Back to Journals » Neuropsychiatric Disease and Treatment » Volume 10
The safety and effectiveness of open-label extended-release carbamazepine in the treatment of children and adolescents with bipolar I disorder suffering from a manic or mixed episode
Authors Findling R ![]() , Ginsberg L
, Ginsberg L
Received 6 June 2014
Accepted for publication 21 July 2014
Published 27 August 2014 Volume 2014:10 Pages 1589—1597
DOI https://doi.org/10.2147/NDT.S68951
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Robert L Findling,1,2 Lawrence D Ginsberg3
1Division of Child and Adolescent Psychiatry, Department of Psychiatry and Behavioral Sciences, Johns Hopkins University, 2Kennedy Krieger Institute, Baltimore, MD, USA; 3Red Oak Psychiatry Associates, PA, Houston, TX, USA
Objective: To assess the safety and effectiveness of open-label treatment with extended-release carbamazepine (ERC) in pediatric subjects suffering from bipolar I disorder.
Method: Medically healthy youths aged 10–17 years suffering from an acute manic or mixed episode were eligible. After screening for study eligibility, the youths began a 5-week titration period in which doses of ERC were adjusted in order to optimize benefit whilst minimizing adverse events, at doses between 200–1,200 mg/day. Thereafter, subjects could continue to receive treatment during a subsequent 21-week period. Safety measures included spontaneously reported adverse events (AEs) and laboratory assessments. The primary efficacy measure was the Young Mania Rating Scale (YMRS).
Results: A total of 60 children (ages 10–12) and 97 adolescents (ages 13–17), with an overall average age of 13.4 years (standard deviation [SD] 2.0 years) received ERC. The mean duration of study participation was 109.6 days (SD 70.2 days), with 66 (42%) completing the entire study. At end of study participation (end point), the most prevalent dose of ERC was 1,200 mg: 31.7% of children and 24.7% of adolescents reached the 1,200 mg dose. The YMRS decreased from a mean of 28.6 (SD 6.2) at baseline to a mean of 13.8 (SD 9.4) (P<0.0001) at end point. A total of 26 subjects discontinued study participation because of AEs, the most common of which were rash (n=6), white blood cell count decreased (n=5), nausea (n=3), and vomiting (n=3). No deaths were reported. The most commonly reported AEs were headache (n=41), somnolence (n=30), nausea (n=22), dizziness (n=21), and fatigue (n=19).
Conclusions: Open-label administration of ERC might be a safe and effective intervention in this subject population. More definitive studies are warranted.
Keywords: bipolar disorder, children, adolescents, treatment, carbamazepine
Introduction
Safe and effective treatments are needed for pediatric bipolar disorder (BD) as pediatric BD is a serious and potentially lethal condition.1–3 Presently, lithium, a benchmark treatment for bipolar illness in adults, is approved by the US Food and Drug Administration (FDA) for use in youths suffering from bipolarity, primarily based upon studies in adults.4 More recently, risperidone, olanzapine, quetiapine, and aripiprazole have received regulatory approval for use in youths, based on results of multisite clinical trials.5–8
An extended-release formulation of carbamazepine (ERC) (Equetro®) is currently FDA-approved for the treatment of BD in adults but not in adolescents or children. Although clinical trials have shown that ERC has efficacy in adults with BD,9,10 there are very little data pertaining to the use of carbamazepine in youths. What data exist are generally limited to case series and modestly sized, single-site, open-label trials.11–14
In 2006, the pharmaceutical company Shire Development, Inc., who owned the compound at that time, initiated the present study in youths aged 10–17 years old with bipolar I disorder (BD-1) in a manic or mixed state, in order to examine whether or not ERC might have a role in the treatment of BD in youths. This multisite, open-label clinical trial was conducted in order to determine whether appropriately powered, definitive efficacy studies should be undertaken. In 2007, Validus Pharmaceuticals LLC (the current sponsor) purchased Equetro from Shire, gathered and organized the data, and contracted for the statistical analysis of the newly centralized data. We report on the results of this study.
Method
This study was conducted at 30 sites in the United States, Mexico, and Argentina, from July 2006 to October 2007. The subject’s parent or legally authorized representative provided written, informed consent, and the subjects gave assent before any study-related procedures were performed.
Participants
Medically healthy outpatient children (aged 10–12) and adolescents (aged 13–17) meeting unmodified diagnostic symptom criteria15 for BD-1 in a mixed or manic state were eligible. The diagnosis was confirmed using the Schedule for Affective Disorders and Schizophrenia for School-Age Children – Present and Lifetime Version (K-SADS-PL).16 In order to be eligible, youths were required to: 1) have suffered from affective symptoms for at least 2 months at study entry; 2) have a Young Mania Rating Scale (YMRS)17 score of ≥16 and 3) have a Clinical Global Impressions – Severity (CGI-S)18 scale score of ≥4 (indicative of at least moderate severity).
