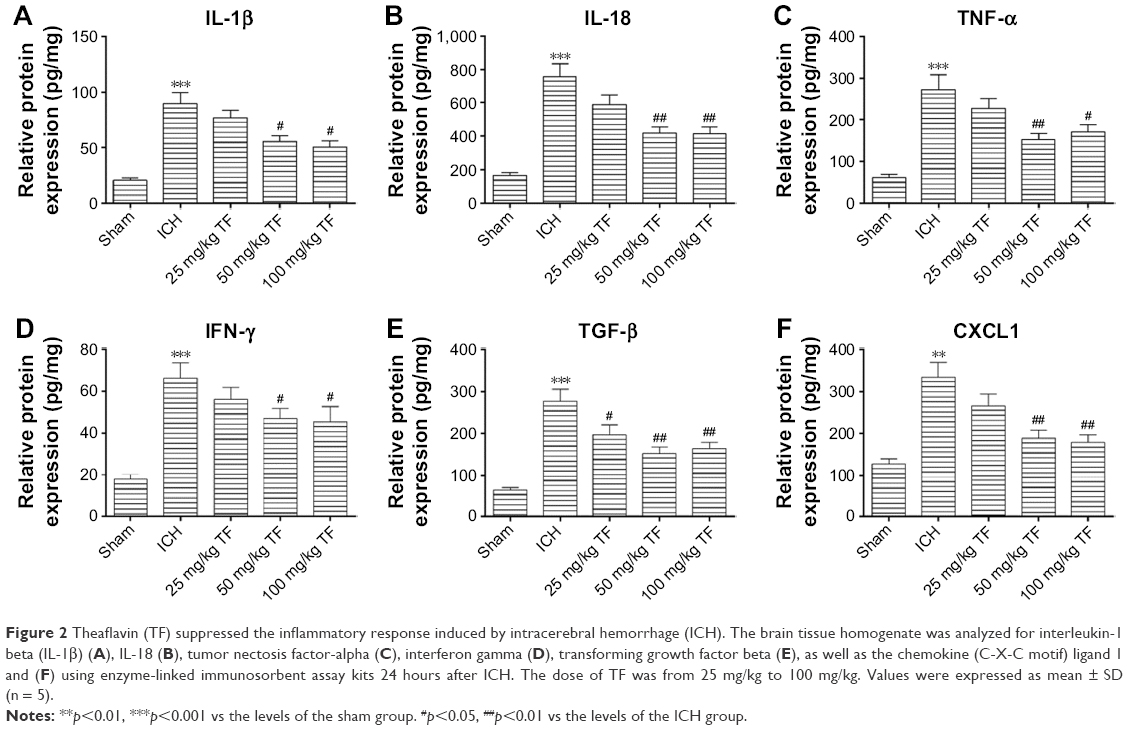 | Figure 2 Theaflavin (TF) suppressed the inflammatory response induced by intracerebral hemorrhage (ICH). The brain tissue homogenate was analyzed for interleukin-1 beta (IL-1β) (A), IL-18 (B), tumor nectosis factor-alpha (C), interferon gamma (D), transforming growth factor beta (E), as well as the chemokine (C-X-C motif) ligand 1 and (F) using enzyme-linked immunosorbent assay kits 24 hours after ICH. The dose of TF was from 25 mg/kg to 100 mg/kg. Values were expressed as mean ± SD (n = 5). |
TF protected neural cells from ICH-induced death via inhibiting the NF-κβ-related pathway
In order to verify the effect of TF on neuron death, we carried out Nissl staining and TUNEL experiment 24 hours post ICH. Nissl staining displayed an apparent loss of Nissl bodies in the ICH group compared to the sham group while showing a remarkable increase in pretreated with TF (50 mg/kg) compared with the ICH group (Figure 3A). TUNEL staining appeared to obviously elevate TUNEL-positive cell numbers in the ICH group compared with the sham group while showing a significant decrease in receiving TF administration (Figure 3B). In order to explore whether the anti-apoptosis mechanism of TF is associated with NF-κβ-p65-related inflammatory response, we made qRT-PCR and Western blot analysis. The mRNA levels of NF-κβ-p65, IL-1β, and caspase-1 were obviously upregulated after ICH attack but downregulated by pretreating TF (Figure 3C). As shown as Figure 3D, the protein levels of NF-κβ-p65, IL-1β, and caspase-1 showed similar trends to the mRNA levels, indicating that TF could protect neurons from ICH-induced apoptosis by inhibiting NF-κβ-related inflammatory response.
