Back to Journals » Risk Management and Healthcare Policy » Volume 18
The Saudi National Policy and Protocol for Epidermolysis Bullosa
Authors Alheggi A ![]() , Alhashem A, Alshihry HMH, Al-khenaizan S, Alshammrie FF, AlEissa MM, Almudaiheem HY, Shehata N
, Alhashem A, Alshihry HMH, Al-khenaizan S, Alshammrie FF, AlEissa MM, Almudaiheem HY, Shehata N ![]() , Alshingetti N, Almudeer AH, Alshammari IF
, Alshingetti N, Almudeer AH, Alshammari IF ![]() , Alsefri MF, Al-jedai A
, Alsefri MF, Al-jedai A ![]() , Eshmawi MT
, Eshmawi MT
Received 7 May 2025
Accepted for publication 11 October 2025
Published 31 October 2025 Volume 2025:18 Pages 3521—3538
DOI https://doi.org/10.2147/RMHP.S532321
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Gulsum Kaya
Ashjan Alheggi,1,* Amal Alhashem,2,3,* Hind Mohammad H Alshihry,4,* Sultan Al-khenaizan,5,* Fawwaz Freih Alshammrie,6,* Mariam M AlEissa,7– 10,* Hajer Y Almudaiheem,11,* Nancy Shehata,12,* Naemah Alshingetti,13,* Ali Husein Almudeer,14,* Ibtesam Fawaz Alshammari,15,* Mohammed Faraj Alsefri,16,* Ahmed Al-jedai,11,17,* Maysa Tariq Eshmawi1,12,*
1Department of Dermatology, College of Medicine, Imam Mohammad Ibn Saud Islamic University (IMSIU), Riyadh, Saudi Arabia; 2Department of Genetic and Metabolic, King Fahad Specialist Hospital, Dammam, Saudi Arabia; 3Department of Anatomy, College of Medicine, Al-Faisal University, Riyadh, Saudi Arabia; 4Department of Dermatology, Prince Sultan Military Medical City, Riyadh, Saudi Arabia; 5Department of Dermatology, King Abdulaziz Medical City, Riyadh, Saudi Arabia; 6Department of Dermatology, University of Hail, Hail, Saudi Arabia; 7Department of Medicine, College of Medicine, Al-Faisal University, Riyadh, Saudi Arabia; 8Department of Research, King Khaled Eye Specialist Hospital (KKESH) Research Centre, Riyadh, Saudi Arabia; 9Department of Molecular Genetics, Public Health Lab, Public Health Authority, Riyadh, Saudi Arabia; 10Computational Sciences Department at the Centre for Genomic Medicine (CGM), King Faisal Specialist Hospital & Research Center, Riyadh, Saudi Arabia; 11The Saudi Society of Clinical Pharmacy, Riyadh, Saudi Arabia; 12Department of Dermatology, King Abdullah Medical Complex, Jeddah, Saudi Arabia; 13Obstetrics & Gynecology Department, King Salman Hospital, Riyadh First Cluster, Riyadh, Saudi Arabia; 14Department of Pediatric, King Saud University Medical City, Riyadh, Saudi Arabia; 15Obstetrics & Gynecology Department, REI&MIS, King Saud Medical City, Cluster One, Riyadh, Saudi Arabia; 16Hospital Affairs, Ministry of Health, Riyadh, Saudi Arabia; 17Colleges of Medicine and Pharmacy, Alfaisal University, Riyadh, Saudi Arabia
*These authors contributed equally to this work
Correspondence: Maysa Tariq Eshmawi, Department of Dermatology, College of Medicine, Imam Mohammad Ibn Saud Islamic University, Riyadh, Saudi Arabia, Tel +966 55 535 2819, Email [email protected]
Abstract: This protocol’s objective is to offer evidence-based suggestions for the identification, treatment, and management of epidermolysis bullosa (EB) in Saudi Arabia. EB is a rare genetic condition that results in blistering and skin fragility. Depending on the subtype, it can produce a variety of consequences. The four primary kinds of EB are EB Simplex, Junctional EB, Dystrophic EB, and Kindler EB. This guideline provides a thorough understanding of EB and describes diagnostic techniques including genetic testing and immunofluorescence mapping. The significance of an interdisciplinary team (IDT) approach in treating severe instances is also covered, guaranteeing integrated treatment across wound care, nutrition, dermatology, genetics, and psychosocial support. In addition, the guideline discusses the role of new treatments for EB, such as Beremagene geperpavec (B-VEC), and stresses the necessity of creating a National Saudi EB Registry to maximize patient care and guarantee fair access to treatment. The guideline also emphasizes the importance of genetic counseling for prenatal diagnosis, family planning, and parental screening, as well as the need to address the psychological difficulties that people living with EB and their families face. In summary, this guideline offers a structured framework for physicians and healthcare providers to guarantee the best possible care for people living with EB in Saudi Arabia, with the goal of improving clinical outcomes, quality of life, and long-term management of the disease.
Keywords: epidermolysis bullosa, management, interdisciplinary care, genetic counseling, psychosocial considerations
Introduction
Epidermolysis bullosa (EB) is a group of rare inherited blistering disorders characterized by fragility of the skin and mucous membranes. The hallmark of these disorders is blister formation and scarring, which result from skin layer separation in response to mild mechanical trauma.1 EB is classified based on the skin cleavage plane into four main types: EB simplex (EBS), Junctional EB (JEB), Dystrophic EB (DEB), and Kindler EB (KEB).2 These disorders result from mutations in genes that encode structural proteins present in various layers of the skin and are critical for maintaining skin integrity and adhesion between skin layers.2,3
EB is currently linked to over 40 different genes and more than 30 unique subtypes. EB is highly heterogeneous with a wide range of variability in symptom severity, blisters distribution, and presence of certain cutaneous and extracutaneous features.2,3 The prevalence of EB varies widely across countries. The estimated incidence of EB was 67.8 and 19.6 per million live births respectively.4 Based on data from US and England national EB registries, EB Simplex was the most common type, followed by DEB, and JEB.5 In Saudi Arabia, the exact prevalence is unknown; however, there are observed increased reports of severe types of EB including DEB and JEB may reflect the frequency of consanguineous marriages.6
Most EB types are present at birth or infancy, while a few subtypes exhibit a delayed onset.5 Mild EB types generally result in painful blistering that is limited to a few regions of the body or the extremities. On the other hand, severe forms of EB are associated with blisters that affect multiple areas of the body, including internal organs.7 Among the various factors influencing symptom severity, abnormal, reduced, or absent protein expression are significant contributors. Mild to intermediate EB types result from abnormal proteins or reduced expression, while severe types result from the absence of protein expression.8 Life expectancy is dependent on the severity of symptoms which can be mostly unaffected in milder EB types. On the other hand, severe types can cause premature death in infancy and significantly reduce life expectancy in adults due to the high frequency of life-threatening complications such as metastatic squamous cell carcinoma (SCC).7
Management of EB is focused on skin and wound care, optimizing nutrition, improving quality of life through symptoms and pain relief, as well as preventing the development of further complications. Recently, advances in EB therapeutics in targeting the primary genetic abnormalities, and/or reducing the secondary inflammation in EB tissues have been made. This offers hope for improved quality of life, and better clinical management for people living with different forms of EB.9,10
The Need for National Saudi EB Registry and Interdisciplinary Team
In May 2023, the US Food and Drug Administration (FDA) granted approval for the first topical gene therapy designed for patients aged six months and older with dominant or recessive dystrophic epidermolysis bullosa (DEB) caused by mutations in the COL7A1 gene. Beremagene geperpavec (B-VEC) utilizes an inactivated herpes simplex virus type 1 vector to restore collagen VII, which is deficient in the skin of DEB patients.11,12 Like many novel gene therapies, the cost of B-VEC therapy will be high.13 Thus, there is an urgent need to establish the Saudi National EB registry, and to restrict gene therapy access to eligible patients.
