Back to Journals » OncoTargets and Therapy » Volume 13
The Use of Molecular Subtypes for Precision Therapy of Recurrent and Metastatic Gastrointestinal Stromal Tumor
Authors Liu P, Tan F, Liu H ![]() , Li B, Lei T
, Li B, Lei T ![]() , Zhao X
, Zhao X
Received 6 December 2019
Accepted for publication 10 March 2020
Published 24 March 2020 Volume 2020:13 Pages 2433—2447
DOI https://doi.org/10.2147/OTT.S241331
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Takuya Aoki
Peng Liu,1,* Fengbo Tan,1,* Heli Liu,1 Bin Li,2 Tianxiang Lei,1 Xianhui Zhao1
1Department of Gastrointestinal Surgery, Xiangya Hospital, Central South University, Changsha, Hunan 410008, People’s Republic of China; 2Department of Oncology, Xiangya Hospital, Central South University, Changsha 410008, Hunan, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Heli Liu
Department of Gastrointestinal Surgery, Xiangya Hospital, Central South University, Changsha 410008, Hunan, People’s Republic of China
Tel +86 13874967285
Email [email protected]
Abstract: Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumor in the digestive tract. Tyrosine kinase inhibitors (TKIs), represented by imatinib, sunitinib, and regorafenib, have become the main treatment for recurrent and metastatic GISTs. With the wide application of mutation analysis and the precision medicine, molecular characteristics have been determined that not only predict the prognosis of patients with recurrent and metastatic GISTs, but also are closely related to the efficacy of first-, second- and third-line TKIs for GISTs, as well as other TKIs. Despite the significant effects of TKIs, the emergence of primary and secondary resistance ultimately leads to treatment failure and tumor progression. Currently, due to the signal transmission of KIT/PDGFRA during onset and tumor progression, strategies to counteract drug resistance include the replacement of TKIs and the development of new drugs that are directed towards carcinogenic mutations. In addition, it is also the embodiment of precision medicine for GISTs to explore new carcinogenic mechanisms and develop new drugs relying on new biotechnology. Surgery can benefit specific patients but its major purpose is to diminish the resistant clones. However, the prognosis of recurrent and metastatic patients is still unsatisfactory. Therefore, it is worth paying attention to how to maximize the benefits for patients.
Keywords: gastrointestinal stromal tumor, tyrosine kinase inhibitor, precise medicine
Introduction
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumor in the digestive tract with an incidence of 10–15 new cases per million each year.1,2 GISTs originate from interstitial cells of Cajal (ICC) in the nerve plexus of the intestinal wall which control the gastrointestinal peristalsis.3 Before the availability of TKIs, the patients with metastatic diseases had a poor prognosis whose median survival ranged from 10 to 20 months and 5-year survival was less than 10% due to resistance to conventional chemotherapy.4 However, the elaboration of carcinogenesis and emergency of TKIs significantly changed the therapeutic mode and improved the prognosis. Like carcinomas, molecular subtypes determine the biological behavior of tumors, and they also can affect the prognosis and efficacy of TKIs. Therefore, it is necessary to summarize the relationship between molecular subtypes and the efficacy and prognosis of recurrent and metastatic GISTs to guide personalized medicine.
Molecular Pathogenesis of GISTs
82%–87% of GISTs harbor activating mutations in KIT or PDGFRA.5 They are both the type III receptor tyrosine kinase with high homology and similar downstream signaling pathways, but mutually exclusive.6,7 Mutations in KIT mainly occur in exon 11, which can activate kinase by destroying the self-inhibitory function of the juxtamembrane domain, and followed by exon 9 which can also activate the kinase.8 Although the mutations in exon 13 and exon 17 are very low, they are common in secondary mutations and have important clinical significance in secondary drug resistance.7,9-11 The PDGFRA mutations often occur in exon 18, which further stabilizes the active status of kinase, but it is rare in exon 12 and exon 149 (Figure 1). The KIT or PDGFRA mutations lead to receptor constitutive and ligand-independent activation, which then activates the downstream signaling pathways, including the MAP kinase pathway (RAF, MEK, ERK), the STAT pathway, and the PI3K/AKT pathway.12,13 However, there are considerable differences in the activation of downstream signal pathways in different mutant types of GIST which explains variation in biological behavior of tumors12 (Figure 2A).
 |
Figure 1 Type and frequency of activating mutations in KIT and PDGFRA. Abbreviations: SCF, stem cell factor; PDGF, platelet-derived growth factor. |
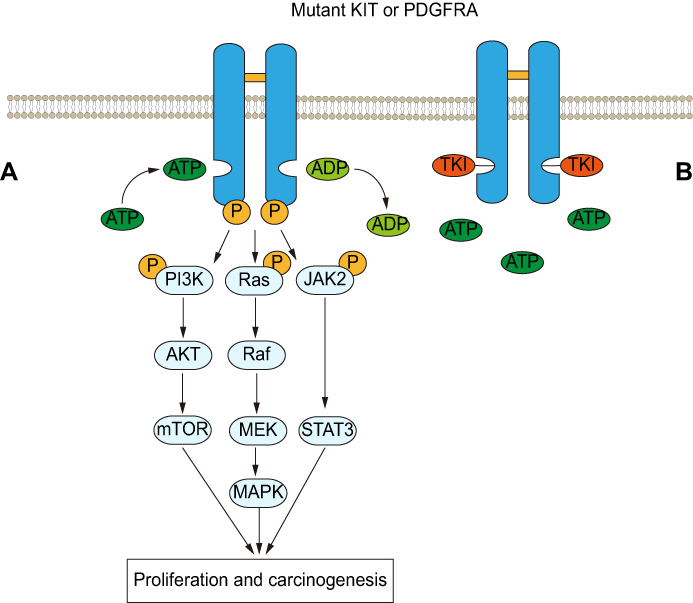 |
Figure 2 (A) Major downstream signaling pathways in GISTs. Mutations in KIT/PDGFRA cause receptors to homodimerize on the cell surface and activate the tyrosine kinase domain which phosphorylates the tyrosine residue by transfer phosphate (P), activates the downstream signal pathway and then promotes the occurrence and development of GIST. (B) Tyrosine kinase inhibitor (TKI) blocks phosphorylation of downstream pathways and inhibits the progression of GIST by competitively binding KIT/PDGFRA with ATP. Abbreviations: PI3K, phosphoinositide 3-kinase; AKT, protein kinase B; mTOR, mammalian target of rapamycin; RAF, RAF proto-oncogene serine/threonine-protein kinase; MEK, mitogen-activated protein kinase; MAPK, mitogen-activated protein kinase; JAK2, Janus kinase 2; STAT3, signal transducer and activator of transcription 3. |
It has been reported that approximately 10%-15% of GISTs have no KIT or PDGFRA mutation, and these are commonly referred to as KIT/PDGFRA wild-type GISTs (WT-GISTs).2 However, these tumors contain alternative signal mutations such as BRAF/KRAS, NF1, or the succinate dehydrogenase (SDH) complex.7,14 Besides, more and more molecular alterations related to the pathogenesis of WT-GIST have been found (Table 1).