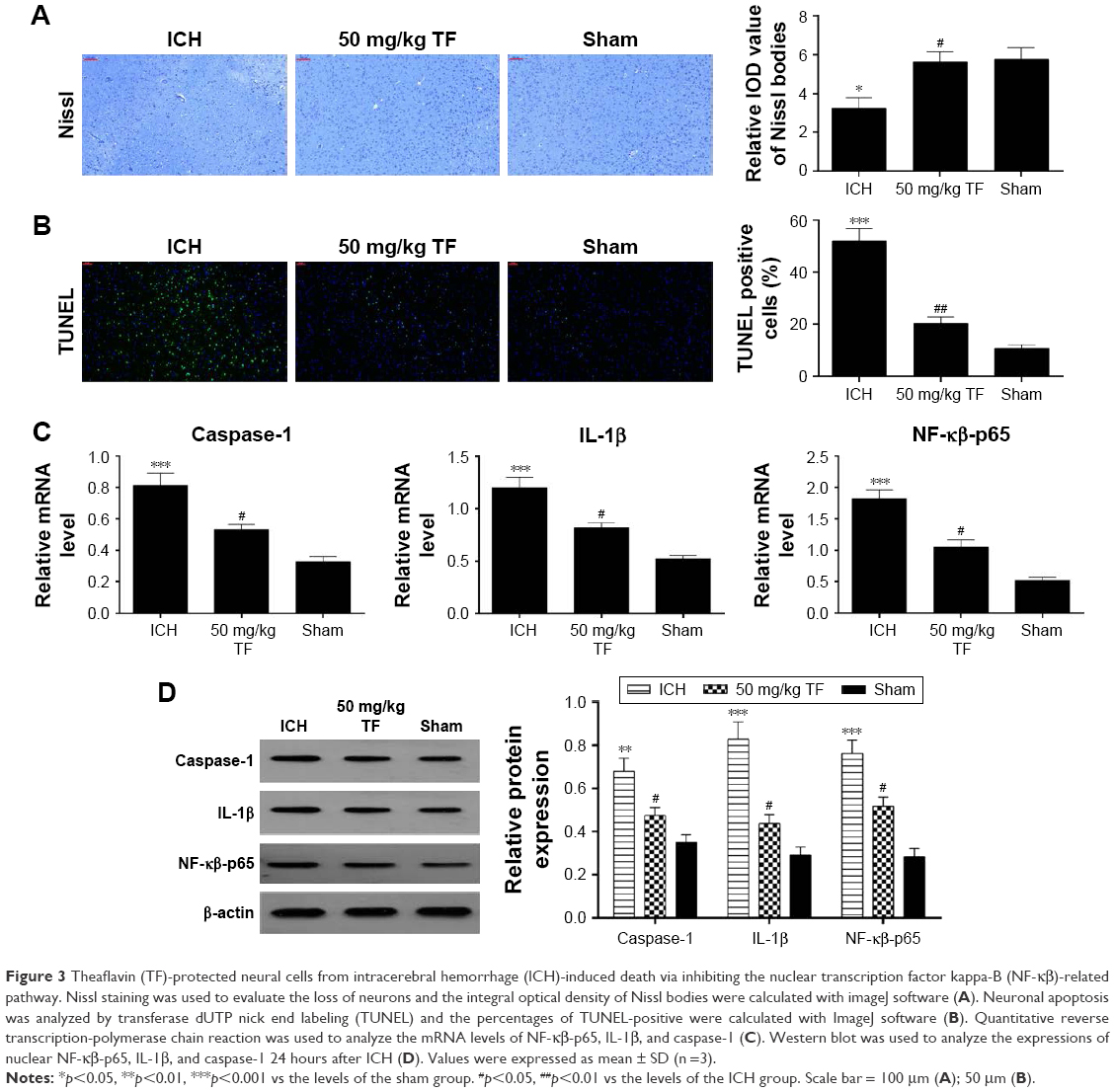 | Figure 3 Theaflavin (TF)-protected neural cells from intracerebral hemorrhage (ICH)-induced death via inhibiting the nuclear transcription factor kappa-B (NF-κβ)-related pathway. Nissl staining was used to evaluate the loss of neurons and the integral optical density of Nissl bodies were calculated with imageJ software (A). Neuronal apoptosis was analyzed by transferase dUTP nick end labeling (TUNEL) and the percentages of TUNEL-positive were calculated with ImageJ software (B). Quantitative reverse transcription-polymerase chain reaction was used to analyze the mRNA levels of NF-κβ-p65, IL-1β, and caspase-1 (C). Western blot was used to analyze the expressions of nuclear NF-κβ-p65, IL-1β, and caspase-1 24 hours after ICH (D). Values were expressed as mean ± SD (n =3). |
PMA attenuated the positive effect of TF on ICH-induced neural deficits and brain injury
In order to further explore the relationship of TF and NF-κβ, we treated the ICH rats with PMA as a nonspecific activator of NF-κβ-p65 after TF administration. All the rats survived after PMA treatment. As shown as Figure 4A and B, the rats treated with PMA got a higher corner test score and a lower paw placement score than those pretreated with only TF, suggesting that PMA aggravated the ICH rats’ behavioral deficits. The ICH rats also showed an obvious increase of brain water content (in the right hemisphere) and Evans blue extravasation after PMA injection (Figure 4C and D). It was worth reminding that there were no significant differences in the brain water contents between the groups in the left hemisphere. These results indicated that PMA increased BBB permeability and promoted the formation of brain edema even if in the case of TF.