Since the disease is uncommon, many healthcare professionals have limited experience to offer the best possible standard of comprehensive care. To achieve the best results, several experts from different fields must be involved in severe cases of EB. Patients with EB should be treated in an interdisciplinary team (IDT) environment, where experts from different specialties collaborate to offer all-encompassing care and support, to ensure the best quality of care.13,14
Diagnostic Approach to Epidermolysis Bullosa
Laboratory Testing for the Diagnosis of EB
Genetic testing should be performed in cases where clinical signs appear to indicate EB in order to correctly identify the gene abnormality and confirm the diagnosis. To enable quick diagnosis, prognosis, and treatment, a newborn may get skin biopsy for immunofluorescence mapping (IFM) in addition to peripheral blood sample for genetic testing, depending on what is available at the designated diagnostic laboratory.15
Available Diagnostic Tests for People Living with EB
Molecular Genetic Tests
Sanger sequencing (SS) for candidate gene testing, next-generation sequencing (NGS) targeted gene panels, and comprehensive genomic testing (whole-exome sequencing, WES, or whole genome sequencing, depending on the situation) are all examples of molecular genetic testing techniques. Therefore, genetics consultation prior to testing is recommended.
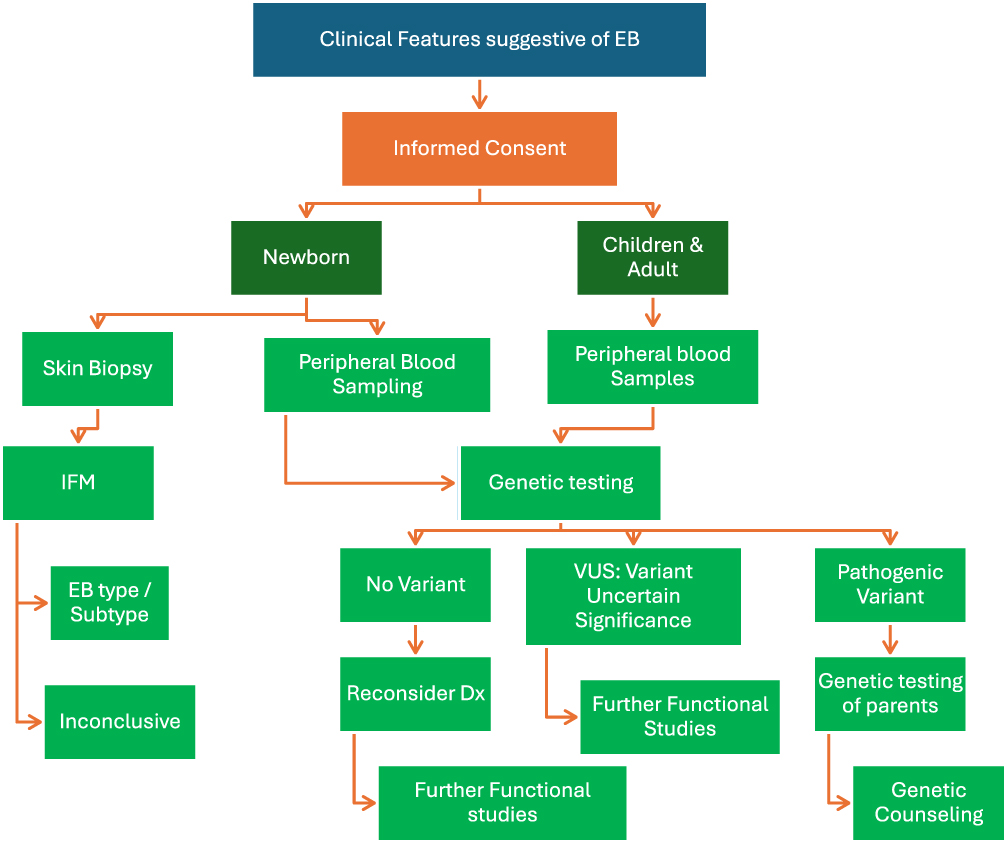 |
Figure 1 Diagnostic Tests for People living with EB. |
Carrier Screening
Recurrence prevention and diagnosis are part of segregation analysis. As EB is inherited, genetic counseling is crucial. Family members should be offered carrier screening to assess their risk of carrying the disease-causing gene. It is important to discuss the chance of recurrence and look into preventative measures like prenatal diagnosis or preimplantation genetic diagnosis (PGD) (Figure 1).15
Interdisciplinary Care
Advantages of an Interdisciplinary Approach in the Management of EB
To guarantee that patients with EB receive the highest quality of care, a interdisciplinary approach is essential. Because severe cases of EB are multi-systemic, multiple specializations must be included. Additionally, the lack of qualified medical personnels with specific knowledge of this illness and the new therapeutic treatments for EB necessitate action to change the current system of care into one that is more integrated. With the use of suitable molecular diagnostic assays, the IDT assists in providing an accurate diagnosis of EB. Improved symptom management, foreseeing complications, precise prognostic information, successful genetic counseling for families, enhanced quality of life for patients and their families, and active involvement in research initiatives are all considered advantages of IDT.16
IDT could have additional beneficial roles:
- Ensure rapid turnaround time for skin biopsy and molecular screening.
- Regular screening to monitor the development of SCC and offer appropriate oncological care.
- Support adolescents in transitioning from pediatrics to adult EB teams.
- Develop the experience, knowledge and skills among team members.
Note: If any health sector wants to initiate an EB center, an interdisciplinary approach must be considered.
Patient Categories
The variety of EB IDT settings is thought to include patients with all types of EB as well as those with other severe genetic skin fragility diseases. Depending on the degree of their illness and their medical requirements, patients are divided into two groups: mild and severe.
- Mild patients - Patients with milder forms require input from the basic EB team only (dermatologist, pediatrician, EB clinical nurse, podiatrist).
- Severe patients - Patients with severe disease require input from the basic EB team and additional EB team specialists. Severity should be reassessed after school entry, at adulthood, and at any point if deemed medically necessary.
The EB IDT Service
The EB IDT service can be divided into outpatients, daycare, inpatients, and virtual clinics as well as teaching and training.
- Outpatients - A baseline EB team is also required in outpatient EB IDT clinics. If necessary, patients with severe forms of EB should be assessed by a larger IDT, especially if they have a history of skin cancer. Equipment for performing skin biopsies and changing dressings should be available in the clinics. Outpatient appointment frequency is decided on an individual basis.
- Day cases - Minor treatments including blood or iron infusions, a biopsy of questionable skin lesions, or daycare attendance may be necessary for certain people living with EB.
- Inpatient, planned elective or emergency admission - Inpatient facilities where patients with EB can be admitted for elective procedures like esophageal dilatation and squamous cell carcinoma excision should be available to the EB IDT service. Emergency hospitalization may be required if a patient with EB experiences significant medical consequences such as kidney issues, severe dehydration, or septic shock.17
IDT Members (Key Core Disciplines for People Living with EB)
Consultant dermatologist, consultant pediatrician (for children), wound care or EB clinical nurse, dietician, physiotherapist, occupational therapist, ophthalmologist, dentist, psychologist, interventional radiologist, gastroenterologist, palliative care/pain clinician, plastic/hand surgeon, podiatrist, administrative support, and service coordinator are among the primary members of the interdisciplinary team.
If more interdisciplinary team members are required for the EB patient population, they will be included in the larger EB care group. Ear, nose, and throat (ENT) surgeons, cardiologists, endocrinologists, speech and language therapists, dental hygienists, urologists, orthotists, and other specialists and therapists should be included in these broader fields.
Tasks including auditing, letter writing, activity recording, Email management, assisting with service development, and liaising with patients and EB wound care nurses fall within the purview of administrative staff.