Exclusion criteria included any comorbid psychiatric or substance abuse disorders except attention-deficit/hyperactivity disorder (ADHD) or mild–moderate oppositional defiant disorder. Patients with a general medical condition that could reduce subject safety, such as an active cardiovascular disorder, hepatic disease, thyroid dysfunction, seizure disorder, unstable asthma, a history of aplastic anemia, or bone marrow depression were also excluded. Patients with a history of hypersensitivity or intolerance to carbamazepine were not enrolled. Females who were pregnant, lactating, or less than 6 months postpartum, or who could not be relied upon to use adequate birth control, were excluded. Youths who had failed prior treatment with carbamazepine or oxcarbazepine, or who had a 50% or greater decrease in their YMRS scores from the screening to the baseline visit were also excluded. Subjects who had a history of a past suicide attempt requiring medical intervention or those who, in the treating Investigator’s opinion, might not safely participate as outpatients, owing to the potential of harming themselves or others, were not eligible.
Study periods
Screening visit
During the screening visit, which was to occur from 14 to 3 days before baseline, a psychiatric and general medical history was obtained, and a physical examination was conducted. In addition, KSADS-PL, CGI-S, and YMRS were administered to confirm study eligibility. In order to assess depressive symptomatology, a Children’s Depression Rating Scale-Revised (CDRS-R)19 was also performed. In addition, physical assessments and laboratory tests (described below), as well as a 12-lead electrocardiogram (ECG), were obtained.
Subjects were permitted to take lorazepam, up to 2 mg/day as needed, during screening and during weeks 1 and 2 of the titration period and up to 1 mg/day during weeks 3, 4, and 5 of the titration period. Hydroxyzine was permitted, up to 50 mg/day, as needed, for screening and for weeks 1 and 2 of the titration period and up to 25 mg/day for weeks 3, 4, and 5 of the titration period. A minimum of 8 hours was required between the last dose of lorazepam or hydroxyzine, and the administration of the efficacy scales. These adjunctive drugs were not permitted during the maintenance period.
During the study, nonpsychopharmacologic drugs with psychotropic effects were permitted if they were well established prestudy (4 weeks). Nonpsychopharmacologic drugs without psychotropic effects were permitted. However, the use of investigational drugs, antidepressants, lithium, psychostimulants, atomoxetine, and other medications or herbal preparations used to treat psychiatric disorders were not allowed during study participation as treatment with these agents could have interfered with the interpretation of study results.
Titration and maintenance periods
Subjects were evaluated seven times during the initial 5-week titration period – at the beginning, middle, and end of the first week, and at the end of weeks 2, 3, 4, and 5. The subsequent maintenance period was 21 weeks long, with the first visit occurring 2 months after the initiation of ERC and subsequent visits occurring at monthly intervals. Finally, there was a 30-day follow-up period after the last dose of ERC was administered so that information about any new or ongoing adverse events (AEs) or serious adverse events (SAEs) could be collected.
Medication dosing
Subjects had ERC initiated at a dose of 200 mg/day in the evening. Doses could then be titrated upward in 200 mg/day increments, with the ERC given in twice-daily divided doses. If the subject remained symptomatic, subsequent dose increases, up to a maximum daily dose of 1,200 mg/day, was permitted every 3 to 7 days. If necessary, in order to improve tolerability, the treating Investigator was permitted to decrease a subject’s ERC dose by 200 mg. If a subject’s ERC was reduced, retitration upward was permitted. Ideally, dose adjustments were to occur at a scheduled visit. However, if dose adjustments were required between visits, these were done after either a telephone call or an additional office-based consultation with a study physician. Subjects were permitted to be treated with lorazepam or hydroxyzine, for anxiety during this period.
Dose adjustments were permitted during the maintenance period. In addition, starting at the first visit of the maintenance period, if the subject, in the opinion of the treating Investigator, had a partial response to ERC yet required additional treatment, a concomitant medication (lithium, valproate, or an atypical antipsychotic [aripiprazole, olanzapine, quetiapine, risperidone, or ziprasidone])20 could be initiated. Permissible concomitant medications were selected on the basis of the extant data available when this study was initiated.
Assessments
Efficacy
The primary outcome measure was the YMRS, an eleven-item scale that assesses symptoms of mania, with scores ranging from 0–60. Secondary efficacy measures included the CDRS-R, the CGI-S, and the CGI – Improvement (CGI-I)18 scale. All of these scales are clinician-completed. The CDRS-R is a 17-item instrument, with scores that range between 17 and 113, that measures symptoms of depression. Both CGI scales consist of one item and are scored from 1–7. On the YMRS, the CDRS-R, and the CGI-S, higher scores are associated with greater degrees of symptomatology. The CGI-I was administered after initiation of treatment. Subjects who are assigned a score of “1” are considered very much improved when compared with the baseline time point, and those who are “much improved” are given a score of “2”.