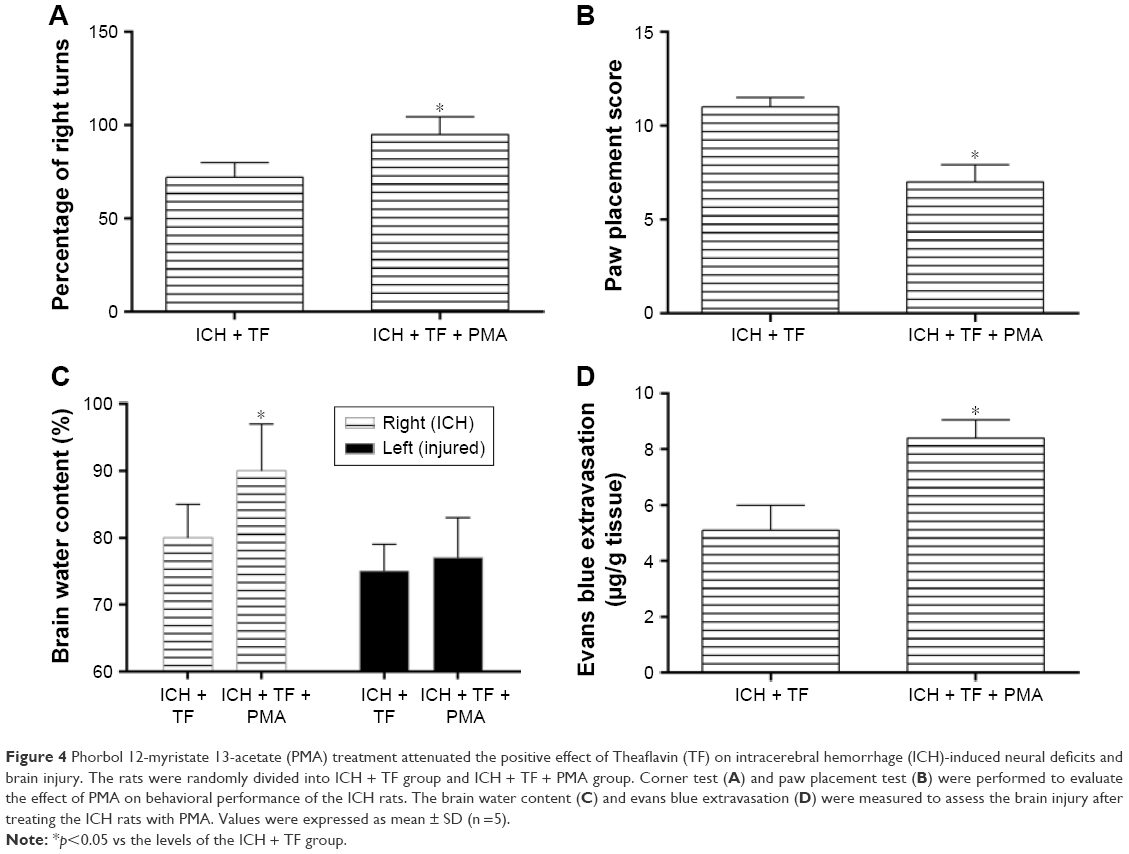 | Figure 4 Phorbol 12-myristate 13-acetate (PMA) treatment attenuated the positive effect of Theaflavin (TF) on intracerebral hemorrhage (ICH)-induced neural deficits and brain injury. The rats were randomly divided into ICH + TF group and ICH + TF + PMA group. Corner test (A) and paw placement test (B) were performed to evaluate the effect of PMA on behavioral performance of the ICH rats. The brain water content (C) and evans blue extravasation (D) were measured to assess the brain injury after treating the ICH rats with PMA. Values were expressed as mean ± SD (n =5). |
PMA limited the anti-apoptosis activity of TF on ICH rats by upregulating NF-κβ-related signaling pathway
At last, we explored the effect of PMA on neuronal apoptosis. As shown in Figure 5A, compared to the TF group, the Nissl bodies dropped off markedly and the TUNEL-positive cells increased significantly (Figure 5B) after PMA administration. qRT-PCR and Western blot analysis showed that the expressions of nuclear NF-κβ-p65, caspase-1, and IL-1β were upregulated as well post PMA treatment (Figure 5C and D). These findings suggested that PMA exacerbated neuronal apoptosis in the ICH rats even in the case of TF by upregulating the nuclear expression of NF-κβ-p65 and activating caspase-1 as well as IL-1β.