Core nursing priorities are described in Table 1.
 |
Table 1 Description of Core Nursing Priorities |
EB Clinical Care Nurses
In the EB IDT service, EB clinical nurses are essential team members. They provide both inpatient and outpatient care for patients with EB.
All national EB facilities should adhere to the following EB wound care nursing principles:
- Only patients who are enrolled and active at their nationally designated center will be treated by nurses.
- Every patient must be registered at a national EB center (ideally the nearest center, but all patients must be given the option to choose their center)
- A patient is deemed active if they see the EB team in person at least once a year, which may include an EB nurse or wound care nurse. The 12-month review period may be extended for some patients, but it cannot be extended past 14 months.
- For the most efficient use of resources, patients should be contacted by the nearest EB centers.
- All activities categorized as key nursing priorities must be documented and reported by providers. In designated centers, EB nurses are responsible for making sure that every activity is documented and reported as needed.17
Home Health Care Nurses
When necessary, home visits may be required for patients with severe conditions to help with dressing changes, perform comprehensive skin examinations, take pictures (which may not always be feasible in a hospital setting), and identify suspicious skin areas that need additional examination and biopsy in the hospital. The IDT service should provide home visit support through the wound home health care nurses. In order to guarantee that EB skills and information are shared and that patients receive proper support, it is imperative to establish relationships with local care professionals.
Initial Assessment
- Neonates - The EB IDT service is notified when a newborn exhibits blistering or skin fragility. The appropriate EB IDT service reviews clinical pictures and history. Skin biopsy can be performed in a setting where immunofluorescence is available, and if EB is thought to be highly likely, a decision should be made regarding whether to transfer the child to the EB IDT service center right away or to provide initial care in a safe and comfortable manner without transferring them too soon (transporting a youngster too soon could be dangerous). The local neonatal unit staff and the EB IDT service can work together to control the blistering and lower the chance of further skin damage. When appropriate, arrangements should be made for follow-up with the EB IDT service if the child is not transferred. After being discharged from the hospital, the EB IDT should stay in touch with the family and the neighborhood pediatric team to make sure the child is receiving the right care at home, to handle any crises, and to help with end-of-life care if necessary. This will save the family from having to drive a seriously unwell child considerable distances to the EB IDT service, which could worsen their condition by causing respiratory distress or skin damage.17
- Pediatrics - For older babies and children, the initial assessment will take place as an outpatient or day case as an outpatient or day case at the EB IDT service.17
Discharge Criteria
Some patients may be discharged from the service when:
- With assistance from nearby providers, they are capable of self-management. In these situations, the service will communicate with nearby providers to offer guidance on the handling and sharing of clinical data.
- Repeated absences from more than two straight clinic appointments, as determined by the eb center. Unless there is a medical or social justification for postponing appointments for more than a year, patients who are not seen annually must be sent back to their general practitioner.
- Patients may transfer to another national center at any time during their treatment. Only when there is a clear indication that care is no longer needed or is provided elsewhere, and after consultations with the patient’s general practitioner and parents or legal guardians have been completed, should pediatric patients be released from the hospital.17
Clinical Management and Recent Advances in EB Treatment
Symptomatic Management
- Wound Care - People living with EB need specific wound care and specific dressing.18 The ideal dressing should:
- be acceptable depending on wound type.
- provide an appropriate environment to encourage wound healing
- be non-adherent to the wound bed and surrounding skin
- be painless on removal
- minimize the need for frequent dressing changes
- be able to absorb exudate
- be comfortable and remain in place
- be easily used and available
- be available in different sizes
- Pain Management - For patients with all types of EB, optimal pain treatment is essential and involves both non-pharmacological and pharmacological approaches. While opioids and anxiolytics are required for severe pain related to dressing changes, simple analgesics like paracetamol and ibuprofen may be adequate to handle minor pain.19
- Preventing & Management of Complications:
Preventing Complications:
- For newborns with EB, avoid oral blisters by using soft nipples (The Habermann Nipple, The Mead Johnson Cleft Palate Nurse or The Pigeon Feeder).
- Take off garment tags and labels before wearing, and stay away from anything with abrasive buttons, snaps, zippers, or tight elastic bands.
- Prevent corneal abrasions by offering comfort measures including lubricating eye drops, avoiding strong light, and taking mild painkillers.
- Avoiding irritants like rough textiles, eating a healthy diet to aid in healing, keeping nails short and skin protected to minimize scratching, staying hydrated and taking care of your skin to prevent dryness, and staying out of the heat.
- In the case of corneal disease, an ophthalmologist’s quick assessment is necessary to stop the formation of irreversible corneal scarring and vision impairment.
- If a wound refuses to heal, consult a dermatologist.20
Management of Complications - Careful monitoring is required for patients with EB subtypes known to be most vulnerable to certain complications, and appropriate interventions (medical, surgical, dental, nutritional, psychological, and other) must be implemented before the affected tissues sustain significant damage.20
- Cardiac Myopathy
- Early identification and use of medications can improve heart function.
- Regular monitoring with heart ultrasounds for early detection and intervention.18
- Renal Complications and Urinary Tract Involvement:
- Blistering or ulcers can cause scarring, obstruction, hydronephrosis, and eventual kidney failure.
- Recurrent UTIs exacerbate risks; circumcision may help reduce strictures.18
- Kidney Damage:
- Chronic infections and urinary issues can lead to renal scarring and failure (~12% by age 35 in RDEB patients).
- Annual urine analysis and kidney function tests are recommended.
- Blood pressure management and protective medications are beneficial.18
- Anemia Management
- Nutritional support with iron, folic acid, and vitamins (E, D, K, B12).
- Oral iron supplements with vitamin C for better absorption.
- If oral supplements fail, intravenous (IV) iron with erythropoietin may be required.
- Blood transfusion for severe symptoms.18
- Bleeding and Clotting Issues
- Loss of proteins disrupts clotting balance.
- Diagnosis via blood tests; treatment with fresh plasma or clotting proteins.18
- Skin Cancer Prevention and Treatment
- Regular skin inspections every 3–6 months for high-risk individuals.
- Surgical removal of cancer, with more extensive procedures for advanced cases.18
- Bone Problems
- Risks include osteoporosis, fractures, and pain, exacerbated by immobility.
- Prevention includes walking, taking vitamin D and calcium supplements, and good nutrition.
- Annual bone density monitoring (DEXA scan).18
- Nail and Hair Issues
- Nail blistering and chronic sores can cause deformities, infections, and loss.
- Hair follicle damage on the scalp can lead to permanent hair loss and increased fragility.18
- Eye Complications
- Eyelid issues: Blisters, ectropion (outward turning), and entropion (inward turning), may require surgery.
- Conjunctiva: Blisters, edema, injection, or symblepharon may need surgical intervention.
