Back to Journals » Drug Design, Development and Therapy » Volume 13
The TwistDock workflow for evaluation of bivalent Smac mimetics targeting XIAP
Authors Huang Q ![]() , Peng Y, Peng Y, Wei D, Wei Y, Feng S
, Peng Y, Peng Y, Wei D, Wei Y, Feng S ![]()
Received 13 November 2018
Accepted for publication 5 February 2019
Published 26 April 2019 Volume 2019:13 Pages 1373—1388
DOI https://doi.org/10.2147/DDDT.S194276
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Qingsheng Huang,1 Yin Peng,2 Yuefeng Peng,1,3 Dan Wei,4 Yanjie Wei,1 Shengzhong Feng1
1Joint Engineering Research Center for Health Big Data Intelligent Analysis Technology and Center for High Performance Computing, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, Shenzhen, Guangdong, People’s Republic of China; 2Department of Pathology, Shenzhen University School of Medicine, Shenzhen, Guangdong, People’s Republic of China; 3Center for Drug Evaluation and Research (CDER), Food and Drug Administration (FDA), Silver Spring, MD 20903, USA; 4School of Computer Science and Technology, Hangzhou Dianzi University, Hangzhou, People’s Republic of China
Purpose: Mimetics based on Smac, the native inhibitor of XIAP, are promising drug-candidates for the treatment of cancer. Bivalent Smac mimetics inhibit XIAP with even higher potency than monovalent mimetics, but how to optimize the linker that tethers the two monovalent binding motifs remains controversial.
Methods: To construct an ensemble of bivalent complex structures for evaluating various linkers, we propose herein a workflow, named TwistDock, consisting of steps of monovalent docking and linker twisting, in which the degrees of freedom are sampled focusing on the rotation of single bonds of the linker.
Results: The obtained conformations of bivalent complex distribute randomly in the conformational space with respect to two reaction coordinates introduced by the linker, which are the distance of the two binding motifs and the dihedral angle of the two planes through the linker and each of the binding motifs. Molecular dynamics starting from 10 conformations with the lowest enthalpy of every complex shows that the conformational tendency of the complex participated by compound 9, one of the compounds with the largest binding affinity, is distinct from others. By umbrella sampling of the complex, we find its global minimum of the free energy landscape. The structure shows that the linker favors a compact conformation, and the two BIR domains of XIAP encompass the ligand on the opposite sides.
Conclusion: TwistDock can be used in fine-tuning of bivalent ligands targeting XIAP and similar receptors dimerized or oligomerized.
Keywords: molecular modeling, docking, molecular dynamics simulation, MMPBSA, umbrella sampling
Introduction
Apoptosis, or programmed cell death, is critical for multicellular organisms to maintain homeostasis. The IAP (Inhibitor of Apoptosis Protein) family, especially XIAP (X chromosome-linked IAP), inhibits apoptosis of cancer cells.1 However, the inhibition by XIAP can be neutralized by Smac (Second mitochondrial activator of caspases).2 The pair of antagonists, XIAP and Smac, regulates apoptosis precisely. XIAP is a 497 amino acid-length protein consisting of three BIR (Baculovirus IAP Repeat) domains and a RING-type domain. BIR2 and BIR3 have similar structures,3 and both have a surface groove as a binding site for Smac.2,4 Smac has a specific IAP-binding motif, built by its N-terminal tetrapeptide AVPI (Ala-Val-Pro-Ile). Mediated by the AVPI sequence, dimerized Smac interacts with BIR2 and BIR3 preventing XIAP from blocking apoptosis.5,6 Since malfunction of apoptosis may induce various pathological conditions and diseases such as cancer,7 XIAP becomes an attractive target for drug design.8–17 Most antagonists of XIAP are designed mimicking the AVPI sequence,18,19 in order to balance out the over-expression of XIAP in tumor cell and overwhelm the apoptosis resistance.20,21 Monovalent Smac mimetics have a single binding motif, which generally binds to BIR3 with a high affinity.19 Bivalent Smac mimetics have two binding motifs binding to BIR2 and BIR3 simultaneously.6 Bivalent Smac mimetics induced apoptosis with even higher activity than that of native Smac and monovalent Smac mimetics.8,10–15,17
The reason that bivalent Smac mimetics acquire even higher pro-apoptotic activity resides not only in the general strategy to increase the binding affinity, but also in the regulation mechanism of XIAP to apoptosis. Apoptosis is initiated through two pathways: the intrinsic pathway and the extrinsic pathway.17,22 In the intrinsic pathway, mitochondria releases cytochrome c and initiates the pathway, often due to chemotherapeutics or radiation stimulation, and then an initiator caspase-9 is activated.23 The extrinsic pathway is triggered through the binding of death receptor and death ligand, like CD95 ligand, TNF and TRAIL, and the signaling is passed by caspase-8.18,24 The two pathways finally converge on effectors caspase-3/7 at downstream. Caspase-3 and -7 determine the apoptosis of cell through cleavage of critical cellular substrates such as poly(ADP-ribose) polymerase and lamins.17,25 The BIR2 domain of XIAP with the linker to its N-terminal inhibits caspase-3 and caspase-7, while the BIR3 domain targets caspase-9.1,26 Over-expression of XIAP in some tumor cell lines blocks the apoptosis pathways and diminishes the efficacy of chemotherapy and radiotherapy.2,4,21 Monovalent Smac mimetics compete with only caspase-9 for XIAP and ignore the interaction of BIR2 with caspase-3/-7; thus, they are generally less potent than bivalent Smac mimetics.6,17,19,27 Attributed to the simultaneous inhibition of caspase-3, -7 and -9,28,29 bivalent Smac mimetics become attractive. On one hand, some of these mimetics have been proved to possess high binding affinity to XIAP and dramatically increased anticancer activity. Among these, a series of bivalent Smac mimetics with different linkers has been studied by Peng et al. experimentally to evaluate the impacts of linkers on the binding affinity and the anticancer activity.28,29 These studies provide biochemical and cellular biological evidence that they are promising lead compounds. On the other hand, a study performed with gene-knock-out cells and mice suggested that the increased binding affinity to XIAP may be not always beneficial, ie, it introduces toxicity to animals.30 To fine-tune the activity of antagonists to XIAP, structural insights into the relationship of linkers and binding affinity can rationalize the design of the linker, help us exploit the properties of the linker and aid virtual screening.
A bivalent ligand has two binding motifs to its targeted receptor, which is usually a protein with multiple domains or dimerized protein. Introducing bivalency in ligand design provides new tunable parameters for optimization of the drug itself and its interaction with targets. For example, bivalency is a strategy to increase the binding affinity, especially through reducing the binding enthalpy.31 Designing a bivalent ligand faces, in addition to the design of its monovalent binding motifs, two new issues concerning the linker tethering the two binding motifs: the selection of a suitable tethering site and the design of the linker with optimal length and other properties.28 However, there is yet no agreement on the design principle of the linker. Although the length of the linker is intuitively of high importance, independent studies on bivalent Smac mimetics showed that the length of the linker may have different effect, from high to only modest, on binding affinity.28,29 Besides the length of the linker, twisting or torsion of the linker, which may determine the orientation of the two binding motifs, should be also an important factor affecting the binding affinity. Twisting of the linker imposes limitation on the arrangement of binding domains of receptors. Different arrangements of the binding domains introduce various steric hindrance and electrostatic interaction between them, affecting the binding affinity. Unfortunately, the importance of this factor has yet been discussed by few studies, especially on the computational modeling. Actually, as far as we know, there are no published or commercially available docking software that constructs an ensemble of bivalent complex and provides structural insights into the roles of the linker.