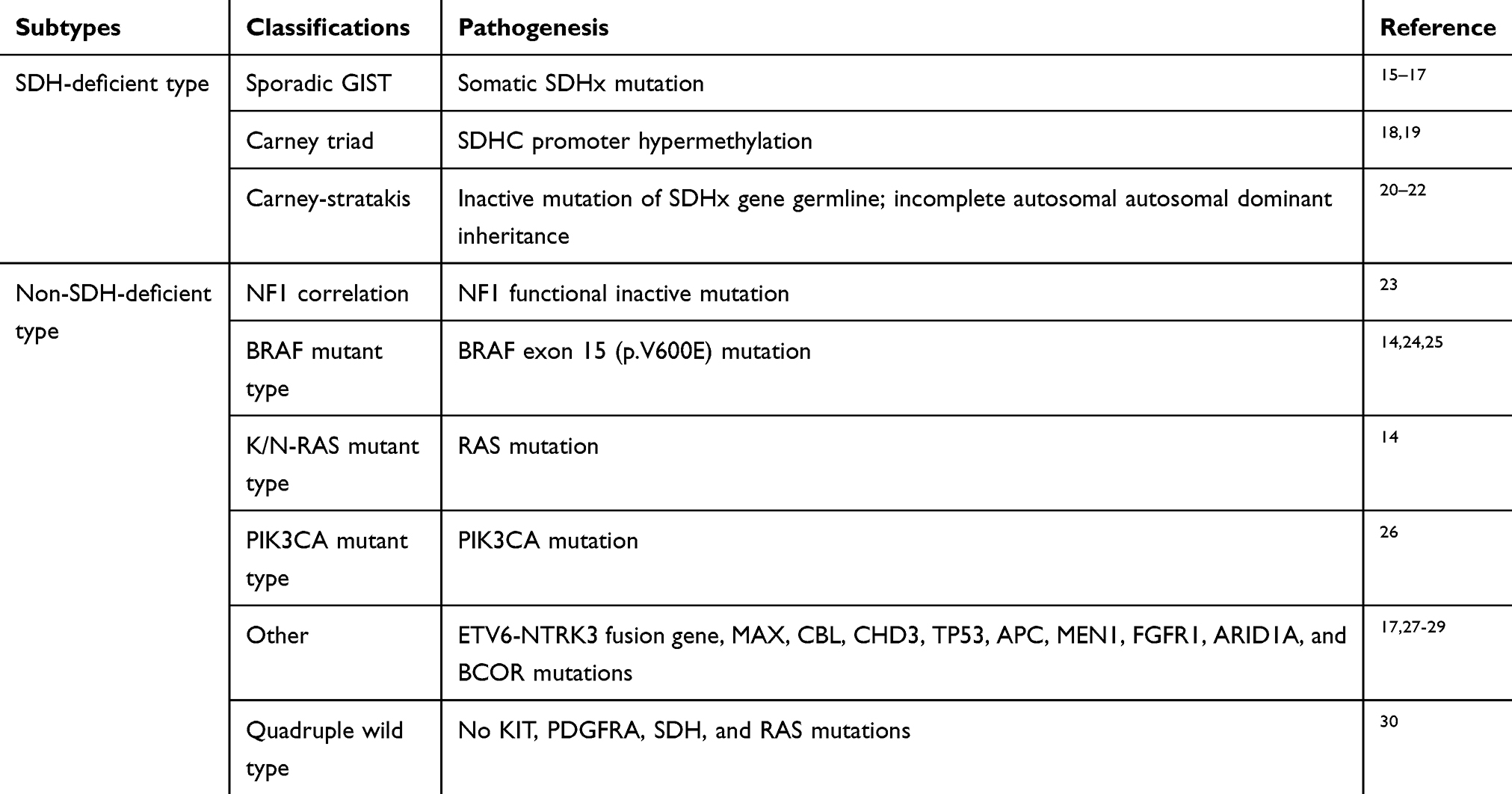 |
Table 1 Molecular Mutations Associated with WT-GIST |
Application of Molecular Subtypes in Treatment of Recurrent and Metastatic GISTs and Drug Resistance
Activating mutations in KIT/PDGFRA are the theoretical basis for the treatment of GISTs with tyrosine kinase inhibitors (TKIs) and also for the development of new drugs (Figure 2B). Imatinib, sunitinib, and regorafenib are representative first-line, second-line, and third-line TKIs, respectively, that have been approved for clinical use.31 A large number of clinical studies have proved that the therapeutic effect of TKIs is closely related to the molecular characteristics of patients with recurrent and metastatic GISTs. Therefore, combined with the molecular subtypes or dynamic monitoring of mutations in patients, targeted drug selection or a timely change in a patient’s treatment plan will result in patients obtaining the best prognosis.
Imatinib (IM)
IM is a small-molecule tyrosine kinase inhibitor with strong efficacy for inhibiting KIT and PDGFRA.32 It has become the first-line treatment for recurrent and metastatic GISTs, and the recommended standard dose is 400 mg/d. Clinical trials have proved that IM can benefit up to 85% of patients with recurrent and metastatic GISTs, and the median overall survival (OS) is 50 months.33,34 However, some studies have shown that increasing the dose of IM to 800 mg/d seems to result in more significant benefit for some patients, with better prognosis, although the results have been controversial.35–38 Further analysis of the above results indicates that the reason for this controversy is the different sensitivities of patients to different molecular subtypes to IM.
The European Organization for Research and Treatment of Cancer (EORTC) Phase III and North American phase III studies analyzed the mutation status of tumor specimens from 377 and 428 patients with recurrent and metastatic GISTs before treatment, respectively, aiming to explore the correlation of molecular subtypes with IM efficacy and prognosis.39,40 In the EORTC study, patients with the KIT exon 11 mutation had a higher response rate to IM treatment, and their 2-year cumulative response rate (69%) was higher than those with the KIT exon 9 mutation (34%) and WT-GIST (25%). Moreover, patients with the KIT exon 11 mutation also had increased progression-free survival (PFS) and OS, and the relative risk of tumor progression was significantly reduced.39 The North American and EORTC studies also reported similar results, consisting of patients with the KIT exon 11 mutation exhibiting a higher complete remission/partial remission (CR/PR) rate as a result of IM treatment, with increased OS and PFS. There was no significant difference in prognosis or IM objective response between patients with KIT exon 9 mutation and WT-GISTs.39,40
At the same time, the relationship between molecular subtypes and IM at different dose levels (400 mg/d and 800 mg/d) was further analyzed. In the EORTC phase III study, PFS in patients with the KIT exon 9 mutation increased after the administration of high-dose IM (P = 0.0013), and relative risk was also reduced by 61%. PFS of patients with KIT exon 11 mutations or WT-GISTs was not affected by dose differences. No significant dose effect was observed for OS in all three subgroups.39 In the North American study, it was also found that patients with the KIT exon 9 mutation exhibited a higher objective response rate (CR/PR 67% vs 17%; P = 0.02) under IM treatment of 800 mg/d. However, a dose difference associated with PFS for patients with the KIT exon 9 mutant was not observed in this study, which might be due to the fact that patients with tumor progression were allowed to receive crossover treatment from 400 mg/d to 800 mg/d, thus obscuring the effect of dose difference.40 A meta-analysis indicated that the KIT exon 9 mutation was the only predictor of the impact of high-dose IM treatment on PFS. In patients with the KIT exon 9 mutation, high-dose IM treatment resulting in higher objective response rate and significantly prolonged PFS, reducing the risk of progression or death by 42%, but it produced no such advantage in OS.41 After longer-term follow-up, it was found that although there were differences in tumor biology among different mutation types in KIT exon 11, there was no significant difference in OS after IM treatment.42
Although the above study confirmed the guiding significance of molecular subtypes in patients with recurrent and metastatic GISTs, the study subjects were limited to those with KIT exon 9 and 11 mutations and WT-GISTs, while other molecular subtypes could not be further analyzed due to the small number of patients. However, in the EORTC study, patients with KIT exon 13/17 mutations were found to have objective response after IM treatment, although the objective response rate was relatively low.39 Corless et al also confirmed that PDGFRA exon 12/14 exhibited sensitivity to IM in vitro.13 Moreover, these studies only analyzed the mutations in tumor specimens before treatment, without considering the influence of IM resistance, especially secondary resistance, on the results. Above all, it was evident that IM 400 mg/d is sufficient for most patients, but for those with the KIT exon 9 mutation, IM 800 mg/d can be used in advance, instead of increasing the dose after disease progression.
Imatinib-Resistant, Recurrent and Metastatic GISTs
IM can control or even alleviate most recurrent and metastatic GISTs, but 85%–90% of patients usually experience disease progression within 20–24 months, which is called IM resistance.43 IM resistance is a very difficult problem in the treatment of recurrent and metastatic GISTs and is also a key factor affecting prognosis. According to the mechanisms, drug resistance can be divided into primary drug resistance (tumor progression in the early stage of treatment) and secondary drug resistance (tumor progression after initial stability or response to IM). According to statistics, approximately 10% of patients exhibit primary IM resistance, while 40%–50% exhibit secondary IM resistance. The median progression time is approximately 24 months.44,45 The occurrence of drug resistance is not random, but is closely related to gene mutation. The study of its correlation can assist in ensuring that patients avoid excessive and inadequate treatment.
The characteristic of primary drug resistance is that the same mutation appears before and after IM treatment, and rarely is there secondary mutation.2,44,46 It usually occurs in specific molecular subtypes: PDGFRA D842V mutation, KIT exon 9 mutation and WT-GIST.44 The PDGFRA D842V mutation is the most common cause of primary drug resistance.47 The mechanism may be that IM can only bind to the inactive conformation of KIT/PDGFRA, while the mutation of D842V results in a change in the kinase activating loop, which results in kinase alteration so that it assumes its active conformation.13 In vivo and in vitro studies and clinical practice have proved that the D842V mutation is responsible for the primary drug resistance to IM.13,48 The median survival time of GIST patients with the D842V mutation is only 12.8 months compared with the mean survival time of 48–60 months for GISTs treated with IM.7 Therefore, the D842V mutation has become a candidate for clinical research with the aim of developing new molecular targeted drugs and is expected to become a new breakthrough in clinical treatment in the future.
There has been extended controversy regarding whether KIT exon 9 is resistant to IM, because the poor effect of IM treatment in these patients is partly due to inadequate IM dosage. Compared with the KIT exon 11 mutation, patients with exon 9 mutation exhibited a lower response rate to the standard dose (400 mg/d) IM.39,49 However, by increasing the IM dose, patients can obtain a response rate equivalent to that obtained by patients with the KIT exon 11 mutation, with increased PFS.39–41 In WT-GIST patients, a mutation of the alternative signaling pathway is the main mechanism of drug resistance.17 For example, BRAF and RAS proteins are part of the MAPK signal cascade, which leads to KIT-independent growth stimulation, and a lack of inhibitory effect by IM when it acts upon KIT for this type of tumor. Multiple clinical trials have confirmed that these tumors exhibit primary resistance to IM.7 Fortunately, because these tumors are often inert, patient mortality is decreased.50
Unlike primary drug resistance, secondary drug resistance is often associated with new secondary mutations of KIT/PDGFRA.46 A B2222 Phase II study showed one or more secondary mutations in 67% of recurrent and metastatic GISTs.51 Secondary mutations are most common in patients with primary mutations in KIT exon 11, followed by those with mutations in KIT exon 9, which rarely occur in WT-GISTs. The reason for this may be that patients with mutations in KIT exon 11 receive extended treatment with IM, which leads to screening of drug-resistant mutations.11,51 Secondary mutations are usually clustered in the ATP binding pocket (exon 13 and exon 14) and the kinase activating loop (exon17 and exon18) of the kinase domain. These mutations are always found in the same allele as that of the primary KIT mutation.52 The mutations change the configuration of the KIT protein, which affects the binding efficiency of targeted drugs and eventually leads to disease progression.53 Although there are advantages in using molecular typing to predicting drug resistance, it also has some limitations because of the obvious heterogeneity of the secondary mutations. Ligel et al found that 83% of patients had secondary mutations in KIT, 67% of them had 2–5 different secondary mutations in separate metastases, and even 34% of them with the same metastasis exhibited two different secondary mutations in KIT.54 Therefore, because of the heterogeneity of secondary mutations, it is difficult to understand the overall mutation of patients through molecular detection so that treatment can be accurately guided.