- Cornea: Abrasions are treated with antibiotics, patches, or bandage lenses. Pannus (scar tissue) may require surgical removal18
- Larynx
A serious and potentially life-threatening extracutaneous complication of EB is laryngeal involvement. Airway complications such as strictures, partial or complete luminal obstruction from granulation tissue, and stenosis may affect the glottic, subglottic, or supraglottic regions. Patients often present with symptoms such as shortness of breath, stridor, dysphagia, and/or cervicothoracic retraction.21 The presence of these symptoms requires further evaluation. All airway devices and adjuncts should be thoroughly lubricated. For airway management, careful fiber-optic intubation using a well-lubricated small endotracheal tube (5.0–6.0 mm internal diameter) is recommended, as this approach minimizes the risk of airway trauma.22 Early tracheostomy should be considered in these patients to secure the airway in the acute phase and minimize additional laryngeal trauma from endotracheal intubation.23
Rehabilitation - The focus is on improving various functional abilities and skills across different domains:
- Fine Motor and Life Skills Development: enhancing the ability to control small hand and finger movements, which is crucial for physical, mental, and behavioral growth. This involves strengthening the sensory processing system to help individuals participate in daily activities.24
- Oral Feeding Skills: improving the ability to eat, swallow, and feed for individuals with various diagnoses related to eating and swallowing difficulties across all ages.24
- Self-Care Support: assisting individuals in performing essential self-care activities, including bathing, dressing, toileting, feeding, grooming, mobility, and other tasks crucial for personal well-being and survival.24
- Instrumental Activities for Daily Living: helping individuals manage more complex tasks like caring for others, maintaining a home, financial management, community mobility, meal preparation, safety, shopping, and participating in activities such as education, work, and leisure.24
- Hand Function Support: focusing on non-surgical methods to maintain and enhance hand movement, strength, coordination, and precision to improve daily task performance.24 However, surgical correction of severe hand deformities significantly enhances a patient’s quality of life.25
Gene Therapy
- Topical treatment: The US Food and Drug Administration approved topical gene therapy (beremagene geperpavec), which is not SFDA registered, in May 2023 to treat wounds in patients aged six months and up who have dystrophic epidermolysis bullosa (DEB) caused by variations in the type VII collagen alpha 1 chain gene (COL7A1). The functional form of the COL7A1 gene is delivered directly to skin cells via the topical application of B-VEC, a modified herpes simplex virus 1 vector that is replication-defective and nonintegrating. B-VEC treatment aims to help individuals with DEB produce more type VII collagen.26
- Currently, a number of experimental techniques are being investigated for potential therapeutic applications. These include transplanting bone marrow-derived stem cells, infusing recombinant protein (ie, type VII collagen for RDEB), and ex vivo gene substitution transplanting allogeneic fibroblasts (in RDEB, to give a supply of normal type VII collagen) for autosomal recessive forms of EB.26
- Investigations into ways to either downregulate the dominant negative gene or compensate for its presence by upregulating other genes whose products might at least partially provide improved structural stability to the skin, thereby overriding the effect of the underlying mutation, are being conducted for autosomal dominantly transmitted EB.26
- Additional clinical trials are currently being conducted to examine potential ways to improve wound healing. One such experiment is evaluating the probable effectiveness of topically applying thymosin β4, a tiny molecular weight protein, to open wounds.
- Several new promising therapies are currently being developed.
Protein Replacement Therapies
Synthetic versions of proteins like recombinant collagen VII for DEB and Laminin-322 for JEB27 are promising in pre-clinical studies but requires more clinical trials.
Cell-Based Therapies
Mesenchymal Stem Cells (MSCs) and Hematopoietic Stem Cells (HSCs): In terms of improving the safety of cell-based treatments for epidermolysis bullosa (EB), bone marrow-derived mesenchymal stem cells (BM-MSCs) have demonstrated encouraging outcomes. They facilitate quicker wound healing, lessen blister development, and lower the chance of serious adverse outcomes.28
Hematopoietic stem cell transplantation (HCT) has recently emerged as an alternative treatment option, showing potential in addressing the systemic nature of the disease. However, its application in treating recessive dystrophic epidermolysis bullosa (RDEB) remains largely unexplored, with limited knowledge about possible side effects specific to RDEB.29
Pathogenesis-Based Therapies
- SFDA-registered Oleogel-S10 (birch triterpenes) is one medication that promotes wound healing. Oleogel-S10 received approval from the European Medicines Agency in June 2022 to treat partial-thickness wounds in patients aged six months and up that are linked to junctional epidermolysis bullosa (JEB) and DEB. Additionally, Oleogel-S10 received approval from the US Food and Drug Administration in December 2023 and the UK Medicines and Healthcare Products Regulatory Agency in September 2022. A sterile gel for topical application, Oleogel-S10 contains 10% birch triterpenes and is made with sunflower oil. It works at several points during the wound-healing process, such as regulating inflammatory mediators, promoting keratinocyte migration, and stimulating differentiation.30
- Anti-inflammatory drugs: When traditional treatments for dystrophic epidermolysis bullosa (DEB) fail, subcutaneous infusion of G-CSF has demonstrated promise in encouraging wound healing. Interestingly, no negative side effects were noted.31
- Patients with generalized severe epidermolysis bullosa simplex (EBS) have shown great potential in responding well to topical diacerein, a prodrug which is metabolized to rhein. Clinical studies have demonstrated that it promotes healing and lessens blister development by blocking important inflammatory pathways and guarding against collagen degradation.32
- Antifibrotic Therapy: After balloon dilation (BD), the antifibrotic drug losartan was shown to be safe and effective in children with dystrophic epidermolysis bullosa (DEB). Its use was linked to improved nutritional status and a lower incidence of restenosis.33
Surgical Procedures, Considerations & Anesthesia
Surgical Procedures:
- Several surgical interventions/ procedures are undertaken and include the following:
- Esophageal dilatation
- Insertion of gastrostomy tube
- Surgery to manage contractures eg of hands
- Excision of skin cancers/amputation/regional lymph node dissection
- Insertion of central venous access
- Tracheostomy
N.B: Operating team must be informed about patient medical situation (EB) to prepare instruments in a special way.34
Surgical Considerations for People Living with EB
- Operating Room Preparation: Warm the room to prevent heat loss due to skin lesions and low body mass index. Extensively pad the operating table using foam padding or “egg crate” material to reduce trauma.34
- Positioning: To support strained joints, place your extremities in neutral positions with adequate padding. By setting up padded surfaces beforehand or permitting patient-assisted transfers, you can prevent repetitive motions.34
- Airway Management: Lubricate all airway equipment and use endotracheal tubes that are smaller than expected. For dental treatments, opt for nasal intubation; for elderly patients, take fiberoptic aid into consideration. Steer clear of oral airways since they could blister; deep extubate to avoid trauma; and utilize well-lubricated equipment.34
- Monitoring and Adhesives: Steer clear of adhesive ECG leads and use non-adhesive (clip-on) pulse oximeters; silicone-based substitutes are recommended. Adjust the inflation intervals and place a layer of padding beneath the blood pressure cuffs.34
- Eye Protection: Use methylcellulose-based lubricants without preservatives and moistened gauze to protect the eyes. Avoid petroleum-based products.34
- Intravenous Access: Utilize ultrasound for difficult IV access and secure lines with non-adhesive dressings or sutures.34
- Intraoperative Anesthesia: Induce anesthesia gently to prevent agitation and skin damage. Maintain euthermia (>35.5°C) and high FiO2 (>0.6) during procedures. Follow infection control protocols with timely antibiotic administration.34
- Regional Anesthesia: Consider regional anesthesia for procedures like hand releases to avoid airway management complications.34
- Anesthesia Procedures
- Unless the center agrees differently, all “severe” patients should have their surgeries performed at the EB center. However, in certain “mild” situations, people living with EB could be sent to nearby healthcare professionals for these operations along with guidance on how to help them during the process.