In this study, we propose a high-efficiency workflow, named TwistDock, to implement the bivalent docking job, ie, to construct reasonable structures of bivalent complex. Exploiting the structural feature that the two BIR domains of XIAP are loosely concatenated by a peptide chain of 34 amino acids long and each of the two is bound independently by one of the binding motifs of the bivalent ligand,11,17,28,32 the workflow neglects the concatenating peptide and allows moving of the BIR2 and BIR3 domains. By twisting the linker and constraining the two binding interfaces, possible arrangements of the BIR2 and BIR3 domains are enumerated. The enumerated conformations are then clustered and the redundant conformations are excluded. In this manner, the sampled degrees of freedom are mainly related to the twisting of the linker of the bivalent ligand, avoiding the atomic-level details that are expensive to sample.33 The excluding of redundant conformations guarantees that the remaining conformations are unique and distant from each other in the conformational space. We constructed a series of complex formed by the BIR2 and BIR3 domains of XIAP and bivalent Smac mimetics of different linkers but the same binding motif. The structures are further equilibrated by molecular dynamics (MD) simulation, to introduce possible interactions between the BIR2 and BIR3 domains. Sampled conformations of one of the complexes, whose ligand is compound 9, are also subjected to umbrella sampling and the weighted histogram analysis method (WHAM) for the free energy landscape. With the structures, we clarify the role of the linker and discuss how the length and the twisting modulate the binding affinity of bivalent Smac mimetics.
Materials and methods
Construction of the bivalent complexes
The BIR2 domain used in the paper corresponds to residues E163 to V230 of XIAP, while the BIR3 domain corresponds to residues Y265 to L330 of XIAP. The structures of BIR2 and BIR3 were extracted from PDB entries 4J4627 and 1G73,34 respectively. Eight bivalent Smac mimetics, whose binding affinities to XIAP were measured by Peng et al.28, are studied in this paper (Figure 1A). Such a mimetic consists of two binding motifs derived from AT-406, a potential drug being tested in clinical trials,19 and a linker tethers the two through the amide group in the eight-membered ring of AT-406. Structures of 8 bivalent Smac mimetics were generated with Simplified Molecular Input Line Entry Specification (SMILES)35 representations according to the structures reported by Peng et al.28, via the distance geometry approach provided by RDKit.36,37 Each generated bivalent Smac mimetic structure was energy minimized for 500 steps with MMFF94 forcefield.38 Because in some of the bivalent Smac mimetic structures the two binding motifs were distorted, these parts of the structures were refined by a substitution with the AT-406 structure obtained from Selleck Chemicals.39 Docking for the two monovalent complexes, AT-406/BIR2 and AT-406/BIR3, was performed separately using AutoDockVina.40 The docking was limited to the groove surface of BIR2 or BIR3 for binding AVPI, with the exhaustiveness set to 100 and the number of result binding poses set to 20.
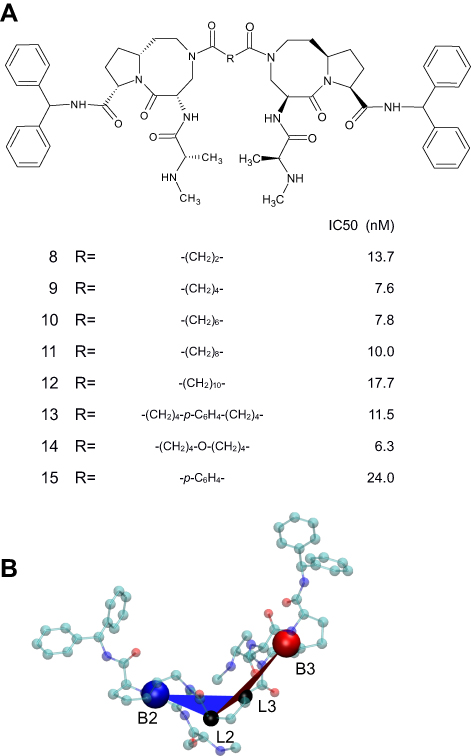 | Figure 1 Chemical structure of bivalent Smac mimetics. (A) 8 bivalent Smac mimetics studied in this paper and their binding affinities to XIAP.28 (B) The reaction coordinates associated with compound 9 shown in CPK representation, where C atoms are colored cyan, N blue and O red. The reaction coordinate ξ is the distance B3-B2, and the reaction coordinate α is the dihedral B3-L3-L2-B2. Points B3 (the large red ball) and B2 (the large blue ball) are the centers of mass of the (8,5)-bicyclic cores of the AT-406 parts, and points L3 and L2 (the two black balls) are the terminal atoms of the linker chain connected to the AT-406 parts. |
For the monovalent complex of AT-406/BIR3, the structure with the highest docking score had a similar binding mode to the complex in PDB entry 1G73 (RMSD 1.056 Å). This structure was subjected to a 25 ns MD simulation (Figure S1), where the ligand (AT-406) was parameterized with GAFF forcefield and AM1-BCC method using Antechamber41 and the receptor (BIR3) was parameterized with Amberff14SB forcefields. Solvation and ionization was done in LEaP.42 In the receptor, the zinc divalent cation, which is far away from the binding site, coordinated with four residues, was parameterized through the CaDA (Cationic Dummy Atom) approach.43 The receptor was capped with neutral termini, ie, acetyl N-terminus (ACE) and methylamine C-terminus (NME). The solvation box was 58×57×58 Å3. With NAMD,44 we ran 50,000 steps minimization of hydrogen atoms and water; 50,000 steps minimization of the receptor sidechains (except the four residues coordinating the zinc ion); 30 ps heating the system to 300 K; 5 ns equilibration in NpT ensemble; 2 ns equilibration in NVT ensemble; and finally 25 ns free dynamics, ie, MD simulation in NVT ensemble at 300 K and without any constraints. The density of the simulation system was watched and we detect no stable vacuum bubble formed.
For the monovalent complex of AT-406/BIR2 (Figure S2), the structure with the third highest docking score was selected for subsequent MD simulation, because it has a binding mode most similar to the complex formed by the AVPI sequence and the BIR2 domain in PDB entry 4J46 (RMSD 1.123 Å). Parameterization and setup of the MD simulation were the same as the complex of AT-406/BIR3. The solvation box was 63×54×56 Å3.
After the MD simulations, the two monovalent complexes were linked together with a chemical linker (shown in Figure 1A) tethering the two AT-406 molecules, by aligning the AT-406 molecules in monovalent complexes with the two AT-406 groups in the bivalent mimetic.