In addition, with further application of molecular biotechnology and second-generation sequencing, other mechanisms of drug resistance have been revealed, such as KIT/PDGFRA gene amplification, IGF1R overexpression in wild-type GISTs, and low IM plasma level, and an understanding of other drug-resistance mechanisms can supplement the guiding role of molecular typing.55–58
Sunitinib and Regorafenib
With the continuous application of new therapies and drugs in clinical treatment, there is an increasing number of options for the treatment of recurrent and metastatic GISTs. In the process of IM treatment with standard doses, if the tumor continues to progress, and the treatment dose of IM can be increased to control the disease.2,38,59,60 However, based on the central role of KIT secondary mutations in IM resistance and tumor progression, the multi-target tyrosine kinase inhibitors (sunitinib and regorafenib) have become standard second-line and third-line treatment options, respectively, after first-line treatment failure.61
Sunitinib (SU)
Sunitinib exhibits not only anti-KIT and PDGFR activity, but also anti-VEGFR ability to inhibit tumor angiogenesis.62–64 Similar to IM, the efficacy of SU is closely related to the primary mutation of GISTs. It has been noted that SU has significant activity against KIT exon 11 in vitro, but most patients treated with SU had been previously treated with IM and usually harbored secondary mutations, and therefore, it was determined that SU is less active against KIT exon 11 mutants in vivo than exon 9 mutants, which have a low incidence of secondary mutation.65 Henrich et al proved that compared with the KIT exon 11 mutation, patients with the KIT exon 9 mutation not only had a higher response rate to SU, but also obtained significantly longer median PFS and OS.66 This may have occurred to the low rate of secondary mutations in KIT exon 9. Subsequent studies have also indicated that there is a greater benefit for SU therapy when KIT exon 9 mutations are present as compared to the presence of the KIT exon 11 mutations.67,68 For patients with advanced GISTs, SU treatment or IM escalation as a second-line therapy has been controversial, because the two strategies can benefit patients with advanced GISTs. Therefore, a direct comparison in 2014 between SU treatment and IM escalation as the first choice strategy of second-line treatment in 2014 showed that the molecular subtypes had guiding significance in strategy selection.69 In this study, although there was no significant difference in response rate (RR=CR+PR), tumor control rate (TCR=CR+PR+SD), PFS and OS between the IM escalation group and SU treatment group, subgroup analysis indicated that different molecular subtypes should be considered in the selection of second-line treatment. In patients with KIT exon 9 mutations, IM escalation tends to favor PFS and OS. In contrast, in patients without KIT exon 9 mutation, SU treatment can achieve better prognosis.
However, unlike IM, the efficacy of SU is not only related to the primary mutations, but it is also affected by the secondary mutations.11 In vitro experiments showed that sunitinib effectively inhibited KIT exon 9, WT-GIST, and KIT exon 11 mutants, and also significantly inhibited the phosphorylation of KIT double mutants in the secondary mutation of ATP binding pocket (exon 13/14). However, the effect of sunitinib on KIT double mutants in the secondary mutation of the activation loop (exon 17/18) was relatively poor, which was also the mechanism of SU resistance.11 Clinical studies have shown that clinical benefit (CB) can be observed in 65% of patients with secondary exon 13 or 14 mutations after sunitinib treatment, but in only 9% of patients with secondary exon 17 or 18 (P = 0.006).66 In conclusion, the choice of second-line therapy for late GISTs should not only consider primary mutations, but also consider the influence of secondary mutations.
Regorafenib (RE)
Regorafenib also blocks the activity of several protein kinases, including KIT, PDGFRA, VEGFR, and BRAF.72 Clinical studies have shown that RE is effective as a third-line treatment for patients with advanced GISTs. In 2013, a multicenter randomized, double-blind, placebo-controlled phase III clinical study on advanced GISTs after failure of RE in IM and SU treatment showed that compared with placebo, regorafenib increased PFS by more than five times, and reduce the risks of progression and death by 73%.73
The therapeutic effect of RE is closely related to tumor mutation. In a long-term follow-up multicenter phase II trial that enrolled in patients with metastatic or unresectable GISTs after failure of treatment with IM and SU, the KIT exon 11 mutation and SDH-deficient GISTs conferred long-term clinical benefits (median PFS: 13.4 months and 10 months, respectively).74 Moreover, there is evidence indicating that RE has a significant anti-tumor effect on patients with KIT exon 17 mutations after exposure to multiple TKIs.75 RE significantly prolonged PFS in patients with exon 17 mutation, with a disease control rate of 93.3% at 16 weeks.76
Wild-Type Stromal Tumors: Controversy and Rationale
Although the treatment of GISTs guided by molecular subtypes can be used as a model of precision medicine for cancer, there is no similar consensus for WT-GISTs. The diagnosis of WT-GISTs is only based on the detection of six exons. The results show a possibility of a “pseudowild-type,” and KIT phosphorylation can be detected in some patients, which indicates that tumor growth still depends on KIT activation.77 This also explains why some patients benefit from IM therapy.39,40 In addition, there is evidence indicating that in SDH-deficient GISTs, the inactivation of succinate dehydrogenase leads to the accumulation of HIF-1α and the formation of transcription factors to induce angiogenesis and glycolysis-related gene expression (including expression of VEGF).74,78 Therefore, there are increased benefits from the use of SU and RE (multiple targeted kinase inhibitors and anti-VEGF activity) than from the use of IM. In a phase I/II study of SU for treatment of recurrent and metastatic GISTs, the median PFS and OS for WT-GISTs reached 19 months and 30.5 months, respectively.11 In a clinical study in which Janeway et al used SU to treat GIST (usually WT) in children, 6/7 of them showed PR or stable disease (SD), and SU delayed tumor progression to a greater extent as compares to previous IM treatment.79 After RE treatment, these patients obtained a tumor response or SD for at least 16 weeks.74 Given the fact that SU and RE are more effective than IM for WT-GIST, especially in the SDH-deficient type, whether to bypass the use of IM and use SU or RE directly for these patients required clinical confirmation.
Metastasectomy
Although recurrent and metastatic diseases, surgery can be considered as another therapeutic strategies. However, its role is limited and only regarded as the adjuvant therapy of TKIs. The main purpose is to eliminate drug-resistant clones as much as possible and control the progression of disease and deal with tumor-related complications such as obstruction and hemorrhage.