- Pulse oximetry is the ideal method for monitoring anesthesia in order to minimize skin friction. Because mucocutaneous blistering can happen, the average length of anesthesia should be fewer than two hours.22
Dental Procedures & Preventions
- Minimal Contact During Treatment: Apply lubricants to preserve intraoral soft tissues and commissures. Soft tissues should not be compressed; instead, use flat, bendable retractors and tiny cotton rolls lubricated for isolation. Steer clear of high vacuum suction to lower the risk of injuries.35
- Oral Hygiene Instructions: When it comes to everyday oral care, parents should help their children who have low manual dexterity. Make use of a gentle nylon toothbrush whose bristles have been pliable by warm water. Use mouthwash with 0.12% chlorhexidine and fluoride rinses without alcohol twice a day for two weeks every three months.35
- Preventive Dental Care: Schedule clinical appointments every three months for the topical application of fluoride varnish.35
Monitoring
- Patients with generalized forms of JEB and RDEB and in selected EB subsets should be monitored by serial DEXA scans for possible osteoporosis or osteopenia.26
- Basic investigations like hematological, renal and other diagnostic tests like (echocardiogram) should also be serially monitored or performed.26
Genetic Counseling for Parental Screening, Family Planning and Prenatal Diagnosis
Genetic counselors (GC) provide essential information regarding the inheritance pattern, risk assessment, prevention, early intervention and family planning, and further discuss diagnostic and prevention options.36,37 As an example, recommending Preimplantation Genetic Diagnosis (PGD), which provides pre-selection of the embryos free from EB mutations before implantation, reduces the risk of giving birth to an affected or a carrier newborn.36 The prenatal detection of pregnant mothers for EB can be done by prenatal diagnosis techniques like chorionic villus sampling (CVS) and amniocentesis.38,39 The success rate of prenatal detection is more than 98% making it reliable for decision making for families at risk.38 Parental Screening implicates family planning and provides better management through the early identification of genetic factors.40 The GC can also prepare and educate families on the potential outcome of having an affected child and the caring strategies.37
Prenatal Diagnosis
- Prenatal Testing and Safe Conception Options - Prenatal testing options include preimplantation genetic testing (PGT), chorionic villus sampling (CVS), and amniocentesis, depending on the pregnancy stage. The ability to diagnose prenatal conditions depends on identifying the mutations involved, which may require DNA banking for future testing. While prenatal diagnosis aids in making informed decisions, it is not always straightforward and may not be available in all centers.40
- Preimplantation Genetic Testing (PGT) and In Vitro Fertilization (IVF) - Safe conception options such as in vitro fertilization (IVF) combined with preimplantation genetic testing (PGT) help reduce the risk of transmitting genetic diseases. PGT is a specialized procedure performed during IVF to screen embryos for genetic mutations and chromosomal abnormalities before implantation. PGT includes:
- PGT-M (for monogenic disorders): Detects specific genetic mutations, such as those causing diseases like dystrophic epidermolysis bullosa (RDEB).
- PGT-A: Screens for chromosomal abnormalities to improve embryo selection.
- Challenges of PGT:
- Risk of false negatives or allele dropout.
- Technical limitations, such as insufficient DNA from single cells.
- Gene Identification and IVF - If a mutation is identified and classified as pathogenic, PGT-M is recommended. The primary doctor in medical genetics should evaluate the family and determine the specific genetic contribution, as multiple genes (and not just one) may be involved. Once the specific gene is identified, IVF becomes a feasible option. Therefore, it is important to test the affected family members for the causative gene in order to assess their eligibility for IVF and PGT-M. Overall, combining PGT-M for genetic mutations with PGT-A for chromosomal analysis optimizes outcomes in IVF procedures.41
- Why is PGT Important in IVF? PGT helps ensure the transfer of healthy embryos, reduces genetic disease transmission, and increases IVF success rates. This advanced screening technique enhances reproductive outcomes and provides a safer approach for couples at risk of passing on genetic disorders.41
- Referral and Counseling - Beyond diagnosis, families need support and access to specialized services. Genetic counselors should guide families to support groups and specialists (eg, wound care, physical therapy) as needed. Counseling involves more than explaining recurrence risks; it’s about offering long-term support and guidance throughout the patient’s life.42
Prevention Strategies
- Confirming the Diagnosis and Prenatal Diagnosis: Genetic testing is essential for diagnostic confirmation. A sample of the fetus’s chorionic villi or amniotic fluid can be examined to check for the familial mutation in order to make a prenatal diagnosis. If the variant is known to be pathogenic or likely to be pathogenic, this crucial testing should be carried out. A board-certified genetic counselor or geneticist should conduct a genetic counseling session with expectant mothers thinking about a prenatal diagnosis. A panel consisting of three experienced and qualified specialists who examine each case separately should decide on prenatal diagnosis and pregnancy termination (in accordance with the relevant Fatwa in Saudi Arabia).43 This decision must be taken only by trusted physicians specializing in Maternal and Fetal medicine, dermatology and genetics. To guarantee that results are obtained before 19 weeks, amniocentesis or chorionic villus sampling should be carried out early in pregnancy (134 days from the first day of the last menstrual period). Termination of pregnancy is prohibited if results are received after this period.
- Carrier Screening: Carrier screening for siblings and at-risk family members is essential for family planning. Premarital screening is an important component of this strategy.
- Consider carrier screening for an unaffected, unrelated partner based on individual circumstances and national regulations.
As shown in Figure 2
- If one parent is affected and passes the altered gene on to their child. There is a 50% chance that any child of theirs would be born with EB.44
- If both parents are unaffected but are carriers of EB, for any child of theirs to be born with EB, the child would have to inherit the disease-causing variant from both parents. There is a 25% chance of this occurring.44
- If a disease-causing variant happens spontaneously for the first time in a person. In this situation, having a second child or more with EB from the same parents is very rare. On the other hand, the person carrying a de novo variant has a 50% chance of passing it (and EB) on to their children.44
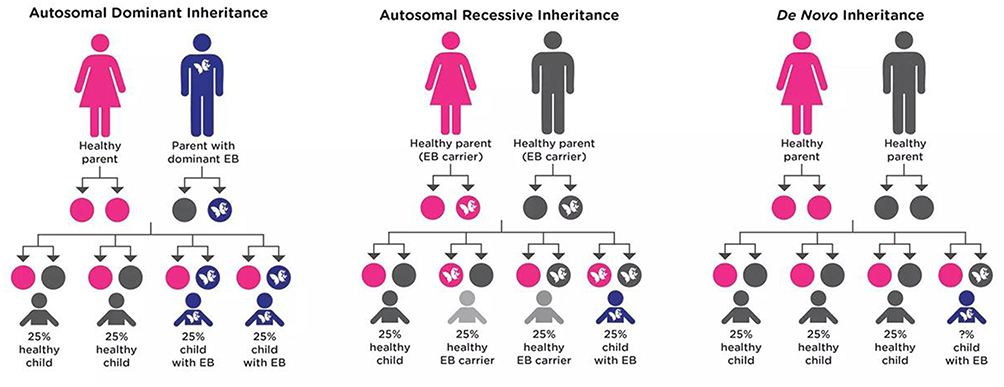 |
Figure 2 Inheritance pattern for autosomal recessive genes. |
Pregnancy Management
- Antenatal Care Monitoring: an interdisciplinary team of a midwife, dermatologist, obstetrician, and anesthetist must closely follow pregnant women with EB. Pregnancy appears to have little effect on the underlying skin problem, even with close observation.45
- Complications: compared to the general population, the majority of reported pregnancies do not have any increased prenatal problems. However, particularly in women with EB-related esophageal stenosis, there is a greater requirement for blood pressure monitoring and a risk of complications like anemia and gestational diabetes.45
- Blood Pressure: it is important to monitor blood pressure regularly with padded cuffs to prevent skin trauma. Cardiotocography should only be used when clinically necessary to avoid blistering.45
- Renal Function: renal function monitoring is recommended, particularly in those with dystrophic EB, due to risks of renal impairment.45
Labor and Delivery
- Delivery Mode: although vaginal birth is usually recommended, there is a chance of soft tissue injury and mucosal blisters. These dangers may rise with instrumental deliveries. At the site of the incision, caesarean sections may cause blisters and scarring.45
- Anesthesia Choices: while epidermolysis bullosa (EB) presents challenges for both general and regional anesthesia, these techniques can still be used in people living with EB.
- General Anesthesia: if necessary, general anesthesia should be carefully managed with precautions to avoid bullae formation, particularly around the face mask and intubation sites.
- Regional Anesthesia: this method is preferred, when possible, as it avoids airway complications. Careful inspection of the puncture site for infection is crucial before proceeding; nonetheless, there is a chance that bullae will form at the puncture site.