Conformational sampling of bivalent complex
The core step of the TwistDock workflow implements the conformation sampling by twisting the linker tethering the two IAP binding motifs of a bivalent Smac mimetic. Through interfaces of the IAP-binding motifs to the BIR2 and BIR3 domains, the twisting of the linker ultimately determines the global conformation of the entire bivalent complex. A flowchart shows the steps for building the bivalent complex and the conformational sampling of bivalent complex in Figure 2.
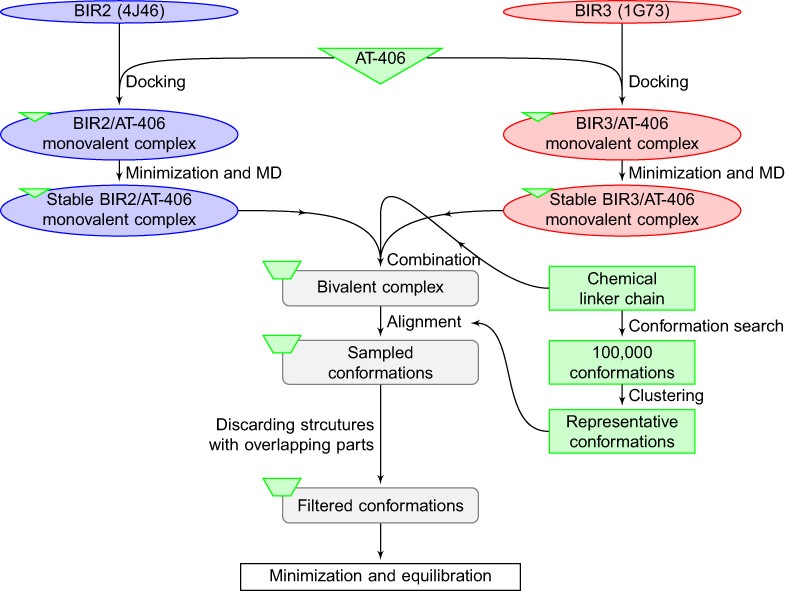 | Figure 2 Flowchart of the TwistDock workflow, including the preparation of ligand (bivalent Smac mimetic) and construction of bivalent complex. |
We applied the workflow to a series of bivalent Smac mimetics, 8 compounds having the same IAP-binding motifs but different linkers, shown in Figure 1A. Compounds 8 to 12 have a linear alkane chain of 2–10 carbon atoms; compounds 13 and 14 have a linker chained by two linear alkane groups of 4 carbon atoms and, respectively, a phenylene group and an oxygen atom; and compound 15 has a phenylene ring as the linker chain. Twisting of the linker was achieved by rotation of C–C and C–O single bonds of a bared linker chain capped with N,N-Dimethylformamide groups on two ends of the linker. With the distance geometry approach36 provided by RDKit, 100,000 chain conformations for each linker chain were randomly generated. Each generated structure was energy minimized for 500 steps with MMFF94 forcefield.38 These conformations were clustered using the quality threshold (QT) algorithm45 implemented in VMD46 (parameters detailed in Table S1 and S2 and Figure S3). All but the first conformation in a cluster were excluded, so that the remaining conformations were distant from each other in the conformational space. A chain conformation replaced the linker part within a bivalent complex structure, by aligning the termini of the chain to the amide group in the eight-membered ring of the AT-406 part. The alignment allowed translation and rotation of the AT-406/BIR2 and the AT-406/BIR3 parts, but the two monovalent parts themselves were moved entirely as rigid bodies. Bivalent complexes whose structures were invalid, ie, the AT-406/BIR2 part overlapped with the AT-406/BIR3 part, were discarded. At the end, an ensemble of conformations of every bivalent complex was obtained.
Minimization and equilibration of the bivalent complex structures
Each structure in an ensemble obtained by our TwistDock workflow was relaxed by a 1,000-step minimization. For compounds 8, 9 and 15, because their linker chains are less flexible, RDKit produced few unique conformations (Table S1) even before clustering. Since more independent structures are needed by the subsequent analysis, we ran more parallel trajectories of minimization to compensate the small number of valid bivalent complex structures (Table S1). Subsequently, MD simulation consisted of a 40 ps run with Cα-atom restraints and two 10 ps runs of equilibration was carried out with PMEMD provided in the AMBER package.42 The minimization and MD were all performed in implicit solvent condition using the generalized Born model with a counterion concentration of 0.1 mol/L.
Extracted from the last 10 ps equilibration, 10 frames of every 1 ps were evaluated for the enthalpy of complex (Hcomplex) using the MM-GBSA method with MMPBSA.py provided in the AMBER package.47 The hydrophobic contribution to the enthalpy was calculated by the LCPO method.48 The salt concentration was 0.1 M. The receptor was set to the BIR2 and BIR3 domains as a whole, and the ligand was set to the bivalent Smac mimetic. We only compared the complex enthalpy among structures of an ensemble of the same complex, instead of calculating the enthalpy of the ligand and receptor parts and reporting the difference. We obtained 10 bivalent complexes with the lowest complex enthalpy for each of the eight bivalent Smac mimetics (Figure S4). In total, 80 complexes were subject to a 2 ns MD. Parameterization of the bivalent complex for MD simulation was similar to the monovalent complex described above, using GAFF for ligand, Amber forcefields for proteins, the CaDA approach for zinc ions and the coordinating residues, and the ACE and NME termini.
Umbrella sampling
To demonstrate the effects of length and twisting of the linker, we project the conformation space of a bivalent complex on a plane of two reaction coordinates: distance B3-B2 (denoted by ξ) and dihedral B3-L3-L2-B2 (denoted by α) (Figure 1B). The points B3 and B2 are the centers of mass of the (8,5)-bicyclic cores of the AT-406 parts bound to, respectively, BIR2 and BIR3. The distance B3-B2 (ξ) reflects the length of the linker and is also related to the distance between the BIR2 and BIR3 domains. The points L3 and L2 are defined as the terminal atoms of the linker chain connected to the AT-406 parts bound to, respectively, BIR3 and BIR2. The dihedral B3-L3-L2-B2 (α) reflects the twisting of the linker and is also related to the rotation of the BIR2 and BIR3 domains. The arrangement of the BIR2 and BIR3 domains in a bivalent complex is uniquely described by ξ and α.