Multiple studies had examined the efficacy of surgery and demonstrated that cytoreduction is more beneficial for patients with response to TKIs than those with drug-resistance or without TKI treatment.80,81 In 2018, there is a large retrospective study involved 400 operations on 323 advanced patients. Prior to the surgery, all of them were developed metastatic diseases and received treatment with TKIs.82 The same as the previous finding, radiographic response to TKIs at the time of surgery was predictive of outcome. Patients with responsive disease experienced a better median PFS (31 months) than those with stable disease, unifocal progressive disease and multifocal progressive disease (19, 10 and 5 months, respectively, P<0.001) after surgery, and it was the same in OS. Interestingly, further subgroup analysis showed that radiographic response influenced the prognosis in patients on IM but not in patients on SU. Besides, patients with IM treatment had a prolonged PFS and OS compared to those treated by SU after cytoreduction which reflects that the effect of surgery in these patients was limited. Multi-site metastasis and high mitotic index in metastasis were also the independent predictors of decreased PFS on multivariate analysis. Therefore, it is feasible that cytoreduction can be performed in patients who exhibit response, stable diseases or unifocal progression after treatment with IM. Moreover, it is crucial to decide when to have the operation among TKI responders. There is suggestion that the timing of surgery ranged from 6 to 24 months which related to the duration of efficacy of IM and secondary resistance.4
Other Treatment Strategies
Novel Molecular Targeted Therapy
Currently, the presence of multi-TKIs-resistant clones in patients reduces the efficacy of molecular targeted therapies. However, even if the treatment fails, tumor progression still depends on the signal transmission of KIT/PDGFRA, which provides the basis for the development of new drugs. The response of some new TKIs, such as sorafenib, pazopanib and dasatinib, has been tested for their effectiveness against recurrent and metastatic GISTs, and some therapeutic effects have been observed.83–85
Sorafenib is a multi-kinase inhibitor. Its biochemical activity has been confirmed in a study of human GIST cell lines with clinically representative primary and secondary mutations of KIT. In this study, sorafenib effectively inhibited the mutation of KIT exon 11, and its effect was greater than or equal to that of IM and SU.86 Moreover, sorafenib was also effective against secondary mutations involving the ATP binding pocket and activating loop, but had no significant resistance against D816V and D842V.86 Given the effectiveness of sorafenib, Park et al reported the phase II results of sorafenib as a third-line treatment, with DCR of 36% at 24 weeks, and median PFS and OS of 4.9 and 9.7 months, respectively. In addition, the PFS of patients with KIT exon 11 mutations was significantly better than that of patients with other mutation types (P = 0.0341). Therefore, sorafenib may be a rescue treatment for GIST patients with mutation of KIT exon 11.83
Ponatinib, which is a multi-target TKI similar to sorafenib, has a wide range of inhibitory effects on KIT, and its activity level has also been evaluated in vitro and in vivo.87 However, ponatinib is different from sorafenib in that it has a strong effect on secondary mutations of the activating loop and T670I gatekeeper of exon 14, but not on secondary mutations of exon 13 (V654A).87 A phase II clinical study with ponatinib that treated late GIST patients with the KIT exon 11 mutation also confirmed its validity. In this trial, the 16-week clinical benefit rate (CBR 37%) of KIT exon 11 mutation was significantly higher than that of non-KIT exon 11 mutation (14%), and the CBR for KIT exon 11 patients who experienced failure of two TKI treatments was higher (44%), possibly due to additional secondary mutations.88
The presence of multi-kinase inhibitors improves the prognosis and increases the survival rate of patients with advanced GIST resistance to IM. However, even the most active TKIs do not achieve a 10% overall response rate, and median PFS does not reach 6 months after IM treatment failure.43 Inhibition of specific mutations, especially drug-resistance mutations, is another strategy to solve TKI resistance and has resulted in some breakthroughs. Avapritinib (BLU-285) is a new type of kinase inhibitor with high selectivity for KIT, PDGFRA and other targets, especially for activation loop mutation (D842V/D816V).89 The PDGFRA D842V mutation is resistant to most TKIs, but under the treatment of BLU285, the response rate of patients to the treatment was as high as 86%, while the response rate of other non-D842V patients was only 20%-26%.90 These data indicate that the drug effectively overcomes the problem of D842V resistance. Crenolanib effectively inhibits the D842V mutation, has been proven to inhibit GIST cell lines containing the D842V mutation in vitro. Currently, a phase III clinical trial for crenolanib is in the process of recruitment (NCT02847429).91 Ripretinib (DCC-2618) is a broad-spectrum, switch-controlled kinase inhibitor targeting cancer and drug-resistant KIT and PDGFRA mutations. It can transform the kinase into an inactive conformation, and inhibit KIT exon 13, 14, 17 and 18 mutations, including KIT D816V and PDGFRA D842V mutations with multiple drug resistance, and shows considerable efficacy in preclinical models.92,93 A clinical phase III trial about this agent is undergoing (NCT03673501).94 In addition, many drugs have been proved to have anti-GIST effects in vitro or by clinical trials, although for some trials, the number of patients was small, and only a limited analysis on molecular subtypes and efficacy was conducted (Table 4).
 |
Table 2 Comparison in Efficacy of SU Treatment and IM Escalation Based on Molecular Subtypes |
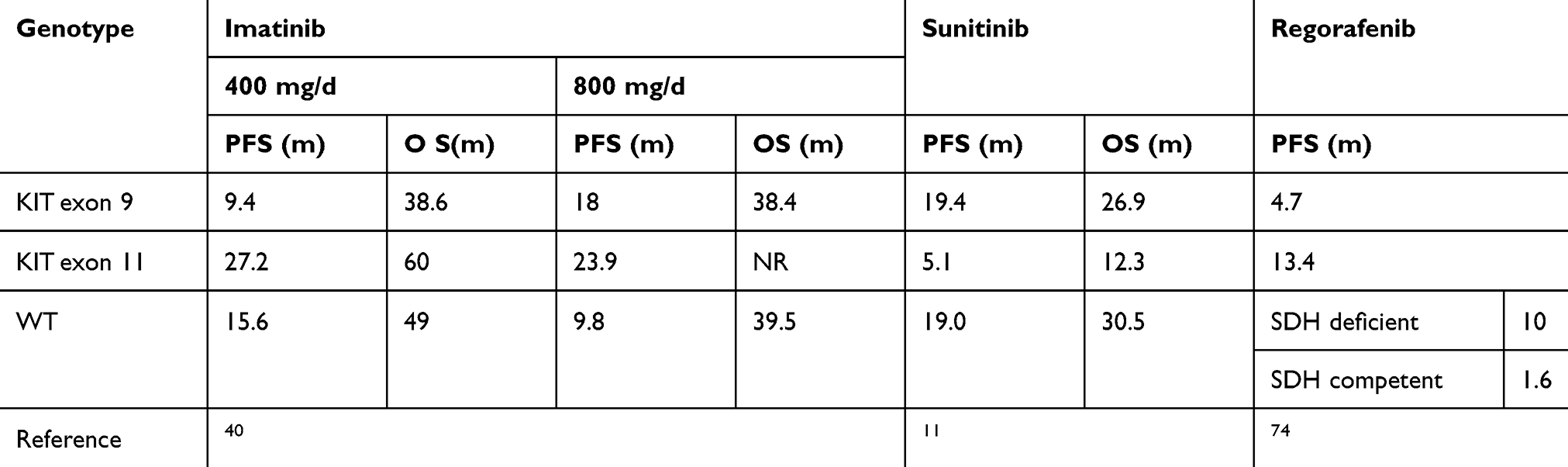 |
Table 3 Relationship of Molecular Subtypes with IM, SU and RE Efficacy |
 |
Table 4 Relationship Between New Drugs and Sensitive Molecular Subtypes |
Drug Combination
Because of the heterogeneity of GISTs and the polyclonality of secondary mutations, it is necessary to study possible combination therapies based on existing drugs for the purpose of prolonging the antitumor effect. Given the complementarity of SU and RE in the inhibition of KIT-resistant mutations, Serrano et al reported a clinical Phase I study on the treatment of TKI-refractory GISTs with a combination of SU and RE. The results showed that rapid administration of the alternative combination of SU and RE effectively overcame multiclonal resistance, and 29% of patients exhibited SD with a median PFS of 1.9 months.109 Drug combination therapy was previously limited to the combinations of TKIs, but has gradually expanded to include combinations of TKIs with other types of drugs, including KIT downstream pathway inhibitors and immunosuppressants and has achieved preliminary results.43
As the understanding of the pathogenesis of GISTs continues to deepen, there has been an expansion of the precision medicine for GISTs. Previously, GIST treatment was limited to the targeting of conventional oncogene mutations, but the research direction has shifted to other fields, including the metabolism of GIST tumor cells, cell cycle regulation, target kinase protein stability and immunotherapy, and these have been described in detail in the relevant literature.31,43 However, the efficacy of these agents requires clinical verification, and may produce positive results (Table 5).
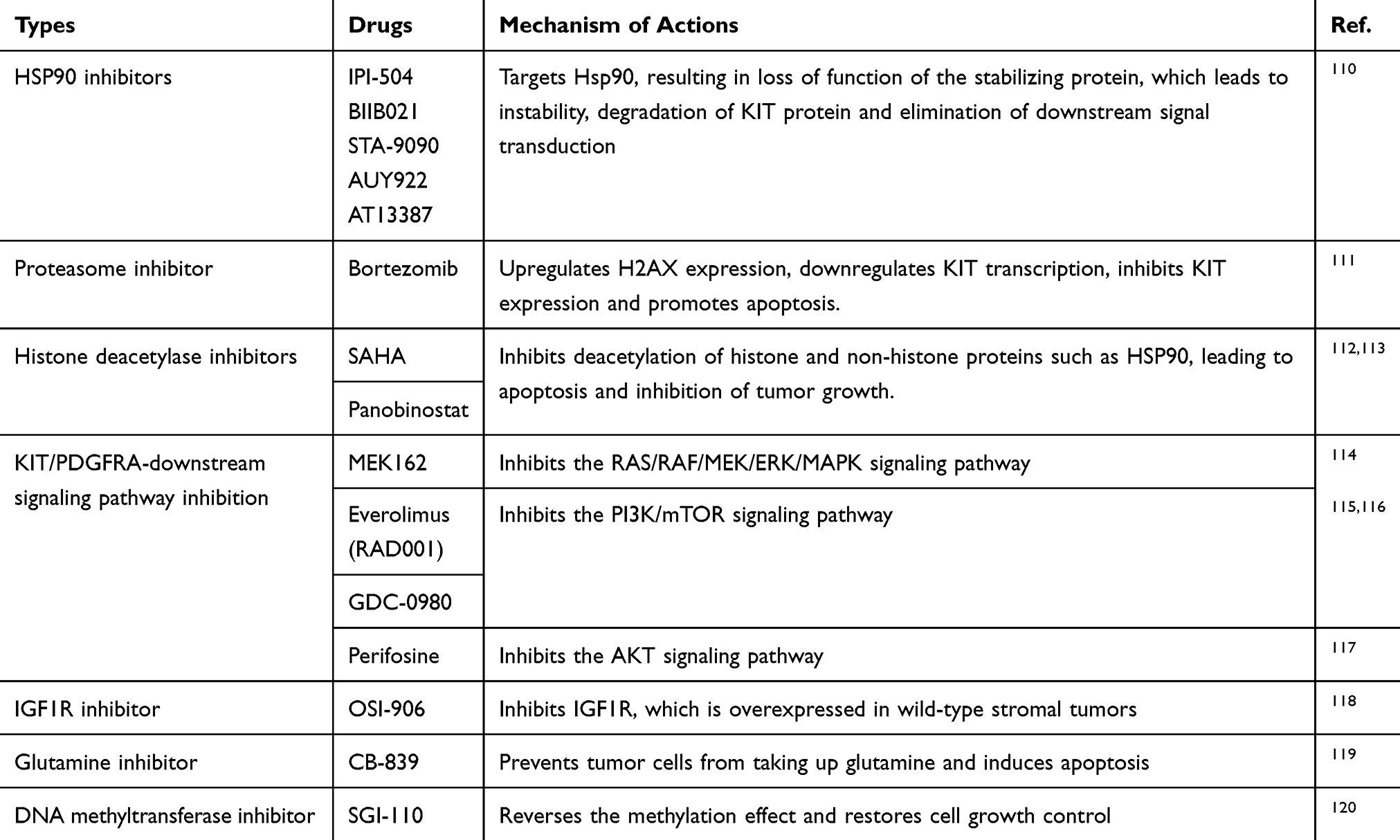 |
Table 5 Precise Medicines for GISTs and Their Mechanism of Action |
Novel Molecular Mechanism of GIST
Currently, the exploration of novel mechanism of pathogenesis is also promising in GIST. In 2010, Chi et al found that ETV1 and KIT had a synergistic effect on the development and progression of GISTs.121 Recently, Pang et al found that DEPDC5 was a GIST-specific tumor suppressor and TKI treatment response regulator, and was closely related to the development and progression of GISTs.122 Sequencing of the entire exon showed that DEPDC5 reduced cell proliferation and induced cell cycle arrest through the mTORC1 signaling pathway, but it could lead to the progression of GISTs when it lost function due to inactivation mutation. In addition, DEPDC5 is not only a potential therapeutic target, but also can regulate the sensitivity of GISTs to KIT inhibitors. Combining KIT inhibitors with mTOR inhibitors results in a stronger antitumor effect in the inactivated mutations of GISTs by DEPDC5, which requires confirmation in clinical studies.