- Precautions: the delicate nature of EB skin should be explained to all surgical staff, and friction should be kept to a minimum to avoid the development of bullae. Avoid items that can cause friction, such as hard medical instruments and anything with an adhesive.45
Postnatal Care
- Complications: there are few reported postnatal complications for women with EB, and their hospital stays are typically similar to those of non-EB-affected patients. Compression stockings should be avoided due to the risk of skin trauma.45
- Breastfeeding: breastfeeding is possible, though nipple blistering is common. Women should be educated on proper positioning to avoid blister formation, and nipple shields should be used to reduce the risk of further damage.45
- Future Considerations:
- Genetic Counseling: women with EB should receive clinical genetics counseling before pregnancy to understand the risks of passing on EB. Prenatal diagnosis via chorionic villus sampling and preimplantation genetic diagnosis can offer more options for families.
- Long-Term Follow-Up: neonates born to women with EB, especially those affected by the condition, should receive long-term dermatological follow-up to manage their skin issues.45
Genetic Counseling
The teams are required to coordinate with appropriate genetic services in line with the shared care model. Patients should be referred to genetic services to get the appropriate support.
- Role of Genetic Counseling - Medical genetics specialists, such as doctors, geneticists, or dermatologists with training, frequently offer genetic counseling in EB. Because of their proficiency in EB diagnosis and treatment, dermatologists are often involved. A referral to a medical genetics’ clinic is advised for individuals who are unable to devote the necessary time to this laborious endeavor. The patient’s family is the main focus of counseling, which recognizes the possible risks to other family members.42
- Core Components of Genetic Counseling - Effective genetic counseling involves:
- Accurate Diagnosis: this forms the basis for all further data, such as molecular testing and recurrence risks. Determining the mechanism of inheritance, directing molecular testing, and comprehending prognosis all depend on an accurate diagnosis. Ineffective testing and inaccurate recurrence risk estimates result from misdiagnosis. Clinical judgment and molecular testing are used to make the diagnosis, which validates rather than proves it. The necessity for accuracy is highlighted by the difficulties associated with allelic and locus heterogeneity, as well as the limitations of molecular testing. After the diagnosis is verified, genetic counseling starts, providing families with clarity and comfort.40,42
- Natural History and prognosis: knowing how EB develops is crucial, but it should be explained gradually, taking into consideration the family’s capacity for information intake. Setting reasonable expectations is essential. Counselors must strike a balance between providing information and recognizing the wide range of ways that diseases manifest. Risks of respiratory arrest and cancer surveillance may be discussed in cases that are more severe, like recessive dystrophic EB. Counselors ought to address families’ worries and offer advice on handling, skincare, and difficulties while adjusting information to the family’s preparedness and needs.40,42
- Treatment Options: counselors must provide information on management, potential interventions, and experimental therapies.
- Mode of Inheritance: clarity on how EB is inherited helps families understand recurrence risks.
- Recurrence Risks and Prenatal Diagnosis: accurate genetic testing and counseling can guide family planning.
- Referrals: Families may require additional resources or specialists for comprehensive care.
- Counseling Approach: Genetic counseling needs to be patient-centered, adaptable, and nonjudgmental. In order to answer inquiries, offering assistance, and reiterate important facts, it takes several sessions. Families require information on gene therapy, experimental trials, and current research as well as advice on how to manage EB. Coordination between intensivists, geneticists, and dermatologists guarantees comprehensive treatment.42
Psychosocial Considerations and Mental Health
Psychosocial Impact of EB - EB affects individuals’ emotional well-being, body image, and social interactions. The condition, being both rare and disfiguring, may lead to isolation, which can result in psychological distress. The visible nature of the disease often leads to discrimination, which intensifies the mental and emotional burden.46
Psychosocial Support for Patients with EB
- Access to Expertise and Training - Healthcare professionals must receive adequate training and support to manage the complexities of EB. Although most studies focus on children, there is a need for broader training for all age groups. While online platforms can be helpful for information, physical medical examination by specialists remains crucial.47
- Collaborative Patient-Professional Relationships - EB care should be a partnership between patients and healthcare professionals. Patients, especially adults, often see themselves as experts in managing their condition. Professionals must encourage self-efficacy while ensuring patients have access to expert care when needed. Training in managing assertive and disengaged patients is essential.47
- Support for Healthcare Professionals - Clinicians should have access to clinical supervision and team support to address the emotional and psychological challenges of working with EB. Working in isolation can harm their mental health, so open discussions and shared support systems are necessary to maintain their well-being.47
Psychologists as Part of the IDT
- Psychological and Social Support: Psychological support is integrated into the interdisciplinary team to address the emotional and mental well-being of the patient and family, recognizing that chronic diseases like EB can have significant psychological impacts. This team-based approach creates a compassionate environment where patients and their families feel supported, helping to alleviate anxiety and improve overall quality of life.48
- Education and Empowerment: Therapeutic patient education, a core aspect of interdisciplinary care, empowers patients and their families to take charge of disease management. Training families and caregivers in managing wounds, recognizing early signs of complications, and performing necessary interventions is a fundamental component of this approach. This continuous education, as part of the team’s role, ensures better disease control, reducing complications and hospital visits, and improving patient outcomes.48
- Family support for families of people living with EB
- Impact on Families: Parents, siblings, and family members often take on caregiving roles, which require immense emotional, physical, and psychological effort. Families may struggle with the stress and burden of managing the disease, which can be compounded by diagnostic delays, lack of specialist centers, and the complexities of disease management. Education for families about the disease process, genetic counseling, and potential complications is crucial, as is emotional support, including access to therapy or palliative care teams.49
- Siblings’ Challenges: Siblings of children with EB also face unique challenges, such as feeling neglected due to the focus on the ill child, missing out on social activities, and coping with societal stigma and teasing. These children may experience emotional distress and difficulties with school performance. Awareness of the sibling’s experience and providing them with support through therapy or support groups is important for their well-being.49
- Coping and resilience strategies:
- Coping Strategies in Children
- Acceptance: Children who are able to accept their condition, such as recognizing that their illness is part of their life, tend to have better emotional and social functioning. Acceptance helps them adapt to the reality of their disease and reduces emotional distress, allowing them to cope more effectively with the physical challenges of EB.50
- Distancing: Distancing involves emotionally detaching from the illness, which can help children manage feelings of fear, sadness, or frustration. Children who use distancing as a strategy report better overall functioning in terms of their emotional and social well-being.50
- Emotional Reactions: Children who frequently express emotional reactions, such as anger, sadness, or the feeling that their illness is unfair, tend to experience poorer emotional and social functioning. Emotional reactions, while natural, can exacerbate the negative impact of the illness and make coping more difficult.50
- Wishful Thinking: Some children engage in wishful thinking, hoping for their disease to disappear or improve despite the chronic nature of EB. This can provide temporary comfort, but it does not necessarily improve long-term coping and may lead to feelings of anxiety or sadness.50
- Cognitive-Palliative Strategies: These strategies involve trying to mentally “cope” with the disease by using distractions or reframing the situation. However, these strategies were found to be less frequently associated with improved quality of life for children with EB in this study.50
- Coping Strategies in Parents
- Acceptance: Parents who accept the chronic nature of their child’s illness tend to have better physical functioning, as they are more likely to engage in daily care routines without excessive emotional strain. However, frequent acceptance in parents was also linked to lower social functioning, indicating that emotional detachment may affect social interactions and support networks.50
- Emotional Reactions: Parents who frequently express emotional reactions, such as crying or feelings of helplessness, tend to have lower quality of life, especially in terms of psychosocial well-being. Emotional reactions can contribute to increased stress, which in turn negatively impacts both the parent and the child’s quality of life.50
- Avoidance: Interestingly, parents tend to avoid using avoidance strategies as much as children. This may be because, as primary caregivers, they cannot ignore their child’s needs. However, avoidance by children was found to be linked to a lower level of anger among parents, suggesting that children who detach from the illness might reduce parental emotional strain.50
- Coping Strategies in Children
Conclusion
To sum up, the National Saudi Policy and Protocol for Epidermolysis Bullosa (EB) is an important step in improving the treatment, care, and management of those who suffer from this uncommon and complicated illness in Saudi Arabia. This procedure guarantees that medical practitioners across can provide consistent, high-quality care for patients with EB by offering a thorough framework founded on the best available scientific data, expert input, and ethical criteria.Enhancing treatment protocols, improving early diagnosis, and providing patients and their families with long-term support are the goals of this guideline. It demonstrates Saudi Arabia’s dedication to improving medical procedures and fostering the welfare of those with long-term illnesses like epidermolysis bullosa.