We performed the umbrella sampling with PMEMD in the AMBER package42 for the bivalent complex formed by compound 9 and the BIR2 and BIR3 domains. The ensemble of this complex obtained by our TwistDock workflow consists of 31 structures. Each structure was relaxed by a 1,000-step minimization and equilibrated with a 40 ps run with Cα-atom restraints and a 10 ps equilibration, in which each last frame was used as the starting point of 8 independent biased MD. Totally, 248 biased MD simulations of 8 ns long were carried out. Restraints were set up along ξ and α. Along ξ, the restrain window was centered on the value of ξ in the structure of the starting point. Along α, the restrain window was centered on one of eight values from {0, 15, 30, …, 345°} that are within ±60° near the value of the dihedral α in the structure of the starting point. The two reaction coordinates were restraint by harmonic potentials of 0.01 kcal/(mol·Å2) and 1 kcal/(mol·rad2), respectively. Structures of every 0.1 ps in the last 2 ns of these trajectories were collected and combined to form the free energy surface by WHAM.49
Results
TwistDock workflow building a conformation ensemble of bivalent complex
The system of a bivalent complex formed by XIAP and a bivalent Smac mimetic has a huge number of degrees of freedom. However, since the BIR2 and BIR3 domains of XIAP are loosely linked by a peptide chain of 34 amino acids, the degrees of freedom that significantly affect the interaction of the receptor and ligand reside in the arrangement of the BIR2 and BIR3 domains. The bivalent ligand has two IAP-binding motifs each of which is bound to one of the BIR2 and BIR3 domains. If we neglect the peptide chain, the arrangement of the BIR2 and BIR3 domains is defined by only the conformation of the linker of the bivalent ligand. Our TwistDock workflow can enumerate the conformation of the linker and based on it construct a conformation ensemble of the bivalent complex. For each of the 8 linkers (Figure 1A), we obtained 100,000 conformers. The number should be sufficient, because even for compound 12 which has the longest linker, we find most of the conformers are redundant and only 11,507 are unique (Table S1). For compounds 8 and 15, which have short linkers, the numbers of unique conformers are even much smaller (Table S1). To save computational resource in subsequent steps, the conformations were clustered and only one representative conformation of a cluster was kept. We tried three sets of cutoff for clustering and found that the distributions of clusters were insensitive to different cutoff, suggesting the cutoff we used was small enough to distinguish significantly different conformation (Table S2 and Figure S3). The clustering step makes the samples uniformly distribute over the conformational space, more regularly than random samples, following the idea of quasi Monte Carlo method.50
The coverage of the sampling can be visualized by projecting the conformational space of bivalent complexes on the two reaction coordinates ξ and α (Figure 3). For all of the complexes, the conformations distribute randomly with respect to the two reaction coordinates, while the range along ξ is limited by the length and possibly flexibility of the linkers, in contrast to the range along α, which has no limitation. The two reaction coordinates are independent, except that small value of ξ is only permitted when α is near 0°. Complexes involving ligands with short linkers have fewer unique conformation samples, indicating their small conformational spaces.
 | Figure 3 Conformational spaces of 8 bivalent complexes sampled by TwistDock, projected on reaction coordinates distance B3-B2 (ξ) and dihedral B3-L3-L2-B2 (α). Red crosses denote invalid conformations that have serious structural clashes, while green circles denote valid conformations. The bivalent complex is formed by the BIR2 and BIR3 domains and one of the bivalent Smac mimetics (A to H correspond to compounds 8 to 15). (Compound 9 has the compact conformational space, which is viable to explore). |
Many of the conformation samples are invalid structure, since they contain overlapping heavy atoms in the bivalent complex. The distribution of valid structure is even more limited, but still covers the available space. Valid structures with small ξ tend to have α away from 0°, meaning that they keep the BIR2 and BIR3 domains away from each other.
Conformational tendency of bivalent complex with low free energy
With the conformation ensemble, we aim at finding the structure that has the lowest free energy and is most close to the native stable conformation. First, we performed parallel runs consisting of minimization and short MD for all structures sampled by our TwistDock workflow to relax the structures and to explore the conformational space. Every structure was evaluated for the enthalpy averaged in its short MD. We compared the complex enthalpy among the parallel runs within an ensemble (Figure S4).
The end frames of trajectories of 10 structures with the lowest enthalpy were further analyzed with long MD. During the MD, the dihedral B3-L3-L2-B2 (α) fluctuated around a mean value in every trajectory of bivalent complexes, and the narrow range indicates that the system stays close to the structure of the local minimum of energy. In trajectories of bivalent complexes formed by XIAP and compounds 8 or 15, the mean values of α are closed to ±180° (Figure 4, A and H). These two bivalent complexes prefer the conformation in which the BIR2 and BIR3 domains stay on opposite sides of the bivalent Smac mimetic. Because the linkers of the two compounds are short but the BIR2 and BIR3 domains are large and both negatively charged, such a conformation allows the two domains to keep a long distance so as to minimize the free energy of the bivalent complex. In trajectories of bivalent complexes involving compounds 10, 11, 12, 13 and 14, the mean values of α are also closed to ±180°, but the circular distributions show that they have no conformation preference (Figure 4, C, D, E, F and G). The linkers of these compounds are long enough so that the BIR2 and BIR3 domains can keep far away from each other with any value of α. In trajectories of bivalent complex formed by the BIR2 and BIR3 domains and compound 9, the circular distribution of α is different to that of other bivalent complexes (Figure 4B). The value is close to 0°, instead of close to ±180°, and its overall distribution is less uniform than that of compounds 10, 11, 12, 13 and 14. The preference of the reaction coordinate α may be a structural feature that results in the high potency of compound 9 (Figure 1A).
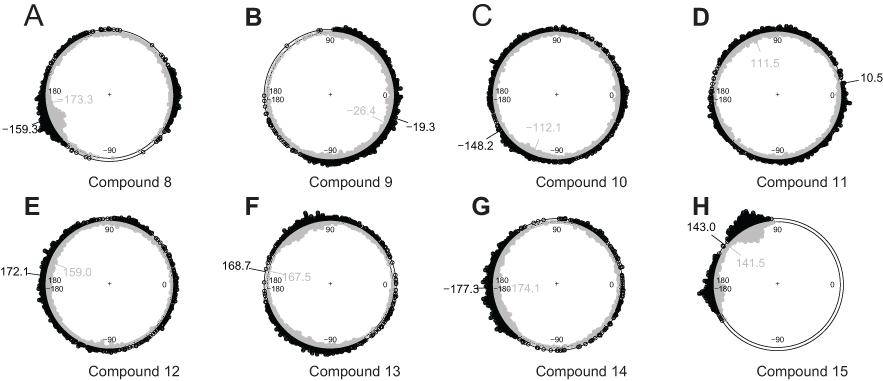 | Figure 4 Circular distribution of the dihedral B3-L3-L2-B2 (α) denoting the global conformation of the bivalent complex. The bivalent complex is formed by the BIR2 and BIR3 domains and one of the bivalent Smac mimetics (A to H correspond to compounds 8 to 15). Samples from first 1.2 ns equilibration (gray, inner circle) and then 800 ps equilibration (black, outer circle) are shown as stacked points, and the circular mean values are shown. |
The free energy landscape of the bivalent complex formed by compound 9 and the BIR2 and BIR3 domains
For a bivalent complex, the degrees of freedom that can be controlled by design of the linker in bivalent ligands are the distance B3-B2 (ξ) and the dihedral B3-L3-L2-B2 (α).