Epigenetic changes during the onset of GISTs also play a crucial role. SDH-deficient GISTs have the characteristics of SDH deletion and overall DNA hypermethylation, but how they cause cancer is still unknown, and this limits the treatment strategy selection for such patients.123,124 However, a landmark study has obtained additional data regarding the pathogenesis of SDH-deficient GISTs, linking hypermethylation with changes in genome topology.125 In SDH-deficient GISTs, hypermethylation can destroy the insulator between the carcinogenic gene FGF4 and the enhancer, thus allowing abnormal interaction between the enhancer and the oncogene, and up-regulating the expression of FGF4. The PDX model based on SDH-deficient GISTs shows that tumors are more sensitive to FGFR and KIT inhibitors, thus providing a new method for treatment of such patients.125
Biomedical Technology and Precision Medicine
The invention and application of biomedical technology can enhance the diagnosis and treatment of many tumors, including GISTs, hasten the development towards precise medicine, and promote the application of basic tumor research in the clinic. These technologies include single-cell technology, PDX and organoid technology.
The subclonal state during the process of tumor cell expansion is the main cause of tumor heterogeneity and drug resistance.126 However, single-cell technology can be used to study tumors at the single-cell level, analyze intratumoral heterogeneity, reveal tumor metastasis mechanisms and study epigenetic changes.127 Moreover, drug-resistance mutations are a major cause of GIST tumor progression. Single-cell technology can predict the drug sensitivity of tumor cells to TKIs, timely identify resistance mutations, analyze resistance mechanism and overcome resistance.127–129 It has been reported that cell surface vimentin-positive macrophage-like circulating tumor cells, as a new biomarker of metastatic GISTs, can predict the risk of GIST metastasis.130 This technology can provide patients with overall and comprehensive tumor information, and elucidate the differences between individuals, so that clinical decision-making and individualized drug use can be a guide for tumor treatment.131 PDX and organoids are used to construct tumor models in vitro, which play an important role in basic tumor research, as well as in the research and development of drugs, and individualized use of drugs, but there are still some limitations to be solved132–135 (Table 6).
 |
Table 6 The Applications and Limitations of Biomedical Technology |
Conclusions
Currently, the molecular subtype of KIT and PDGFRA is crucial for the selection of therapeutic strategies and can maximize the benefit of recurrent and metastatic patients, as summarized in Tables 2 and 3. However, WT-GIST and drug-resistance are very tough and should be resolved as soon as possible. In addition to novel anti-tyrosine kinase agents as listed in Table 4, exploration of new mechanism and application of biomedical technology are very promising, but further research should be required. On this basis, surgery should also be considered as part of targeted therapy.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Soreide K, Sandvik OM, Soreide JA, Giljaca V, Jureckova A, Bulusu VR. Global epidemiology of gastrointestinal stromal tumours (GIST): a systematic review of population-based cohort studies. Cancer Epidemiol. 2016;40:39–46. doi:10.1016/j.canep.2015.10.031
2. Demetri GD, von Mehren M, Antonescu CR, et al. NCCN task force report: update on the management of patients with gastrointestinal stromal tumors. J Natl Compr Canc Ne. 2010;8:S1–S43. doi:10.6004/jnccn.2010.0116
3. van Roggen JFG, van Velthuysen MLF, Hogendoorn PCW. The histopathological differential diagnosis of gastrointestinal stromal tumours. J Clin Pathol. 2001;54(2):96–102. doi:10.1136/jcp.54.2.96
4. Keung EZ, Fairweather M, Raut CP. The role of surgery in metastatic gastrointestinal stromal tumors. Oncol RCJCtoi. 2016;17(2):8.
5. Szucs Z, Thway K, Fisher C, et al. Molecular subtypes of gastrointestinal stromal tumors and their prognostic and therapeutic implications. Future Oncol. 2017;13(1):93–107. doi:10.2217/fon-2016-0192
6. Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299(5607):708–710.
7. Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011;11(12):865–878. doi:10.1038/nrc3143
8. vM M. Gastrointestinal stromal tumors. J Oncol. 2018;36(2):136–143.
9. Lasota J, Miettinen M. Clinical significance of oncogenic KIT and PDGFRA mutations in gastrointestinal stromal tumours. Histopathol. 2008;53(3):245–266. doi:10.1111/j.1365-2559.2008.02977.x
10. Lasota J, Corless CL, Heinrich MC, et al. Clinicopathologic profile of gastrointestinal stromal tumors (GISTs) with primary KIT exon 13 or exon 17 mutations: a multicenter study on 54 cases. Modern Pathol. 2008;21(4):476–484. doi:10.1038/modpathol.2008.2
11. Heinrich MC, Maki RG, Corless CL, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol. 2008;26(33):5352–5359. doi:10.1200/JCO.2007.15.7461
12. Corless CL, Heinrich MC. Molecular pathobiology of gastrointestinal stromal sarcomas. Annu Rev Pathol-Mech. 2008;3:557–586. doi:10.1146/annurev.pathmechdis.3.121806.151538
13. Corless CL, Schroeder A, Griffith D, et al. PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol. 2005;23(23):5357–5364. doi:10.1200/JCO.2005.14.068
14. Miranda C, Nucifora M, Molinari F, et al. KRAS and BRAF mutations predict primary resistance to imatinib in gastrointestinal stromal tumors. Clin Cancer Res. 2012;18(6):1769–1776. doi:10.1158/1078-0432.CCR-11-2230
15. Pantaleo MA, Astolfi A, Urbini M, et al. Analysis of all subunits, SDHA, SDHB, SDHC, SDHD, of the succinate dehydrogenase complex in KIT/PDGFRA wild-type GIST. Eur J Hum Genet. 2014;22(1):32–39. doi:10.1038/ejhg.2013.80
16. Oudijk L, Gaal J, Korpershoek E, et al. SDHA mutations in adult and pediatric wild-type gastrointestinal stromal tumors. Modern Pathol. 2013;26(3):456–463. doi:10.1038/modpathol.2012.186
17. Boikos SA, Pappo AS, Killian JK, et al. Molecular subtypes of KIT/PDGFRA wild-type gastrointestinal stromal tumors a report from the national institutes of health gastrointestinal stromal tumor clinic. JAMA Oncol. 2016;2(7):922–928. doi:10.1001/jamaoncol.2016.0256
18. Killian JK, Miettinen M, Walker RL, et al. Recurrent epimutation of SDHC in gastrointestinal stromal tumors. Sci Transl Med. 2014;6:268. doi:10.1126/scitranslmed.3009961
19. Haller F, Moskalev EA, Faucz FR, et al. Aberrant DNA hypermethylation of SDHC: a novel mechanism of tumor development in carney triad. Endocr-Relat Cancer. 2014;21(4):567–577. doi:10.1530/ERC-14-0254
20. McWhinney SR, Pasini B, Stratakis CA. Familial gastrointestinal stromal tumors and germ-line mutations. New Engl J Med. 2007;357(10):1054–1056. doi:10.1056/NEJMc071191
21. Dwight T, Benn DE, Clarkson A, et al. Loss of Sdha expression identifies sdha mutations in succinate dehydrogenase-deficient gastrointestinal stromal tumors. Am J Surg Pathol. 2013;37(2):226–233. doi:10.1097/PAS.0b013e3182671155
22. Pasini B, McWhinney SR, Bei T, et al. Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur J Hum Genet. 2008;16(1):79–88. doi:10.1038/sj.ejhg.5201904
23. Maertens O, Prenen H, Wozniak A, et al. Molecular pathogenesis of multiple gastrointestinal stromal tumors in NF1 patients. Hum Mol Genet. 2006;15(6):1015–1023. doi:10.1093/hmg/ddl016
24. Huss S, Elges S, Trautmann M, Sperveslage J, Hartmann W, Wardelmann E. Classification of KIT/PDGFRA wild-type gastrointestinal stromal tumors: implications for therapy. Expert Rev Anticanc. 2015;15(6):623–628. doi:10.1586/14737140.2015.1032941
25. Agaram NP, Wong GC, Guo T, et al. Novel V600E BRAF mutations in imatinib-naive and imatinib-resistant gastrointestinal stromal tumors. Gene Chromosome Canc. 2008;47(10):853–859. doi:10.1002/gcc.20589
26. Lasota J, Felisiak-Golabek A, Wasag B, et al. Frequency and clinicopathologic profile of PIK3CA mutant GISTs: molecular genetic study of 529 cases. Modern Pathol. 2016;29(3):275–282. doi:10.1038/modpathol.2015.160
27. Brenca M, Rossi S, Polano M, et al. Transcriptome sequencing identifies ETV6-NTRK3 as a gene fusion involved in GIST. J Pathol. 2016;238(4):543–549. doi:10.1002/path.4677
28. Shi E, Chmielecki J, Tang CM, et al. FGFR1 and NTRK3 actionable alterations in “wild-type” gastrointestinal stromal tumors. J Transl Med. 2016;14.