We realize that in order to continually enhance the care practices outlined in this work, more research, cooperation, and teaching are required. In addition to being a useful tool for medical professionals, we believe that these guidelines will promote additional developments in the knowledge and treatment of EB, which will ultimately improve the lives of impacted patients and their families.
Declaration
In order to guarantee the best possible treatment and management of patients with epidermolysis bullosa (EB) in Saudi Arabia, we thus announce that the National Saudi Policy and Protocol for EB was created in compliance with the best available scientific findings and expert consensus. In order to guarantee consistency in medical practice and enhance patient outcomes, this policy and protocol seek to provide standardized recommendations for the diagnosis, treatment, and long-term care of those impacted by EB. A careful review of national and international recommendations, professional consultations, and feedback from major stakeholders in the healthcare and research sectors all helped to produce this strategy. We declare that the entire process complies with the highest standards of medical practice and that all pertinent ethical issues have been taken into consideration.
Abbreviations
BD, Balloon Dilation; B-VEC, Beremagene Geperpavec; COL7A1, Collagen Type VII Alpha 1 Chain Gene; CVS, Chorionic Villus Sampling; DEB, Dystrophic Epidermolysis Bullosa; DEXA, Dual-Energy X-ray Absorptiometry (DEXA) Scan; EB, Epidermolysis Bullosa; EBS, EB Simplex; ENT, Ear, Nose, and Throat; FDA, Food and Drug Administration; GC, Genetic Counselors; GCSF, Granulocyte Colony-Stimulating Factor Injections; HCT, Hematopoietic Stem Cell Transplantation; HSCs, Hematopoietic Stem Cells; IFM, Immunofluorescence Mapping; IV, Intravenous; IVF, In Vitro Fertilization; JEB, Junctional EB; KEB, Kindler EB; IDT, Interdisciplinary Team; MSCs, Mesenchymal Stem Cells, NGS, Next-Generation Sequencing; NSAIDs Non-Steroidal Anti-Inflammatory Drugs; PGD, Preimplantation Genetic Diagnosis; RDEB, Recessive Dystrophic Epidermolysis Bullosa; SCC, Squamous Cell Carcinoma; SFDA, Saudi Food and Drug Administration; SS, Sanger Sequencing; VUS, Variant of Uncertain Significance; WES, Whole-Exome Sequencing.
Acknowledgment
The authors would like to express their sincere gratitude to Riyadh Home Company and its dedicated team members for their support in medical writing. Their expertise and dedication were invaluable in the successful completion of this manuscript.
Our deepest appreciation goes to Dr. Khaled Mohajer, Dr. Mohammed Alajlan, Dr. Mishal Almebayadh, Dr. Lulu Almubarak, Dr. Khalid Alattas, and Dr. Maha Alsahli for their invaluable expertise, dedication, and collaborative efforts throughout this project. Their commitment to evidence-based practice has been instrumental in shaping this guideline.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design and execution or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Fine JD, Bruckner-Tuderman L, Eady RA, et al. Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Academy Dermatol. 2014;70(6):1103–1126. doi:10.1016/j.jaad.2014.01.903
2. Fine JD, Eady RA, Bauer EA, et al. The classification of inherited epidermolysis bullosa (EB): report of the third international consensus meeting on diagnosis and classification of EB. J Am Acad Dermatol. 2008;58(6):931–950. doi:10.1016/j.jaad.2008.02.004
3. Has C, Bauer JW, Bodemer C, et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br J Dermatol. 2020;183(4):614–627. doi:10.1111/bjd.18921
4. Korte EWH, Baardman R, Pool G, Duipmans JC, van den Akker PC, Bolling MC. Navigating the diagnostic journey of epidermolysis bullosa: a qualitative study of the lived experiences and needs of parents and patients. British J Dermatol. 2024;191(5):737–745. doi:10.1093/bjd/ljae242
5. Fine JD. Epidemiology of inherited epidermolysis bullosa based on incidence and prevalence estimates from the national epidermolysis bullosa registry. JAMA Dermatol. 2016;152(11):1231–1238. doi:10.1001/jamadermatol.2016.2473
6. Shehata NA, Shaik NA, Irfan Thalib H. Genetic implications and management of epidermolysis bullosa in the Saudi Arabian population. Cureus. 2024. doi:10.7759/cureus.66678
7. Fine JD, Mellerio JE. Extracutaneous manifestations and complications of inherited epidermolysis bullosa. J Am Acad Dermatol. 2009;61(3):367–384. doi:10.1016/j.jaad.2009.03.052
8. Kotalevskaya YY, Stepanov VA. Molecular genetic basis of epidermolysis bullosa. Vavilovskii Zhurnal Genet Selektsii. 2023;27(1):18–27. doi:10.18699/VJGB-23-04
9. Hou PC, Del Agua N, Lwin SM, Hsu CK, McGrath JA. Innovations in the treatment of dystrophic epidermolysis bullosa (DEB): current landscape and prospects. Ther Clin Risk Manag. 2023;19:455–473. doi:10.2147/TCRM.S386923
10. Hou PC, Wang HT, Abhee S, Tu WT, McGrath JA, Hsu CK. Investigational treatments for epidermolysis bullosa. Am J Clin Dermatol. 2021;22(6):801–817. doi:10.1007/s40257-021-00626-3
11. Guide SV, Gonzalez ME, Bağcı IS, et al. Trial of beremagene geperpavec (B-VEC) for dystrophic epidermolysis bullosa. New England Journal of Medicine. 2022;387(24):2211–2219. doi:10.1056/NEJMoa2206663
12. Khan A, Riaz R, Ashraf S, Akilimali A. Revolutionary breakthrough: FDA approves Vyjuvek, the first topical gene therapy for dystrophic epidermolysis bullosa. Ann Med Surg. 2023;85(12):6298–6301. doi:10.1097/MS9.0000000000001422
13. Raymakers AJN, Kesselheim AS, Mostaghimi A, Feldman WB. Estimated spending on beremagene geperpavec for dystrophic epidermolysis bullosa. JAMA Dermatol. 2024;160(3):297–302. doi:10.1001/jamadermatol.2023.5857
14. El Hachem M, Zambruno G, Bourdon-Lanoy E, et al. Multicentre consensus recommendations for skin care in inherited epidermolysis bullosa. Orphanet J Rare Dis. 2014;9(1):76. doi:10.1186/1750-1172-9-76
15. Has C, Liu L, Bolling MC, et al. Clinical practice guidelines for laboratory diagnosis of epidermolysis bullosa. Br J Dermatol. 2020;182(3):574–592. doi:10.1111/bjd.18128
16. Badger KS, O’Haver J, Price H. Recommendations for a comprehensive management plan for the child diagnosed with epidermolysis bullosa. J Dermatol Nurses Assoc. 2013;5(2):72–78. doi:10.1097/JDN.0b013e31828866fa
17. Epidermolysis Bullosa Service (all ages). Available from: https://www.england.nhs.uk/wp-content/uploads/2018/08/Epidermolysis-bullosa-service-all-ages.pdf.
18. Skin and wound care guidance for adults with eb and their carers, DEBRA International. Available at: https://www.debra-international.org/skin-and-wound-care-in-eb.
19. Goldschneider KR, Good J, Harrop E, et al. Pain care for patients with epidermolysis bullosa: best care practice guidelines. BMC Med. 2014;12(1):178. doi:10.1186/s12916-014-0178-2
20. Section of Dermatology at the Hospital for Sick Children with the support of DEBRA Canad. Epidermolysis bullosa a handbook for EB patients and families.