The YMRS and the CGI-S were administered at screening, at baseline, and at each postbaseline visit. The CDRS-R was given at screening, baseline, at 2 weeks, at 2 and 4 months, and at end of study (EOS). The CGI-I was administered at every visit after baseline.
Safety
AEs were assessed with open-ended queries at each study visit. In addition, vital signs were obtained at each study visit. Weight was measured at baseline, at week 5, and at every subsequent visit during the study. Height was measured at baseline, at month 3, and at EOS. Laboratory tests (hematology, chemistry, and urinalysis) were done at screening, at 2 and 5 weeks during titration, at 2 and 4 months during maintenance treatment, and at EOS. An ECG was obtained at screening, at week 5 and at 3 months of therapy, and at EOS. A physical examination was completed at screening and EOS. A serum pregnancy test was completed at screening, and subsequent urine pregnancy tests were completed at baseline, at week 4, and at every visit during the maintenance period (months 2, 3, 4, 5, and 6 [EOS]).
Statistical analyses
Descriptive statistics were used to summarize the YMRS total score, the CDRS-R total score, and the CGI-S score at each postbaseline visit and at end point, by subject age group (children vs adolescents), as well as by the intent-to-treat (ITT) population, using the mean, standard deviation (SD), median, minimum, and maximum of the distribution. The ITT population was defined as all subjects who had a baseline primary efficacy measurement (YMRS), received at least one dose of ERC, and had at least one postbaseline YMRS score recorded. Differences in scores at baseline and at subsequent time points for efficacy and safety parameters were analyzed by means of one-sample t-tests.
The number and percent of the following types of responses were summarized at each postbaseline visit and at end point, by subject age group and by the ITT population: 1) “treatment responder”, defined as YMRS total score decreased by at least 50% from the baseline total score; and, 2) “remission”, defined as YMRS score ≤12 at a given postbaseline visit.
Safety parameters were analyzed for the safety population, defined as all subjects who received study medication. Treatment-emergent AEs (TEAEs) were tabulated. Other safety parameters were summarized for the safety population and for each subject age group. Reference ranges supplied by the laboratory at the clinical sites or by a central laboratory were used by the investigators to classify the results as below, within, or above the normal range and to assess the results for clinical significance and/or out-of-range pathological changes. The treating Investigator also determined the clinical significance of the ECG results. Outlier criteria for laboratory parameters, vital signs, and ECGs were identified in the Statistical Analysis Plan for the study.
Although the planned enrollment for the study was 225 subjects so as to achieve 75 completed subjects, the difficulty of enrolling young subjects meeting the required diagnostic criteria for a study duration of 6 months prevented the anticipated enrollment from being achieved.
Results
Subjects
There were 161 eligible subjects enrolled in this study. Four discontinued before receiving study medication. Two of the 157 treated subjects did not have a postbaseline YMRS assessment; therefore, they were not included in the efficacy analyses (but all 157 were included in the safety analyses). Subject demographics and disposition are summarized in Table 1. Overall, 66 of the 157 (42%) subjects completed the study. The overall mean duration of treatment was 109.6 days (SD 70.2 days).