We performed umbrella sampling for the complex formed by compound 9 and the BIR2 and BIR3 domains to find its global minimum of the free energy landscape projected on reaction coordinates ξ and α. By WHAM, we obtained the free energy landscape (Figure 5). The global minimum of the free energy is near ξ=9.25 Å, α=−25°. Besides the global minimum, there are two major local minima, which are near ξ=9.25 Å, α=−75° and ξ=12.25 Å, α=165°, and several minor local minima near the global minimum and the two major local minima. The entire low energy area is wedge-shaped, whose tapered side points to ξ=6 Å, α=0°.
 | Figure 5 The free energy landscape of bivalent complex formed by compound 9 and the BIR2 and BIR3 domains of XIAP, projected on two reaction coordinates, the distance B3-B2 (ξ) and the dihedral B3-L3-L2-B2 (α). |
Structure of the bivalent complex formed by compound 9 and the BIR2 and BIR3 domains
By umbrella sampling for the complex participated by compound 9, we detect the free energy minimum near the reaction coordinates ξ=9.25 Å, α=−25°. To visualize a complex near the free energy minimum in the conformational space, we examine the 10 trajectories of 2 ns equilibration. Three structures at time 1.625, 1.710, 1.780 ns from one of the 8 independent trajectories have reaction coordinates ξ≈9 Å, and respectively, α≈−24, −22.5, −26.0° (Figure 6), which should be very close to the native structure of the complex. In these structures, the linker of compound 9 adapts a compact conformation, and the two binding motifs encompass the ligand on the opposite sides. Such a conformation resembles the complexes formed by two BIR domains and other bivalent ligands (Figure S5).15,51 The distance between BIR2 and BIR3 domains becomes much shorter than that of the two domains in unbound XIAP.12,15,20,52
 | Figure 6 Three structures of the bivalent complex formed by BIR2, BIR3 and compounds 9. The bivalent ligand (colored in green) has two binding motifs interacting with BIR2 (ice blue) and BIR3 (pink). Residues surrounding the binding sites are shown in the Licorice and colored by their charge (red for negative, blue for positive and white for neutral). Residues that maintain interactions between the BIR2 and BIR3 domains are labeled with residue names and numbers. The N-terminals of BIR2 and BIR3 are highlighted with blue balls, while the C-terminals, red balls. |
During the MD simulation, binding sites of the ligand kept a similar pattern of the starting structure of the MD simulation, in which the binding motifs of compound 9 were surrounded by residues Lys206, Leu207, Lys208, Asn209, Glu211, His223 and Phe224 of BIR2 and residues Gly306, Leu307, Thr308, Asp309, Lys311, Trp323 and Tyr324 of BIR3. Residues near the binding sites of the ligand are still able to interact with the residues belonging to the other BIR domain. Residue Lys208 of BIR2 forms salt bridge to Asp309 of BIR3 during the equilibration. Although the electrostatic interaction of this residue pair may contribute to the stability of the bivalent complex, each of the two BIR domains independently interact with one of the IAP-binding motifs of the ligand.
Discussion
A bivalent Smac mimetic consists of two binding motifs bound to BIR2 or BIR3 domain, and a linker tethering the two binding motifs. For designing a bivalent ligand, a short and rigid linker may result in spatial mismatch between the two binding motifs and the two domains, diminishing the gain of binding enthalpy of the second binding site, whereas a long and flexible linker raises the entropic cost for the binding and possibly shields the binding motifs.31,53–55 Taking advantage of the flexibility of the linker peptide between BIR2 and BIR3 domains,32,52 the arrangement of the BIR2 and BIR3 domains is almost free and may simultaneously fit to a bivalent ligand. Some bivalent Smac mimetics with short and rigid linkers and various binding motifs show binding activity to XIAP.13,15,28,51,56 Comparing with the compounds with a long linker, homologs with a short linker do not always benefit from the reduced entropic effect for binding, as shown in experiments.28 As have been shown, the BIR2 and BIR3 domains have an optimized arrangement maximizing the binding affinity for a bivalent Smac mimetic. This study proposed the TwistDock workflow to find out such an arrangement and to assist the design of a bivalent Smac mimetic. Our analysis demonstrates that a bivalent complex in general favors a bivalent ligand that the linker allows the two BIR domains stay far away, for example, a short linker that maintains the dihedral α about ±180°. Astonishingly, a further optimized bivalent ligand is the one that allows the BIR2 and BIR3 domains forming favorable interactions with each other, resulting in compact structure (Figures 6 and S5). According to this structure feature, we have designed and synthesized 5 novel inhibitors. A WST-based cell growth assay suggests two of them are higher potent than compound 9 (unpublished data: IC50 of HL-60 cell is 0.058 and 0.028 μmol/L, compared to 0.088 μmol/L of compound 9; IC50 of SK-OV-3 cell is 0.096 and 0.034 μmol/L, compared to 0.084 μmol/L of compound 9). For drug design, recent studies performed with gene-knock-out cells and mice suggest that the binding affinity to XIAP is not the only key feature. Efficiency of Smac mimetics at antagonizing XIAP, compared to cIAP1/2, is critical for determining if the drug is tolerated in animals.30,57 Antagonists with decreased ability to inhibit XIAP may improve the tolerability of animals, thus may be more promising drug candidates.30 For testing a tunable linker in such a bivalent Smac mimetic as anti-cancer drugs, the TwistDock workflow may help virtual screening and optimization. There are other antagonists discovered by alternative approaches that are not based on Smac, such as fragment-based screening and macrocycle library screening.9,16 Though the benefit of this type of antagonists has not been fully documented,11 these drugs may be further optimized with our approach. For fine-tuning the activity of a bivalent antagonist, various modifications of the linker chain result in different flexibility of the conformation and change their affinity to the receptor. The TwistDock workflow makes the conformational sampling viable, and together with energy-based evaluation method, like umbrella sampling or MMPBSA, predicts desired properties. The TwistDock workflow is suitable to XIAP whose two binding domains are loosely connected, and may be applied to studies on bivalent ligands targeting other receptors that are usually dimerized or oligomerized.31,54,55,58
Conclusion
We developed the TwistDock workflow to construct reasonable structures of bivalent complex formed by XIAP and a bivalent Smac mimetic. Conformations sampled by the method provide good starting points for subsequent modeling study. The results show that in the bivalent complex formed by compound 9, the BIR2 and BIR3 domains stay on the opposite sides of the ligand while the two domains form beneficial interaction, which is the possible reason that compound 9 achieves high binding affinity. The TwistDock workflow can also be deployed for optimization of other bivalent ligands.
Acknowledgments
This work is supported by the National Key Research and Development Program of China under grant number 2016YFB0201305, the National Natural Science Foundation of China under grant numbers 61702494, 31601028 and U1813203, the Shenzhen Basic Research Fund under grant numbers JCYJ20160331190123578, JCYJ20170413093358429 and GGFW2017073114031767, and the Zhejiang Provincial Natural Science Foundation of China under grant number LQ16F020006. We would also like to thank the funding support from the Shenzhen Discipline Construction Project for Urban Computing and Data Intelligence, Youth Innovation Promotion Association, Chinese Academy of Sciences, to Yanjie Wei.
Author contributions
Qingsheng Huang, Yin Peng and Dan Wei contributed to the conception and design of the study, acquisition of data, and analysis and interpretation of data. Yuefeng Peng, Yanjie Wei, and Shengzhong Feng contributed to the conception and design of the study and analysis and interpretation of data. All authors were involved in drafting or critically revising the article for intellectual content, provided approval for the final version to be published and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that they have no competing interests or conflicts of interest in this work.