29. Mei L, Smith SC, Faber AC, et al. Gastrointestinal stromal tumors: the gist of precision medicine. Trends Cancer. 2018;4(1):74–91. doi:10.1016/j.trecan.2017.11.006
30. Nannini M, Astolfi A, Urbini M, et al. Integrated genomic study of quadruple-WT GIST (KIT/PDGFRA/SDH/RAS pathway wild-type GIST). BMC Cancer. 2014;14.
31. Lim KT, Tan KY. Current research and treatment for gastrointestinal stromal tumors. World J Gastroentero. 2017;23(27):4856–4866. doi:10.3748/wjg.v23.i27.4856
32. Savage DG, Antman KH. Drug therapy: imatinib mesylate - a new oral targeted therapy. New Engl J Med. 2002;346(9):683–693. doi:10.1056/NEJMra013339
33. J H, H P. Gastrointestinal stromal tumour. Lancet CCJ. 2013;382(9896):973–983. doi:10.1016/S0140-6736(13)60106-3
34. Szucs Z, Thway K, Fisher C, et al. Promising novel therapeutic approaches in the management of gastrointestinal stromal tumors. Future Oncol. 2017;13(2):185–194. doi:10.2217/fon-2016-0194
35. Verweij J, Casali PG, Zalcberg J, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet. 2004;364(9440):1127–1134. doi:10.1016/S0140-6736(04)17098-0
36. Casali PG, Verweij J, Kotasek D, et al. Imatinib mesylate in advanced gastrointestinal stromal tumors (GIST): survival analysis of the intergroup EORTC/ISG/AGITG randomized trial in 946 patients. EJC Suppl. 2005;3(2):201–202.
37. Blanke CD, Rankin C, Demetri GD, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26(4):626–632. doi:10.1200/JCO.2007.13.4452
38. Rankin C, Von Mehren M, Blanke C, et al. Dose effect of imatinib (IM) in patients (pts) with metastatic GIST - phase III sarcoma group study S0033. J Clin Oncol. 2004;22(14):819s–819s. doi:10.1200/jco.2004.22.14_suppl.9005
39. Debiec-Rychter M, Sciot R, Le Cesne A, et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer. 2006;42(8):1093–1103. doi:10.1016/j.ejca.2006.01.030
40. Heinrich MC, Owzar K, Corless CL, et al. Correlation of kinase genotype and clinical outcome in the north american intergroup phase iii trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: calgb 150105 study by cancer and leukemia group B and southwest oncology group. J Clin Oncol. 2008;26(33):5360–5367. doi:10.1200/JCO.2008.17.4284
41. Van Glabbeke M. MetaGIST. comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors: a meta-analysis of 1640 patients. J Clin Oncol. 2010;28(7):1247–1253.
42. Heinrich MC. Correlation of long-term results of imatinib in advanced gastrointestinal stromal tumors with next-generation sequencing results: analysis of Phase 3 SWOG intergroup trial S0033 (vol 3, pg 944, 2017). JAMA Oncol. 2017;3(7):1002. doi:10.1001/jamaoncol.2016.6728
43. Serrano C, George S, Valverde C, et al. Novel insights into the treatment of imatinib-resistant gastrointestinal stromal tumors. Target Oncol. 2017;12(3):277–288. doi:10.1007/s11523-017-0490-9
44. Gramza AW, Corless CL, Heinrich MC. Resistance to tyrosine kinase inhibitors in gastrointestinal stromal tumors. Clin Cancer Res. 2009;15(24):7510–7518. doi:10.1158/1078-0432.CCR-09-0190
45. Van Glabbeke M, Verweij J, Casali PG, et al. Initial and late resistance to imatinib in advanced gastrointestinal stromal. tumors are predicted by different prognostic factors: a European organisation for research and treatment of cancer-Italian sarcoma group-australasian gastrointestinal trials group study. J Clin Oncol. 2005;23(24):5795–5804. doi:10.1200/JCO.2005.11.601
46. Benjamin RS, Debiec-Rychter M, Le Cesne A, et al. Gastrointestinal stromal tumors II: medical oncology and tumor response assessment. Semin Oncol. 2009;36(4):302–311. doi:10.1053/j.seminoncol.2009.06.003
47. Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21(23):4342–4349. doi:10.1200/JCO.2003.04.190
48. Wang WL, Conley A, Reynoso D, et al. Mechanisms of resistance to imatinib and sunitinib in gastrointestinal stromal tumor. Cancer Chemoth Pharm. 2011;67:15–24. doi:10.1007/s00280-010-1513-8
49. Yuzawa S, Opatowsky Y, Zhang ZT, Mandiyan V, Lax I, Schlessinger J. Structural basis for activation of the receptor tyrosine kinase KIT by stem cell factor. Cell. 2007;130(2):323–334. doi:10.1016/j.cell.2007.05.055
50. Rege TA, Wagner AJ, Corless CL, Heinrich MC, Hornick JL. “Pediatric-type” gastrointestinal stromal tumors in adults: distinctive histology predicts genotype and clinical behavior. Am J Surg Pathol. 2011;35(4):495–504. doi:10.1097/PAS.0b013e31820e5f7d
51. Heinrich MC, Corless CL, Blanke CD, et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol. 2006;24(29):4764–4774. doi:10.1200/JCO.2006.06.2265
52. Li GZ, Raut CP. Targeted therapy and personalized medicine in gastrointestinal stromal tumors: drug resistance, mechanisms, and treatment strategies. Oncotargets Ther. 2019;12:5123–5133. doi:10.2147/OTT.S180763
53. Wardelmann E, Merkelbach-Bruse S, Pauls K, et al. Polyclonal evolution of multiple secondary KIT mutations in gastrointestinal stromal tumors under treatment with imatinib mesylate. Clin Cancer Res. 2006;12(6):1743–1749. doi:10.1158/1078-0432.CCR-05-1211
54. Liegl B, Kepten I, Le C, et al. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J Pathol. 2008;216(1):64–74. doi:10.1002/path.v216:1
55. Miselli F, Casieri P, Negri T, et al. c-Kit/PDGFRA gene status alterations possibly related to primary imatinib resistance in gastrointestinal stromal tumors. Clin Cancer Res. 2007;13(8):2369–2377. doi:10.1158/1078-0432.CCR-06-1745
56. Tarn C, Rink L, Merkel E, et al. Insulin-like growth factor 1 receptor is a potential therapeutic target for gastrointestinal stromal tumors. Proc Natl Acad Sci USA. 2008;105(24):8387–8392. doi:10.1073/pnas.0803383105
57. Agaram NP, Laquaglia MP, Ustun B, et al. Molecular characterization of pediatric gastrointestinal stromal tumors. Clin Cancer Res. 2008;14(10):3204–3215. doi:10.1158/1078-0432.CCR-07-1984
58. Demetri GD, Wang YF, Wehrle E, et al. Imatinib plasma levels are correlated with clinical benefit in patients with unresectable/metastatic gastrointestinal stromal tumors. J Clin Oncol. 2009;27(19):3141–3147. doi:10.1200/JCO.2008.20.4818
59. Hislop J, Mowatt G, Sharma P, et al. Systematic review of escalated imatinib doses compared with sunitinib or best supportive care, for the treatment of people with unresectable/metastatic gastrointestinal stromal tumours whose disease has progressed on the standard imatinib dose. J Gastrointest Cancer. 2012;43(2):168–176. doi:10.1007/s12029-011-9325-6
60. Zalcberg JR, Verweij J, Casali PG, et al. Outcome of patients with advanced gastro-intestinal stromal tumours crossing over to a daily imatinib dose of 800 mg after progression on 400 mg. Eur J Cancer. 2005;41(12):1751–1757. doi:10.1016/j.ejca.2005.04.034
61. Casali PG, Abecassis N, Bauer S, et al. Gastrointestinal stromal tumours: ESMO-EURACAN clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2018;29:68–78. doi:10.1093/annonc/mdy095
62. KL O, DE H, A F, F Y, S Y. The receptor tyrosine kinase inhibitor SU11248 impedes endothelial cell migration, tubule formation, and blood vessel formation in vivo, but has little effect on existing tumor vessels. Angiogenesis GLJ. 2004;7(3):225–233. doi:10.1007/s10456-004-3149-y
63. Abrams TJ, Lee LB, Murray LJ, Pryer NK, Cherrington JM. SU11248 inhibits KIT and platelet-derived growth factor receptor beta in preclinical models of human small cell lung cancer. Mol Cancer Ther. 2003;2(5):471–478.