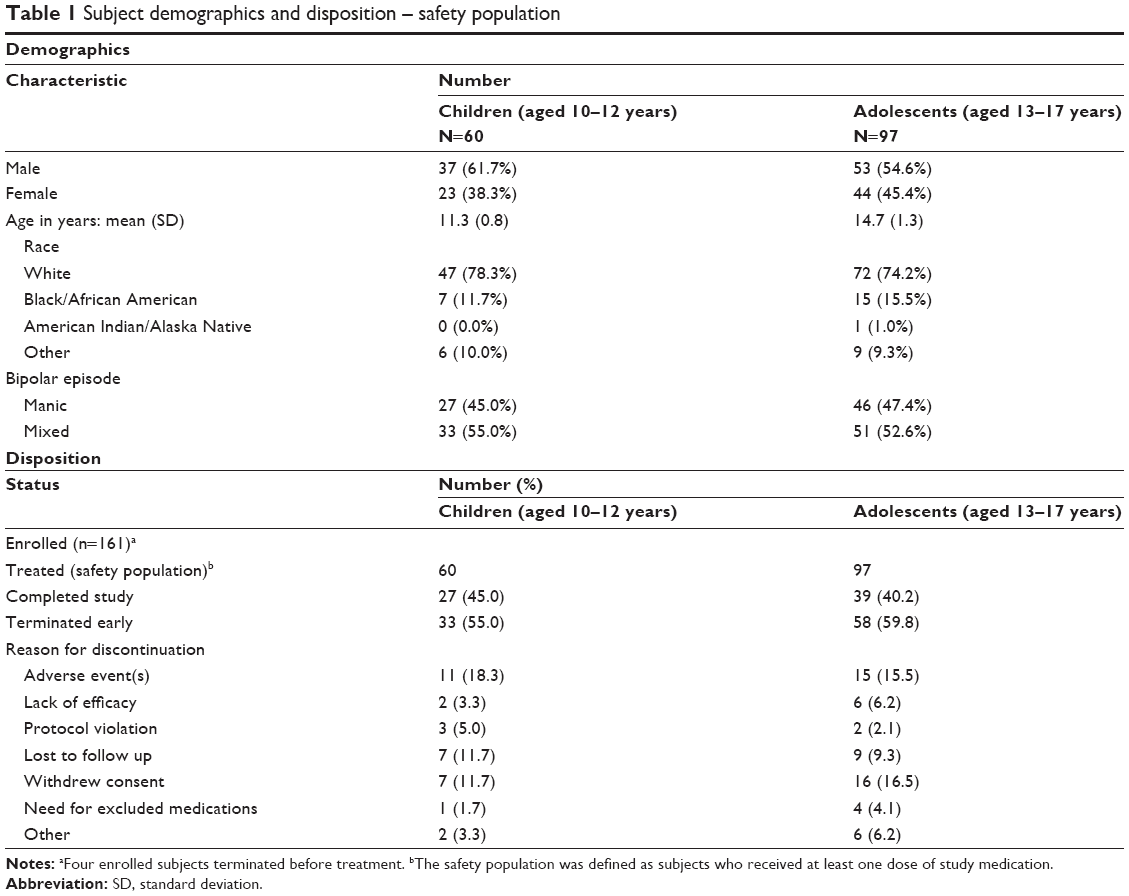 | Table 1 Subject demographics and disposition – safety population |
The final ERC daily dose for study subjects is found in Table 2. Approximately 32% of children and 25% of adolescents reached the 1,200 mg dose at end point.
 | Table 2 Final ERC daily dose and overall total exposure |
During the study, concomitant medications taken by more than one subject were risperidone (nine subjects); hydrocodone/paracetamol and ziprasidone hydrochloride (five subjects each), quetiapine fumarate (four subjects), sodium chloride, Thomapyrin® (acetylsalicylic acid/paracetamol/caffeine), and Tussin DM® (dextromethorphan bromide/guaifenesin) (two subjects each). Valproate semisodium and trazodone were taken by one subject each.
Efficacy
Overall efficacy results are summarized in Table 3.
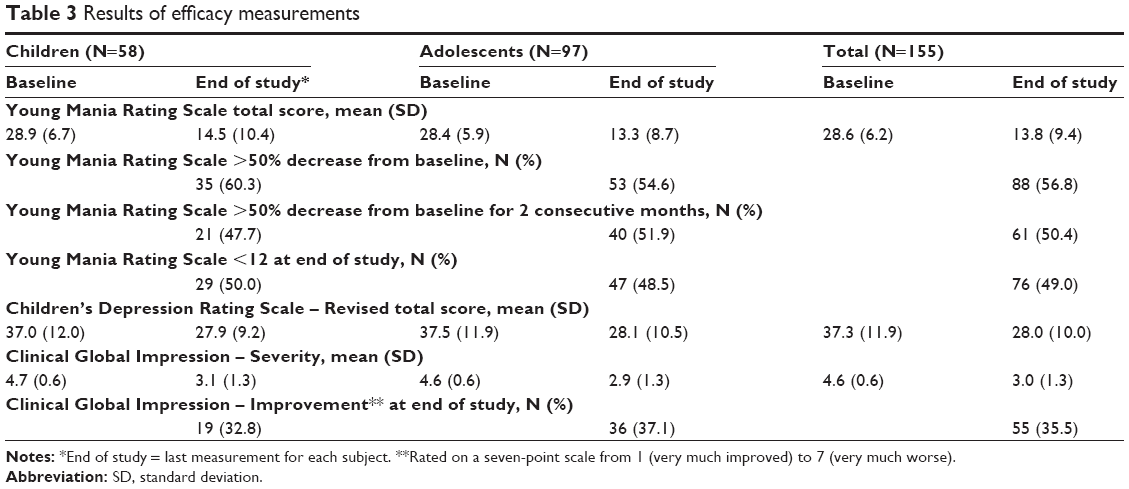 | Table 3 Results of efficacy measurements |
YMRS
ERC treatment was associated with highly significant reductions (P<0.0001) in the YMRS at all study visits when visit-wise data from the children and adolescents were analyzed together. When children and adolescents were evaluated separately, significant reductions (P≤0.0001) in the YMRS was present at all visits (visit-wise data), starting at day 4. YMRS scores over time, by visit, are summarized in Figure 1.