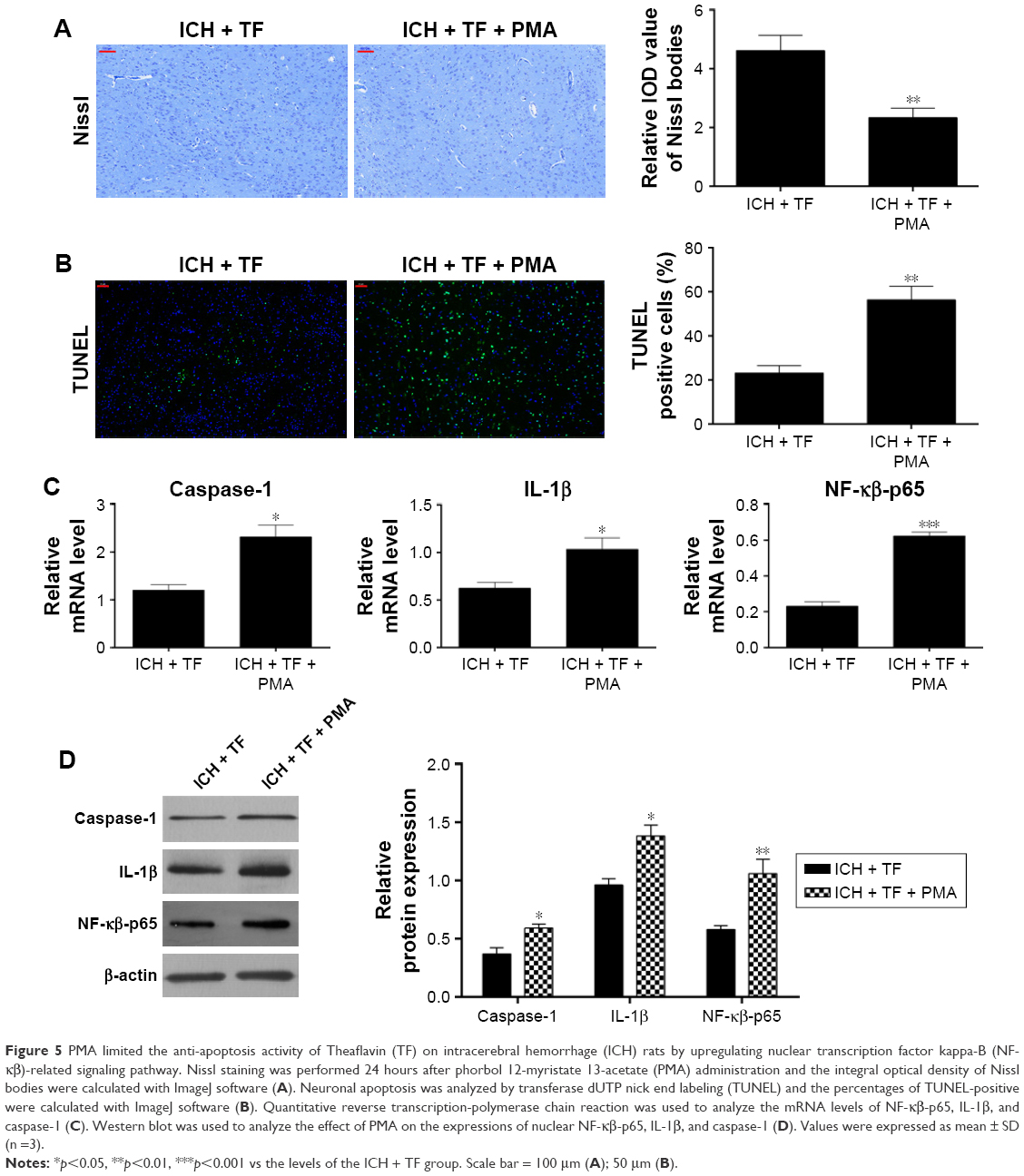 | Figure 5 PMA limited the anti-apoptosis activity of Theaflavin (TF) on intracerebral hemorrhage (ICH) rats by upregulating nuclear transcription factor kappa-B (NF-κβ)-related signaling pathway. Nissl staining was performed 24 hours after phorbol 12-myristate 13-acetate (PMA) administration and the integral optical density of Nissl bodies were calculated with ImageJ software (A). Neuronal apoptosis was analyzed by transferase dUTP nick end labeling (TUNEL) and the percentages of TUNEL-positive were calculated with ImageJ software (B). Quantitative reverse transcription-polymerase chain reaction was used to analyze the mRNA levels of NF-κβ-p65, IL-1β, and caspase-1 (C). Western blot was used to analyze the effect of PMA on the expressions of nuclear NF-κβ-p65, IL-1β, and caspase-1 (D). Values were expressed as mean ± SD (n =3). |
Discussion
As is well known, TF has a variety of health care functions.34 It was reported that TF could ameliorate ionizing radiation-induced hematopoietic injury as well as cerebral ischemia-reperfusion injury via anti-inflammatory response.21,35 In this study, we found that TF could improve the neural functions and alleviate brain injury as well as inflammatory response via suppressing NF-κβ-p65-related pathway in the ICH rats.