64. Mendel DB, Laird AD, Xin XH, et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003;9(1):327–337.
65. Somaiah N, von Mehren M. New therapeutic approaches for advanced gastrointestinal stromal tumors. Hematol Oncol Clin North Am. 2009;23(1):139–150.
66. Heinrich MC, Maki RG, Corless CL, et al. Sunitinib (SU) response in imatinib-resistant (IM-R) GIST correlates with KIT and PDGFRA mutation status. J Clin Oncol. 2006;24(18):520s–520s. doi:10.1200/jco.2006.24.18_suppl.9502
67. Li J, Gao J, Hong JL, Shen L. Efficacy and safety of sunitinib in Chinese patients with imatinib-resistant or -intolerant gastrointestinal stromal tumors. Future Oncol. 2012;8(5):617–624. doi:10.2217/fon.12.29
68. Reichardt P, Demetri GD, Gelderblom H, et al. Correlation of KIT and PDGFRA mutational status with clinical benefit in patients (pts) with gastrointestinal stromal tumor (GIST) treated with sunitinib (SU) in a worldwide treatment-use (TU) trial. J Clin Oncol. 2014;32:15. doi:10.1200/jco.2014.32.15_suppl.10549
69. Hsu CC, Wu CE, Chen JS, et al. Imatinib escalation or sunitinib treatment after first-line imatinib in metastatic gastrointestinal stromal tumor patients. Anticancer Res. 2014;34(9):5029–5036.
70. Bertucci F, Goncalves A, Monges G, et al. Acquired resistance to imatinib and secondary KIT exon 13 mutation in gastrointestinal stromal tumour. Oncol Rep. 2006;16(1):97–101.
71. Casali PG. Successes and limitations of targeted cancer therapy in gastrointestinal stromal tumors. Prog Tumor Res. 2014;41:51–61.
72. Wilhelm SM, Dumas J, Adnane L, et al. Regorafenib (BAY 73-4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer. 2011;129(1):245–255. doi:10.1002/ijc.v129.1
73. Demetri GD, Reichardt P, Kang YK, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):295–302. doi:10.1016/S0140-6736(12)61857-1
74. Ben-Ami E, Barysauskas CM, Von Mehren M, et al. Long-term follow-up results of the multicenter phase II trial of regorafenib in patients with metastatic and/or unresectable GI stromal tumor after failure of standard tyrosine kinase inhibitor therapy. Ann Oncol. 2016;27(9):1794–1799. doi:10.1093/annonc/mdw228
75. Serrano-Garcia C, Heinrich MC, Zhu MJ, et al. In vitro and in vivo activity of regorafenib (REGO) in drug-resistant gastrointestinal stromal tumors (GIST). J Clin Oncol. 2013;31:15. doi:10.1200/jco.2013.31.15_suppl.10510
76. Yeh CN, Chen MH, Chen YY, et al. A phase II trial of regorafenib in patients with metastatic and/or a unresectable gastrointestinal stromal tumor harboring secondary mutations of exon 17. Oncotarget. 2017;8(27):44121–44130.
77. Duensing A, Joseph NE, Medeiros F, et al. Protein kinase C theta (PKCtheta) expression and constitutive activation in gastrointestinal stromal tumors (GISTs). Cancer Res. 2004;64(15):5127–5131.
78. Pollard PJ, Briere JJ, Alam NA, et al. Accumulation of krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet. 2005;14(15):2231–2239.
79. Janeway KA, KH A, AD VDA, et al. Sunitinib treatment in pediatric patients with advanced GIST following failure of imatinib. Pediatr Blood Cancer. 2009;52(7):767–771.
80. Raut CP, Posner M, Desai J, et al. Surgical management of advanced gastrointestinal stromal tumors after treatment with targeted systemic therapy using kinase inhibitors. J Clin Oncol. 2006;24(15):2325–2331.
81. An HJ, Ryu MH, Ryoo BY, et al. The effects of surgical cytoreduction prior to imatinib therapy on the prognosis of patients with advanced GIST. Ann Surg. 2013;20(13):4212–4218.
82. Fairweather M, Balachandran ÃV, Li GZ, et al. Cytoreductive surgery for metastatic gastrointestinal stromal tumors treated with tyrosine kinase inhibitors: a 2-institutional analysis. J Surg Oncol. 2018;268(2):296–302.
83. Park SH, Ryu MH, Ryoo BY, et al. Sorafenib in patients with metastatic gastrointestinal stromal tumors who failed two or more prior tyrosine kinase inhibitors: a phase II study of korean gastrointestinal stromal tumors study group. Invest New Drug. 2012;30(6):2377–2383. doi:10.1007/s10637-012-9795-9
84. Mir O, Cropet C, Toulmonde M, et al. Pazopanib plus best supportive care versus best supportive care alone in advanced gastrointestinal stromal tumours resistant to imatinib and sunitinib (PAZOGIST): a randomised, multicentre, open-label Phase 2 trial. Lancet Oncol. 2016;17(5):632–641. doi:10.1016/S1470-2045(16)00075-9
85. Schuetze SM, Bolejack V, Thomas DG, et al. Association of dasatinib with progression-free survival among patients with advanced gastrointestinal stromal tumors resistant to imatinib. JAMA Oncol. 2018;4(6):814–820.
86. Heinrich MC, Marino-Enriquez A, Presnell A, et al. Sorafenib inhibits many kinase mutations associated with drug-resistant gastrointestinal stromal tumors. Mol Cancer Ther. 2012;11(8):1770–1780. doi:10.1158/1535-7163.MCT-12-0223
87. Garner AP, Gozgit JM, Anjum R, et al. Ponatinib inhibits polyclonal drug-resistant kit oncoproteins and shows therapeutic potential in heavily pretreated gastrointestinal stromal tumor (GIST) patients. Clin Cancer Res. 2014;20(22):5745–5755. doi:10.1158/1078-0432.CCR-14-1397
88. Heinrich MC, von Mehren M, Demetri GD, et al. Ponatinib efficacy and safety in patients (pts) with advanced gastrointestinal stromal tumors (GIST) after tyrosine kinase inhibitor (TKI) failure: results from a phase 2 study. J Clin Oncol. 2015;33:15. doi:10.1200/jco.2015.33.15_suppl.10535
89. Falkenhorst J, Hamacher R, Bauer S. New therapeutic agents in gastrointestinal stromal tumours. Curr Opin Oncol. 2019;31(4):322–328. doi:10.1097/CCO.0000000000000549
90. Evans EK, Hodous BL, Gardino AK, et al. BLU-285, the first selective inhibitor of PDGFRα D842V and KIT Exon 17 mutants. Exp Mol Ther. 2015;75(15 Supplement):791.
91. Heinrich MC, Griffith D, McKinley A, et al. Crenolanib inhibits the drug-resistant PDGFRA D842V mutation associated with imatinib-resistant gastrointestinal stromal tumors. Clin Cancer Res. 2012;18(16):4375–4384.
92. Smith BD, Hood MM, Wise SC, et al. DCC-2618 is a potent inhibitor of wild-type and mutant KIT, including refractory Exon 17 D816 KIT mutations, and exhibits efficacy in refractory GIST and AML xenograft models. Cancer Res. 2015;75.
93. Smith BD, Kaufman MD, Lu WP, et al. Ripretinib (DCC-2618) is a switch control kinase inhibitor of a broad spectrum of oncogenic and drug-resistant KIT and PDGFRA variants. Cancer Cell. 2019;35(5):738. doi:10.1016/j.ccell.2019.04.006
94. N J, B S, JY B, et al. Intrigue: phase III study of ripretinib versus sunitinib in advanced gastrointestinal stromal tumor after imatinib. Future Oncol. 2019;16(1):4251–4264.
95. G T, NP A, GC W, et al. Sorafenib inhibits the imatinib-resistant KITT670I gatekeeper mutation in gastrointestinal stromal tumor. Clin Cancer Res. 2007;13(16):4874–4881.
96. G T, A S, P L, et al. Sorafenib inhibits imatinib-resistant KIT and platelet-derived growth factor receptor beta gatekeeper mutants. Clin Cancer Res. 2007;13(11):3363–3369.
97. Gebreyohannes YK, Van Looy T, Wozniak A, et al. Anti-tumor effects of dovitinib, a multi-target kinase inhibitor, in patient-derived gastrointestinal stromal tumor (GIST) xenograft models. Cancer Res. 2015;75.