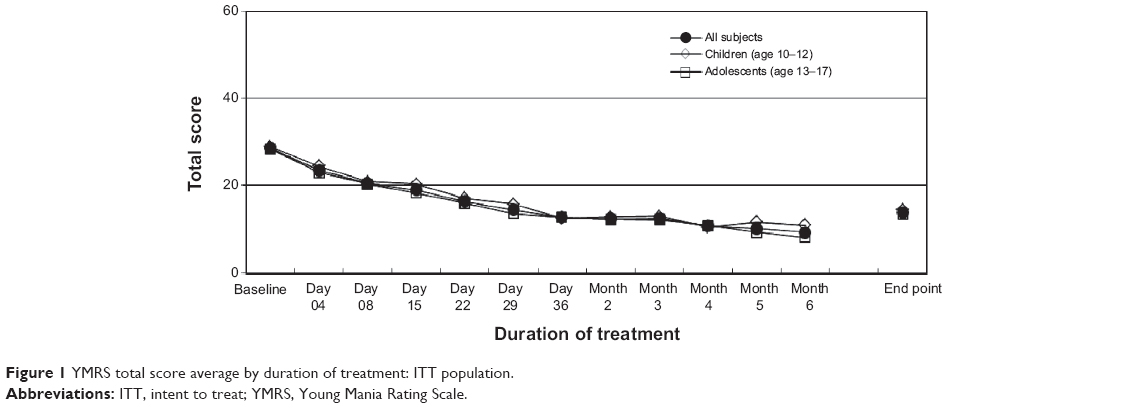 | Figure 1 YMRS total score average by duration of treatment: ITT population. |
At study end point, 56.8% of the subjects had YMRS scores 50% or less than baseline, thereby meeting the criterion for response. Similarly, at EOS participation, 49.0% of subjects had YMRS scores <12, thus achieving the criterion for remission.
CDRS-R
The mean baseline CDRS-R score for the ITT population was 37.3. At EOS, it was 28.0. The changes (visit-wise data) were statistically significant (P<0.0001) at each postbaseline visit, for both children and adolescents.
CGI
The CGI-S scores (visit-wise data) had a statistically significant decrease (P<0.0001), from a mean of 4.7 at baseline to an end point score of 3.0. At the EOS participation, the CGI-I score indicated that 35.5% of the subjects were “much improved” or “very much improved.”
Safety
AEs
AE data are summarized in Table 4. Most subjects experienced at least one AE during study participation. No deaths or completed suicides occurred in any subjects while they were enrolled in the study.
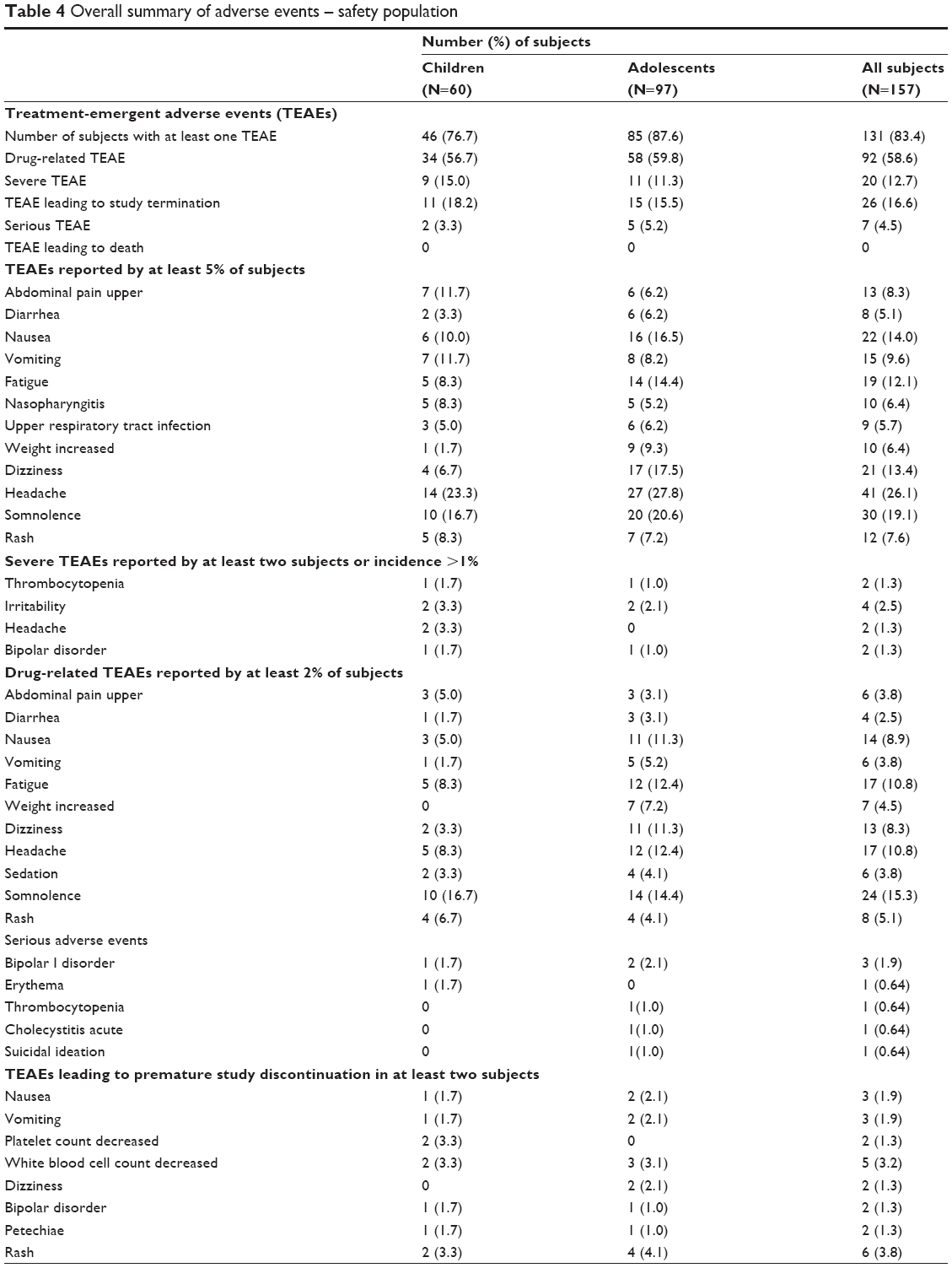 | Table 4 Overall summary of adverse events – safety population |
Twenty AEs were considered “severe” (incapacitating; subject was unable to work or to complete usual daily activities) by the treating Investigator. Nine of these SAEs were in children. The other AEs that were reported were either “mild” (easily tolerated and did not interfere with daily activity) or “moderate” (interfered with daily activity, but subject was able to function) in intensity.
There were 26 TEAEs, eleven of which occurred in children and which led to study discontinuation. A TEAE was an AE that occurred on or after the first dosing date and before or at the same time as the last dosing date plus 7 days, or if there was an increase in severity during that time frame. The most common TEAEs leading to study discontinuation are summarized in Table 4.