21. Alotaibi SM, Zahlan A, AlAteeq M, et al. Supraglottic laryngeal manifestation of epidermolysis bullosa in a pediatric population: a literature review with four case reports. Otolaryngol Case Rep. 2023;27:100516. doi:10.1016/j.xocr.2023.100516
22. Mittal BM, Goodnough CL, Bushell E, Turkmani-Bazzi S, Sheppard K. Anesthetic management of adults with epidermolysis bullosa. Anesthesia Analgesia. 2022;134(1):90–101. doi:10.1213/ANE.0000000000005706
23. Lyos AT, Malpica A, Levy ML, Sulek M. Laryngeal involvement in epidermolysis bullosa. Ann Otol Rhinol Laryngol. 1994;103(7):542–546. doi:10.1177/000348949410300707
24. Chan JM, Weisman A, King A, et al. Occupational therapy for epidermolysis bullosa: clinical practice guidelines. Orphanet J Rare Dis. 2019;14(1):129. doi:10.1186/s13023-019-1059-8
25. Lembo F, Parisi D, Cecchino LR, Ciancio F, Innocenti A, Portincasa A. Release of pseudosyndactyly in recessive dystrophic epidermolysis bullosa using a dermal regeneration template glove: the Foggia experience. Orphanet J Rare Dis. 2021;16(1):52. doi:10.1186/s13023-021-01697-5
26. Fine JD. Inherited epidermolysis bullosa. Orphanet J Rare Dis. 2010;5(1):12. doi:10.1186/1750-1172-5-12
27. Chung HJ, Uitto J. Type VII collagen: the anchoring fibril protein at fault in dystrophic epidermolysis bullosa. Dermatol Clin. 2010;28(1):93–105. doi:10.1016/j.det.2009.10.011
28. Agustin M, Mahadewi A, Danarti R. Bone marrow transplantation and bone marrow-derived mesenchymal stem cell therapy in epidermolysis bullosa: a systematic review. Pediatr Dermatol. 2024;41(4):599–605. doi:10.1111/pde.15591
29. Vanden Oever M, Muldoon D, Mathews W, Tolar J. Fludarabine modulates expression of type VII collagen during haematopoietic stem cell transplantation for recessive dystrophic epidermolysis bullosa*. British J Dermatol. 2021;185(2):380–390. doi:10.1111/bjd.19757
30. Torres Pradilla M, Álvarez E, Novoa M, Lozano I, Trujillo M. Oleogel-S10 in dystrophic epidermolysis bullosa: a case series evaluating the impact on wound burden over two years. Adv Ther. 2024;41(2):867–877. doi:10.1007/s12325-023-02749-x
31. Fine JD, Manes B, Frangoul H. Systemic granulocyte colony-stimulating factor (G-CSF) enhances wound healing in dystrophic epidermolysis bullosa (DEB): results of a pilot trial. J Am Acad Dermatol. 2015;73(1):56–61. doi:10.1016/j.jaad.2015.04.015
32. Limmer AL, Nwannunu CE, Shah R, et al. Topical diacerein ointment for epidermolysis bullosa simplex: a review. Skin Therapy Lett. 2019;24(3):7–9.
33. Oldakovskiy V, Murashkin N, Lokhmatov M, et al. Our experience of using Losartan for esophageal stenosis in children with dystrophic form of congenital epidermolysis bullosa. J Pediatr Surg. 2023;58(4):619–623. doi:10.1016/j.jpedsurg.2022.11.001
34. Goldschneider K, Lucky AW, Mellerio JE, Palisson F, Del Carmen Viñuela Miranda M, Azizkhan RG. Perioperative care of patients with epidermolysis bullosa: proceedings of the 5th international symposium on epidermolysis bullosa. Pediatric Anesthesia. 2010;20(9):797–804. doi:10.1111/j.1460-9592.2010.03359.x
35. Dağ C, Bezgin T, Özalp N. Dental management of patients with epidermolysis bullosa. Oral Health Dent Manag. 2014;13(3):623–627.
36. Izmiryan A, Hovnanian A. Prenatal Diagnosis of Epidermolysis Bullosa: Current Aspects and Perspectives in Blistering Diseases. Berlin, Heidelberg: Springer; 2015:239–248.
37. Sánchez-Jimeno C, Escámez MJ, Ayuso C, Trujillo-Tiebas MJ, Del Río M. Del Río M, en representación de la Cátedra de la Fundación Jiménez Díaz de Medicina Regenerativa y Bioingeniería Tisular DE y de otros profesionales sanitarios. Genetic diagnosis of epidermolysis bullosa: recommendations from an expert Spanish research group. Actas Dermosifiliogr. 2018;109(2):104–122. doi:10.1016/j.ad.2017.08.008
38. Pfendner EG, Nakano A, Pulkkinen L, Christiano AM, Uitto J. Prenatal diagnosis for epidermolysis bullosa: a study of 144 consecutive pregnancies at risk. Prenat Diagn. 2003;23(6):447–456. doi:10.1002/pd.619
39. Christiano AM, Pulkkinen L, Mcgrath JA, Uitto J. Mutation-based prenatal diagnosis of herlitz junctional epidermolysis bullosa. Prenat Diagn. 1997;17(4):343–354. doi:10.1002/(SICI)1097-0223(199704)17:4<343::AID-PD73>3.0.CO;2-7
40. Sybert VP, Holbrook KA. Prenatal diagnosis and genetic screening for epidermolysis bullosa. In: Epidermolysis Bullosa. New York: Springer New York; 1992:235–251.
41. Trieutien S, Vu Van T, Tran Ngoc Thao M, et al. Preimplantation genetic diagnosis for DEB by detecting a novel family-specific COL7A1 mutation in Vietnam. Appl Clin Genet. 2021;14:467–472. doi:10.2147/TACG.S344107
42. Sybert VP. Genetic counseling in epidermolysis bullosa. Dermatol Clin. 2010;28(2):239–243. doi:10.1016/j.det.2009.12.004
43. Fatwa of the senior scholars – fatwa on abortion – 1432 hijri.
44. Genetics of EB. DEBRA international. [cited March 19, 2025]. Available from: https://www.debra-international.org/genetics-of-eb#:~:text=One%20parent%20is%20affected%20and,causing%20variant%20from%20both%20parents.
45. Bolt LA, O’Sullivan G, Rajasingham D, Shennan A. A review of the obstetric management of patients with epidermolysis bullosa. Obstet Med. 2010;3(3):101–105. doi:10.1258/om.2010.100009
46. Dures E, Morris M, Gleeson K, Rumsey N. The psychosocial impact of epidermolysis bullosa. Qual Health Res. 2011;21(6):771–782. doi:10.1177/1049732311400431
47. Martin K, Geuens S, Asche JK, et al. Psychosocial recommendations for the care of children and adults with epidermolysis bullosa and their family: evidence-based guidelines. Orphanet J Rare Dis. 2019;14(1):133. doi:10.1186/s13023-019-1086-5
48. Retrosi C, Diociaiuti A, De Ranieri C, et al. Multidisciplinary care for patients with epidermolysis bullosa from birth to adolescence: experience of one Italian reference center. Ital J Pediatr. 2022;48(1):58. doi:10.1186/s13052-022-01252-3
49. Chateau AV, Blackbeard D, Aldous C. The impact of epidermolysis bullosa on the family and healthcare practitioners: a scoping review. Int J Dermatol. 2023;62(4):459–475. doi:10.1111/ijd.16197
50. Mauritz PJ, Bolling M, Duipmans JC, Hagedoorn M. The relationship between quality of life and coping strategies of children with EB and their parents. Orphanet J Rare Dis. 2021;16(1):53. doi:10.1186/s13023-021-01702-x
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.