References
1. Scott FL, Denault JB, Riedl SJ, Shin H, Renatus M, Salvesen GS. XIAP inhibits caspase-3 and -7 using two binding sites: evolutionarily conserved mechanism of IAPs. Embo J. 2005;24(3):645–655. doi:10.1038/sj.emboj.7600544
2. Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102(1):33–42.
3. Chai JJ, Shiozaki E, Srinivasula SM, et al. Structural basis of caspase-7 inhibition by XIAP. Cell. 2001;104(5):769–780.
4. Verhagen AM, Ekert PG, Pakusch M, et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102(1):43–53.
5. Chai JJ, Du CY, Wu JW, Kyin S, Wang XD, Shi YG. Structural and biochemical basis of apoptotic activation by Smac/DIABLO. Nature. 2000;406(6798):855–862. doi:10.1038/35022514
6. Huang YH, Rich RL, Myszka DG, Wu H. Requirement of both the second and third BIR domains for the relief of X-linked inhibitor of apoptosis protein (XIAP)-mediated caspase inhibition by Smac. J Biol Chem. 2003;278(49):49517–49522. doi:10.1074/jbc.M310061200
7. Lowe SW, Lin AW. Apoptosis in cancer. Carcinogenesis. 2000;21(3):485–495.
8. Benetatos CA, Mitsuuchi Y, Burns JM, et al. Birinapant (TL32711), a bivalent SMAC mimetic, targets TRAF2-associated cIAPs, abrogates TNF-induced NF-kB activation, and is active in patient-derived xenograft models. Mol Cancer Ther. 2014;13(4):867–879. doi:10.1158/1535-7163.MCT-13-0798
9. Chessari G, Buck IM, Day JEH, et al. Fragment-based drug discovery targeting inhibitor of apoptosis proteins: discovery of a non-alanine lead series with dual activity against cIAP1 and XIAP. J Med Chem. 2015;58(16):6574–6588. doi:10.1021/acs.jmedchem.5b00706
10. Gao Z, Tian Y, Wang J, et al. A dimeric Smac/diablo peptide directly relieves caspase-3 inhibition by XIAP. Dynamic and cooperative regulation of XIAP by Smac/Diablo. J Biol Chem. 2007;282(42):30718–30727. doi:10.1074/jbc.M705258200
11. Hird AW, Aquila BM, Hennessy EJ, Vasbinder MM, Yang B. Small molecule inhibitor of apoptosis proteins antagonists: a patent review. Expert Opin Ther Pat. 2015;25(7):755–774. doi:10.1517/13543776.2015.1041922
12. Lecis D, Mastrangelo E, Belvisi L, et al. Dimeric Smac mimetics/IAP inhibitors as in vivo-active pro-apoptotic agents. Part II: structural and biological characterization. Bioorg Med Chem. 2012;20(22):6709–6723. doi:10.1016/j.bmc.2012.09.041
13. Li L, Thomas RM, Suzuki H, De Brabander JK, Wang XD, Harran PG. A small molecule Smac mimic potentiates TRAIL- and TNF alpha-mediated cell death. Science. 2004;305(5689):1471–1474. doi:10.1126/science.1098231
14. Lu J, Rong S, Sun H, et al. A potent and highly efficacious bivalent Smac mimetic APG-1387 in Phase I clinical development. Eur J Cancer. 2014;50(50):89. doi:10.1016/S0959-8049(14)70394-6
15. Nikolovska-Coleska Z, Meagher JL, Jiang S, et al. Interaction of a cyclic, bivalent Smac mimetic with the X-linked inhibitor of apoptosis protein. Biochemistry-Us. 2008;47(37):9811–9824. doi:10.1021/bi800785y
16. Seigal BA, Connors WH, Fraley A, et al. The discovery of macrocyclic XIAP antagonists from a DNA-programmed chemistry library, and their optimization to give lead compounds with in vivo antitumor activity. J Med Chem. 2015;58(6):2855–2861. doi:10.1021/jm501892g
17. Sun HY, Nikolovska-Coleska Z, Lu JF, et al. Design, synthesis, and characterization of a potent, nonpeptide, cell-permeable, bivalent smac mimetic that concurrently targets both the BIR2 and BIR3 domains in XIAP. J Am Chem Soc. 2007;129(49):15279–15294. doi:10.1021/ja074725f
18. Fulda S, Vucic D. Targeting IAP proteins for therapeutic intervention in cancer. Nat Rev Drug Discov. 2012;11(2):109–124. doi:10.1038/nrd3627
19. Wang SM. Design of small-molecule smac mimetics as IAP antagonists. Curr Top Microbiol. 2011;348:89–113.
20. Speer KF, Cosimini CL, Splan KE. Characterization of a heterodimeric Smac-based peptide that features sequences specific to both the BIR2 and BIR3 domains of the X-linked inhibitor of apoptosis protein. Biopolymers. 2012;98(2):122–130. doi:10.1002/bip.21732
21. Tamm I, Kornblau SM, Segall H, et al. Expression and prognostic significance of IAP-family genes in human cancers and myeloid leukemias. Clin Cancer Res. 2000;6(5):1796–1803.
22. Salvesen GS, Duckett CS. IAP proteins: blocking the road to death’s door. Nat Rev Mol Cell Bio. 2002;3(6):401–410. doi:10.1038/nrm830
23. Reubold TF, Eschenburg S. A molecular view on signal transduction by the apoptosome. Cell Signal. 2012;24(7):1420–1425. doi:10.1016/j.cellsig.2012.03.007
24. Crowder RN, El-Deiry WS. Caspase-8 regulation of TRAIL-mediated cell death. Exp Oncol. 2012;34(3):160–164.
25. Cohen GM. Caspases: the executioners of apoptosis. Biochem J. 1997;326(Pt 1):1–16.
26. Obexer P, Ausserlechner MJ. X-linked inhibitor of apoptosis protein – a critical death resistance regulator and therapeutic target for personalized cancer therapy. Front Oncol. 2014;4:197. doi:10.3389/fonc.2014.00197
27. Lukacs C, Belunis C, Crowther R, et al. The structure of XIAP BIR2: understanding the selectivity of the BIR domains. Acta Crystallogr D. 2013;69:1717–1725. doi:10.1107/S0907444913016284
28. Peng YF, Sun HY, Lu JF, et al. Bivalent Smac mimetics with a diazabicyclic core as highly potent antagonists of XIAP and cIAP1/2 and novel anticancer agents. J Med Chem. 2012;55(1):106–114. doi:10.1021/jm201072x
29. Sun H, Liu L, Lu J, et al. Potent bivalent Smac mimetics: effect of the linker on binding to inhibitor of apoptosis proteins (IAPs) and anticancer activity. J Med Chem. 2011;54(9):3306–3318. doi:10.1021/jm101651b
30. Condon SM, Mitsuuchi Y, Deng YJ, et al. Birinapant, a Smac-Mimetic with improved tolerability for the treatment of solid tumors and hematological malignancies. J Med Chem. 2014;57(9):3666–3677. doi:10.1021/jm500176w
31. Krishnamurthy VM, Estroff LA, Whitesides GM. Multivalency in ligand design. In: Jahnke W and Erlanson DA, editors. Fragment-Based Approaches in Drug Discovery. Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA; 2006:11–53.