A SAE was defined as an AE that resulted in death, was life-threatening, required hospitalization or prolongation of existing hospitalization, resulted in persistent or significant disability or incapacity, or was a congenital anomaly or birth defect. SAEs occurred in two children and five adolescents. Only two of these SAEs, one case of thrombocytopenia and one case of erythema multiforme, were considered by the treating Investigator to be related to treatment with ERC.
Physical parameters and laboratory tests
Weight gain was noted in both children and adolescents; seven adolescents and no children reported weight gain as an AE. A total of 55 children and 91 adolescents had both a screening and an end point weight measurement. Mean (SD) weight gain in children was 4.1 kg (4.6) from baseline to EOS (P<0.0001). Mean (SD) weight gain in adolescents during trial participation was 3.0 kg (7.1) (P=0.0001). For all 146 subjects, mean (SD) weight gain was 3.4 kg (6.2) (P<0.0001).
For laboratory tests, the mean changes from the screening visit (used as baseline) to end point for the majority of tests were relatively small and within ±10% of the screening values and not clinically relevant. For hematology, consistent with ERC prescribing information,21 platelet counts showed the maximum relative mean change from screening – a decrease of approximately 7%. For the chemistry tests, alkaline phosphatase increased 13.3% from screening (baseline) to end point, and total bilirubin decreased 50% from baseline to end point. All pregnancy test results were negative.
For vital signs, the mean changes from baseline were small and not clinically meaningful (ie, greater than 15%). Vital signs were evaluated according to the percentages of subjects with values outside (above or below) the normal range at baseline, end point, day 29, and month 6. Values above the normal range occurred in 1.6% to 4.5% of subjects, for pulse; in 3.2% to 4.7%, for systolic blood pressure (BP); and in 3.2% to 4.5%, for diastolic BP. Values below the normal range occurred in 0% to 0.6% of subjects, for pulse; in 0.8% to 6.4%, for systolic BP; and in 0.8% to 1.9%, for diastolic BP. There was no apparent trend between baseline and end point in the percentages of subjects with vital sign values above or below the normal range. Less than 10% of subjects had values either above or below the normal range at any postbaseline visit.