ICH, closely associated with hypertension, cerebral amyloid angiopathy, brain tumors, and so on,36,37 has a mortality rate as high as 40% at one month, of which most survivors have some sequelae with different degrees, such as dyskinesia, logopathy, cognitive disorder, dysphagia.38,39 Here, we used collagenase to induce ICH model and found that the rats showed a setback in the behavioral performance and an increase of BBB permeability as well as brain water content 24 hours after ICH, consistent with the results by others.22,23,32 However, pretreating the ICH rats with TF got an improvement of behavioral ability with a decrease of BBB permeability and brain water content (Figure 1A–D). It has been known that the leukocytes immediately invading into brain after blooding contributed to disruption of BBB followed by brain edema formation.37 So we considered that TF could protect BBB from leukocyte invasion to inhibit the information of brain edema, which further enhanced the neural functions of the ICH rats.
It has been found that ROS stimulates numerous signal transduction pathways that are important in maintaining cellular homeostasis in the neuron.40 For instance, we discovered along with generation of ROS, the nuclear NF-κβ-p65 also showed an upregulated expression after ICH in rats, indicating a close relationship between ROS and NF-κβ- p65 (Figures 1E and 2C). However, pretreatment with TF markedly reduced the release of ROS. In agreement with our study, Yuan et al16 found that the ICH model mice had a significant increase in ROS generation; while treatment with silymarin, an antioxidant from plant, significantly decreased the generation of ROS and lipid peroxidation. These results suggested that maybe scavenging the over produced ROS could be considered as a method for alleviating the ICH injury, consistent with the point that ROS could be a potential target for ICH therapy.7
Numerous studies have shown that inflammatory response plays an important role in ICH-induced brain injury.4,37 Li et al32 found that the expression levels of inflammatory cytokines including TNF-α, IL-1β, and IL-18, regulated by Foxo1/TLR4/NF-κβ signaling pathway, were significantly increased at 12 hours post ICH compared with the sham operation group, which was also demonstrated by Yuan et al.16 TNF-α and IL-1β, as the classic pro-inflammatory cytokines, have been proposed to exacerbate ICH-induced brain injury and could induce each other mutually and activate a positive feedback of cellular activation.7,41 IFN-γ, released mainly by activated T lymphocytes and natural killer cells, also has an upregulated expression in the ICH mice42 and could interact with microglia producing a number of inflammatory cytokines (such as TNF and IL-1) after cerebral ischemia reperfusion, which further aggravated the inflammation.43 Decades ago, Krupinski et al44 found the expression of TGF-β was increased in brain tissue after ischemic stroke in humans. However, in contrast with TNF-α and IL-1β as well as IFN-γ, TGF-β was considered as a neuroprotective factor or an anti-inflammatory cytokine. Study by Zhu et al demonstrated that TGF-beta1 could protect cultured hippocampal neurons against apoptosis by efficiently inhibiting caspase-3 activation.45 In the cerebral ischemic stroke rats, TGF-β1 may activate the expression of Bcl-2 via Smad3 to suppress the neuronal apoptosis.46 Similar with TGF-β, CXCL1 was also considered as an anti-inflammatory factor and was found to be upregulated in the ICH mouse.47,48 TF has been demonstrated to have an anti-inflammatory property. For example, Ukil et al20 found that treatment frequency and duration guidelines treatment significantly decreased the expressions of TNF-α, IL-12, IFN-γ, as well as inducible nitric oxide synthase and improved TNBS-induced colitis. We analyzed the protein expressions of IL-1β, IL-18, TNF-α, IFN-γ, TGF-β, and CXCL1 in the ICH brain homogenates finding that TF obviously decreased the levels of all these proteins compared to the ICH animals without TF treatment, indicating TF could alleviate ICH-induced inflammatory response (Figure 2). These pro-inflammatory cytokines and anti-inflammatory factors interacted with each other as a feedback to the ICH attack.
Neuronal death (including apoptotic and necrotic neurons) at the site of the hematoma occurs rapidly after ICH has happened.49,50 TUNEL-positive cells were observed at 6 hours in the ICH model and peaking at 3 days but not in the saline control brains.51 Recent research showed that the human neuronal apoptosis after ICH was closely related with NF-κβ-p65, IL-1β, and TNF-α expressions.52 Others showed that the formation of mature IL-1β and IL-18 was closely associated with activated caspase-1,53 which belongs to inflammatory caspase, also known as IL-converting enzyme, and could be activated by NLRP3 inflammasome.54 Here, we detected that pretreating the ICH rats with TF could effectively decrease the loss of neuron as well as apoptosis and the expressions of nuclear NF-κβ-p65, caspase-1, and IL-1β on the mRNA and protein levels (Figure 3). So, we supposed that TF played the protective effects by inhibiting the activation of NF-κβ-p65 and the downstream responses, namely the generation of cleaved caspase-1 and IL-1β.