32. Cossu F, Milani M, Vachette P, et al. Structural insight into inhibitor of apoptosis proteins recognition by a potent divalent Smac-mimetic. PLoS One. 2012;7(11). doi:10.1371/journal.pone.0049527.
33. Zhang H, Xi W, Hansmann UH, Wei Y. Fibril–barrel transitions in cylindrin amyloids. J Chem Theory Comput. 2017;13(8):3936–3944. doi:10.1021/acs.jctc.7b00383
34. Wu G, Chai JJ, Suber TL, et al. Structural basis of IAP recognition by Smac/DIABLO. Nature. 2000;408(6815):1008–1012. doi:10.1038/35050012
35. Weininger D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J Chem Inf Comput Sci. 1988;28(1):31–35.
36. Blaney JM, Dixon JS. Distance geometry in molecular modeling. In: Lipkowitz KB, Boyd DB, editors. Reviews in Computational Chemistry, Volume 5. New York: VCH Publishers, Inc.; 2007:299–335.
37. Landrum G and Penzotti J.RDKit: Open-Source Cheminformatics Software [Computer Program]. Available from:
38. Halgren TA. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J Comput Chem. 1996;17(5–6):490–519. doi:10.1002/(SICI)1096-987X(199604)17:5/6<>1.0.CO;2-C
39.
40. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. doi:10.1002/jcc.21334
41. Wang JM, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25(9):1157–1174. doi:10.1002/jcc.20035
42. Case DA, Babin V, Berryman JT, et al. AMBER 14 [Computer Program]. San Francisco: University of California; 2014.
43. Pang YP, Xu K, Yazal JE, Prendergas FG. Successful molecular dynamics simulation of the zinc-bound farnesyltransferase using the cationic dummy atom approach. Protein Sci. 2000;9(10):1857–1865.
44. Phillips JC, Braun R, Wang W, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26(16):1781–1802. doi:10.1002/jcc.20289
45. Heyer LJ, Kruglyak S, Yooseph S. Exploring expression data: identification and analysis of coexpressed genes. Genome Res. 1999;9(11):1106–1115.
46. Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph Model. 1996;14(1):33–38. doi:10.1016/0263-7855(96)00018-5
47. Miller BR, McGee TD, Swails JM, Homeyer N, Gohlke H, Roitberg AE. MMPBSA.py: an efficient program for end-state free energy calculations. J Chem Theory Comput. 2012;8(9):3314–3321. doi:10.1021/ct300418h
48. Weiser J, Shenkin PS, Still WC. Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO). J Comput Chem. 1999;20(2):217–230. doi:10.1002/(ISSN)1096-987X
49. Roux B. The calculation of the potential of mean force using computer simulations. Comput Phys Commun. 1995;91(1–3):275–282. doi:10.1016/0010-4655(95)00053-I
50. Lemieux C. Preface. In: Monte Carlo and Quasi-Monte Carlo Sampling. Springer Series in Statistics Vol. 2009. New York: Springer Science+Business Media, LLC; 2009:vii.
51. Cossu F, Milani M, Mastrangelo E, et al. Structural basis for bivalent smac-mimetics recognition in the IAP protein family. J Mol Biol. 2009;392(3):630–644. doi:10.1016/j.jmb.2009.04.033
52. Splan KE, Allen JE, McLendon GL. Biochemical basis for enhanced binding of peptide dimers to X-linked inhibitor of apoptosis protein. Biochemistry-Us. 2007;46(42):11938–11944. doi:10.1021/bi061938t
53. Jahromi AH, Fu Y, Miller KA, et al. Developing bivalent ligands to target CUG triplet repeats, the causative agent of myotonic dystrophy type 1. J Med Chem. 2013;56(23):9471–9481. doi:10.1021/jm400794z
54. Joce C, White R, Stockley PG, Warriner S, Turnbull WB, Nelson A. Design, synthesis and in vitro evaluation of novel bivalent S-adenosylmethionine analogues. Bioorg Med Chem Lett. 2012;22(1):278–284. doi:10.1016/j.bmcl.2011.11.017
55. Shan M, Bujotzek A, Abendroth F, et al. Conformational analysis of bivalent estrogen receptor ligands: from intramolecular to intermolecular binding. Chembiochem. 2011;12(17):2587–2598. doi:10.1002/cbic.201100529
56. Sheng R, Sun H, Liu L, et al. A potent bivalent Smac Mimetic (SM-1200) achieving rapid, complete, and durable tumor regression in mice. J Med Chem. 2013;56(10):3969–3979. doi:10.1021/jm400216d
57. Silke J, Vaux DL. IAP gene deletion and conditional knockout models. Semin Cell Dev Biol. 2015;39:97–105. doi:10.1016/j.semcdb.2014.12.004
58. Zhang A, Liu ZL, Kan Y. Receptor dimerization – rationale for the design of bivalent ligands. Curr Top Med Chem. 2007;7(4):343–345.