ECGs were evaluated at baseline, day 36, and month 6/EOS. Mean changes in ECG parameters from baseline to end point were small, ranging from increases of 1.6 beats per minute, for heart rate; 4.4 msec, for PR interval; and small decreases, ranging from -7.3 to -3.5 msec, for QT/QTc intervals. There were no QT/QTc interval changes ≥60 msec from the screening (baseline) visit at follow-up assessments for any subject. Abnormal ECG results occurred in 5.2% of subjects at screening, in 1.5% at treatment end point, and in 1.5% and 1.6%, respectively, after 36 days and 6 months of treatment.
Conclusion and discussion
Overall, the incidence of AEs, SAEs, and AEs leading to treatment discontinuation was relatively low in this 6-month study, especially after 8 weeks of treatment. The occurrence of AEs was similar between children and adolescents. The findings were consistent with the common side effects and laboratory test abnormalities contained in the ERC prescribing information21 for the treatment of BDs in adults. It should be noted that several of the side effects observed in this patient population could be serious. As such, careful monitoring is recommended if carbamazepine is prescribed to this group of youngsters.
On all measures of efficacy, and for the total efficacy population (both children and adolescents), ERC treatment resulted in statistically significant and clinically meaningful improvement over baseline measurements at end point. For example, the YMRS total score decreased from 28.6 (SD 6.2) at baseline to 13.8 (SD 9.4) at end point (P<0.0001). A >50% decrease from baseline in the YMRS was noted in 56.8% of children and adolescents combined, and the 50% decrease was maintained for 2 consecutive months in 50.4%. On both the CDRS-R and the CGI-S, highly significant (P<0.0001) improvement was also noted for the overall efficacy population. At EOS participation, the CGI-I score indicated that 35.5% of the subjects were “much improved” or “very much improved.”
This study was the first substantive prospective clinical trial to provide information to prescribing physicians on the long-term safety and effectiveness of ERC in this population. However, as with all clinical trials, this study had limitations. Perhaps most noteworthy was the open-label design. However, despite this fact, the study design was strengthened by the 6-month duration and the relatively large number of youths treated under the auspices of a clinical trial. It should be noted that despite these positive features, the number of patients enrolled were still relatively modest when compared to the number of youths suffering BD-1. Similarly, 6 months is a reasonably brief epoch when considering that pharmacotherapy for this patient population may be indicated for several years. Also, youths with comorbid medical and psychiatric conditions were not eligible for participation in this study. For that reason, these results may not be applicable to all patients in this age group suffering from a manic or mixed episode with BD-1.
Carbamazepine levels were not routinely obtained during the study. As carbamazepine levels are often used in clinical practice, hypotheses about relationships between carbamazepine levels and dose, as well as about carbamazepine levels and efficacy/tolerability, cannot be generated based on these data.
Despite these limitations, this study does substantively contribute to the medical literature as it notably adds to what is known about the use of ERC in the treatment of children and adolescents suffering from BD-1.
In summary, open-label treatment with ERC in the range of 200 mg to 1,200 mg per day in divided doses may be effective and safe for the long-term treatment of children and adolescents with manic and mixed episodes of BD-1. However, a randomized, placebo-controlled study would confirm the efficacy of prescribing ERC in this subject population.
Acknowledgments
This study concerned a medication use that has not been approved by the US Food and Drug Administration.
Patricia A Blaine, RRT, EdM, medical writer/consultant, President of Medical Writing Corporation, provided editorial assistance for this paper, under the direction of the authors.
Disclosure
Validus Pharmaceuticals, LLC provided funding for the study.
Dr Findling receives or has received research support, acted as a consultant, received royalties from, and/or served on a speaker’s bureau for Abbott, Addrenex, Alexza, American Psychiatric Press, AstraZeneca, Biovail, Bristol-Myers Squibb, Cognition Group, Dainippon Sumitomo Pharma, Forest, GlaxoSmithKline, Guilford Press, Johns Hopkins University Press, Johnson & Johnson, KemPharm Lilly, Lundbeck, Merck, National Institutes of Health, Neuropharm, Novartis, Noven, Organon, Otsuka, Pfizer, Physicians’ Post-Graduate Press, Rhodes Pharmaceuticals, Roche, Sage, Sanofi-Aventis, Schering-Plough, Seaside Therapeutics, Sepracore, Shionogi, Shire, Solvay, Stanley Medical Research Institute, Sunovion, Supernus Pharmaceuticals, Transcept Pharmaceuticals, Validus, WebMD, and Wyeth.
In the last 12 months, Dr Ginsberg receives or has received research support, acted as a consultant, and/or served on a speaker’s bureau for AstraZeneca, Bristol Myers Squibb, Forest, Jaymac, Merck, Novartis, Noven, Otsuka, Pamlab, Pfizer, Seaside Therapeutics, Shionogi, Shire, Sunovion, and Validus.