98. Joensuu H, Blay J-Y, Comandone A, et al. Dovitinib in patients with gastrointestinal stromal tumour refractory and/or intolerant to imatinib. Br J Cancer. 2017;117(9):1278–1285. doi:10.1038/bjc.2017.290
99. Hung HT, Ong R, Chow P. Dovitinib exerts potent antitumor effects in gastrointestinal stromal tumors. Cancer Res. 2015;75.
100. Dubreuil P, Letard S, Ciufolini M, et al. Masitinib (AB1010), a potent and selective tyrosine kinase inhibitor targeting KIT. PLoS One. 2009;4(9):9. doi:10.1371/journal.pone.0007258
101. Adenis A, Blay JY, Bui-Nguyen B, et al. Masitinib in advanced gastrointestinal stromal tumor (GIST) after failure of imatinib: a randomized controlled open-label trial. Ann Oncol. 2014;25(9):1762–1769. doi:10.1093/annonc/mdu237
102. Cullinane C, Natoli A, Hui Y, et al. Preclinical evaluation of nilotinib efficacy in an imatinib-resistant KIT-driven tumor model. Mol Cancer Ther. 2010;9(5):1461–1468. doi:10.1158/1535-7163.MCT-09-1181
103. Blay J-Y, Shen L, Kang Y-K, et al. Nilotinib versus imatinib as first-line therapy for patients with unresectable or metastatic gastrointestinal stromal tumours (ENESTg1): a randomised phase 3 trial. Lancet Oncol. 2015;16(5):550–560. doi:10.1016/S1470-2045(15)70105-1
104. Hsueh YS, Lin CL, Chiang NJ, et al. Selecting tyrosine kinase inhibitors for gastrointestinal stromal tumor with secondary KIT activation-loop domain mutations. PLoS One. 2013;8(6):6. doi:10.1371/journal.pone.0065762
105. Schittenhelm MM, Shiraga S, Schroeder A, et al. Dasatinib (BMS-354825), a dual SRC/ABL kinase inhibitor, inhibits the kinase activity of wild-type, juxtamembrane, and activation loop mutant KIT isoforms associated with human malignancies. Cancer Res. 2006;66(1):473–481. doi:10.1158/0008-5472.CAN-05-2050
106. Wagner AJ, Kindler H, Gelderblom H, et al. A phase II study of a human anti-PDGFRα monoclonal antibody (olaratumab, IMC-3G3) in previously treated patients with metastatic gastrointestinal stromal tumors. Ann Oncol. 2017;28(3):541–546.
107. Falchook GS, Trent JC, Heinrich MC, et al. BRAF mutant gastrointestinal stromal tumor: first report of regression with BRAF inhibitor dabrafenib (GSK2118436) and whole exomic sequencing for analysis of acquired resistance. Oncotarget. 2013;4(2):310–315. doi:10.18632/oncotarget.v4i2
108. Glod J, Arnaldez F, Wiener L, et al. A phase II trial of vandetanib (ZD6474) in children and adults with wild-type gastrointestinal stromal tumors. J Clin Oncol. 2016;34(15_suppl):15. doi:10.1200/JCO.2016.34.15_suppl.11009
109. Serrano C, L A, K Y, et al. Phase I study of rapid alternation of sunitinib and regorafenib for the treatment of tyrosine kinase inhibitor refractory gastrointestinal stromal tumors. Clin Cancer Res. 2019;25(24):7287–7293.
110. Bauer S, Lynn KY, Demetri GD, Fletcher JA. Heat shock protein 90 inhibition in imatinib-resistant gastrointestinal stromal tumor. Research FJJC. 2006;66(18):9153–9161.
111. Bauer S, Parry JA, Muehlenberg T, et al. Proapoptotic activity of bortezomib in gastrointestinal stromal tumor cells. Cancer Res. 2010;70(1):150–159. doi:10.1158/0008-5472.CAN-09-1449
112. Muhlenberg T, Zhang YX, Wagner AJ, et al. Inhibitors of deacetylases suppress oncogenic KIT signaling, acetylate HSP90, and induce apoptosis in gastrointestinal stromal tumors. Cancer Res. 2009;69(17):6941–6950. doi:10.1158/0008-5472.CAN-08-4004
113. Floris G, Debiec-Rychter M, Sciot R, et al. High efficacy of panobinostat towards human gastrointestinal stromal tumors in a xenograft mouse model. Clin Cancer Res. 2009;15(12):4066–4076. doi:10.1158/1078-0432.CCR-08-2588
114. Ran LL, Sirota I, Cao Z, et al. Combined inhibition of map kinase and KIT signaling synergistically destabilizes ETV1 and suppresses GIST tumor growth. Cancer Discov. 2015;5(3):304–315. doi:10.1158/2159-8290.CD-14-0985
115. S P, R P, JY B, et al. A Phase I-II study of everolimus (RAD001) in combination with imatinib in patients with imatinib-resistant gastrointestinal stromal tumors. Ann Oncol. 2010;21(10):1990–1998.
116. Dolly SO, Wagner AJ, Bendell JC, et al. Phase I study of apitolisib (GDC-0980), dual phosphatidylinositol-3-kinase and mammalian target of rapamycin kinase inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2016;22(12):2874–2884. doi:10.1158/1078-0432.CCR-15-2225
117. Conley AP, Araujo D, Ludwig J, et al. A randomized phase II study of perifosine (P) plus imatinib for patients with imatinib-resistant gastrointestinal stromal tumor (GIST). J Clin Oncol. 2009;27(15_suppl):10563.
118. Belinsky MG, Rink L, Cai KQ, et al. The insulin-like growth factor system as a potential therapeutic target in gastrointestinal stromal tumors. Cell Cycle. 2008;7(19):2949–2955. doi:10.4161/cc.7.19.6760
119. Gross MI, Demo SD, Dennison JB, et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther. 2014;13(4):890–901. doi:10.1158/1535-7163.MCT-13-0870
120. Issa JPJ, Roboz G, Rizzieri D, et al. Safety and tolerability of guadecitabine (SGI-110) in patients with myelodysplastic syndrome and acute myeloid leukaemia: a multicentre, randomised, dose-escalation Phase 1 study. Lancet Oncol. 2015;16(9):1099–1110. doi:10.1016/S1470-2045(15)00038-8
121. Chi P, Chen Y, Zhang L, et al. ETV1 is a lineage survival factor that cooperates with KIT in gastrointestinal stromal tumours. Nature. 2010;467(7317):849–U117. doi:10.1038/nature09409
122. Pang Y, X F, C H, et al. DEPDC5 Mutational inactivation of mTORC1 repressor gene in human gastrointestinal stromal tumors. Proc Natl Acad Sci. 2019;116(45):22746–22753.
123. Janeway KA, Kim SY, Lodish M, et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. P Natl Acad Sci USA. 2011;108(1):314–318. doi:10.1073/pnas.1009199108
124. Killian JK, Kim SY, Miettinen M, et al. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov. 2013;3(6):648–657. doi:10.1158/2159-8290.CD-13-0092
125. Flavahan WA, Drier Y, Johnstone SE, et al. Altered chromosomal topology drives oncogenic programs in SDH-deficient GISTs. Nature. 2019;575(7781):229. doi:10.1038/s41586-019-1668-3
126. Van Loo P, Voet T. Single cell analysis of cancer genomes. Curr Opin Genet Dev. 2014;24:82–91. doi:10.1016/j.gde.2013.12.004
127. Liang SB, Fu LW. Application of single-cell technology in cancer research. Biotechnol Adv. 2017;35(4):443–449. doi:10.1016/j.biotechadv.2017.04.001
128. Laughney AM, Kim E, Sprachman MM, et al. Single-cell pharmacokinetic imaging reveals a therapeutic strategy to overcome drug resistance to the microtubule inhibitor eribulin. Sci Transl Med. 2014;6:261. doi:10.1126/scitranslmed.3009318
129. Ravegnini G, Sammarini G, Serrano C, et al. Clinical relevance of circulating molecules in cancer: focus on gastrointestinal stromal tumors. Ther Adv Med Oncol. 2019;11.
130. Li HM, Meng QH, Noh H, et al. Cell-surface vimentin-positive macrophage-like circulating tumor cells as a novel biomarker of metastatic gastrointestinal stromal tumors. Oncoimmunology. 2018;7:5. doi:10.1080/2162402X.2017.1420450
131. Ding L, Wendl MC, McMichael JF, Raphael BJ. Expanding the computational toolbox for mining cancer genomes. Nat Rev Genet. 2014;15(8):556–570. doi:10.1038/nrg3767
132. Xu HX, Jiao Y, Qin S, Zhao WH, Chu Q, Wu KM. Organoid technology in disease modelling, drug development, personalized treatment and regeneration medicine. Exp Hematol Oncol. 2018;7.
133. Xu HX, Lyu XD, Yi M, Zhao WH, Song YP, Wu KM. Organoid technology and applications in cancer research. J Hematol Oncol. 2018;11.
134. Lai YX, Wei XR, Lin SH, Qin L, Cheng L, Li P. Current status and perspectives of patient-derived xenograft models in cancer research. J Hematol Oncol. 2017;10.
135. Jung J, Seol HS, Chang S. The generation and application of patient-derived xenograft model for cancer research. Cancer Res Treat. 2018;50(1):1–10. doi:10.4143/crt.2017.307
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.