NF-κβ existed as a dimer mainly with p50 and p65. In the case of resting, NF-κβ is colocated in the cytoplasm with IκBs as an inactive trimer. Once stimulated, such as through PMA treatment,55 IκB was phosphorylated and released from the trimer leading to translocation of NF-κβ to the nucleus.10 PMA, usually used as a protein kinase C activator, could phosphorylate IκB and elevate p65 nuclear translocation.56,57 We used PMA as a nonspecific activator of NF-κβ-p65 and found that treating ICH rats with PMA after pretreating with TF aggravated behavior degradation, BBB permeability, as well as the brain edema and exacerbated the neuron death with anabatic expressions of NF-κβ-p65, caspase-1, and IL-1β (Figures 4 and 5), suggesting that TF does alleviate ICH-induce brain injury by inhibiting NF-κβ-related pathway.
Conclusion
TF, as an anti-inflammatory factor, could alleviate the ICH-induced inflammatory response and brain injury in rats via inhibiting NF-κβ-related pathway, which provided a new way for the therapy of ICH. In addition to the anti-inflammatory activity, maybe TF played the protective effect on ICH by other ways, for example, the antioxidant pathway, for we detected TF could scavenge the ROS induced by ICH. So next, we need to work more for exploring the targets for TF in ICH.
Acknowledgment
This study was supported by the medical Scientific Research Foundation of Guangdong Province (A2015464) and the Science and Technology Program of Guangzhou (NO: 201607010367).
Disclosure
The authors report no conflicts of interest in this work.
References
Flower O, Smith M. The acute management of intracerebral hemorrhage. Curr Opin Crit Care. 2011;17(2):106–114. | ||
Wang J. Preclinical and clinical research on inflammation after intracerebral hemorrhage. Prog Neurobiol. 2010;92(4):463–477. | ||
Zhou Y, Wang Y, Wang J, Anne Stetler R, Yang QW. Inflammation in intracerebral hemorrhage: from mechanisms to clinical translation. Prog Neurobiol. 2014;115:25–44. | ||
Aronowski J, Hall CE. New horizons for primary intracerebral hemorrhage treatment: experience from preclinical studies. Neurol Res. 2005;27(3):268–279. | ||
Keep RF, Hua Y, Xi G. Intracerebral haemorrhage: mechanisms of injury and therapeutic targets. Lancet Neurol. 2012;11(8):720–731. | ||
Qureshi AI, Mendelow AD, Hanley DF. Intracerebral haemorrhage. Lancet. 2009;373(9675):1632–1644. | ||
Wang J, Doré S. Inflammation after intracerebral hemorrhage. J Cereb Blood Flow Metab. 2006;27(5):894–908. | ||
Aronowski J, Zhao X. Molecular pathophysiology of cerebral hemorrhage: secondary brain injury. Stroke. 2011;42(6):1781–1786. | ||
Hickenbottom SL, Grotta J, Strong R, Denner LA, Aronowski J. Nuclear factor-kappab and cell death after experimental intracerebral hemorrhage in rats. Stroke. 1999;30(11):2472–2477. | ||
Zhao X, Zhang Y, Strong R, Zhang J, Grotta JC, Aronowski J. Distinct patterns of intracerebral hemorrhage-induced alterations in NF-κB subunit, iNOS, and COX-2 expression. J Neurochem. 2006;101(3):652–663. | ||
Wang YX, Yan A, Ma ZH, et al. Nuclear factor-κB and apoptosis in patients with intracerebral hemorrhage. J Clin Neurosci. 2011;18(10):1392–1395. | ||
Facchinetti F, Dawson VL, Dawson TM. Free radicals as mediators of neuronal injury. Cell Mol Neurobiol. 1998;18(6):667–682. | ||
Zhang J, Wang X, Vikash V, et al. ROS and ROS-mediated cellular signaling. Oxid Med Cell Longev. 2016;2016(5782):1–18. | ||
Chen YC, Chen CM, Liu JL, Chen ST, Cheng ML, Chiu DT. Oxidative markers in spontaneous intracerebral hemorrhage: leukocyte 8-hydroxy-2′-deoxyguanosine as an independent predictor of the 30-day outcome. J Neurosurg. 2011;115(6):1184–1190. | ||
Alexandrova ML, Danovska MP. Serum C-reactive protein and lipid hydroperoxides in predicting short-term clinical outcome after spontaneous intracerebral hemorrhage. J Clin Neurosci. 2011;18(2):247–252. | ||