The authors report no other conflicts of interest.
References
Pavuluri MN, Birmaher B, Naylor MW. Pediatric bipolar disorder: a review of the past 10 years. J Am Acad Child Adolesc Psychiatry. 2005; 44(9):846–871. | ||
Youngstrom EA, Birmaher B, Findling RL. Pediatric bipolar disorder: validity, phenomenology, and recommendations for diagnosis. Bipolar Disord. 2008;10(1 Pt 2):194–214. | ||
Goldstein BI. Recent progress in understanding pediatric bipolar disorder. Arch Pediatr Adolesc Med. 2012;166(4):362–371. | ||
Findling RL, Frazier JA, Kafantaris V, et al. The Collaborative Lithium Trials (CoLT): specific aims, methods, and implementation. Child Adolesc Psychiatry Ment Health. 2008;2(1):21. | ||
Tohen M, Kryzhanovskaya L, Carlson G, et al. Olanzapine versus placebo in the treatment of adolescents with bipolar mania. Am J Psychiatry. 2007;164(10):1547–1556. | ||
Findling RL, Nyilas M, Forbes RA, et al. Acute treatment of pediatric bipolar I disorder, manic or mixed episode, with aripiprazole: a randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2009; 70(10):1441–1451. | ||
Haas M, Delbello MP, Pandina G, et al. Risperidone for the treatment of acute mania in children and adolescents with bipolar disorder: a randomized, double-blind, placebo-controlled study. Bipolar Disord. 2009;11(7):687–700. | ||
Pathak S, Findling RL, Earley WR, Acevedo LD, Stankowski J, Delbello MP. Efficacy and safety of quetiapine in children and adolescents with mania associated with bipolar I disorder: a 3-week, double-blind, placebo-controlled trial. J Clin Psychiatry. 2013;74(1): e100–e109. | ||
Weisler RH, Kalali AH, Ketter TA; SPD417 Study Group. A multicenter, randomized, double-blind, placebo-controlled trial of extended-release carbamazepine capsules as monotherapy for bipolar disorder patients with manic or mixed episodes. J Clin Psychiatry. 2004;65(4): 478–484. | ||
Weisler RH, Keck PE, Swann AC, Cutler AJ, Ketter TA, Kalali AH; SPD417 Study Group. Extended-release carbamazepine capsules as monotherapy for acute mania in bipolar disorder: a multicenter, randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2005;66(3):323–330. | ||
Hsu LK. Lithium-resistant adolescent mania. J Am Acad Child Psychiatry. 1986;25(2):280–283. | ||
Woolston JL. Case study: carbamazepine treatment of juvenile-onset bipolar disorder. J Am Acad Child Adolesc Psychiatry. 1999;38(3): 335–338. | ||
Kowatch RA, Suppes T, Carmody TJ, et al. Effect size of lithium, divalproex sodium, and carbamazepine in children and adolescents with bipolar disorder. J Am Acad Child Adolesc Psychiatry. 2000;39(6): 713–720. | ||
Joshi G, Wozniak J, Mick E, et al. A prospective open-label trial of extended-release carbamazepine monotherapy in children with bipolar disorder. J Child Adolesc Psychopharmacol. 2010;20(1):7–14. | ||
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision: DSM-IV-TR. Arlington, VA: American Psychiatric Association; 2000. | ||
Kaufman J, Birmaher B, Brent D, et al. Schedule for Affective Disorders and Schizophrenia for School-Age Children-Present and Lifetime Version (K-SADS-PL): initial reliability and validity data. J Am Acad Child Adolesc Psychiatry. 1997;36(7):980–988. | ||
Young RC, Biggs JT, Ziegler VE, Meyer DA. A rating scale for mania: reliability, validity and sensitivity. Br J Psychiatry. 1978;133: 429–435. | ||
National Institute of Mental Health. Clinical Global Impressions. Psychopharmacology Bulletin. 1985;21(4):839–843. | ||
Poznanski EO, Freeman LN, Mokros HB. CDRS-R (Children’s Depression Rating Scale, Revised). Psychopharmacology Bulletin. 1985;21(4): 979–989. | ||
Kowatch RA, Fristad M, Birmaher B, Wagner KD, Findling RL, Hellander M; Child Psychiatric Workgroup on Bipolar Disorder. Treatment guidelines for children and adolescents with bipolar disorder. J Am Acad Child Adolesc Psychiatry. 2005;44(3):213–235. | ||
Equetro® (Carbamazepine) [prescribing information]. Parsippany, NJ: Validus Pharmaceuticals, LLC; 2012. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.