Supplementary materials
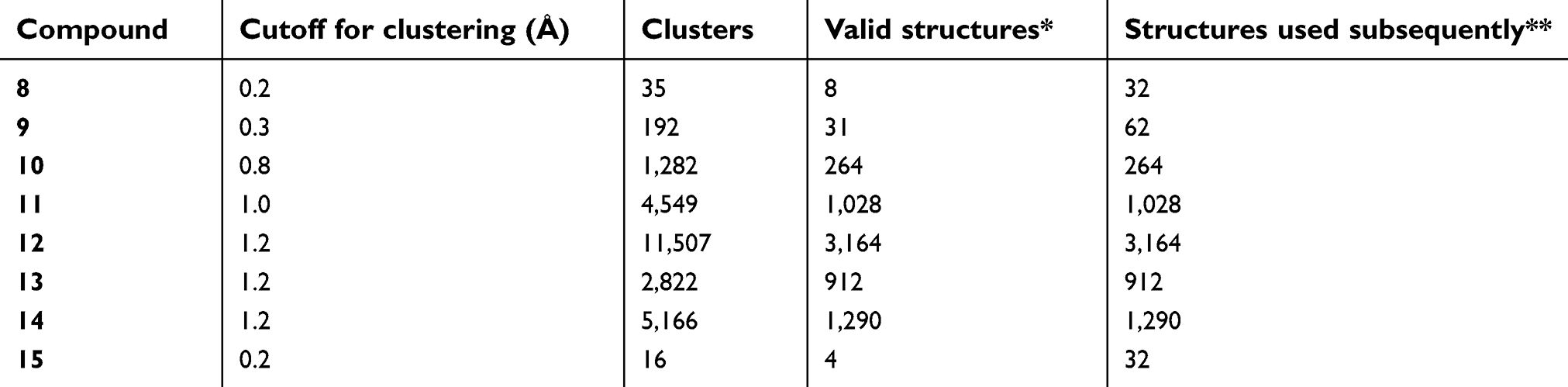 | Table S1 The clustering results of the linker conformations |
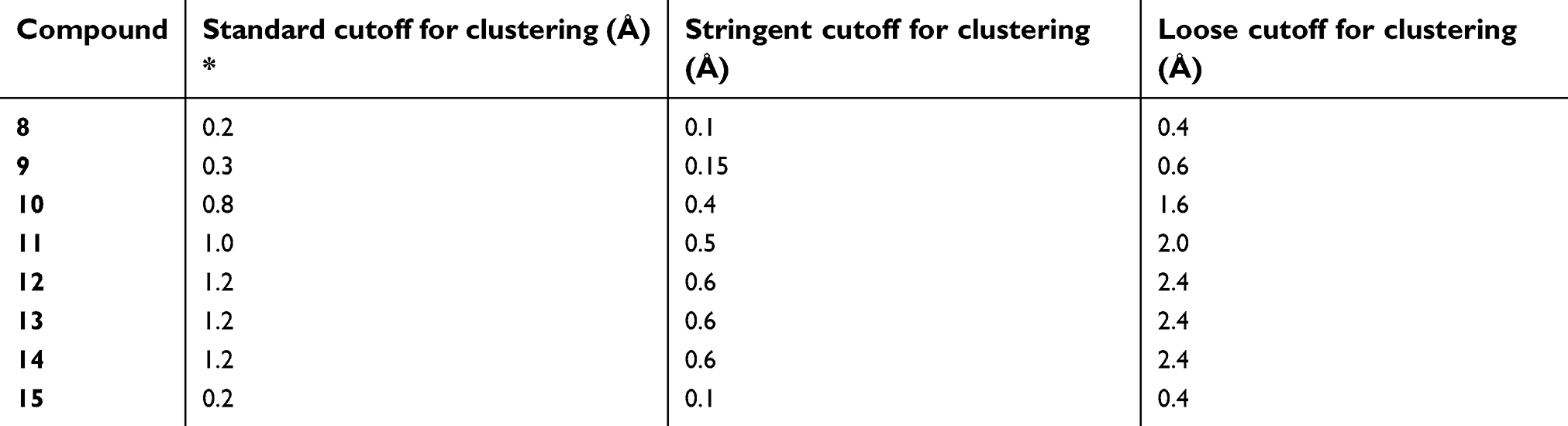 | Table S2 Three sets of cutoff for clustering |
 | Figure S1 Structures of the monovalent complex formed by BIR3 and AT-406. The docking structure (A) with the highest docking score was selected and further subjected to a 25 ns MD. This structure has a binding mode similar to the complex in PDB entry 1G73, and the RMSD between backbone heavy atoms of AVPI in 1G73 and the corresponding atoms of AT-406 is 1.056Å (calculated by VMD). The structure (B) after 25 ns MD was used to construct the bivalent complex. The structures of BIR3 and AVPI in PDB entry 1G73 are shown in the pink NewCartoon and the yellow CPK representations, respectively, while the docked AT-406 is shown in the green Licorice representation. The structure of BIR3 after 25 ns MD is shown in ochre NewCartoon representation. The zinc atom (the gray ball) is not involved in the binding site. |
 | Figure S2 Structures of the monovalent complex formed by BIR2 and AT-406. Among the top three docking structures (A, B and C), the third structure (C) was selected and subjected to a 25 ns MD run, since it has a binding mode most similar (RMSD 1.123Å) to the complex formed by the AVPI sequence and the BIR2 domain in PDB entry 4J46. The RMSD is calculated between backbone heavy atoms of AVPI in 4J46 and the corresponding atoms of AT-406. The structure (D) after the 25 ns MD was used to construct the bivalent complex. In contrast, for (A) and (B) the corresponding RMSDs are 1.866Å and 1.130Å. The structures of BIR2 and AVPI in PDB entry 4J46 are shown in the iceblue NewCartoon and the yellow CPK representations, while the docked AT-406 is shown in the green Licorice representation. The structure of BIR2 after the MD run is shown in the ochre NewCartoon representation in (D). The zinc atom (the gray ball) is not involved in the binding site. |
 | Figure S3 Distributions of clusters are insensitive to the clustering cutoff. For a bivalent complex, distributions are obtained in three sets of cutoff for clustering, where “standard” is the condition set used to produce the structures for the subsequent minimization and MD, “stringent” is the condition set resulting more structures and “loose” is the condition set resulting fewer structures. The names of the compounds that participate the formation of bivalent complexes are shown on the top line. In each distribution histogram, the x-axis denotes the value of (A) radius of gyration of the bivalent complex (Rgyr) or (B) the dihedral B2-L2-L3-B3, while the y-axis denotes the number of structures. |
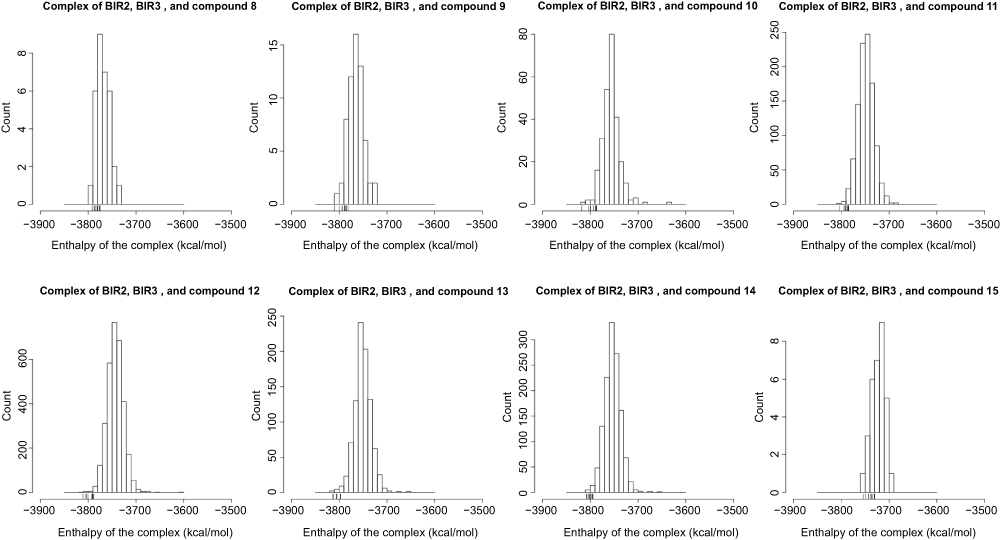 | Figure S4 Distribution of the complex enthalpy. We only compared the complex enthalpy among structures of an ensemble of the same complex. Small verticle lines near the x-axis (a rug representation) indicate the 10 lowest values of the enthalpy of conformations of a complex. |
 | Figure S5 Comparison of a bivalent complex formed by BIR2, BIR3 and compounds 9, and a complex formed by two BIR3 domains and a bivalent Smac mimetic (PDB entry 2VSL). The structure of the complex formed by BIR2, BIR3 and compound 9, respectively, shown in iceblue and pink NewCartoon, and green Licorice, is the same one shown in Figure 6A. In this structure, the arrangement of the BIR2 and BIR3 domains is compact, similar to PDB entry 2VSL, whose BIR3 domains are shown in ochre NewCartoon representation and the ligand in yellow CPK representation. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.