Yuan R, Fan H, Cheng S, et al. Silymarin prevents NLRP3 inflammasome activation and protects against intracerebral hemorrhage. Biomed Pharmacother. 2017;93:308–315. | ||
Roberts EAH, Cartwright R, Oldschool M. The phenolic substances of manufactured tea. I.-fractionation and paper chromatography of water-soluble substances. J Sci Food Agr. 1957;8:72–80. | ||
He HF. Research progress on theaflavins: efficacy, formation, and preparation. Food Nutr Res. 2017;61(1):1344521. | ||
Kim S, Joo YE. Theaflavin inhibits LPS-induced IL-6, MCP-1, and ICAM-1 expression in bone marrow-derived macrophages through the blockade of NF-κB and MAPK signaling pathways. Chonnam Med J. 2011;47(2):104–110. | ||
Ukil A, Maity S, Das PK. Protection from experimental colitis by theaflavin-3,3′-digallate correlates with inhibition of IKK and NF-κB activation. Br J Pharmacol. 2006;149(1):121–131. | ||
Cai F, Li CR, Wu JL, et al. Theaflavin ameliorates cerebral ischemia-reperfusion injury in rats through its anti-inflammatory effect and modulation of STAT-1. Mediators Inflamm. 2006;2006(5):1–9. | ||
Xi T, Jin F, Zhu Y, et al. MicroRNA-126-3p attenuates blood-brain barrier disruption, cerebral edema and neuronal injury following intracerebral hemorrhage by regulating PIK3R2 and Akt. Biochem Biophys Res Commun. 2017;494(1–2):144–151. | ||
Guo S, Zhen Y, Wang A. Geranylgeranylacetone exerts neuroprotective roles through medicating the phosphatidylinositol-3 kinase/Akt signaling pathway in an intracerebral hemorrhage rat model. Int J Neurosci. Epub 2018 Feb 22:1–6. | ||
Takada Y, Mukhopadhyay A, Kundu GC, Mahabeleshwar GH, Singh S, Aggarwal BB. Hydrogen peroxide activates NF-κB through tyrosine phosphorylation of IκBα and serine phosphorylation of p65. J Biol Chem. 2003;278(26):24233–24241. | ||
Manna SK, Aggarwal RS, Sethi G, Aggarwal BB, Ramesh GT. Morin (3,5,7,2′,4′-Pentahydroxyflavone) abolishes nuclear factor-B activation induced by various carcinogens and inflammatory stimuli, leading to suppression of nuclear factor-B-regulated gene expression and up-regulation of apoptosis. Clin Cancer Res. 2007;13(7):2290–2297. | ||
Sandur SK, Ichikawa H, Sethi G, Ahn KS, Aggarwal BB. Plumbagin (5-Hydroxy-2-methyl-1,4-naphthoquinone) suppresses NF-κB activation and NF-κB-regulated gene products through modulation of p65 and IκBα kinase activation, leading to potentiation of apoptosis induced by cytokine and chemotherapeutic agents. J Biol Chem. 2006;281(25):17023–17033. | ||
Ohnishi M, Monda A, Takemoto R, et al. Sesamin suppresses activation of microglia and p44/42 MAPK pathway, which confers neuroprotection in rat intracerebral hemorrhage. Neuroscience. 2013;232:45–52. | ||
Chang P, Dong W, Zhang M, et al. Anti-necroptosis chemical necrostatin-1 can also suppress apoptotic and autophagic pathway to exert neuroprotective effect in mice intracerebral hemorrhage model. J Mol Neurosci. 2014;52(2):242–249. | ||
Meng Y, Chen JX, Sun SG. Effect of VEGF165 on the permeability of blood brain barrier of intracerebral hemorrhage. Stroke Nerv Dis (In Chinese). 2006;13(2):79–81. | ||
Zhang Y, Gong K, Zhou W, et al. Involvement of subtypes γ and ε of protein kinase C in colon pain induced by formalin injection. Neurosignals. 2011;19(3):142–150. | ||
Wu B, Wang S, Qin G, et al. Protein kinase C γ contributes to central sensitization in a rat model of chronic migraine. J Mol Neurosci. 2017;63:131–141. | ||
Li Z, He Q, Zhai X, et al. Foxo1-mediated inflammatory response after cerebral hemorrhage in rats. Neurosci Lett. 2016;629:131–136. | ||
Tang Y, Han S, Asakawa T, et al. Effects of intracerebral hemorrhage on 5-hydroxymethylcytosine modification in mouse brains. Neuropsychiatr Dis Treat. 2016;12:617–624. | ||
Lorenz M. Cellular targets for the beneficial actions of tea polyphenols. Am J Clin Nutr. 2013;98(6):1642S–1650S. | ||
Han X, Zhang J, Xue X, et al. Theaflavin ameliorates ionizing radiation-induced hematopoietic injury via the NRF2 pathway. Free Radic Biol Med. 2017;113:59–70. | ||
Aguilar MI, Freeman WD. Spontaneous intracerebral hemorrhage. Semin Neurol. 2010;30(5):555–564. | ||
Xi G, Keep R, Hoff JT. Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 2006;5(1):53–63. | ||
van Asch CJ, Luitse MJ, Rinkel GJ, van der Tweel I, Algra A, Klijn CJ. Incidence, case fatality, and functional outcome of intracerebral haemorrhage over time, according to age, sex, and ethnic origin: a systematic review and meta-analysis. Lancet Neurol. 2010;9(2):167–176. | ||
Feigin VL, Barker-Collo S, McNaughton H, Brown P, Kerse N. Long-term neuropsychological and functional outcomes in stroke survivors: current evidence and perspectives for new research. Int J Stroke. 2008;3(1):33–40. | ||
Crack PJ, Taylor JM. Reactive oxygen species and the modulation of stroke. Free Radic Biol Med. 2005;38(11):1433–1444. | ||
Turrin NP, Plata-Salamán CR. Cytokine-cytokine interactions and the brain. Brain Res Bull. 2000;51(1):3–9. | ||
Liu W, Yuan J, Zhu H, et al. Curcumin reduces brain-infiltrating T lymphocytes after intracerebral hemorrhage in mice. Neurosci Lett. 2016;620:74–82. | ||
Stoll G, Jander S, Schroeter M. Cytokines in CNS disorders: neurotoxicity versus neuroprotection. J Neural Transm Suppl. 2000;59:81–89. | ||
Krupinski J, Kumar P, Kumar S, Kaluza J. Increased expression of TGF-beta 1 in brain tissue after ischemic stroke in humans. Stroke. 1996;27(5):852–857. | ||
Zhu Y, Ahlemeyer B, Bauerbach E, Krieglstein J. TGF-beta1 inhibits caspase-3 activation and neuronal apoptosis in rat hippocampal cultures. Neurochem Int. 2001;38(3):227–235. | ||
Zhu H, Gui Q, Hui X, et al. TGF-β1/Smad3 signaling pathway suppresses cell apoptosis in cerebral ischemic stroke rats. Med Sci Monit. 2017;23:366–376. | ||
Sansing LH, Harris TH, Welsh FA, Kasner SE, Hunter CA, Kariko K. Toll-like receptor 4 contributes to poor outcome after intracerebral hemorrhage. Ann Neurol. 2011;70(4):646–656. | ||
Askenase M, Sansing L. Stages of the inflammatory response in pathology and tissue repair after intracerebral hemorrhage. Semin Neurol. 2016;36(03):288–297. | ||
Qureshi AI, Ling GS, Khan J, et al. Quantitative analysis of injured, necrotic, and apoptotic cells in a new experimental model of intracerebral hemorrhage. Crit Care Med. 2001;29(1):152–157. | ||
Qureshi AI, Suri MF, Ostrow PT, et al. Apoptosis as a form of cell death in intracerebral hemorrhage. Neurosurgery. 2003;52(5):1041–1047. | ||
Gong C, Boulis N, Qian J, Turner DE, Hoff JT, Keep RF. Intracerebral hemorrhage-induced neuronal death. Neurosurgery. 2001;48(4):875–882. | ||
Zhang Z, Liu Y, Huang Q, et al. NF-κB activation and cell death after intracerbral hemorrhage in patients. Neurol Sci. 2014;37(5):1097–1102. | ||
Li P, Allen H, Banerjee S, et al. Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock. Cell. 1995;80(3):401–411. | ||
Ma Q, Chen S, Hu Q, Feng H, Zhang JH, Tang J. NLRP3 inflammasome contributes to inflammation after intracerebral hemorrhage. Ann Neurol. 2014;75(2):209–219. | ||
Traenckner EB, Pahl H, Henkel T, Schmidt KN, Wilk S, Baeuerle PA. Phosphorylation of human IKB-ox on serines 32 and 36 controls IKB-a proteolysis and NF-KB activation in response to diverse stimuli. EMBO J. 1995;14(12):2876–2883. | ||
Chang MS, Chen BC, Yu MT, Sheu JR, Chen TF, Lin CH. Phorbol 12-myristate 13-acetate upregulates cyclooxygenase-2 expression in human pulmonary epithelial cells via Ras, Raf-1, ERK, and NF-kappaB, but not p38 MAPK, pathways. Cell Signal. 2005;17(3):299–310. | ||
Zheng J, Kong C, Yang X, Cui X, Lin X, Zhang Z. Protein kinase C-α (PKCα) modulates cell apoptosis by stimulating nucleartranslocation of NF-kappa-B p65 in urothelial cell carcinoma of the bladder. BMC Cancer. 2017;17(1):432. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.